
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Depth-dependent transformation of ZnO and Ag nanoparticles in sulfate-reducing sediments tracked using scanning transmission electron microscopy†
L.
Stetten‡
 *a,
R.
Kaegi
b,
T.
Hofmann
a and
F.
von der Kammer
*a
*a,
R.
Kaegi
b,
T.
Hofmann
a and
F.
von der Kammer
*a
aDepartment of Environmental Geosciences, Centre for Microbiology and Environmental Systems Science, University of Vienna, Josef-Holaubeck Platz 2, 1090, Vienna, Austria. E-mail: luciestetten@gmail.com; frank.von.der.kammer@univie.ac.at
bEawag, Swiss Federal Institute of Aquatic Science and Technology, Überlandstrasse 133, 8600 Dübendorf, Switzerland
First published on 14th December 2023
Abstract
Studies on the transformation of engineered nanomaterials (ENMs) based on relevant environmental exposure scenarios are scarce. In this context, we investigated the use of Transmission Electron Microscopy (TEM) grids to expose minute amounts of ZnO and Ag nanoparticles (NPs) to artificial and natural aqueous media and follow their transformation using scanning transmission electron microscopy coupled with energy dispersive X-ray spectroscopy. Short-term experiments conducted with inorganic sulfides confirmed the potential of using TEM grids to monitor the transformation of ENMs at the single particle level. After 30 min, Ag NPs transformed into Ag-sulfides, with Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S ratios ≈ 2:1, while ZnO NPs showed little evidence of Zn-sulfides precipitation after 6 hours. Ag NPs and ZnO NPs were also exposed to depth-dependent pore water concentration gradients in freshwater sediment columns. After three days and four weeks, all the Ag NPs observed were transformed into Ag-sulfides with various morphologies and Ag:S ratios (1 ≤ Ag/S ≤ 2), depending on the depth and duration of the exposure. Furthermore, a depth-dependent transformation was observed for ZnO NPs. At low sulfide concentration, in the first millimeters below the water–sediment interface, ZnO NPs were completely transformed into ZnS harboring empty shell structures, together with smaller particles in their vicinity. By contrast, ZnO cores persisted in the deeper layers, indicating that ZnO NPs dissolution was inhibited at high sulfide concentrations. Our results demonstrate the advantage of experimental and analytical strategies adapted to study the transformation of ENMs under environmentally relevant conditions, to unravel transformation rates and products not yet considered in risk assessment studies.
:S ratios ≈ 2:1, while ZnO NPs showed little evidence of Zn-sulfides precipitation after 6 hours. Ag NPs and ZnO NPs were also exposed to depth-dependent pore water concentration gradients in freshwater sediment columns. After three days and four weeks, all the Ag NPs observed were transformed into Ag-sulfides with various morphologies and Ag:S ratios (1 ≤ Ag/S ≤ 2), depending on the depth and duration of the exposure. Furthermore, a depth-dependent transformation was observed for ZnO NPs. At low sulfide concentration, in the first millimeters below the water–sediment interface, ZnO NPs were completely transformed into ZnS harboring empty shell structures, together with smaller particles in their vicinity. By contrast, ZnO cores persisted in the deeper layers, indicating that ZnO NPs dissolution was inhibited at high sulfide concentrations. Our results demonstrate the advantage of experimental and analytical strategies adapted to study the transformation of ENMs under environmentally relevant conditions, to unravel transformation rates and products not yet considered in risk assessment studies.
Environmental significanceThe development of experimental procedures adapted to investigate the transformation of engineered nanomaterials (ENMs) under environmentally relevant conditions is essential to predict the fate of ENMs in the environment, and advance OECD objectives on ENM safety. We demonstrate the potential of using transmission electron microscopy grids to expose nanoparticles (NPs) to aqueous media and follow their transformations using electron microscopy techniques. Experiments performed with Ag and ZnO NPs in sediment columns highlighted the formation of various transformation products along pore water depth gradients, suggesting complex transformation paths and kinetics. This work emphasizes the advantage of tailored experimental strategies to unravel environmentally relevant transformation rates and products, not yet considered in risk assessment studies. |
1. Introduction
In the last two decades, engineered nanomaterials (ENMs) have been extensively produced to meet the needs of industrial, medical, agricultural, and domestic applications.1–3 The resulting releases of ENMs into the environment were stressed by predictive models,4–6 and numerous field investigations confirmed the presence of nanoparticles (NPs) from anthropogenic origins in rain, urban runoffs,7 surface waters,8,9 soils,10 and sediments.11 Once released into the environment, ENMs may undergo physical, chemical, and biological transformations which will affect their fate, bioavailability and toxicity.12,13 For example, the transformation of Ag NPs and ZnO NPs into less soluble metal sulfides is known to reduce the release of toxic Ag+ and Zn2+ ions and therefore mitigate NP toxicity.14,15 Organic compounds, ubiquitous in the environment, have also been shown to significantly affect the transformation, aggregation, and surface properties of NPs, and ultimately their toxicity.16 Understanding the transformations of ENMs in the different environmental compartments is therefore essential to predict their fate and risk to the ecosystem.The transformation of ENMs in aqueous media has been extensively studied, and is reviewed in previous publications.12,13 In general, the transformations of ENMs are monitored through laboratory-controlled experiments designed to target specific transformation pathways and geochemical conditions recognized to control the behavior of ENMs in the environment (e.g., pH, redox conditions, etc.). For example, the dissolution of ENMs was studied in the presence of inorganic and organic ligands, to better predict the release of soluble, potentially toxic species in aquatic environments.17 The sulfidation of chalcophile metal nanoparticles such as Ag NPs and ZnO NPs, expected to occur in sulfate-reducing environments, was investigated in inorganic sulfide (Na2S) solutions, and dissolution of the resulting end-products was assessed.18,19 It has been shown that the sulfidation of Ag NPs, which requires the oxidation of Ag0 to Ag+, occurs via a solid-state or a dissolution pathway.19,20 By contrast, the sulfidation of ZnO NPs does not involve redox reactions and is controlled by the solubility of ZnO following a dissolution–precipitation pathway.18 Although these studies provide in-depth insights into the mechanisms of transformation, they may lead to mispredictions of the fate of ENMs, mainly due to their lack of complexity, neglecting environmental parameters that may affect transformation paths, kinetics, and products. Studies on the transformation of ENMs in complex media such as wastewaters, soils, and sediments, allow a more realistic representation of ENM fate in the environment. They are well suited to unravel the formation of realistic end products,21–25 and provide confirmation of the theorized mechanisms.26 Nevertheless, the study of ENM fate in soils and sediments usually requires working with concentrations of ENMs higher than the expected environmental concentrations,4–6 and is not adapted to follow chemical and morphological changes at the single particle level. A few studies have overcome such challenges and investigated the transformation of ENMs using specific experimental designs allowing to expose realistic concentrations of ENMs into full-scale wastewater treatment plants,26 freshwater streams27 and sediments.28 However, no experimental procedures or guidance have been proposed for testing the transformation of ENMs in aquatic environments, although the need to develop environmentally relevant test procedures has been outlined by international regulatory bodies such as the Organization for Economic Cooperation and Development (OECD).29,30
The aim of this study was to investigate the transformation of Ag NPs and ZnO NPs for a realistic environmental exposure scenario. For this purpose, a novel experimental approach has been explored using TEM grids as probes to expose minute amounts of Ag NPs or ZnO NPs in aqueous media, and examine their transformation using Scanning Transmission Electron Microscopy coupled with Energy Dispersive X-ray Spectroscopy (STEM-EDXS). In order to assess possible losses of Ag NPs and ZnO NPs from the TEM grids after their exposure to aqueous media, an experiment was carried out using inorganic sulfide solutions. This experiment confirmed that the use of NPs-attached TEM grids was suitable to monitor the transformation of NPs in aquatic environments. Using such an approach, it was then possible to identify the transformation products of Ag NPs and ZnO NPs along pore water depth gradients in incubated sulfate-reducing freshwater sediments.
2. Materials & methods
2.1. Pristine ZnO NPs and Ag NPs samples
Nanosun™ Zinc Oxide P99/30 NPs were purchased from Micronisers (Dandenong, Australia) as a dry powder. It corresponds to uncoated ZnO NPs with a nominal average particle size of 30 nm and an oval geometry. NanoXact™ citrate-coated Ag NPs (JRD0035) were obtained from Nanocomposix (San Diego, CA). It corresponds to spherical 80 nm Ag NPs dispersed in 2 mM sodium citrate solution ([Ag NPs]stock = 0.02 mg mL−1). These NPs were chosen as models to represent manufactured ZnO NPs and Ag NPs because of their commercial availability and previous use in multi-institution international works.31,32ZnO NPs or Ag NPs have been deposited on 300 mesh carbon-coated nickel (Ni) or gold (Au) TEM grids (EM Resolutions Ltd, United Kingdom), preliminary functionalized using 0.1% poly-L-lysine. For Ag NPs, samples were prepared by centrifugation following a procedure described elsewhere.33 Briefly, 1.5 ml of a 0.6 mg L−1 Ag NPs dispersion was centrifuged onto the TEM grid at 1800g during 5 minutes. For ZnO NPs, samples were prepared by depositing the TEM grid on the top of a drop of a ZnO NP suspension (500 mg L−1) for 5 minutes. The ZnO NP suspension was preliminary sonicated for 10 min. This method aimed to limit the attachment of big ZnO NP aggregates on the TEM grids. The Ag NPs and ZnO NPs-TEM grids were then washed with distilled water and air-dried. According to these procedures, a maximum load on the TEM grids of 8.3 × 10−3 μmoles for Ag NPs, and 6.1 × 10−1 μmoles for ZnO NPs was estimated, although lower loadings are expected given that not all the NPs bind to the grids.
2.2. Suitability test with sodium sulfide (Na2S) solutions
An experiment was carried out to evaluate whether the use of NPs-attached TEM grids is suitable to study the transformation of NPs in aqueous media. In this regard, the detachment of Ag NPs and ZnO NPs from the TEM grids following their immersion into inorganic sulfide solutions was assessed. Before the exposure, the pristine NPs-TEM grids were analyzed by STEM-EDXS. Regions of interest (ROIs) of 13 × 13 μm were examined for each sample. For each ROI, smaller zones were selected and EDXS analyses were performed on targeted nanoparticles. Then, the NPs-TEM grid samples were exposed to 8 ml of 500 μM Na2S solutions buffered at pH 7.5 using UltraPure™ Tris-HCl (Invitrogen™, Thermo Fisher) for 30 minutes. After the exposure, the previously selected ROIs were reinvestigated by STEM, and new particles were analyzed by EDXS. Thereafter, the samples were exposed a second time to 500 μM Na2S solutions buffered at pH 7.5 for 5 hours and 30 minutes, and analyzed by STEM-EDXS following the same approach. The experiment was performed in duplicate, namely, Ag_A, Ag_B, ZnO_A and ZnO_B, using Ni-TEM grids. An illustration of the exposure experiment is presented in Fig. SI-1a.† Briefly, the NPs-TEM grids were fixed by self-closing tweezers and immersed into open glass vials containing the exposure media. After the exposure, the samples were carefully removed, washed with deionized water, and air-dried (the excess of water was wiped off with wipers). To minimize Ag NP changes during the storage of the samples, all the STEM-EDXS analyses were carried out 1 to 2 hours after the removal of the samples from the exposure media. Furthermore, to evaluate possible changes of the Ag NPs during the manipulation and storage of the samples, a pristine Ag NPs-TEM grid, which was not exposed to the Na2S solutions but prepared in parallel to the exposed samples, was analyzed after one day of storage.The sulfide concentrations in the batches were monitored on 500 μL aliquots collected over the course of the experiment from the exposure solutions. The initial concentration of sulfide, t = 0, was determined on a separate control batch. Total sulfide (H2S, HS− and S2−) concentrations were determined by spectrophotometry using the methylene blue method developed by Cline et al. (1968).34 Absorbance of the methylene blue was measured at 670 nm using a Cary 60 UV/vis spectrometer (Agilent technologies AG, Basel, Switzerland).
2.3. Exposure experiments in incubated sediment columns
In order to investigate the transformation of Ag NPs and ZnO NPs under environmentally relevant conditions, experiments have been performed in freshwater sediment columns incubated in the laboratory. For this purpose, undisturbed sediment cores were collected from the lake Neusiedl See (Podersdorf am See, Austria), in a location close to the shoreline, where fine sediments accumulate. Four sediment cores (C1, C2, C3, C4) were sampled using 5 cm diameter and 20 cm long PVC core liners. The cores were closed using rubber stoppers, transported within a few hours to the laboratory in a cool box (∼4 °C), and immediately incubated into a tank containing the surface water of the lake Neusiedl collected the same day. The tank measured 35 cm (l) × 25 cm (L) × 35 cm (H) and was closed on the top. The water of the tank was oxygenated via the injection of air using an air pump. The walls of the tank were covered with aluminum foil from the bottom to about 10 cm from the top. Two sediment cores were used for the three days exposure experiment (C1_Ag NPs_3d and C3_ZnO NPs_3d) and two sediment cores were used for the four weeks exposure experiment (C2_Ag NPs_4w and C4_ZnO NPs_4w).Due to the high affinity of Ni and Cu to precipitate with sulfides, Au-TEM grids were preferred for these experiments, to ensure more stability of the grids in the sediment.35,36 To avoid damaging the TEM grids, the samples were enclosed between two carved Teflon plates covered with a nylon mesh filter (pore size of 5 μm) (Fig. SI-1c†). Furthermore, the NPs-TEM grid samples were not investigated by STEM-EDXS before the exposure, to prevent changes of the pristine NPs during the manipulation, analysis, and prolonged storage of the samples. Immediately after the preparation of the pristine Ag NPs and ZnO NPs-TEM grids, the NPs-TEM grids were placed in the TEM grid holders, at 0–3 mm, 7–10 mm, 15–18 mm, 25–30 mm, and 60 mm depth. The devices were tightly closed and carefully inserted into the sediment columns. For the three days experiment, one set of TEM grids was inserted in the sediment columns for each Ag NPs and ZnO NPs. The four weeks exposure experiment was performed in duplicate by inserting two TEM grid holders within the same sediment column. At the end of the experiments, the holders were carefully extracted from the sediment and the NPs-TEM grids were collected, washed in deionized water, and stored in the dark, under vacuum in a TEM grid vacuum desiccator. STEM-EDXS analyses were performed in the following days. For each sample, several zones were observed on the TEM grids and STEM micrographs and EDX spectra were recorded on a subset of particles, representative of the morphologies identified. The number of particles analyzed for each sample is displayed in Tables SI-1 and SI-2.† To address potential changes of the Ag NPs during the storage of the samples, an additional pristine Ag NPs-TEM-grid sample was prepared in parallel to the three days experiment and analyzed after 4.5 days of storage in the TEM grid vacuum desiccator.
Oxygen (O2) concentration, pH and hydrogen sulfide (H2S) concentration depth profiles were acquired below the water–sediment interfaces using Unisense® microelectrodes (UNISENCE A/S, Aarhus, Denmark) coupled with a Unisense® MicroProfilling System. The pH was measured using a 200 μm conventional glass pH microelectrode. O2 concentration was measured at the water–sediment interface using a 50 μm Clark-type amperometric O2 microsensor.37 The O2 microsensor was calibrated at ambient temperature (∼20 °C), using air-saturated deionized water and O2-free deionized water prepared by dissolving sodium sulfite at a concentration of 30 g L−1. H2S concentration was measured using a 200 μm Clark-type amperometric H2S microsensor,37–39 calibrated using H2S-free water and a solution with a known H2S concentration from the Unisense® hydrogen sulfide calibration kit. Depth profiles were acquired at 75 μm intervals for O2 concentration and 500 μm intervals for H2S concentration and pH, down to 6–7 cm below the water–sediment interface. O2 concentration, H2S concentration and pH values were processed using the software SensorTrace Suite v.3.3 (Unisense A/S, Denmark). Extended information on the operation of the microsensors can be found at http://unisense.com. For the three-day experiment, O2 concentration, pH and H2S concentration depth profiles were recorded at the insertion of the TEM grid holders in the sediments. For the four-week experiment, depth profiles were recorded at the insertion of the TEM grid holders, and at the end of the experiment.
2.4. Scanning transmission electron microscopy analyses
Scanning transmission electron microscopy and energy dispersive X-ray spectroscopy analyses were performed using a scanning transmission electron microscope (STEM, HD-2700-Cs, Hitachi, Japan), at an accelerating voltage of 200 kV. Images were recorded using a secondary electron (SE) detector and a high-angle annular dark field (HAADF) detector. Elemental analysis of the particles was performed with an EDX system (EDAX, New Jersey, USA). In addition, elemental distribution mapping was performed using a (S)TEM (Talos F200X, Super-X EDS, 4 detector configurations, FEI) at an accelerating voltage of 200 kV. Electron micrographs and EDX spectra were processed using the Digital Micrograph® software. High-resolution elemental maps were processed using the Velox software and the Hyperspy software.403. Results and discussion
3.1. Suitability of the method: Ag NPs and ZnO NPs in Na2S solutions
The suitability of the experimental approach was assessed by exposing Ag NPs and ZnO NPs-TEM grids to 500 μM Na2S solutions at pH 7.5. No changes were observed in the pristine Ag NPs-TEM grid sample not exposed in the Na2S solution and stored for one day (Fig. SI-3a†), indicating that negligible NP transformation occurred during the manipulation and storage of the samples. After 30 minutes, the dissolved sulfide concentrations decreased from 557 μM to 511 μM (sample Ag_A) and 528 μM (sample Ag_B) for the Ag NPs experiment, and to 514 μM (sample ZnO_A) and 518 μM (sample ZnO_B) for the ZnO NPs experiment. After the second exposure of 5 hours and 30 minutes, the concentrations of dissolved sulfide decreased from 470 μM to 142 μM (sample Ag_A) and 145 μM (sample Ag_B) for the Ag NPs experiment and to 143 μM (sample ZnO_A) and 5 μM (sample ZnO_B) for the ZnO NPs experiment (Fig. SI-2†). Although the decrease in sulfide in the solutions may be due to the sulfidation of Ag NPs and ZnO NPs, and/or its volatilization and oxidation due to the open batch experimental design, the presence of sulfide at the end of the experiment in all batches indicates that a sufficient amount of sulfide was available in the media during the experiment.For each Ag and ZnO NPs-TEM grid sample, 13 × 13 μm ROIs were analyzed before, and after t = 30 min and t = 5 h 30 min of exposure. For both Ag NPs and ZnO NPs-TEM grid samples, the same spatial distribution of NPs was observed before and after exposure, indicating that the NPs remained attached to the TEM grids during the time of the experiment (Fig. 1a and b and 2a and b and SI-4–SI-6†). After 30 minutes, the Ag NPs exhibited various sizes with spheroidal to asymmetric shapes, as well as filamentous structures exiting the particles (Fig. 1 and SI-4†). Of the 10 Ag NPs analyzed by EDXS, 8 particles displayed EDX spectra consistent with the formation of Ag2S (Fig. 1), with an intensity ratio between the Ag Lα and S Kα emission lines (i.e., height of the EDX peaks) ranging between 1.8–2.2, indicating their complete sulfidation. Only two Ag NPs did not exhibit changes in composition, including one particle analyzed prior to the exposure, which may have been subjected to electron beam damage impacting its reactivity. No significant changes were observed after the second exposure, and pristine Ag NPs were not identified in the investigated ROIs. These results are in line with previous kinetic studies showing that sulfidation of Ag NPs with an average size <100 nm occurs within a few minutes or hours.20,41 There are several pathways for Ag NPs sulfidation in aquatic environments.42 In the presence of HS− and dissolved O2, it is suggested that the sulfidation of Ag NPs to Ag2S depends on competing rates between oxidative dissolution and direct oxysulfidation. At high sulfide concentration, it has been shown that Ag NPs transform to Ag2S NPs through a solid–fluid reaction, while at lower sulfide concentration, oxidative dissolution followed by the subsequent precipitation of Ag2S dominates.19,20 The formation of Ag-sulfide filaments in our experiment may result from dissolution–precipitation mechanisms. On the other hand, the presence of sulfidized Ag NPs harboring a shape similar to the pristine Ag NPs (Fig. 1 and SI-4†) suggests that Ag sulfidation also occurred with limited displacement of Ag+. It is therefore likely that multiple pathways of sulfidation occurred in the experiment, none of which resulted in particles detachment and loss.
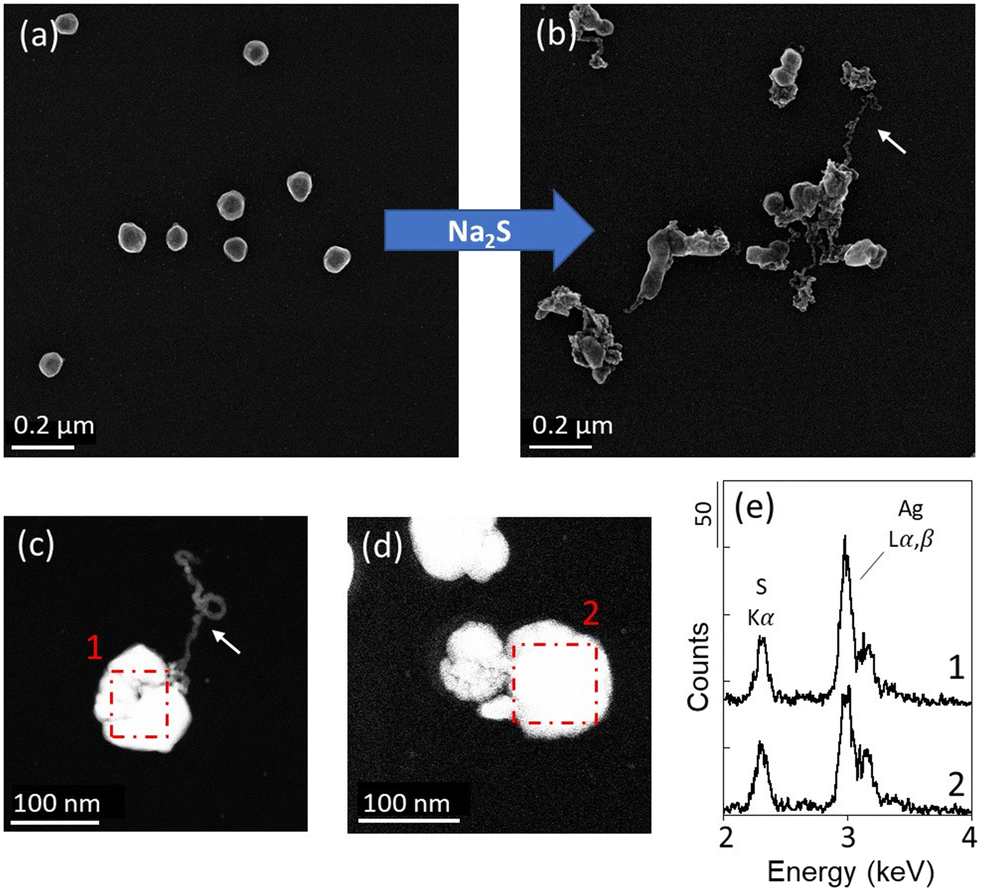 | ||
| Fig. 1 Ag NPs exposed to 500 μM Na2S solutions buffered at pH 7.5 for 30 minutes. (a and b) Representative STEM micrographs acquired with a SE detector of Ag NPs (a) before and (b) after the exposure (sample Ag_A). (c and d) STEM micrographs acquired with a HAADF detector and (e) corresponding EDX spectra (sample Ag_B). The red dotted squares correspond to the scanned areas for the EDXS analyses. The white arrows indicate filamentous structures. | ||
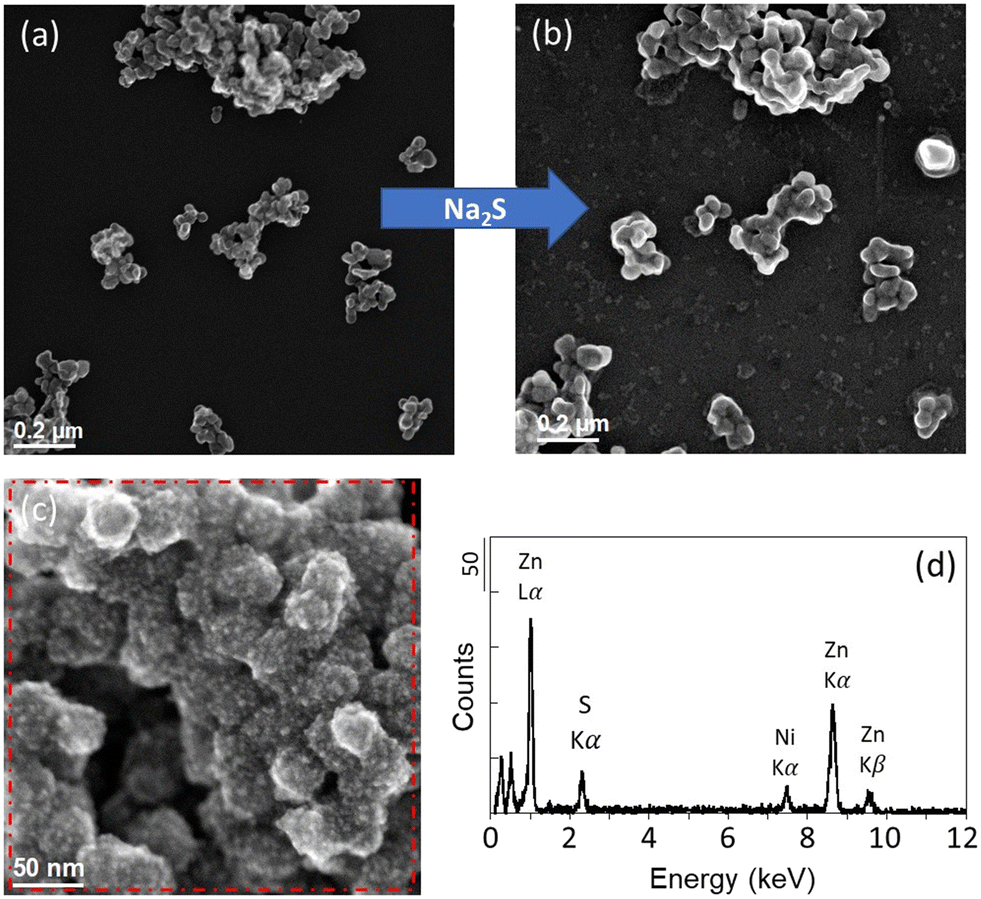 | ||
| Fig. 2 ZnO NPs exposed to 500 μM Na2S solutions buffered at pH 7.5 for 5 hours and 30 minutes. STEM micrographs (a) before and (b) after the exposure (sample ZnO_A). (c) STEM micrograph after the exposure and (d) corresponding EDX spectrum (sample ZnO_B). The micrographs were acquired with a SE detector. The red dotted square corresponds to the scanned area for the EDX analysis. | ||
In contrast to the Ag NPs experiment, no major changes were observed on the ZnO NPs-TEM grids after the exposure. ZnO NPs remained as aggregates with similar oval shapes (Fig. 2 and SI-5 and SI-6†). For this experiment, one duplicate was damaged after the 30 minutes exposure, and therefore a new sample was investigated for the second exposure of 5 hours 30 minutes (sample ZnO_B). After 5 hours and 30 minutes, two ZnO NP aggregates exhibited a coarse surface compared to the smooth surface of the pristine ZnO NPs. For these particles, EDXS analyses revealed the presence of sulfur (S), suggesting the precipitation of ZnS at the surface of the ZnO NPs (Fig. 2). The sulfidation of ZnO NPs is known to be controlled by a dissolution–precipitation pathway.18 The limited sulfidation of the ZnO NPs in this experiment, although a sufficient amount of sulfide was present in the media, could therefore be explained by the limited dissolution of the ZnO NPs. At circumneutral pH, ZnO NP dissolution is expected to be low,43,44 although higher extents of ZnO NP dissolution were observed in experiments using lower concentration of ZnO NPs.45,46 In our experiment, the minor dissolution of ZnO NPs could be explained by the near-neutral pH and the short exposure time. Nevertheless, other factors may have contributed to reducing ZnO NP dissolution, such as surface precipitation of Zn(OH)2, which is known to precipitate at 6 < pH < 9.46,47
The transferability of knowledge gained from laboratory-controlled experiments may sometimes lead to mispredictions of ENM fate in natural environments due to the many physico-chemical parameters known to influence the transformation of NPs, which are often not all represented in artificial media. This has been stressed by a few studies, encouraging the development of experimental work that takes into account more realistic exposure scenarios.14,23,48 In this context, and given the limited loss of Ag NPs and ZnO NPs from the TEM grids following their partial or enhanced transformation in Na2S solution, the use of NPs-TEM grids appears to be a promising approach to expose small amounts of NPs in environmental aqueous media and follow their transformation. Although no impact of the Ni-TEM grids on the transformation of Ag NPs and ZnO NPs was detected in the experiment in Na2S, the use of Au-TEM grids was preferred for further experiments to reduce the reactivity of the grids with the aqueous media.
3.2. O2, pH and H2S concentration in the sediment pore waters
In order to demonstrate the applicability and advantages of the NPs-TEM grids approach for a more complex environmental scenario, experiments were carried out using freshwater sediments. Especially, NPs-TEM grid samples were inserted in incubated, undisturbed sediment columns to investigate Ag NPs and ZnO NPs transformation along sediment pore water depth gradients. For all incubated sediment columns, dissolved oxygen depth profiles showed that the sediments were anoxic after 2–5 mm below the water–sediment interfaces (Fig. SI-7†). In the overlaying water columns and the first millimeter's depth, the pH values ranged between 9 and 9.5, in agreement with the alkaline conditions of Lake Neusiedl.49 Below 2–5 mm depth, the pH values progressively decreased to 8.4 for the core C1, 7.7 for the core C2, and 7.6 for the cores C3 and C4 (Fig. 3). H2S concentration was below the detection limit (<0.5 μM) in the first 13 mm below the water–sediment interface for the C1 core, and in the first 10 mm for the C2 core. Similarly, H2S concentration was below the detection limit in the first 6 mm below the water–sediment interface for the C3 core, and in the first 10 mm for the C4 core. In addition, for all the cores, H2S concentration quickly increased to reach 200–250 μM at ∼70 mm depth (Fig. 3), supporting dominant sulfate-reducing conditions in the sediment.50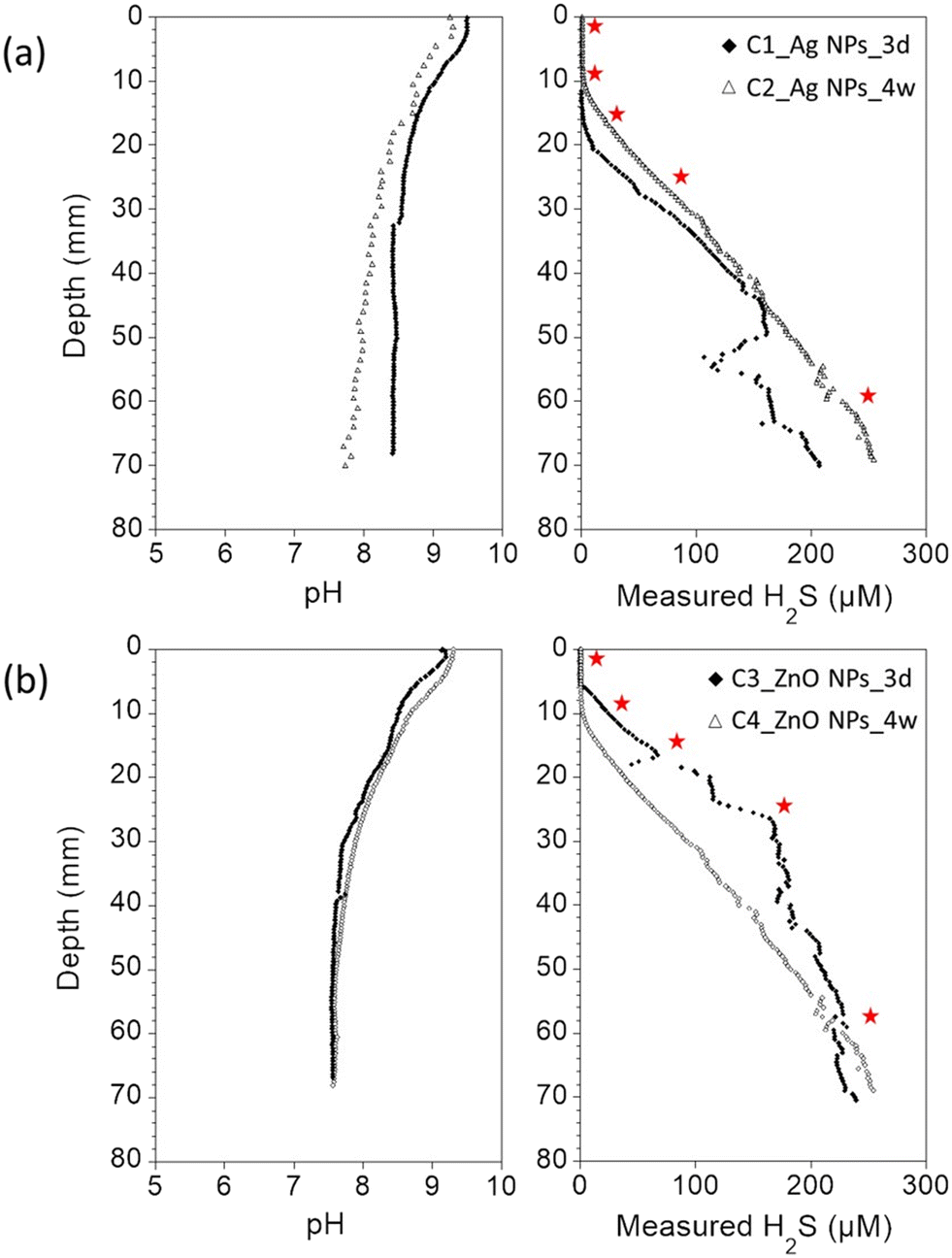 | ||
| Fig. 3 Pore water depth profiles of pH and measured H2S concentration in the incubated sediment cores used to expose (a) Ag NPs and (b) ZnO NPs-TEM grids. The red stars correspond to the depths at which the NPs-TEM grids were inserted in the sediment columns. | ||
3.3. Sulfidation of Ag NPs along sediment pore water depth gradients
The sediment columns C1 and C2 were used to expose the Ag NPs-TEM grid samples. After the exposure, slight physical damage was observed for some of the TEM grids. The attachment of new particles originating from the sediment pore water was also observed (Fig. SI-8†). However, for all samples, intact zones on the TEM grids were found and examined. Moreover, the partial sulfidation of Ag NPs was observed in the Ag NPs-TEM grid sample not inserted into the sediment and stored for 4.5 days (Fig. SI-3b†). This transformation can be explained by the longer storage time of the grid, and may also have been enhanced by the volatilization of sorbed sulfide from the TEM grid samples that were exposed to the sediment and stored with the pristine Ag NPs-TEM grid. Nevertheless, since the Ag NPs-TEM grids were inserted into the sediment immediately after their preparation, it is unlikely that the pristine Ag NPs have undergone any changes prior to the exposure. Furthermore, the exposed Ag NPs-TEM grid samples were analyzed 1.5 days after their extraction from the sediment and were stored dry and under vacuum in a TEM desiccator holder. Eventual changes of the exposed Ag NPs were therefore expected to be minor considering that no changes were observed on Ag NPs after 1 day of storage.For the samples exposed at 0–3 mm depth for three days (Ag_0–3 mm_3d) and four weeks (Ag_0–3 mm_4w_a and Ag_0–3 mm_4w_b), 100% of the Ag NPs investigated by STEM-EDXS were transformed into Ag-sulfides (Fig. 4a–d and Fig. SI-9–SI-13; Table SI-1†), despite the negligible sulfide concentration at this depth (Fig. 3). Khaksar et al. (2015)28 also reported the transformation of citrate-coated Ag NPs into Ag-sulfides near the water–sediment interface of a saltwater sediment subjected to tidal cycling, due to the diffusion of dissolved sulfide from deeper layers to the sediment surface.51 The sediment of Neusiedl Lake harbors strong sulfate-reducing conditions in its upper layer,50 which is also underlined by the substantial increase of the concentration of sulfide below 10–13 mm depth (Fig. 3). Considering the minute amount of Ag NPs on the TEM grids, and the dynamic production of sulfide from the microbial sulfate reduction activity, the presence of undetected, unlimited trace levels of sulfide (H2S concentration <0.5 μM) could explain the sulfidation of Ag NPs at 0–3 mm depth. Moreover, it is also possible that the heterogeneity of the sulfide concentration within the sediment column and/or the sediment disturbance created during the insertion of the sample holders had enhanced the diffusion of sulfide in the surface layer of the sediment, resulting in the formation of a zone transiently enriched in sulfide in the vicinity of the TEM grids. No evidence of H2S (g) released from the deep sediment layers was observed during the insertion of the devices or was suspected by the H2S concentration depth profiles, suggesting that if any, the disturbance of the sediment remained limited.
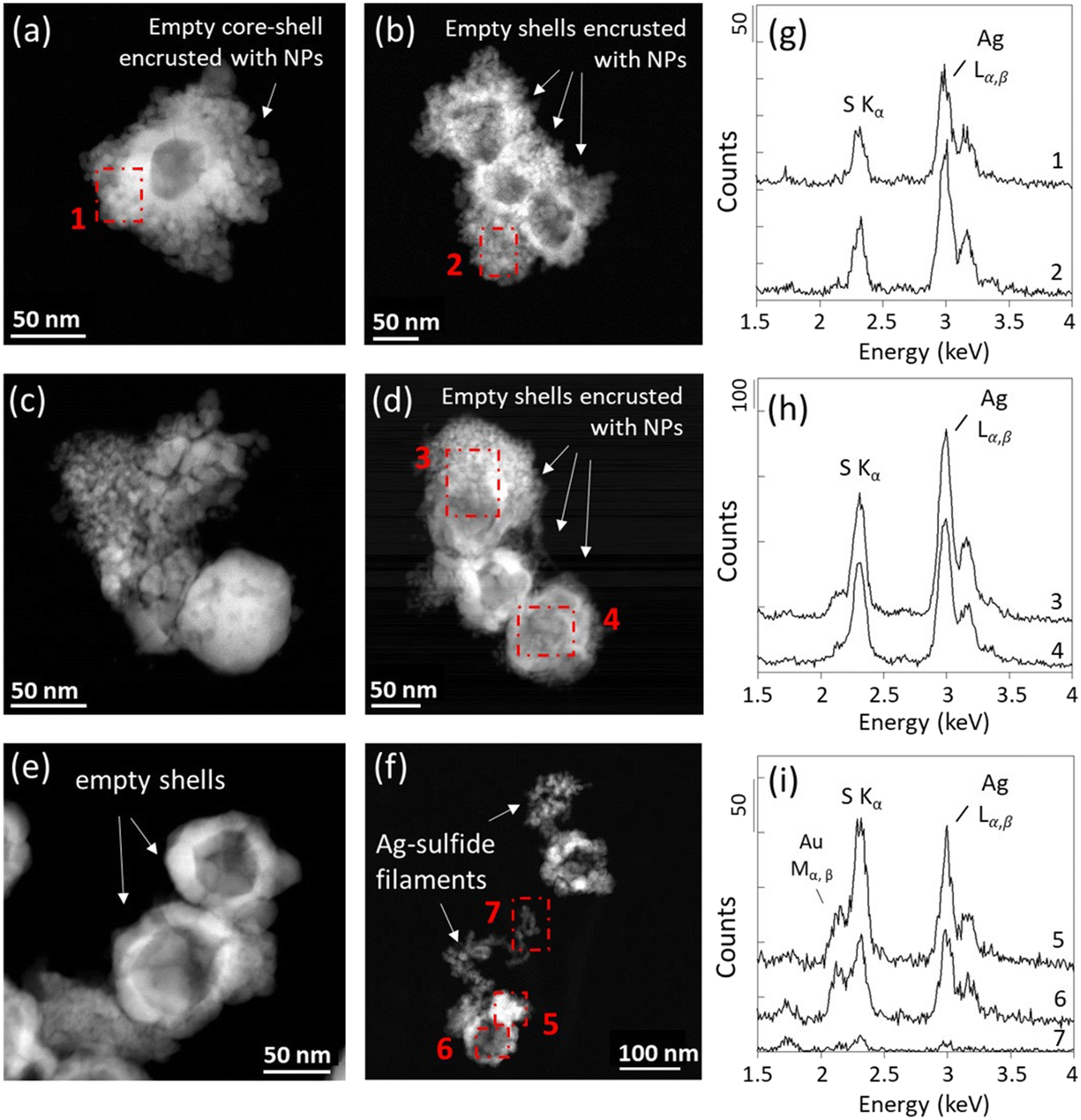 | ||
| Fig. 4 (a–f) STEM micrographs acquired with a HAADF detector and (g–i) corresponding EDX spectra of Ag NPs after (a and b) three days and (c and d) four weeks exposure at 0–3 mm depth, and after (e and f) four weeks at 60 mm depth. The red dotted squares correspond to the scanned areas for the EDX analyses. (a and b) Ag_0–3 mm_3d sample; (c) Ag_0–3 mm_4w_a sample; (d) Ag_0–3 mm_4w_b sample; (e and f) Ag_60 mm_4w_a sample. | ||
In the deeper sediment layers, 100% of the Ag NPs examined by STEM-EDXS were transformed to Ag-sulfides after three days and four weeks (Fig. 4 and SI-9–SI-13; Table SI-1†). For all samples, the transformed Ag NPs exhibited empty core–shell structures encrusted with smaller NPs. Aggregates of small NPs located in their vicinity or replacing the pristine Ag NPs, and extruding filaments were also observed (Fig. 4 and SI-9†). These morphologies are consistent with the morphologies of Ag2S NPs reported in the literature following the direct and/or indirect sulfidation of Ag NPs, including irregular protrusions, chain-like and core–shell structures, and smaller Ag2S particles.52
EDXS analyses of the Ag NPs-TEM grid sample exposed for three days at 0–3 mm depth were consistent with the formation of Ag2S, with intensity ratios of the Ag Lα and S Kα emission lines (Ag/S) similar to the ratio expected for a stoichiometric Ag2S phase (Ag:S ≈ 2:1). By contrast, for the samples exposed for four weeks at 0–3 mm depth, the EDXS analyses indicated a slight excess of sulfur for some particles (Fig. 4c and d and SI-12 and SI-13†). EDXS analyses obtained for the samples exposed at 7–10 mm depth also indicated an excess of sulfur for some precipitates, which is more pronounced in the samples exposed for four weeks (Fig. SI-10–SI-13†). Moreover, a greater abundance of S-rich Ag-sulfide particles was observed for the TEM grids exposed in deeper layers during both three days and four weeks (Fig. 4 and SI-10–SI-13†). For instance, among all particles investigated by EDXS, ∼35% of the particles in the samples exposed for three days at 0–18 mm depth (Ag_0–3 mm_3d; Ag_7–10 mm_3d; Ag_15–18 mm_3d) exhibited an Ag/S ratio < 2 (±0.2). In contrast, for the samples exposed for three days below 18 mm depth (Ag_25–30 mm_3d; Ag_60 mm_3d), ∼85% of the particles exhibited an Ag/S ratio < 2 (±0.2). After four weeks, ∼60% of the particles exposed at 0–18 mm depth (Ag_0–3 mm_4w_a; Ag_0–3 mm_4w_b; Ag_7–10 mm_4w_a; Ag_7–10 mm_4w_b; Ag_15–18 mm__4w_a; Ag_15–18 mm__4w_b) exhibited an Ag/S ratio < 2 (±0.2), and ∼80% of the particles exhibited an Ag/S ratio < 2 (±0.2) in the samples exposed below 18 mm depth (Ag_25–30 mm_4w_a; Ag_25–30 mm_4w_b; Ag_60 mm__4w_a). Particles harboring Ag/S ratios up to 1 were identified in the TEM grid samples exposed at 60 mm depth. For these latter samples, high intensity of the Au Mα,β lines at ∼2.1 and 2.2 keV was also observed, and may have contributed to increase the intensity of the Sα line at 2.3 keV, leading to an overestimation of the S contribution. Nevertheless, extraordinarily low Ag/S ratios were also observed for particles which did not exhibit high Au background in samples exposed for four weeks (Fig. 4 and SI-10–SI-13†).
Ag2S1+x nanoparticles were previously identified in sewage sludges and constructed wetland microcosms.53,54 Laboratory studies also reported the precipitation of S-rich Ag2S phases in the presence of H2S(g) and dissolved sulfide (H2S, HS−, S2−).20,55,56 In particular, Liu et al. (2011)20 showed that dissolved sulfide consumption during Ag NPs sulfidation has two distinct kinetics. A first rapid stage of Ag sulfidation followed by a slower sulfide consumption. The authors hypothesized that the slow sulfide consumption stage is due to the formation of super-stoichiometric Ag-sulfides, and/or particle-catalyzed sulfide oxidation. In the sediment, S-rich Ag-sulfide particles are observed in the samples exposed to high concentrations of sulfide and/or for an extended time. In anoxic environments, polysulfides can form complexes with Ag(I),57 and can induce the sulfidation of Ag NPs.58,59 The excess of S could then be related to the presence of surface-bound polysulfides, as already observed following the reaction between sulfide and ferric oxyhydroxides.60,61 Another pathway which may have contributed to virtually decreasing Ag/S ratios is the interaction of Ag NPs with other divalent metals and/or metal sulfides.57,62 Indeed, EDXS results show the presence of Fe and Cu for particles with low Ag/S ratios (Fig. 5 and SI-14†). The presence of Fe and Cu was identified only for particles exhibiting an excess of S. For instance, in the sample Ag_0–3 mm_3d, neither Fe nor Cu was identified by EDXS (Fig. SI-14†). Furthermore, elemental distribution analysis revealed that the distribution of Cu and Fe was heterogeneous within the particles, and is concomitant with high S and low Ag signal intensity distributions (Fig. 5). These latter observations support the association of other metal-sulfides with the transformed Ag NPs, and/or the occurrence of solid-state exchange reactions with other divalent metals and the formation of Agx–(Cu/Fe)y–Sn phases.57
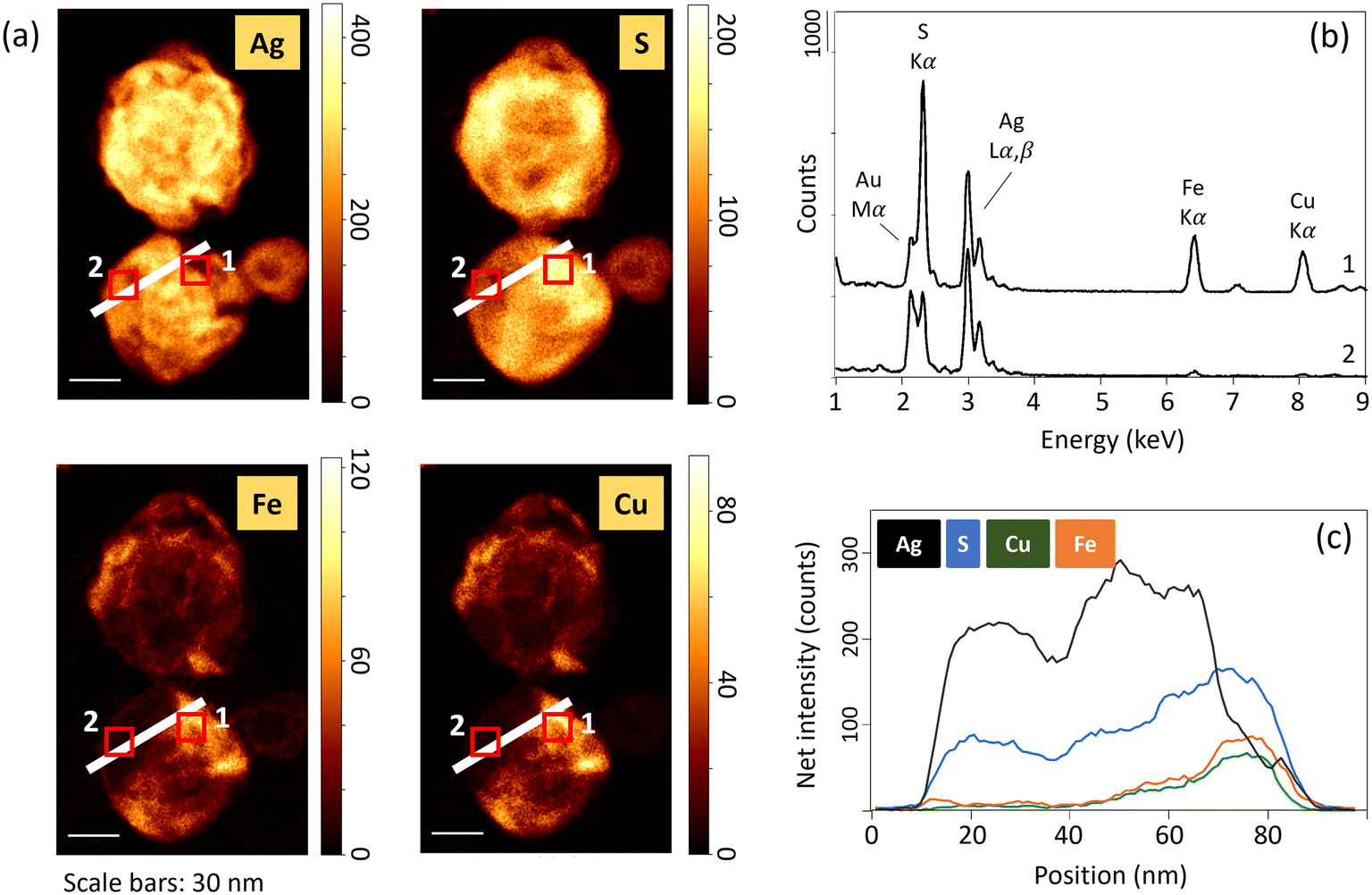 | ||
| Fig. 5 (a) High resolution STEM-EDXS elemental maps of transformed Ag NPs after four weeks exposure at 60 mm depth (Ag_60 mm_4w_a sample). The maps show the distribution of Ag, S, Fe, and Cu. The color bars indicate the signal intensities. (b) EDX spectra extracted from the marked areas (red squares 1 and 2), and (c) net intensity profiles for Ag, S, Cu and Fe, as indicated by the white line in (a). | ||
In laboratory assays, the complete sulfidation of Ag NPs is observed within minutes or hours provided that sufficient sulfide is available.20,41 Although we can't exclude that a minor amount of pristine Ag NPs remains but was not observed on the TEM grids, our results suggest the complete transformation of Ag NPs after three days in the sediment, even at low concentration of sulfide. This result agrees well with a study of Ag NPs exposure in saltwater and freshwater sediments, showing the transformation of Ag NPs into Ag-sulfides after 48 hours at depths where sulfate reduction occurs.28 In paddy soils incubated under anoxic flooded conditions, up to 95% of Ag was also found as Ag2S after two days.63 By contrast, a lower extent of Ag NPs sulfidation was observed in mesocosm wetlands spiked with Ag NPs, even after 18 months, which could be explained by the source of the sulfide and the relatively low S/Ag molar ratio.21 Similarly, lower sulfidation extents were also reported after 28 days in marine sediments64 and in anoxic soils after 30 days.65 Nevertheless, considering the low concentration of ENMs expected in natural environments,4–6 it is likely that sufficient sulfide would be available in sulfate-reducing environments to completely transform Ag NPs into Ag-sulfides.66
3.4. Transformation of ZnO NPs along sediment pore water depth gradients
The sediment columns C3 and C4 were used for the ZnO NPs-TEM grid exposure experiments. After four weeks, neither pristine nor transformed ZnO NPs were identified in the samples inserted at 0–3 mm below the water–sediment interface in the C4 core. In aqueous environments, ZnO NPs dissolution kinetic depends on extrinsic factors such as the pH and the presence of organic ligands.43,44 In a previous study, we showed that 75% of ZnO NPs dissolved within 30 minutes in sediments from the Lake Neusiedl incubated under oxic conditions. ZnO NPs were completely dissolved after 8 hours, followed by the adsorption of Zn onto organic matter and phyllosilicates.23 The absence of ZnO NPs on the TEM grid samples inserted at 0–3 mm can therefore be attributed to the dissolution of ZnO NPs, not followed by the precipitation of secondary Zn-bearing minerals in the vicinity of the pristine ZnO NPs, but rather by Zn adsorption onto sediment particles. By contrast, STEM-EDXS analyses of the sample exposed at 0–3 mm depth for three days in the C3 core reveal the presence of Zn-sulfide precipitates harboring an empty-shell morphology (Fig. SI-15, Table SI-2†). Neither pristine ZnO NPs, nor ZnO cores were identified in this sample. This result indicates the precipitation of ZnS at the surface of the ZnO NPs, followed by the complete dissolution of the ZnO cores. As minute amount of ZnO NPs was loaded onto the TEM grid, trace levels of sulfide naturally present and not detected by the microelectrode could have resulted in the partial sulfidation of ZnO NPs. As for the Ag NPs exposure experiment, the diffusion of sulfide in the surface layer of the sediment during the insertion of the TEM grid holder could also have contributed to the formation of ZnS at this depth. Especially, H2S concentration in the C3 core shows a sharp increase after 6 mm depth and is significantly higher than the concentration measured in the C4 core (Fig. 3), which may have favored the formation of an enriched sulfide zone in the vicinity of the TEM grid. In soils, nanosized ZnS is more soluble than its microsized counterpart.67,68 Moreover, several factors have been shown to influence the dissolution of nanosized ZnS, such as soil properties,69 particle size, and the presence of structural defects.70 Although little is known about the fate of ZnS empty-shell structures in lacustrine sub-oxic sediments, the absence of ZnS in the clay-rich sediment after four weeks exposure at 0–3 mm depth suggests the dissolution of the initially formed ZnS precipitates and/or the ZnO NPs. We also can't exclude the possibility that ZnO NPs and/or ZnS shells detached from the TEM grids after a prolonged exposure time. Nevertheless, NPs were observed in all the other samples exposed for four weeks, reinforcing the hypothesis of a complete dissolution of the NPs at 0–3 mm depth.For the samples exposed at 7–10 mm below the water–sediment interface (ZnO_7–10 mm_3d, ZnO_7–10 mm_4w_a, and ZnO_7–10 mm_4w_b), 100% of the particles identified by STEM-EDXS exhibited empty ZnS shell structures (Fig. 6 and SI-16–SI-19; Table SI-2†). ZnS nanoparticles with a size ranging between 5–50 nm were also observed in the vicinity of the ZnS shells and grouped as clusters, indicating the nucleation and growth of ZnS phases both in the vicinity and on the surface of the ZnO NPs, which completely dissolved after three days. Core–shell structures were also identified in the samples exposed to higher H2S concentration, deeper in the sediment columns (Fig. 6 and SI-16–SI-19†). However, in contrast with the ZnO NPs-TEM grids exposed at 0–3 mm and 7–10 mm depth (i.e., low sulfide concentrations), ZnO cores remained for all the particles identified in the samples exposed at 15–18 mm, 25–30 mm and 60 mm depth (Table SI-2†). Thin and neat shells were observed in the samples exposed at 25–30 mm and 60 mm depth, as compared to the thick and fluffy shells that dominate in the sample exposed at 7–10 mm depth. At 15–18 mm depth, intermediate results are observed, with the persistence of ZnO–ZnS cores–shells and ZnS nanoparticles in their vicinity.
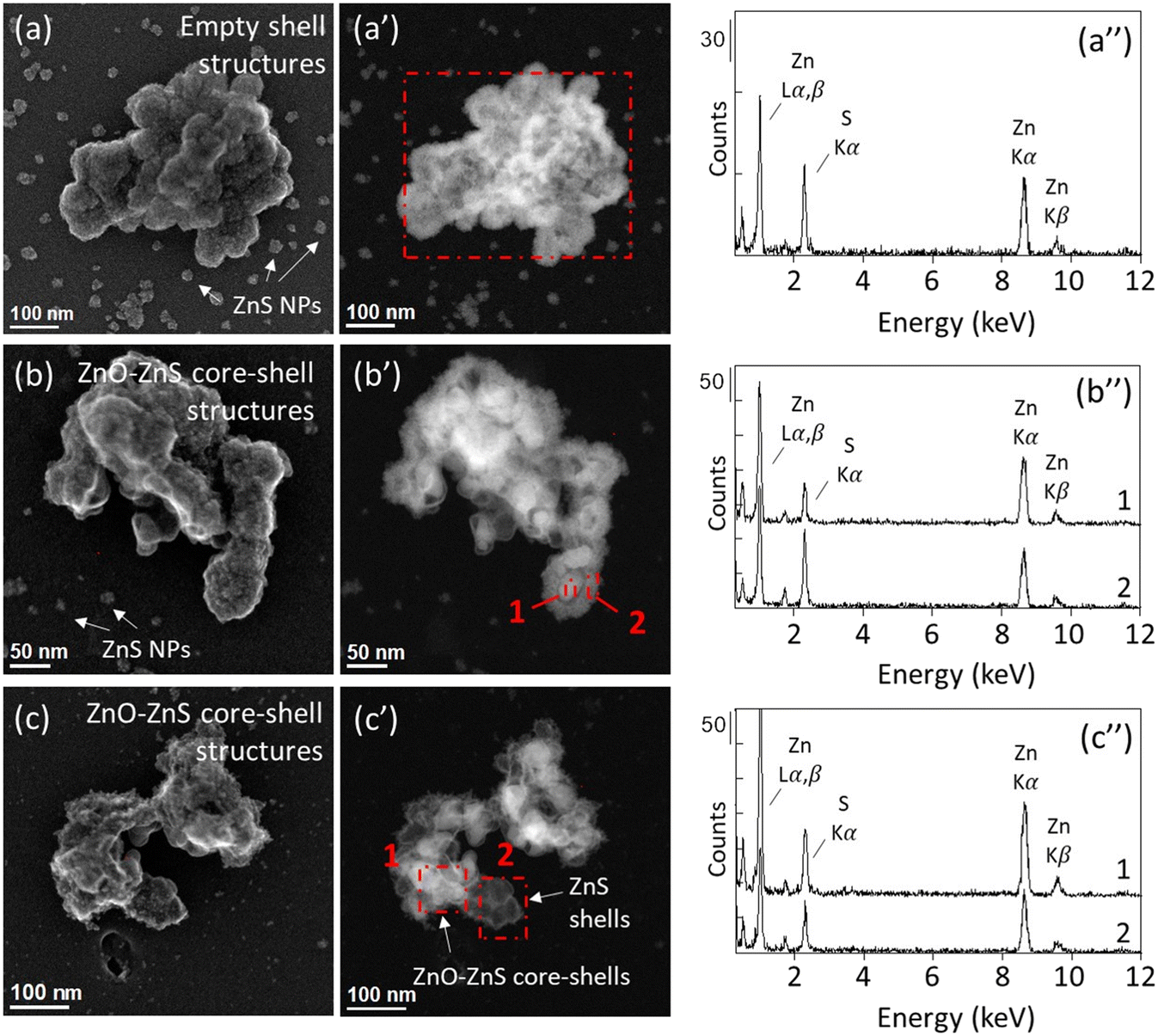 | ||
| Fig. 6 STEM micrographs of ZnO NPs exposed at (a and a′) 7–10 mm depth for three days, (b and b′) at 15–18 mm for four weeks, and (c and c′) at 60 mm for four weeks. (a–c) were acquired in SE mode and (a′–c′) were acquired in HAADF mode. The EDX spectra of the respective aggregates are displayed in (a′′–c′′). The red dotted squares represent the areas scanned for the EDX analyses. (a and a′) ZnO_7–10 mm_3d sample; (b and b′) ZnO_15–18 mm_4w_b sample; (c and c′) ZnO_60 mm_4w_b sample. | ||
Altogether, these results indicate that the sulfidation extent of ZnO NPs is variable along the redox gradient in the sediment. Surprisingly, the lower dissolution and sulfidation extents of ZnO NPs are observed in the deep layers, where sulfide concentration is high. This result contrasts with the kinetic observed in laboratory assays where ZnO NPs were completely transformed into ZnS after 5 days in Na2S solutions, when sulfide was added in excess.18 Nevertheless, the results of this study are consistent with a previous anoxic batch experiment performed with sediments from the Lake Neusiedl, showing that ZnO NP dissolution was inhibited during the first four weeks of incubation, despite available suflide.23 In this latter study, although the high concentration of ZnO NPs likely impacted the proportion of Zn transformed into ZnS, a similar behavior of ZnO NPs was evidenced. However, the morphology of the transformed ZnO NPs and the potential formation of ZnO–ZnS core–shell structures were not elucidated. Here, we provide evidence that the slow dissolution of ZnO NPs is closely related to the concentration of sulfides along the sediment column, and the formation of ZnS shells. At high sulfide concentrations, the dissolution rate of ZnO NPs and subsequent ZnS precipitation are reduced. Considering that a lower saturation index favors particle growth,71 the unlimited availability of sulfide may have led to the precipitation of ZnS and the formation of an impermeable layer of amorphous and/or nanoparticulate ZnS. By contrast, at low sulfide concentration, the nucleation and growth of ZnS particles forming a porous ZnS shell at the surface of the ZnO core may have a limited impact on ZnO dissolution, not preventing Zn diffusion in the aqueous media. Finally, other factors could also have impacted the transformation of ZnO NPs. For example, organic ligands, which are likely to be abundant in the sediment, are known to enhance the dissolution of ZnO NPs43–45 and impact the formation of ZnS precipitates.72 Although the effect of organic compounds on ZnO NP sulfidation kinetic is yet to be determined, the transformation of ZnO NPs could have been impacted by organic ligands, affecting the crystallinity and permeability of the ZnS shells.
4. Conclusions
In this study, we investigated the transformation of Ag NPs and ZnO NPs along sulfate-reducing pore water depth gradients in incubated, undisturbed sediment columns. We show that the transformation of Ag NPs and ZnO NPs varies along the depth gradients and with the exposure time. Especially, we reveal the presence of Ag-sulfides exhibiting various morphologies and Ag:S ratios, and the association of Ag-sulfides with divalent metals (Fe, Cu), indicating complex transformation and interaction mechanisms occurring in the sediment. Moreover, we show that the sulfide concentration and the formation of ZnS shells are important factors controlling the extent of ZnO NP transformation in the sediment. These results are important to consider for the study and prediction of (1) the rate of NP transformation in different aqueous compartments, and (2) the long-term fate of sulfidized Ag and ZnO NPs, yet considered to form poorly soluble Ag2S and ZnS.14,18
Several studies investigated the transformation of NPs in media that are expected to be their main receivers.21,23,69 However, only a few studies investigated the fate of NPs in realistic environmental media at environmentally relevant concentrations of NPs, despite its crucial influence on the transformation rates, nature, and reactivity of the transformation products.27,28,73 In this context, this study demonstrates the advantage of developing experimental methods able to investigate the transformation of NPs under environmentally relevant conditions. In particular, the use of TEM grids to expose NPs to aquatic media is a promising approach. It allows monitoring the chemical and morphological transformation of NPs at the single particle level, for environmentally relevant concentrations of NPs, and for ex situ or in situ exposure scenarios. Ultimately, the results of this work may support the development of OECD guidance and guideline documents for testing the transformation of nanomaterials in aquatic environments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Scientific Center for Optical and Electron Microscopy (ScopeM) of the ETH Zurich for providing access to their microscopes. We thank Brian Sinnet and Wolfgang Obermaier for their support in the laboratories. The three anonymous reviewers are also thanked for their constructive comments on the manuscript. This work is part of the EU-funded project Gov4Nano funded by the European Union's Horizon 2020 Research and Innovation Program under Grant Agreement 814401.References
- A. Becheri, M. Dürr, P. Lo Nostro and P. Baglioni, Synthesis and characterization of zinc oxide nanoparticles: application to textiles as UV-absorbers, J. Nanopart. Res., 2008, 10, 679–689 CrossRef CAS.
- O. V. Salata, Applications of nanoparticles in biology and medicine, J. Nanobiotechnol., 2004, 2, 3 CrossRef PubMed.
- M. D. Newman, M. Stotland and J. I. Ellis, The safety of nanosized particles in titanium dioxide–and zinc oxide–based sunscreens, J. Am. Acad. Dermatol., 2009, 61, 685–692 CrossRef CAS PubMed.
- B. Giese, F. Klaessig, B. Park, R. Kaegi, M. Steinfeldt, H. Wigger, A. von Gleich and F. Gottschalk, Risks, Release and Concentrations of Engineered Nanomaterial in the Environment, Sci. Rep., 2018, 8, 1565 CrossRef PubMed.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Modeled Environmental Concentrations of Engineered Nanomaterials (TiO2, ZnO, Ag, CNT, Fullerenes) for Different Regions, Environ. Sci. Technol., 2009, 43, 9216–9222 CrossRef CAS PubMed.
- N. C. Mueller and B. Nowack, Exposure modeling of engineered nanoparticles in the environment, Environ. Sci. Technol., 2008, 42(12), 4447–4453 CrossRef CAS PubMed.
- J. Wang, M. M. Nabi, S. K. Mohanty, A. N. Afrooz, E. Cantando, N. Aich and M. Baalousha, Detection and quantification of engineered particles in urban runoff, Chemosphere, 2020, 248, 126070 CrossRef CAS PubMed.
- J. Wang, M. M. Nabi, M. Erfani, E. Goharian and M. Baalousha, Identification and quantification of anthropogenic nanomaterials in urban rain and runoff using single particle-inductively coupled plasma-time of flight-mass spectrometry, Environ. Sci.: Nano, 2022, 9, 714–729 RSC.
- A. P. Gondikas, F. von der Kammer, R. B. Reed, S. Wagner, J. F. Ranville and T. Hofmann, Release of TiO2 Nanoparticles from Sunscreens into Surface Waters: A One-Year Survey at the Old Danube Recreational Lake, Environ. Sci. Technol., 2014, 48(10), 5415–5422 CrossRef CAS PubMed.
- A. E. Pradas del Real, H. Castillo-Michel, R. Kaegi, C. Larue, W. de Nolf, J. Reyes-Herrera, R. Tucoulou, N. Findling, E. Salas-Colera and G. Sarret, Searching for relevant criteria to distinguish natural vs. anthropogenic TiO2 nanoparticles in soils, Environ. Sci.: Nano, 2018, 5, 2853–2863 RSC.
- S. Taskula, L. Stetten, F. von der Kammer and T. Hofmann, Platinum Nanoparticle Extraction, Quantification, and Characterization in Sediments by Single-Particle Inductively Coupled Plasma Time-of-Flight Mass Spectrometry, Nanomaterials, 2022, 12, 3307 CrossRef CAS PubMed.
- D. J. Spurgeon, E. Lahive and C. L. Schultz, Nanomaterial Transformations in the Environment: Effects of Changing Exposure Forms on Bioaccumulation and Toxicity, Small, 2020, 16(36), 2000618 CrossRef CAS PubMed.
- J. R. Lead, G. E. Batley, P. J. J. Alvarez, M. N. Croteau, R. D. Handy, M. J. McLaughlin, J. D. Judy and K. Schirmer, Nanomaterials in the environment: Behavior, fate, bioavailability, and effects-An updated review, Environ. Toxicol. Chem., 2018, 37(8), 2029–2063 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown Jr., Environmental transformations of silver nanoparticles: impact on stability and toxicity, Environ. Sci. Technol., 2012, 46(13), 6900–6914 CrossRef CAS PubMed.
- G. Lee, B. Lee and K.-T. Kim, Mechanisms and effects of zinc oxide nanoparticle transformations on toxicity to zebrafish embryos, Environ. Sci.: Nano, 2021, 8, 1690–1700 RSC.
- R. Grillo, A. H. Rosa and L. F. Fraceto, Engineered nanoparticles and organic matter: A review of the state-of-the-art, Chemosphere, 2015, 119, 608–619 CrossRef CAS PubMed.
- S. K. Misra, A. Dybowska, D. Berhanu, S. N. Luoma and E. Valsami-Jones, The complexity of nanoparticle dissolution and its importance in nanotoxicological studies, Sci. Total Environ., 2012, 438, 225–232 CrossRef CAS PubMed.
- R. Ma, C. Levard, F. M. Michel, G. E. Brown Jr. and G. V. Lowry, Sulfidation Mechanism for Zinc Oxide Nanoparticles and the Effect of Sulfidation on Their Solubility, Environ. Sci. Technol., 2013, 47(6), 2527–2534 CrossRef CAS PubMed.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown Jr., Sulfidation Processes of PVP-Coated Silver Nanoparticles in Aqueous Solution: Impact on Dissolution Rate, Environ. Sci. Technol., 2011, 45(12), 5260–5266 CrossRef CAS PubMed.
- J. Liu, K. G. Pennell and R. H. Hurt, Kinetics and Mechanisms of Nanosilver Oxysulfidation, Environ. Sci. Technol., 2011, 45(17), 7345–7353 CrossRef CAS PubMed.
- G. V. Lowry, B. P. Espinasse, A. R. Badireddy, C. J. Richardson, B. C. Reinsch, L. D. Bryant, A. J. Bone, A. Deonarine, S. Chae, M. Therezien, B. P. Colman, H. Hsu-Kim, E. S. Bernhardt, C. W. Matson and M. R. Wiesner, Long-Term Transformation and Fate of Manufactured Ag Nanoparticles in a Simulated Large Scale Freshwater Emergent Wetland, Environ. Sci. Technol., 2012, 46, 7027–7036 CrossRef CAS PubMed.
- J. Wielinski, A. Gogos, A. Voegelin, C. Müller, E. Morgenroth and R. Kaegi, Transformation of Nanoscale and Ionic Cu and Zn during the Incineration of Digested Sewage Sludge (Biosolids), Environ. Sci. Technol., 2019, 53, 11704–11713 CrossRef CAS PubMed.
- L. Stetten, T. Hofmann, O. Proux, G. Landrot, R. Kaegi and F. von der Kammer, Transformation of zinc oxide nanoparticles in freshwater sediments under oxic and anoxic conditions, Environ. Sci.: Nano, 2022, 9, 4255–4267 RSC.
- C. Levard, M. Le Bars, T. Formentini, S. Legros and E. Doelsch, Organic waste-borne ZnS nanoparticles: The forgotten ones, Environ. Pollut., 2022, 308, 119629 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Fate and transformation of silver nanoparticles in urban wastewater systems, Water Res., 2013, 47(12), 3866–3877 CrossRef CAS PubMed.
- R. D. Kent, J. G. Oser and P. J. Vikesland, Controlled Evaluation of Silver Nanoparticle Sulfidation in a Full-Scale Wastewater Treatment Plant, Environ. Sci. Technol., 2014, 48(15), 8564–8572 CrossRef CAS PubMed.
- R. D. Kent and P. J. Vikesland, Dissolution and Persistence of Copper-Based Nanomaterials in Undersaturated Solutions with Respect to Cupric Solid Phases, Environ. Sci. Technol., 2016, 50(13), 6772–6781 CrossRef CAS PubMed.
- M. Khaksar, D. F. Jolley, R. Sekine, K. Vasilev, B. Johannessen, E. Donner and E. Lombi, In Situ Chemical Transformations of Silver Nanoparticles along the Water–Sediment Continuum, Environ. Sci. Technol., 2015, 49(1), 318–325 CrossRef CAS PubMed.
- OECD GD 3018, Guidance document for the testing of dissolution and dispersion stability of nanomaterials and the 554 use of the data for further environmental testing and assessment strategies, 2020, Available at: https://www.oecd.org/chemi-555calsafety/testing/series-testing-assessment-publications-number.htm.
- K. Rasmussen, H. Rauscher, P. r. Kearns, M. González and J. Riego Sintes, Developing OECD test guidelines for regulatory testing of nanomaterials to ensure mutual acceptance of test data, Regul. Toxicol. Pharmacol., 2019, 104, 74–83 CrossRef PubMed.
- OECD (2015a) Dossier on Zinc Oxide, Series on the Safety of Manufactured Nanomaterials, No. 52, OECD, Paris, France, 2015, [(accessed on February 2023)], Available online: https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2015)15/ANN10&docLanguage=En.
- OECD (2015b) Dossier on silver nanoparticles, Series on the Safety of Manufactured Nanomaterials, No. 53, OECD, Paris, France, 2015, [(accessed on February 2023)], Available online: https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2015)16/PART1&docLanguage=En.
- R. Kaegi, A. Voegelin, B. Sinnet, S. Zuleeg, H. Hagendorfer, M. Burkhardt and H. Siegrist, Behavior of Metallic Silver Nanoparticles in a Pilot Wastewater Treatment Plant, Environ. Sci. Technol., 2011, 45, 3902–3908 CrossRef CAS PubMed.
- J. D. Cline, Spectrophotometric determination of hydrogen sulfide in natural waters, Limnol. Oceanogr., 1969, 14, 454–458 CrossRef CAS.
- A. E. Williams-Jones, R. J. Bowell and A. A. Migdisov, Gold in Solution, Elements, 2009, 5(5), 281–287 CrossRef CAS.
- D. Vlassopoulos and S. A. Wood, Gold Speciation in Natural Waters: I. Solubility and Hydrolysis Reactions of Gold in Aqueous Solution, Geochim. Cosmochim. Acta, 1990, 54(1), 3–12 CrossRef CAS.
- N. P. Revsbech, Simple Sensors That Work in Diverse Natural Environments: The Micro-Clark Sensor and Biosensor Family, Sens. Actuators, B, 2021, 329, 129168 CrossRef CAS.
- M. Kühl, C. Steuckart, G. Eickert and P. Jeroschewski, A H2S Microsensor for Profiling Biofilms and Sediments: Application in an Acidic Lake Sediment, Aquat. Microb. Ecol., 1998, 15, 201–209 CrossRef.
- P. Jeroschewski, C. Steuckart and M. Kühl, An Amperometric Microsensor for the Determination of H2S in Aquatic Environments, Anal. Chem., 1996, 68(24), 4351–4357 CrossRef CAS.
- F. de la Peña, E. Prestat, V. T. Fauske, P. Burdet, J. Lähnemann, P. Jokubauskas, T. Furnival, M. Nord, T. Ostasevicius, K. E. MacArthur, D. N. Johnstone, M. Sarahan, J. Taillon, T. Aarholt, Pquinn-Dls, V. Migunov, A. Eljarrat, J. Caron, C. Francis, T. Nemoto, T. Poon, S. Mazzucco, actions-user, N. Tappy, N. Cautaerts, S. Somnath, T. Slater, M. Walls, F. Winkler and H. W. Ånes, Hyperspy/hyperspy: Release V1.7.2, Zenodo, September 18, 2022 Search PubMed.
- B. Thalmann, A. Voegelin, E. Morgenroth and R. Kaegi, Effect of humic acid on the kinetics of silver nanoparticle sulfidation, Environ. Sci.: Nano, 2016, 3(1), 203–212 RSC.
- D. He, S. Garg, Z. Wang, L. Li, H. Rong, X. Ma, G. Li, T. An and T. D. Waite, Silver sulfide nanoparticles in aqueous environments: formation, transformation and toxicity, Environ. Sci.: Nano, 2019, 6(6), 1674–1687 RSC.
- S.-W. Bian, I. A. Mudunkotuwa, T. Rupasinghe and V. H. Grassian, Aggregation and Dissolution of 4 Nm ZnO Nanoparticles in Aqueous Environments: Influence of PH, Ionic Strength, Size, and Adsorption of Humic Acid, Langmuir, 2011, 27, 6059–6068 CrossRef CAS PubMed.
- I. A. Mudunkotuwa, T. Rupasinghe, C.-M. Wu and V. H. Grassian, Dissolution of ZnO Nanoparticles at Circumneutral pH: A Study of Size Effects in the Presence and Absence of Citric Acid, Langmuir, 2012, 28, 396–403 CrossRef CAS PubMed.
- R. F. Domingos, Z. Rafiei, C. E. Monteiro, M. A. K. Khan and K. J. Wilkinson, Agglomeration and dissolution of zinc oxide nanoparticles: role of pH, ionic strength and fulvic acid, Environ. Chem., 2013, 10, 306 CrossRef CAS.
- N. Wang, T. Tong, M. Xie and J.-F. Gaillard, Lifetime and dissolution kinetics of zinc oxide nanoparticles in aqueous media, Nanotechnology, 2016, 27, 324001 CrossRef PubMed.
- S. Yamabi and H. Imai, Growth Conditions for Wurtzite Zinc Oxide Films in Aqueous Solutions, J. Mater. Chem., 2002, 12(12), 3773–3778 RSC.
- M. Bundschuh, J. Filser, S. Lüderwald, M. S. McKee, G. Metreveli, G. E. Schaumann, R. Schulz and S. Wagner, Nanoparticles in the environment: where do we come from, where do we go to?, Environ. Sci. Eur., 2018, 30, 6 CrossRef PubMed.
- D. Fussmann, H. Babková, R. Peticzka, A. Maier, G. Arp, R. Daniel and P. Meister, Authigenic Formation of Ca–Mg Carbonates in the Shallow Alkaline Lake Neusiedl, Austria, Biogeosciences, 2020, 17, 2085–2106 CrossRef CAS.
- A. J. E. von Hoyningen-Huene, D. Schneider, D. Fussmann, A. Reimer, G. Arp and R. Daniel, Bacterial succession along a sediment porewater gradient at Lake Neusiedl in Austria, Sci. Data, 2019, 6, 163 CrossRef PubMed.
- M. Taillefert, S. Neuhuber and G. Bristow, The effect of tidal forcing on biogeochemical processes in intertidal salt marsh sediments, Geochem. Trans., 2007, 8, 6 CrossRef PubMed.
- W. Zhang, B. Xiao and T. Fang, Chemical Transformation of Silver Nanoparticles in Aquatic Environments: Mechanism, Morphology and Toxicity, Chemosphere, 2018, 191, 324–334 CrossRef CAS PubMed.
- B. Kim, C.-S. Park, M. Murayama and M. F. Hochella, Discovery and Characterization of Silver Sulfide Nanoparticles in Final Sewage Sludge Products, Environ. Sci. Technol., 2010, 44, 7509–7514 CrossRef CAS PubMed.
- H. Auvinen, R. Kaegi, D. P. L. Rousseau and G. Du Laing, Fate of Silver Nanoparticles in Constructed Wetlands—a Microcosm Study, Water, Air, Soil Pollut., 2017, 228, 97 CrossRef.
- O. Choi, T. E. Clevenger, B. Deng, R. Y. Surampalli, L. Ross Jr. and Z. Hu, Role of sulfide and ligand strength in controlling nanosilver toxicity, Water Res., 2009, 43, 1879–1886 CrossRef CAS PubMed.
- R. Chen, N. T. Nuhfer, L. Moussa, H. R. Moprris and P. M. Whitmore, Silver sulfide nanoparticle assembly obtained by reacting an assembled silver nanoparticle template with hydrogen sulfide gas, Nanotechnology, 2008, 19(45), 1–11 CrossRef PubMed.
- R. A. Bell and J. R. Kramer, Structural chemistry and geochemistry of silver-sulfur compounds: Critical review, Environ. Toxicol. Chem., 1999, 18(1), 9–22 CAS.
- J. Zeng, J. Tao, D. Su, Y. Zhu, D. Qin and Y. Xia, Selective Sulfuration at the Corner Sites of a Silver Nanocrystal and Its Use in Stabilization of the Shape, Nano Lett., 2011, 11(7), 3010–3015 CrossRef CAS PubMed.
- B. A. D. Thalmann, Transformations of Silver Nanoparticles in Urban Wastewater and Water Systems, PhD dissertation, ETH Zurich, 2015 Search PubMed.
- K. Hellige, K. Pollok, P. Larese-Casanova, T. Behrends and S. Peiffer, Pathways of ferrous iron mineral formation upon sulfidation of lepidocrocite surfaces, Geochim. Cosmochim. Acta, 2012, 81, 69–81 CrossRef CAS.
- M. Wan, A. Shchukarev, R. Lohmayer, B. Planer-Friedrich and S. Peiffer, Occurrence of surface polysulfides during the interaction between ferric (hydr)oxides and aqueous sulfide, Environ. Sci. Technol., 2014, 48, 5076–5084 CrossRef CAS PubMed.
- B. Thalmann, A. Voegelin, B. Sinnet, E. Morgenroth and R. Kaegi, Sulfidation Kinetics of Silver Nanoparticles Reacted with Metal Sulfides, Environ. Sci. Technol., 2014, 48, 4885–4892 CrossRef CAS PubMed.
- M. Li, P. Wang, F. Dang and D.-M. Zhou, The Transformation and Fate of Silver Nanoparticles in Paddy Soil: Effects of Soil Organic Matter and Redox Conditions, Environ. Sci.: Nano, 2017, 4(4), 919–928 RSC.
- H. Wang, K. T. Ho, K. G. Scheckel, F. Wu, M. G. Cantwell, D. R. Katz, D. B. Horowitz, W. S. Boothman and R. M. Burgess, Toxicity, Bioaccumulation, and Biotransformation of Silver Nanoparticles in Marine Organisms, Environ. Sci. Technol., 2014, 48(23), 13711–13717 CrossRef CAS PubMed.
- Y. Hashimoto, S. Takeuchi, S. Mitsunobu and Y.-S. Ok, Chemical Speciation of Silver (Ag) in Soils under Aerobic and Anaerobic Conditions: Ag Nanoparticles vs. Ionic Ag, J. Hazard. Mater., 2017, 322, 318–324 CrossRef CAS PubMed.
- C. H. S. J. Chou, Hydrogen Sulfide: Human Health Aspects, World Health Organization, Geneva, 2003 Search PubMed.
- A. Voegelin, O. Jacquat, S. Pfister, K. Barmettler, A. C. Scheinost and R. Kretzschmar, Time-dependent changes of zinc speciation in four soils contaminated with zincite or sphalerite, Environ. Sci. Technol., 2011, 45(1), 255–261 CrossRef CAS PubMed.
- T. C. Robson, C. B. Braungardt, J. Rieuwerts and P. Worsfold, Cadmium contamination of agricultural soils and crops resulting from sphalerite weathering, Environ. Pollut., 2014, 184, 283–289 CrossRef CAS PubMed.
- M. Le Bars, S. Legros, C. Levard, C. Chevassus-Rosset, M. Montes, M. Tella, D. Borschneck, A. Guihou, B. Angeletti and E. Doelsch, Contrasted fate of zinc sulfide nanoparticles in soil revealed by a combination of X-ray absorption spectroscopy, diffusive gradient in thin films and isotope tracing, Environ. Pollut., 2022, 292, 118414 CrossRef CAS PubMed.
- J. R. Eskelsen, J. Xu, M. Chiu, J.-W. Moon, B. Wilkins, D. E. Graham, B. Gu and E. M. Pierce, Influence of Structural Defects on Biomineralized ZnS Nanoparticle Dissolution: An in-Situ Electron Microscopy Study, Environ. Sci. Technol., 2018, 52(3), 1139–1149 CrossRef CAS PubMed.
- A. Mersmann, Crystallization and precipitation, Chem. Eng. Process.: Process Intesif., 1999, 38(4–6), 345–353 CrossRef CAS.
- A. Deonarine, B. L. T. Lau, G. R. Aiken, J. N. Ryan and H. Hsu-Kim, Effects of Humic Substances on Precipitation and Aggregation of Zinc Sulfide Nanoparticles, Environ. Sci. Technol., 2011, 45(8), 3217–3223 CrossRef CAS PubMed.
- L. Stetten, A. Mackevica, N. Tepe, T. Hofmann and F. von der Kammer, Towards Standardization for Determining Dissolution Kinetics of Nanomaterials in Natural Aquatic Environments: Continuous Flow Dissolution of Ag Nanoparticles, Nanomaterials, 2022, 12(3), 519 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3en00550j |
| ‡ Present address: Biosciences Division, Argonne National Laboratory, Lemont, IL, USA. |
| This journal is © The Royal Society of Chemistry 2024 |