
A fluorinated greenhouse gas sensor based on N-doped tin oxide materials†
Hu
Meng
 ,
Zhiwen
Liu
,
Xiaoxin
Wang
and
Liang
Feng
*
,
Zhiwen
Liu
,
Xiaoxin
Wang
and
Liang
Feng
*
Department of Instrumentation and Analytical Chemistry, CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: fengl@dicp.ac.cn
First published on 14th December 2023
Abstract
Compared with carbon dioxide, fluorinated greenhouse gases (F-type gases), such as hydrofluorocarbons (HFCs), perfluorocarbons (PFCs) and sulfur hexafluoride (SF6), have a stronger greenhouse effect, and their global warming potential can reach thousands or even tens of thousands of times that of carbon dioxide. Thus, the rapid online accurate detection of fluorinated greenhouse gases is highly desired and it can provide a basis for the evaluation of the greenhouse effect and a yardstick for the formulation of emission reduction measures. Unfortunately, these highly noble chemical gas molecules, especially at trace concentration levels, are extremely difficult to detect with conventional rapid screening methods. The use of the most common gas sensing techniques, semiconducting metal oxides, in the detection of HFCs and PFCs has not appeared within the scope of our knowledge. Herein, we report a novel N-doped tin oxide semiconducting metal oxide prepared by using a facile hydrothermal method. The experimental characterization results showed that the as-prepared materials were uniform spherical nanoparticles. The doping ratio of N-doped tin oxide was further optimized, and gas-sensing tests using these materials were carefully conducted. The results showed that the sensor had good detection performance for F-type gases, represented by SF6, C2F6 and C2H2F4, including low working temperature (200 °C), high selectivity, good repeatability, relatively high response, and low limit of detection (LOD ∼7 ppb). To the best of our knowledge, this is the first report on an F-type gas sensor based on semiconductor metal oxide. The ultra-fine particle size and uniform morphology of spherical particles, high concentration of oxygen vacancy defects and N doping contribute to the excellent performance of the sensor.
Environmental significanceCompared with carbon dioxide, fluorinated greenhouse gases (F-type gases), such as hydrofluorocarbons (HFCs), perfluorocarbons (PFCs) and sulfur hexafluoride (SF6), have a stronger greenhouse effect, and their global warming potential can reach thousands or even tens of thousands of times that of carbon dioxide. Thus, the rapid online accurate detection of fluorinated greenhouse gases is highly desired. Unfortunately, these highly noble chemical gas molecules, especially at trace concentration levels, are extremely difficult to detect with conventional rapid screening methods. Herein, we report a fluorinated greenhouse gas sensor based on N-doped tin oxide materials. The sensor had good detection performance for F-type gases represented by SF6, C2F6 and C2H2F4. To the best of our knowledge, this is the first report about an F-type gas sensor based on semiconductor metal oxide. |
1 Introduction
Humans have come to rely on factory-made synthetic gases that have an extremely significant global warming potential. Fluorinated greenhouse gases (hereinafter referred to as “F-type gases”), such as hydrofluorocarbons (HFCs), perfluorocarbons (PFCs) and sulfur hexafluoride (SF6), have been dubbed “super pollutants” and “super greenhouse gases” because of their serious and powerful effects on the climate.1 They are the most potent greenhouse gases known to modern science, with a global warming potential far greater than that of carbon dioxide, some as much as nearly 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times.2 Since 1997, F-type gases have been officially listed in the ranks of greenhouse gases according to the “Kyoto Protocol”. Disturbingly, they are also the fastest growing category of greenhouse gas emissions in the world, especially in developing countries. Studies show that by 2050, nearly 40% of emissions will fall outside the scope of international agreements such as the Paris Agreement, the Montreal Protocol and the Kigali Amendment. Without comprehensive and sustained interventions, the unchecked growth of F-gas emissions could offset the Kyoto Protocol's clean development mechanism and existing cornerstones of international climate governance.1 With the implementation of the national “dual carbon goals”, rapid online accurate detection of F-type gases is increasingly needed, and can provide a basis for the evaluation of the greenhouse effect and a yardstick for the formulation of emission reduction measures. It is also of great significance for regional carbon emissions, mastering source and sink information, and providing data guidance for the implementation of the national “dual carbon goals”.3
000 times.2 Since 1997, F-type gases have been officially listed in the ranks of greenhouse gases according to the “Kyoto Protocol”. Disturbingly, they are also the fastest growing category of greenhouse gas emissions in the world, especially in developing countries. Studies show that by 2050, nearly 40% of emissions will fall outside the scope of international agreements such as the Paris Agreement, the Montreal Protocol and the Kigali Amendment. Without comprehensive and sustained interventions, the unchecked growth of F-gas emissions could offset the Kyoto Protocol's clean development mechanism and existing cornerstones of international climate governance.1 With the implementation of the national “dual carbon goals”, rapid online accurate detection of F-type gases is increasingly needed, and can provide a basis for the evaluation of the greenhouse effect and a yardstick for the formulation of emission reduction measures. It is also of great significance for regional carbon emissions, mastering source and sink information, and providing data guidance for the implementation of the national “dual carbon goals”.3
At present, the analytical methods that can detect F-type gases mainly include gas chromatography4 and Fourier transform infrared spectroscopy.5 Their disadvantage is that the measurement results are easily affected by the environment, and the test process is relatively complicated. Usually, the samples need to be collected and brought to the laboratory for analysis, which is not suitable for online measurement.
Semiconductor metal oxide gas sensors are smaller in size and can be mass-produced using low-cost technologies such as microelectromechanical systems (MEMS).6,7 In addition, semiconductor metal oxide sensors have low power consumption, portable size and high sensitivity.8–11 By using coral-like Zn-doped SnO2 composites, a semiconductor metal oxide gas sensor system for the detection of sulfur hexafluoride vapor was developed based on its catalytic luminescence (CTL) emission.12 Although semiconductor metal oxide gas sensors have these advantages and seem like to be good candidates for F-type gas online measurement, the use of semiconducting metal oxides in gas detection for most other F-type gases, such as HFCs and PFCs, has rarely been reported within the scope of our knowledge. Furthermore, the results of our extensive experiments show that pristine metal oxides such as In2O3, SnO2, WO3, ZnO and Co3O4 all have very poor sensing performance for F-type gases. Since F-type gases such as HFCs and PFCs are mostly inactive, the intrinsic active sites on the surface of pristine metal oxides that may participate in gas-sensing reactions are largely limited, resulting in low sensitivity and lack of selectivity. In order to extend the application of semiconductor gas sensors in the detection of F-type gases, there are still great challenges to be overcome.
Generally, noble metal modification is considered as the most common means for improving the performance of semiconductor metal oxide gas sensors.13–15 We have modified semiconductor metal oxide materials with different noble metals, but the experimental results showed that it cannot meet our needs for efficient and specific detection of F-type greenhouse gases. Non-metal element-doped semiconductor metal oxides have been widely used in catalysis,16–20 and have also been tentatively utilized in gas detection as gas sensing materials. For example, the surface of TiO2 was modified by co-doping with N and F, and three kinds of SF6 decomposition components (SO2, SOF2 and SO2F2) were detected.21 Cl was uniformly doped in flower-like In2O3 nanomaterials, and its significance for the study of the sensing mechanism of photoactivated NO2 gas sensors was investigated.22 A SnO2 material doped with S exhibits ultrahigh sensitivity to NO2 with excellent selectivity at low optimum operating temperature.23 And the preparation of Cl-doped LaFeO3 by the citric acid sol–gel method comprehensively improved its ethanol sensing performance. Also, the obvious gas selectivity of SnS was obtained with different doping of Si, P and Cl towards NH3, SO2, and NO2, respectively,24etc. All these investigations prove that non-metal element doping is also an efficient approach for improving the sensing performance of semiconductor metal oxide gas sensors. However, until now we have not found any application of non-metal element doped metal oxide semiconductor materials for the detection of HFCs and PFCs.
In this work, a non-metal element N was doped in semiconductor metal oxide tin oxide, and its sensing performance for F-type gases, such as SF6, C2F6 and C2H2F4, was carefully evaluated. The sensing mechanism for a representative gas, C2F6, was discussed through FTIR investigation. N-doped tin oxide (N–SnO2) spherical nanoparticles were prepared by a simple hydrothermal method, and the optimal ratio of N/Sn doping was explored. The as-obtained N-doped tin oxide (N–SnO2) is in the rutile phase, and the shape is a spherical particle with a diameter of about 1 μm. The gas sensor based on N-doped tin oxide (N–SnO2) spherical material exhibits ultra-sensitive responses to F-type gases such as SF6, C2F6, and C2H2F4 (30 ppm responses are 3.13, 11.99, and 2.96, respectively). At the same time, good selectivity and reproducibility were achieved (within 35 days of 5 cycles, there is no obvious drop in response). After detecting SF6, C2F6, and C2H2F4 gases at 0.5–30 ppm, the response values showed a good linear relationship with gas concentrations (R2 is 0.98828, 0.97021 and 0.9953, respectively), proving its application prospect in daily detection. To the best of our knowledge, this is the first report about an F-type gas sensor based on semiconductor metal oxide. Its excellent sensing performance towards F-type gases is attributed to the doping of N and the creation of more oxygen vacancies as well as the ultra-fine particle size and uniform morphology of spherical particles, providing more active centers to participate in gas reactions.
2 Experiments
2.1 Experimental reagents and instruments
Tin chloride pentahydrate (SnCl4·5H2O) was purchased from Sinopharm Chemical Reagent Co., Ltd and hexamethylenetetramine (HMT) was purchased from Aladdin Industries (China). All chemicals were analytically pure reagents and used directly without further treatment. The concentrations of SF6, C2H2F4 and C2F6 are all 50 ppm and balanced with N2. Nitrous oxide and carbon dioxide were prepared in the same way as SF6, C2H2F4 and C2F6. Toluene and hexane were prepared by volatilizing the corresponding VOC (volatile organic compound) liquid. The concentration (C) of gas was confirmed by the following equation (eqn (1)): | (1) |
The microstructural information was identified using a field-emission scanning electron microscope (FE-SEM, JSM-7900F, JEOL, Japan) and transmission electron microscope (TEM, Jem-2100F, JEOL, Japan). Energy dispersive spectroscopy (EDS) was conducted with an Ultim Extreme (Oxford Instruments, UK). X-ray diffraction (XRD) was measured with an Empyrean (PANalytical, Netherlands). The scanned scope of 2θ angle ranged from 10–80°. X-ray photoelectron spectroscopy (XPS) spectra were recorded using a Thermo Scientific K-Alpha+ (USA) spectrometer analyzer. And the C 1s peak (284.5 eV) was used as the calibration reference for binding energy. Fourier transform infrared spectroscopy (FTIR) was conducted with a VERTEX 80v (Bruker, USA). The model of the digital multimeter is VICTOR 86E purchased from Shenzhen Victory High Electronic Technology Co.
2.2 Synthesis of pristine tin oxide and N-doped tin oxide (N–SnO2)
The synthesis of pristine tin oxide and N-doped tin oxide (N–SnO2) was achieved by hydrothermal synthesis. Specifically, 170 mg of tin tetrachloride pentahydrate and 280 mg of HMT (hexamethylenetetramine) were weighed (HMT/Sn4+ molar ratio is 4:1). After dissolving crystalline tin tetrachloride in 34 ml of ethanol, the quantitative amount of HMT obtained from the above step was added to the solution, and stirred for 30 min at a speed of 1050 r min−1 to form a uniform suspension. After thorough mixing, the above suspension was transferred to a 45 ml polytetrafluoroethylene liner, a reaction kettle was installed, and the reaction kettle was operated at 180 °C for 24 hours. After the reaction was completed, the above product was washed with deionized water several times and then with absolute ethanol several times, and then the above precipitate was dried in an oven at 80 °C for 4 h.25 As a comparison, we prepared different N-doped tin oxides (N–SnO2) by adjusting the molar ratio of HMT/Sn4+ (2:1, 3:1, 5:1, 6:1) in the same way as the above material.
At the same time, for the synthesis of the pristine tin oxide, aside from HMT not being added in the above reaction steps, the rest of the synthesis conditions are the same.
2.3 Preparation of the sensor and testing of sensing performance
The preparation process of the gas sensor is as follows. First, 5 mg of N–SnO2 powder is dispersed in 0.1 ml of absolute ethanol, and then ultrasonicated for 5 min to form a uniform suspension. A pipette gun was used to pipette 10 μL of the suspension which was spread evenly on a commercial ceramic tube (outer diameter 1.2 mm, inner diameter 0.8 mm, length 4 mm) to form a uniform film. The ceramic tube is printed with gold electrodes on both sides and through the wire connected with the base, the hollow runs through the nickel–chromium alloy heating wire to adjust the working temperature. Finally, in order to improve the long-term stability and repeatability of the sensor, the sensor was aged at 240 °C for 24 h in an air atmosphere.As shown in Fig. S1a,† the commercial ceramic tube has 6 interfaces, and the interfaces of the nickel–chromium alloy heating wire are 2 and 5, which are respectively connected to the DC power supply. The working temperature of the sensor is adjusted by adjusting the output voltage of the power supply; the other four interfaces are connected to a digital multimeter to observe the change of the resistance value in real time and record the data. The interface connected with the multimeter must be connected on both sides respectively. Gas test experiments were performed at high air flow rates. To reduce the influence of airflow on the sensor resistance, a perforated cover is usually placed on the sensor (Fig. S1c†). The test instrument is a self-made 100 mL bottle with a blue cap, three holes are punched and sealed, and an electric wire and a tetrafluoro tube are put in respectively. The DC power supply and the digital multimeter are connected with the sensor through wires. The PTFE tube is used to detect the entry and output of gas.
In this work, in addition to the static method partially used in the selectivity test of the sensor performance, dynamic gas distribution (Fig. S2†) is used in the gas test to test the sensor performance. The multimeter and DC power supply are connected to the air circuit after tightening the bottle cap. By adjusting the output voltage and power of the external DC power supply, the sensor can be adjusted to work at the corresponding temperature. The valve of the air cylinder was opened, and the flow controller was set so that the gas passes through the gas path at a flow rate of 500 mL min−1, and the sensor performs baseline (Ra) adjustment in flowing dry air. After the baseline is completely flat, the total gas flow rate was kept unchanged, the cylinder valve of the test gas was opened, and the concentration of the gas to be tested was adjusted by setting the corresponding flow controller, and timing was started at this time. The final resistance value (Rg) was recorded before the end of the ventilation. After the end of the ventilation, the flow rate of the gas to be tested was adjusted to zero, so that the dry air replaces the residual gas to be tested. At this time, the sensor is in a recovery state. When the baseline returns to the original level, the next round of testing can be performed.
When using the static method for sensor testing, the manufactured sensor is placed in a sealed cavity, and the base of the ceramic tube is connected to a multimeter and a heating power supply respectively. The sensor works at the corresponding temperature by adjusting the external power supply. When the resistance baseline (Ra) in air is stable, the required amount of the liquid to be tested was injected into the heater in the test chamber to completely form the vapor. The detection time was fixed. Before the end of the test, the final resistance value (Rg) was recorded and the chamber lid was opened to allow fresh air to enter, and the resistance of the sensor was restored.
3 Results and discussion
3.1 Microstructure and properties of materials
In the preparation process of N–SnO2 materials, we use HMT as the nitrogen source, SnCl4·5H2O as the tin source, and a small amount of water in SnCl4·5H2O to provide the water needed for the hydrolysis reaction. Using ethanol as a solvent can slow down the hydrolysis reaction of tin tetrachloride, and the solubility of tin oxide in ethanol is low, making its crystal growth slower.26 The cage structure would also promote the coordination of N to the Sn atoms on the tin oxide surface.27 At the same time, HMT reacts with water to generate ammonia, which can further promote nitrogen doping into the material.28 During the solvothermal process, the hydrolysis reaction of SnCl4·5H2O and HMT proceeded simultaneously. The tiny nuclei formed in the supersaturated solution will rapidly grow into smaller nanoparticles and further aggregate spontaneously. The N atoms of the ligands would be adsorbed onto the surface by the smaller nanoparticles, while preventing the further growth of tin oxide particles during the aggregation process.We further performed a series of characterizations on the optimal proportioned N–SnO2 (HMT/Sn4+ HMT/Sn4+ molar ratio is 4:1) material. According to the results of EDS (as shown in Fig. 1), N atoms account for approximately 5.73% of the total atomic number in N–SnO2, and the product is composed of three elements (N, O and Sn). Fig. 1d further proves the existence of N element and that the distribution of N in the obtained product is very uniform, indicating that N has entered the lattice of tin oxide. At the same time, after N element doping, the tin oxide color changes from white to brown, which is in good agreement with an existing report,29 further proving the successful N element doping.
 | ||
| Fig. 1 (a) EDS energy spectrum diagram of N–SnO2, the inset shows the percentage of weight and the percentage of total atoms of each element. (b–d) EDS mapping images of N–SnO2. | ||
The XRD characterization results of our prepared pristine SnO2 and N–SnO2 are shown in Fig. 2. All diffraction peaks of the pristine tin oxide material correspond well to the standard diffraction data (PDF #71-0652) of tin oxide with rutile structure. And no other impurity peaks were detected, which showed that the original tin oxide was successfully prepared and had high purity. In contrast, the diffraction peaks of the N–SnO2 materials broadened significantly, indicating that the as-synthesized N–SnO2 materials had a smaller grain size.
 | ||
| Fig. 2 The XRD result of N–SnO2 and pristine SnO2. | ||
The morphologies and microstructures are investigated by SEM, TEM, and high-resolution TEM (HRTEM). The SEM images in Fig. 3a and d show that the N–SnO2 material is composed of uniformly distributed spherical particles with a size of about 1 μm. TEM images in Fig. 3f reveal that the pristine SnO2 particles are an aggregate of spatial nanocrystals with a diameter of 10–15 nm. The selected-area electron diffraction (SAED) pattern (inset of Fig. 3f) shows distinct bright diffraction spots of pristine SnO2. HRTEM images in Fig. 3e show high resolution lattice lines with lattice spacings of 0.331 nm, 0.268 nm and 0.164 nm, which correspond to the (110), (101) and (211) planes of SnO2, respectively. The TEM image (Fig. 3c) shows that the large particles of N–SnO2 consist of many tiny nanocrystals. The SAED pattern (inset of Fig. 3c) shows the presence of diffuse diffraction rings rather than distinct bright spots, indicating a high degree of polycrystallinity. The HRTEM image (Fig. 3b) further shows that the tiny nanocrystals have diameters of predominantly 2–3 nm, close to the excitation Bohr radius of SnO2 of 2.7 nm. The apparent reduction in the size of the N–SnO2 materials may be due to the introduction of nitrogen atoms into SnO2, which may inhibit the formation of long-range order in the SnO2 lattice. Interlayer spacings of 0.319, 0.262, and 0.163 nm were observed for the nanocrystals, corresponding to the (110), (101), and (211) planes of the tetragonal phase SnO2, respectively. The N–SnO2 crystals have a smaller particle size, which is consistent with our XRD results obtained above.
 | ||
| Fig. 3 (a and d) SEM, (b) HRTEM, and (c) TEM images of N–SnO2; (e) HRTEM and (f) TEM images of pristine SnO2. The insets display the corresponding SAED patterns and interlayer spacing. | ||
In order to determine the chemical states of N, Sn and O in the N–SnO2 nanostructures, XPS measurements were used. As shown in Fig. 4, the XPS survey spectra are dominated by the spectral lines of Sn and O. The XPS spectra of N–SnO2 nanostructures are shown in Fig. 4. The C 1s peak with a binding energy of 284.5 eV was used as a calibration reference. In addition, N–SnO2 exhibits a distinct N 1s characteristic peak. This confirms the N doping effect in SnO2. Fig. 4a shows the N 1s XPS peaks of N–SnO2. The dominant peak at 400.0 eV is assigned to the Sn–N bond, which involves the nitrogen-substitution of O sites in bulk SnO2 and the surface tin oxynitride species at the edge of SnO2. The Sn 3d spectra of pristine SnO2 (Fig. 4b) show the standard Sn 3d5/2 and Sn 3d3/2 peaks of rutile SnO2 at 486.6 and 495.0 eV, respectively, thus implying that Sn exists only in the 4+ state.30 Their difference of 8.4 eV (i.e., the binding energy of the Sn 3d electron) also agrees with the data in the standard database.31 For N–SnO2 materials, the binding energies of both Sn 3d5/2 and 3d3/2 (0.5 eV) are shifted to lower energies, suggesting doping with nitrogen and the formation of oxygen vacancies. The change in the core binding energies of Sn 3d5/2 and 3d3/2 may be due to the fact that N3− is more negative than O2−, which leads to a shift in the energy levels of the 3d orbitals due to a higher Coulomb potential. Fig. 4c shows the O 1s spectra of the pristine SnO2 material. The asymmetric O 1s peak can be decomposed into three contributions of 530.6, 531.2, and 532.2 eV, which can be attributed to lattice oxygen (OL), oxygen vacancies (OV), and chemisorbed oxygen species (OC), respectively.32 Moreover, the O 1s peaks in the N–SnO2 materials can also be decomposed into three curves at 530.2, 530.9, and 531.9 eV in Fig. 4d. The results showed that the peak area of oxygen vacancies increased significantly, indicating that nitrogen doping increased the content of oxygen vacancies in SnO2. Nitrogen doping results in the formation of nitrogen doping sites and oxygen vacancy defects in SnO2.33,34
 | ||
| Fig. 4 Binding energy spectra of as-prepared materials: (a) N 1s (b) Sn 3d of the N–SnO2 materials, (c) O 1s spectrum of pristine SnO2, and (d) O 1s spectrum of N–SnO2. | ||
3.2 Sensing performance test and analysis
The sensors coated with drops of pristine SnO2 materials were marked as N(0). The sensors coated with drops of N-doped material were respectively marked as N(2), N(3), N(4), N(5), N(6) according to the molar ratio of HMT/Sn4+ (2:1, 3:1, 4:1, 5:1, 6:1). As we all know, the operating temperature is an important parameter of semiconductor sensors. Therefore, the best operating temperature of N(4) to 30 ppm SF6, C2F6 and C2H2F4 is tested in the temperature range of 160–400 °C. The results are shown in Fig. 5a. It is obvious that as the temperature increases, the response of the sensor first experiences a climb and then gradually decreases. The sensor presents a “volcanic” feature, and the optimum temperature is 200 °C. This phenomenon can be explained by the thermodynamics and kinetics of gas adsorption and desorption on the surface of semiconducting metal oxides.35 This is because the oxygen adsorbed on the surface of the material has sufficient ability to participate in the reaction at the optimal working temperature. When the working temperature is low, gases such as SF6, C2F6 and C2H2F4 and adsorbed oxygen species have poor reactivity on the surface of the material and cannot achieve a good response. With the increase of temperature, the increase of reactivity is beneficial to the improvement of sensing performance. However, when the temperature is too high, the desorption rate of gases such as SF6, C2F6 and C2H2F4 increases, resulting in a decrease in active sites on the surface of the material and a decrease in the sensing response. And can be seen from Fig. 5b the sensing performance of N(4) is significantly better than that of N(0), N(2), N(3), N(5) and N(6). So the optimum molar ratio of HMT/Sn4+ in the synthesis process is 4:1. After N element doping, the particle size of the material becomes smaller and some pores on the surface are slightly blocked by N to form spherical nanoparticles. As shown in Fig. S4,† when the ratio of N/Sn is smaller, the morphology of the material is amorphous nanoparticle aggregates, which can't form uniform spherical nanoparticles; meanwhile some aggregates are formed on the surface of the spherical nanoparticles when the ratio of N/Sn is larger, so that the surface of the nanoparticles becomes unsmooth.
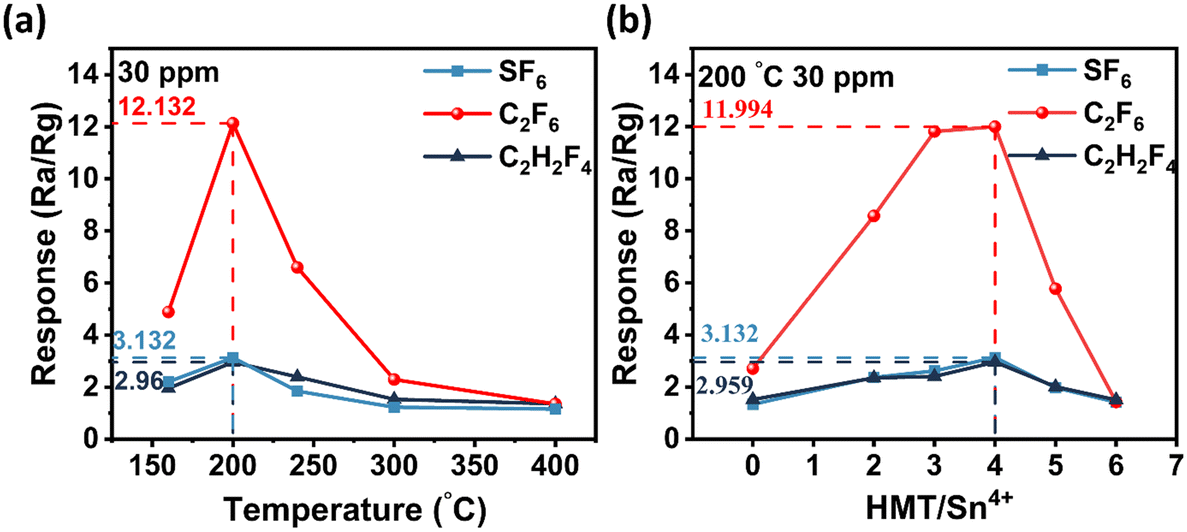 | ||
| Fig. 5 (a) Sensor response of N–SnO2 to 30 ppm C2H2F4 corresponding to the operating temperature (160–400 °C). (b) Sensor response of N(0), N(2), N(3), N(4), N(5) and N(6) sensors for gases such as SF6, C2F6 and C2H2F4. | ||
As shown in Fig. 6, we plotted the typical response/recovery curves of N(0) and N(4) to 30 ppm SF6, C2F6 and C2H2F4 gases at 200 °C (the real resistance response–recovery curves of N(0) and N(4) are shown in Fig. S3†). In this case, the results show that the response values of N-doped SnO2 sensors to gases such as SF6, C2F6 and C2H2F4 are greatly improved compared with the pristine SnO2 sensor. The pristine SnO2 doped with the element N increases its sensitivity to F-type gases by 200% and even for C2F6 by 400%. The response/recovery time is extended and shortened to varying degrees, but it does not affect the real-time rapid response. Comprehensively, compared with the original pristine SnO2 sensor, the sensitivity of the N-doped SnO2 sensor has been greatly improved.
 | ||
| Fig. 6 Dynamic response–recovery curves of N(0) and N(4) to 30 ppm (a) SF6, (b) C2F6 and (c) C2H2F4 at 200 °C. | ||
After that, we continued to conduct sensing tests on the N(4) sensor at 200 °C for gases such as 0.5–30 ppm SF6, C2F6 and C2H2F4. The result is shown in Fig. 7a–c. The N(4) sensor has a fast and sensitive response to gases such as SF6, C2F6, and C2H2F4. When the concentration of SF6, C2F6, C2H2F4 gases is as low as 0.5 ppm, it still has high response values of 1.44, 1.35, and 1.41. The limit of detection (LOD) was calculated in this paper, and the limits of detection of the N(4) sensor for gases such as SF6, C2F6 and C2H2F4 are 44 ppb, 7 ppb, and 48 ppb. Therefore, it is very suitable for low-concentration detection of gases such as SF6, C2F6, C2H2F4. At the same time, the response value of the sensor and the gas concentration of SF6, C2F6 and C2H2F4 show an excellent linear relationship (Fig. 7d–f). The standard deviation of data from multiple parallel experiments is small, indicating the reliability of the N(4) sensor test data.
 | ||
| Fig. 7 The gas sensing performance of the N(4) sensor. The real-time resistance change curve of the N(4) sensor to 0.5–30 ppm (a) SF6, (b) C2F6 and (c) C2H2F4 at 200 °C. (d–f) Responses of the N(4) sensor as a function of various (d) SF6, (e) C2F6 and (f) C2H2F4 concentrations. | ||
The stability and repeatability of the signal output are also important parameters to measure the properties of the sensor. Therefore, the cycle repeatability and long-term stability within 35 days of the N(4) sensor were explored, and the test results are shown in Fig. 8. It can be seen from the figure that the N(4) sensor has carried out 5 cycle tests on 30 ppm SF6, C2F6 and C2H2F4 at 200 °C, and the response values are all kept at a certain value, which shows that the sensing effect has not decreased significantly. By replacing with dry air, the resistance base value of the sensor can be restored to before the test, indicating that SF6, C2F6 and C2H2F4 gases can be desorbed from the surface of the material in a short time, and will not affect the next test of the sensor. At the same time, the long-term stability test results within 35 days can be seen in Fig. 8d. The response value of the N(4) sensor has also remained at around 10.76 within 35 days, with little change. The short-term excellent repeatability and long-term excellent stability of the N(4) sensor mean that this sensor can not only perform multiple tests in time, but also has a long service life, which is expected to be put into use in daily life.
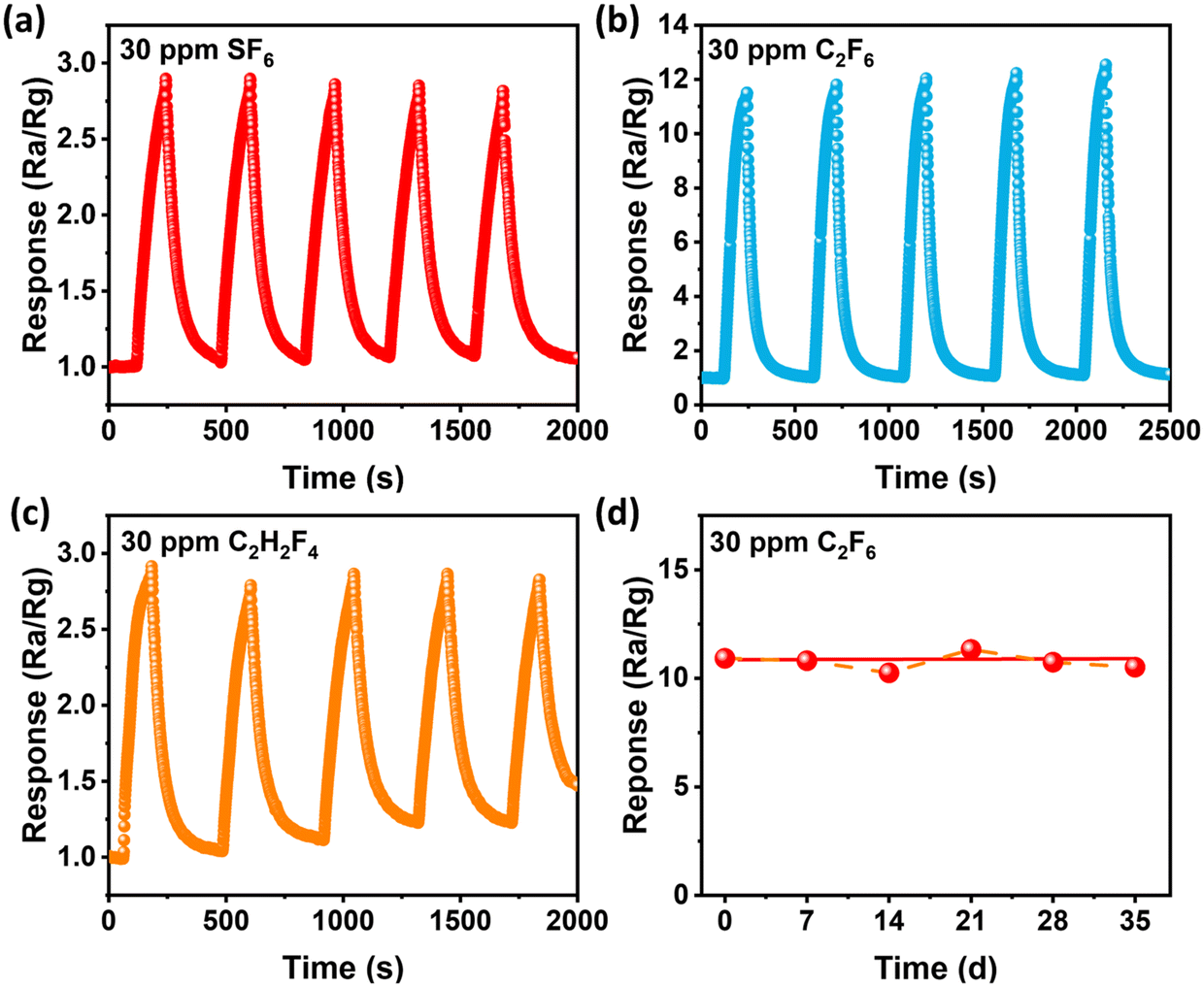 | ||
| Fig. 8 Stability of the N(4) sensor towards 30 ppm (a) SF6, (b) C2F6 and (c) C2H2F4 at 200 °C. (d) Long-term stability of N(4) on successive exposure to 30 ppm C2F6 at 200 °C. | ||
For sensors, the anti-interference ability is another very important parameter besides the sensitivity, the stability and the repeatability of signal output. It indicates whether the sensor can work accurately under the interference of other greenhouse gases in the atmosphere. Therefore, we continued to explore the sensing effect of the N(4) sensor on other atmospheric gases and compared the performance with SF6, C2F6 and C2H2F4 gases. From the results shown in Fig. 9, it can be seen that higher concentrations of hexane, toluene, nitrous oxide and carbon dioxide did not cause a significant change in the resistance of the N(4) sensor at 200 °C, and there is almost no interference effect.
 | ||
| Fig. 9 The effect of other atmospheric gases on sensing performance. | ||
3.3 Exploration of the sensing mechanism
According to existing reports, SnO2 is a typical n-type semiconductor,36 which is explained by the classical electron depletion layer model.37 When gas sensors based on n-type tin dioxide are exposed to air, oxygen molecules adsorb onto the surface and capture electrons from the tin dioxide to form negatively charged ions such as O−, O2−, or O2−. As the amount of electrons decreases, a depletion layer forms near the surface, resulting in an increase in the resistance of the gas sensor. The principle of operation of the sensor is primarily that a redox reaction takes place between the adsorbed oxygen species and the gas to be measured. Specifically, O−, O2− or O2− reacts with SF6, C2F6 and C2H2F4 gases, causing the captured electrons to be released back into the conduction band, which in turn leads to a dramatic change in resistance.38 As a result, the OV centers have a strong gas adsorption capacity and a high electron donor capacity. These defects can react as donors to trap SF6, C2F6, and C2H2F4 to produce chemisorbed species. The XPS experimental results show that N doping is more likely to form O vacancies, suggesting that the improved selectivity and sensitivity can be attributed to the OV centers.The experimental results of SEM and TEM show that the prepared N–SnO2 material consists of spherical particles with ultra-fine particle size and uniform morphology. Its microscopic morphology is uniform and stable, which is a guarantee for the stable operation of the sensor.
In addition, we also believe that the doping of N is one of the dominant factors influencing the specific sensing of the sensor for SF6, C2F6 and C2H2F4. In order to further explore the sensing mechanism, we explored the intermediate product of a surface reaction of C2F6 adsorbed on the surface of N–SnO2 materials by Fourier transform infrared spectroscopy (FTIR),39 and the results are shown in Fig. 10b. Because the concentration of the gas we used is relatively low, the changes of the FTIR spectral signal before and after gas adsorption is weak. So we use a differential spectrum to explain the sensing mechanism. The FTIR spectra before and after adsorption are shown in Fig. 10a. According to the information in Fig. 10b, the peaks at 1660 cm−1 and 1720 cm−1 can correspond to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the disappearance of the peak of the O–H stretching vibration of water at 2635 cm−1. Based on the results, it can be inferred that the following chemical reactions occur during the catalytic process:40
O bond and the disappearance of the peak of the O–H stretching vibration of water at 2635 cm−1. Based on the results, it can be inferred that the following chemical reactions occur during the catalytic process:40
| C2F6 → 2·CF3 | (2) |
 | (3) |
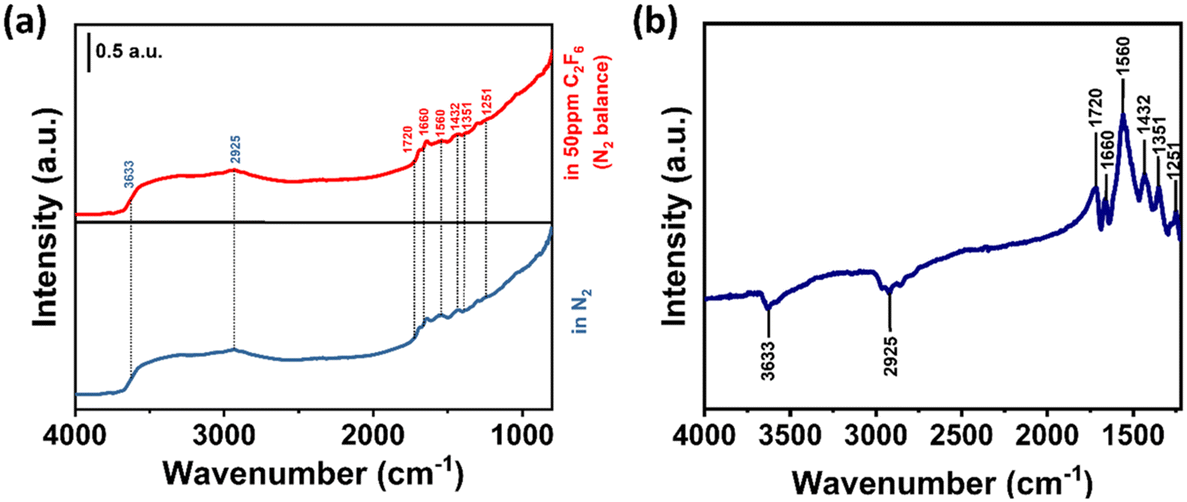 | ||
| Fig. 10 (a) FTIR spectra of N–SnO2 nanoparticles before and after adsorption of 50 ppm C2F6 in nitrogen gas at 200 °C. (b) FTIR difference spectra of N–SnO2 nanoparticles before and after adsorption of 50 ppm C2F6 in nitrogen gas at 200 °C. | ||
4 Conclusion
In summary, we tried to improve the sensing performance of metal oxide semiconductors for inactive F-type gases by doping non-metallic elements and effectively extended the application of semiconductor gas sensors in the rapid online detection of F-type gases in our study. The N-doped SnO2 nanoparticles were achieved by a direct solvothermal process using an environmentally friendly and inexpensive Sn-source and N-source. The doping ratio of N/Sn4+ was optimized and its sensing performance was carefully evaluated at high temperature in the gas path. The as-obtained N-doped tin oxide spherical nanoparticle (N–SnO2) sensor exhibited ultrasensitive responses to F-type gases such as 30 ppm SF6, C2F6 and C2H2F4 at 200 °C, and the responses were 3.13, 11.99 and 2.96, respectively. The sensor also showed excellent selectivity and repeatability. The long-term stability further proved that the sensor has a good application prospect in daily detection. To the best of our knowledge, this is the first report about an F-type gas sensor based on semiconductor metal oxide. Its excellent sensing performance towards F-type gases is attributed to the doping of N and the creation of more oxygen vacancies as well as the ultra-fine particle size and uniform morphology of spherical particles. Our work has pioneered a new strategy for the rapid online detection of F-type gases using metal oxide semiconductor sensors.Author contributions
Hu Meng: conceptualization, methodology, investigation, formal analysis, writing – original draft, review & editing, funding acquisition. Zhiwen Liu: investigation, formal analysis, writing – original draft. Xiaoxin Wang: investigation, formal analysis. Liang Feng: project administration, writing – review & editing, funding acquisition, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by DICP (Grant: DICP I202308, DICP I202331), the Natural Science Foundation of Liaoning Province (2021-MS-020), and the AI S&T Program of Yulin Branch, Dalian National Laboratory For Clean Energy, CAS (Grant No. DNL-YL A202203).References
- B. K. Sovacool, S. Griffiths, J. Kim and M. Bazilian, Climate change and industrial F-gases: A critical and systematic review of developments, sociotechnical systems and policy options for reducing synthetic greenhouse gas emissions, Renewable Sustainable Energy Rev., 2021, 141, 110759 CrossRef CAS.
- S.-J. Lee, I.-S. Ryu, S.-G. Jeon and S.-H. Moon, Emission sources and mitigation of fluorinated Non-CO2 greenhouse gas in registered CDM projects, Greenh. Gases: Sci. Technol., 2017, 7, 589–601 CrossRef CAS.
- B. Zhang, Z. M. Chen, H. Qiao, B. Chen, T. Hayat and A. Alsaedi, China's non-CO2 greenhouse gas emissions: Inventory and input–output analysis, Ecol. Inform., 2015, 26, 101–110 CrossRef.
- R. Ferraz-Almeida, K. A. Spokas and R. C. De Oliveira, Columns and Detectors Recommended in Gas Chromatography to Measure Greenhouse Emission and O2 Uptake in Soil: A Review, Commun. Soil Sci. Plant Anal., 2020, 51, 582–594 CrossRef CAS.
- F. Rauh, M. Schwenk, B. Pejcic, M. Myers, K.-B. Ho, L. Stalker and B. Mizaikoff, A mid-infrared sensor for the determination of perfluorocarbon-based compounds in aquatic systems for geosequestration purposes, Talanta, 2014, 130, 527–535 CrossRef CAS.
- Y. Su, Y. Liu, W. Li, X. Xiao, C. Chen, H. Lu, Z. Yuan, H. Tai, Y. Jiang, J. Zou, G. Xie and J. Chen, Sensing–transducing coupled piezoelectric textiles for self-powered humidity detection and wearable biomonitoring, Mater. Horiz., 2023, 10, 842–851 RSC.
- Y. Su, W. Li, X. Cheng, Y. Zhou, S. Yang, X. Zhang, C. Chen, T. Yang, H. Pan, G. Xie, G. Chen, X. Zhao, X. Xiao, B. Li, H. Tai, Y. Jiang, L.-Q. Chen, F. Li and J. Chen, High-performance piezoelectric composites via β phase programming, Nat. Commun., 2022, 13, 4867 CrossRef CAS PubMed.
- C. Dong, R. Zhao, L. Yao, Y. Ran, X. Zhang and Y. Wang, A review on WO3 based gas sensors: Morphology control and enhanced sensing properties, J. Alloys Compd., 2020, 820, 153194 CrossRef CAS.
- C. Feng, Z. Jiang, J. Wu, B. Chen, G. Lu and C. Huang, Pt-Cr2O3-WO3 composite nanofibers as gas sensors for ultra-high sensitive and selective xylene detection, Sens. Actuators, B, 2019, 300, 127008 CrossRef CAS.
- C. Chen, M. Jiang, X. Luo, H. Tai, Y. Jiang, M. Yang, G. Xie and Y. Su, Ni-Co-P hollow nanobricks enabled humidity sensor for respiratory analysis and human-machine interfacing, Sens. Actuators, B, 2022, 370, 132441 CrossRef CAS.
- C. Chen, G. Xie, J. Dai, W. Li, Y. Cai, J. Li, Q. Zhang, H. Tai, Y. Jiang and Y. Su, Integrated core-shell structured smart textiles for active NO2 concentration and pressure monitoring, Nano Energy, 2023, 116, 108788 CrossRef CAS.
- F. Deng, Y. He, G. Shi, B. Li and X. Wu, Low-temperature cataluminescence sensor for sulfur hexafluoride utilizing coral like Zn-doped SnO2 composite, Sens. Actuators, B, 2016, 237, 120–126 CrossRef CAS.
- J. Liu, L. Zhang, J. Fan, B. Zhu and J. Yu, Triethylamine gas sensor based on Pt-functionalized hierarchical ZnO microspheres, Sens. Actuators, B, 2021, 331, 129425 CrossRef CAS.
- P. Cao, Z. Yang, S. T. Navale, S. Han, X. Liu, W. Liu, Y. Lu, F. J. Stadler and D. Zhu, Ethanol sensing behavior of Pd-nanoparticles decorated ZnO-nanorod based chemiresistive gas sensors, Sens. Actuators, B, 2019, 298, 126850 CrossRef CAS.
- H. Pan, G. Chen, Y. Chen, A. Di Carlo, M. A. Mayer, S. Shen, C. Chen, W. Li, S. Subramaniam, H. Huang, H. Tai, Y. Jiang, G. Xie, Y. Su and J. Chen, Biodegradable cotton fiber-based piezoresistive textiles for wearable biomonitoring, Biosens. Bioelectron., 2023, 222, 114999 CrossRef CAS PubMed.
- S. Huang, Y. Geng, J. Xia, D. Chen and J. Lu, NiCo Alloy Nanoparticles on a N/C Dual-Doped Matrix as a Cathode Catalyst for Improved Microbial Fuel Cell Performance, Small, 2022, 18, e2106355 CrossRef PubMed.
- G. Jiang, T. Cumberland, X. Fu, M. Li, J. Zhang, P. Xu, S. Delaat, M. Xiao, S. Hemmati, J. Li, Z. Mao, H. Jin, A. Yu, S. Wang and Z. Chen, Highly Stable Low-Cost Electrochemical Gas Sensor with an Alcohol-Tolerant N,S-Codoped Non-Precious Metal Catalyst Air Cathode, ACS Sens., 2021, 6, 752–763 CrossRef CAS PubMed.
- A. Wang, J. Ni, W. Wang, X. Wang, D. Liu and Q. Zhu, MOF-derived N-doped ZnO carbon skeleton@hierarchical Bi2MoO6 S-scheme heterojunction for photodegradation of SMX: Mechanism, pathways and DFT calculation, J. Hazard. Mater., 2022, 426, 128106 CrossRef CAS PubMed.
- H. Zangeneh, M. Farhadian and A. A. Zinatizadeh, A reusable visible driven N and C–N doped TiO2 magnetic nanocomposites for photodegradation of direct red 16 azo dye in water and wastewater, Environ. Technol., 2020, 43, 1269–1284 CrossRef PubMed.
- J. Li, G. Xie, J. Jiang, Y. Liu, C. Chen, W. Li, J. Huang, X. Luo, M. Xu, Q. Zhang, M. Yang and Y. Su, Enhancing photodegradation of Methyl Orange by coupling piezo-phototronic effect and localized surface plasmon resonance, Nano Energy, 2023, 108, 108234 CrossRef CAS.
- X. Zhang, J. Zhang and H. Cui, Adsorption mechanism of SF6 decomposition components onto N, F-co-doped TiO2: A DFT study, J. Fluorine Chem., 2018, 213, 18–23 CrossRef CAS.
- H. Jiang, Y. Qu, X. Zhang, R. Gao, X. Cheng, S. Gao, L. Huo, Z. A. Major and Y. Xu, Light-enhanced NO2 sensing performance and sensing mechanism of flower-like Cl uniformly doped In2O3, Appl. Surf. Sci., 2022, 590, 153033 CrossRef CAS.
- K. Xu, S. Tian, J. Zhu, Y. Yang, J. Shi, T. Yu and C. Yuan, High selectivity of sulfur-doped SnO2 in NO2 detection at lower operating temperatures, Nanoscale, 2018, 10, 20761–20771 RSC.
- P. Qiu, Y. Qin, Y. Bai, Q. Xia and A. Zheng, Gas selectivity regulation of monolayer SnS by introducing nonmetallic dopants: A combined theoretical and experimental investigation, Appl. Surf. Sci., 2021, 570, 151155 CrossRef CAS.
- X. Guan, One-step Facile Synthesis of Hierarchically Porous Nitrogen- Doped SnO2 Nanoparticles with Ultrahigh Surface Area for Enhanced Lithium Storage Performance, Int. J. Electrochem. Sci., 2018, 18, 5667–5680 CrossRef.
- J. Zhang, F. Huang and Z. Lin, Progress of nanocrystalline growth kinetics based on oriented attachment, Nanoscale, 2010, 2, 18–34 RSC.
- Y.-H. Cai, Y.-H. Zhang and L.-P. Ren, Synthesis of Co(II) Complex with Hexamethylenetetramine Using Jet Milling and Its Influence on the Thermal Performance of Poly(l-lactic acid), J. Macromol. Sci., Part B: Phys., 2016, 55, 547–558 CrossRef CAS.
- L. Xu, Y. Zhao, J. Lian, Y. Xu, J. Bao, J. Qiu, L. Xu, H. Xu, M. Hua and H. Li, Morphology controlled preparation of ZnCo2O4 nanostructures for asymmetric supercapacitor with ultrahigh energy density, ACS Sens., 2017, 123, 296–304 CAS.
- S. Wei, R. Wu, J. Jian, F. Chen and Y. Sun, Black and yellow anatase titania formed by (H,N)-doping: strong visible-light absorption and enhanced visible-light photocatalysis, Dalton Trans., 2015, 44, 1534–1538 RSC.
- J. Zhang, X. Liu, S. Wu, M. Xu, X. Guo and S. Wang, Au nanoparticle-decorated porous SnO2 hollow spheres: a new model for a chemical sensor, J. Mater. Chem., 2010, 20, 6453 RSC.
- W. Dong, J. Xu, C. Wang, Y. Lu, X. Liu, X. Wang, X. Yuan, Z. Wang, T. Lin, M. Sui, I. W. Chen and F. Huang, A Robust and Conductive Black Tin Oxide Nanostructure Makes Efficient Lithium-Ion Batteries Possible, Adv. Mater., 2017, 29, 1700136 CrossRef.
- M. R. Alenezi, A. S. Alshammari, K. D. Jayawardena, M. J. Beliatis, S. J. Henley and S. R. Silva, Role of the Exposed Polar Facets in the Performance of Thermally and UV Activated ZnO Nanostructured Gas Sensors, J. Phys. Chem. C Nanomater. Interfaces, 2013, 117, 17850–17858 CrossRef CAS PubMed.
- P. Li, C. Ruan, J. Xu and Y. Xie, Supercapacitive performance of CoMoO4 with oxygen vacancy porous nanosheet, Electrochim. Acta, 2020, 330, 135334 CrossRef CAS.
- G. X. Zhou, S. J. Xiong, X. L. Wu, L. Z. Liu, T. H. Li and P. K. Chu, N-doped SnO2 nanocrystals with green emission dependent upon mutual effects of nitrogen dopant and oxygen vacancy, Acta Mater., 2013, 61, 7342–7347 CrossRef CAS.
- S. Shi, F. Zhang, H. Lin, Q. Wang, E. Shi and F. Qu, Enhanced triethylamine-sensing properties of P-N heterojunction Co3O4/In2O3 hollow microtubes derived from metal–organic frameworks, Sens. Actuators, B, 2018, 262, 739–749 CrossRef CAS.
- H.-J. Kim and J.-H. Lee, Highly sensitive and selective gas sensors using p-type oxide semiconductors: Overview, Sens. Actuators, B, 2014, 192, 607–627 CrossRef CAS.
- C. Li, Y. Liu, W. Wan, Y. Li, Y. Ma, J. Zhang, X. Ren and H. Zhao, Hydrothermal synthesis of novel porous butterfly-like hierarchical SnO2 architecture with excellent gas-sensing performance to acetaldehyde, Sens. Actuators, B, 2020, 318, 128209 CrossRef CAS.
- S. Torai, T. Ueda, K. Kamada, T. Hyodo and Y. Shimizu, Effects of Addition of CuxO to Porous SnO2 Microspheres Prepared by Ultrasonic Spray Pyrolysis on Sensing Properties to Volatile Organic Compounds, Chemosensors, 2023, 11, 59 CrossRef CAS.
- V. Platonov, A. Nasriddinov and M. Rumyantseva, Electrospun ZnO/Pd Nanofibers as Extremely Sensitive Material for Hydrogen Detection in Oxygen Free Gas Phase, Polymer, 2022, 14, 3481 CAS.
- M.-I. Baraton and L. Merhari, Investigation of the Gas Detection Mechanism in Semiconductor Chemical Sensors by FTIR Spectroscopy, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 35, 733–742 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3en00679d |
| This journal is © The Royal Society of Chemistry 2024 |