
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCritical insights into data curation and label noise for accurate prediction of aerobic biodegradability of organic chemicals†
Paulina
Körner
 a,
Juliane
Glüge
*a,
Stefan
Glüge
b and
Martin
Scheringer
a
a,
Juliane
Glüge
*a,
Stefan
Glüge
b and
Martin
Scheringer
a
aInstitute of Biogeochemistry and Pollutant Dynamics, ETH Zürich, 8092 Zürich, Switzerland. E-mail: juliane.gluege@usys.ethz.ch
bInstitute for Computational Life Science, ZHAW, 8820 Wädenswil, Switzerland
First published on 26th September 2024
Abstract
The focus of this work is to enhance state-of-the-art Machine Learning (ML) models that can predict the aerobic biodegradability of organic chemicals through a data-centric approach. To do that, an already existing dataset that was previously used to train ML models was analyzed for mismatching chemical identifiers and data leakage between test and training set and the detected errors were corrected. Chemicals with high variance between study results were removed and an XGBoost was trained on the dataset. Despite extensive data curation, only marginal improvement was achieved in the classification model's performance. This was attributed to three potential reasons: (1) a significant number of data labels were noisy, (2) the features could not sufficiently represent the chemicals, and/or (3) the model struggled to learn and generalize effectively. All three potential reasons were examined and point (1) seemed to be the most decisive one that prevented the model from generating more accurate results. Removing data points with possibly noisy labels by performing label noise filtering using two other predictive models increased the classification model's balanced accuracy from 80.9% to 94.2%. The new classifier is therefore better than any previously developed classification model for ready biodegradation. The examination of the key characteristics (molecular weight of the substances, proportion of halogens present and distribution of degradation labels) and the applicability domain indicate that no/not a large share of difficult-to-learn substances has been removed in the label noise filtering, meaning that the final model is still very robust.
Environmental significanceResistance to environmental degradation is one of the characteristics of hazardous substances. Our newly developed yes/no classification model for ready biodegradation is currently the most accurate model that is available for organic chemicals and will enable a better prediction of ready biodegradation. We also present a list of substances that is called “curated_removed” with “noisy labels” (uncertain degradability); these substances should no longer be used to train and test degradation models. Instead, these substances should be tested further experimentally to elucidate their biodegradability behavior. |
1 Introduction
To effectively mitigate exposure to harmful substances, it is crucial to understand the properties of the substances that can reach the environment. Potentially harmful substances can be identified by considering inter alia persistence, aquatic and human toxicity, bioconcentration, mobility, ozone depletion, and global warming potential.1,2 If persistent substances are continuously emitted into the environment, the environmental concentration of these substances increases. Even if these emissions are stopped, the concentration will only slowly decrease.3,4 Therefore, persistent substances raise significant concerns due to their unpredictable long-term effects.5The distinction as to whether a substance is persistent or not is in the first step at a regulatory level often assessed by using ready-biodegradability tests (RBT). Biodegradation is an important degradation mechanism for chemicals in the environment; it refers to the capacity of a substance to be broken down and transformed into simpler compounds by microorganisms.6 If a chemical passes the RBT, it will likely be readily biodegradable (RB) in the environment. Conversely, chemicals that do not pass the RBT are likely to be not readily biodegradable (NRB) in the environment.3,7 However, it has also been shown that the test results depend on several factors, including the test procedure, the initial concentration of the substrate, and the activity and adaptation of the microbial population.7 Additionally, environmental conditions such as temperature, pH, and oxygen levels can impact the test results.7 Nevertheless, RBT are established as screening tests in many regulatory frameworks and form the basis for the assessment of persistence.3,8,9 Models that can predict the biodegradation of substances have also been developed in the past as cheaper and less time-consuming alternatives to experimental studies.1,8,10–21 A summary of the previous work on models on ready biodegradability is provided in Section 2.
Recently, Huang and Zhang20 used a machine learning (ML) approach to build both, a classification and a regression model, to predict the ready biodegradability of organic substances. They gathered the largest dataset so far with 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750 samples for 6032 substances for regression and 6139 substances for classification. The classification dataset was based on the regression data but enhanced with data from Lunghini et al.19 The original dataset was obtained through the eChemPortal, which accesses data from the Japan Chemicals Collaborative Knowledge (J-CHECK) database, Canadian Categorization Results (CCR), the European Chemicals Agency (ECHA) database and the Organisation for Economic Cooperation and Development (OECD) Existing Chemicals Screening Information Data Sets (SIDS).22 Seven molecular fingerprints (FPs) were tested in Huang and Zhang20 as input features describing the chemicals. The addition of features containing information about chemical speciation was also examined. In total, 14 ML algorithms were tested, and the best results were achieved with the Molecular Access System key (MACCS key) as input features and an eXtreme Gradient Boosting (XGBoost) model. The XGBClassifier achieved a balanced accuracy of 84.9%. Adding further features containing information on chemical speciation, meaning whether or not the chemical is charged, improved the balanced accuracy to 87.6%. Huang and Zhang20 used a model-centric approach with an emphasis on feature and model selection and hyperparameter tuning rather than data quality.23–25
750 samples for 6032 substances for regression and 6139 substances for classification. The classification dataset was based on the regression data but enhanced with data from Lunghini et al.19 The original dataset was obtained through the eChemPortal, which accesses data from the Japan Chemicals Collaborative Knowledge (J-CHECK) database, Canadian Categorization Results (CCR), the European Chemicals Agency (ECHA) database and the Organisation for Economic Cooperation and Development (OECD) Existing Chemicals Screening Information Data Sets (SIDS).22 Seven molecular fingerprints (FPs) were tested in Huang and Zhang20 as input features describing the chemicals. The addition of features containing information about chemical speciation was also examined. In total, 14 ML algorithms were tested, and the best results were achieved with the Molecular Access System key (MACCS key) as input features and an eXtreme Gradient Boosting (XGBoost) model. The XGBClassifier achieved a balanced accuracy of 84.9%. Adding further features containing information on chemical speciation, meaning whether or not the chemical is charged, improved the balanced accuracy to 87.6%. Huang and Zhang20 used a model-centric approach with an emphasis on feature and model selection and hyperparameter tuning rather than data quality.23–25
In contrast, the Data-Centric Artificial Intelligence (DCAI) paradigm that has emerged in recent years shifts the focus towards the systematic design, engineering, and continuous improvement of data to build robust and efficient ML models. This strong focus on data quality ensures that ML models generalize better, making them more effective tools for real-world applications.23 Refining data includes enhancing the quality of individual data points and the dataset in total. Even though the model-centric and data-centric approaches are often contrasted, it is important to emphasize their complementary nature. Both paradigms should be combined to build robust ML-based systems.23
With the current paper, we intend to improve the ML model of Huang and Zhang20 by first taking a data-centric and then a model-centric approach. In particular, we want to answer the question of how important correct representations of chemical structures (Simplified Molecular Input Line Entry Specification (SMILES)) are for the model and whether it is possible to bring the model to a balanced accuracy of over 90% by improving SMILES alone. If this is not possible, the aim is to find out what is preventing the model from making better predictions – too much noise in the data labels themselves, features that cannot adequately represent the chemicals, or whether the model cannot generalize well enough. Noisy labels refer here to mislabeled or inaccurately labeled instances in the training and test datasets.26,27 To investigate these points, first, the dataset published by Huang and Zhang20 is analyzed, and all SMILES are assessed to determine whether they match the provided Chemical Abstracts Service Registry Number™ (CAS RN™). Follow-up steps include assessing the data labels and critically investigating the test and training sets of the ML model. Finally, a model-centric approach is applied to examine if other ML algorithms are more suitable for the curated dataset.
2 Previous work
All models with their reported accuracies, sensitivities and specificities are given in Tables 1 and 2.| Model | Dataset size | Balanced accuracy | Sensitivity | Specificity |
|---|---|---|---|---|
| Howard et al. (1992)10 (non-linear) | 264 | |||
| Test set | 7.4% | 88.8%* | — | — |
|
||||
| Boethling et al. (1994)1 (non-linear) | 295 | |||
| Training set | — | 93.2%* | — | — |
|
||||
| Loonen et al. (1999)11 (with fragment interactions) | 894 | |||
| Test set | 25% | 89%* | — | — |
|
||||
| Tunkel et al. (2000)12 (linear) | 884 | |||
| Validation set | 33.3% | 74.9%* | — | — |
|
||||
| Cheng et al. (2012)13 | 1440 | |||
| Test set (GASVM-kNN) | 11.4% | 81.9% | 72.6% | 91.2 |
| External test set (GASVM-kNN) | 27 | 53.8% | 25.0% | 82.6% |
| External test set (consensus model) | 27 | 100% | 100% | 100% |
|
||||
| Mansouri et al. (2013)14 (consensus II) | 1055 | |||
| Test set | 20% | 91%† | 89%† | 94%† |
| External test set | 670 | 87%‡ | 81%‡ | 94%‡ |
|
||||
| Cao and Leung (2014)15 | 1055 | |||
| Test set | 20% | 86.0% | 77% | 93% |
| External test set | 670 | 83.5% | 74% | 93% |
|
||||
| Lombardo et al. (2014)16 | 728 | |||
| Test set | 20% | 82.1% | 87.3% | 76.9& |
| External test set | 874 | 78.4% | 73.1% | 83.6% |
|
||||
| Blay et al. (2016)17 (ANN) | 130 | |||
| Test set | 20% | 91.5% | 94.1% | 88.9% |
|
||||
| Zhan et al. (2017)18 (NBC) | 1055 | |||
| Test set | 20% | 83.8% | 86.1% | 81.5% |
| External test set | 670 | 82.6% | 79.6% | 85.6% |
|
||||
| Lunghini et al. (2020)19 | 3146 | |||
| Test set | 30% | 81%* | — | — |
| External test | 362 | 75%* | 65% | 85% |
|
||||
| Huang and Zhang (2022)20 | 6139 | |||
| Test set | 20% | 84.9% | 89.0% | 80.9% |
| Test set with chemical speciation | 20% | 87.6% | 87.8% | 87.4% |
|
||||
| Yin et al. (2023)21 | 1928 | |||
| Test set | 26% | 87.3% | 94% | 72% |
| Model | Dataset size | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|
| a Signifies balanced accuracy. | ||||
| BIOWIN1 | ||||
| Train | 295 | 89.5% | 97.3% | 76.1% |
| External test set (MITI) | 884 | 65.4% | 92.7% | 44.3% |
| External test set (premanufacture notices (PMN)) | 305 | 54% | 85% | 44% |
|
||||
| BIOWIN2 | ||||
| Train | 295 | 93.2% | 97.3% | 86.2% |
| External test set (MITI) | 884 | 67.5% | 86.0% | 53.3% |
| External test set (PMN) | 305 | 67% | 78% | 63% |
|
||||
| BIOWIN5 | ||||
| Test | 295 | 81.4% | 80.2% | 82.3% |
| External test set (PMN) | 305 | 83% | 82% | 83% |
|
||||
| BIOWIN6 | ||||
| Test set | 295 | 80.7% | 78.6% | 82.3% |
| External test set (PMN) | 305 | 83% | 72% | 87% |
|
||||
| VEGA | ||||
| Test set | 146 | 81.7% | 87.3% | 76.9% |
| External test set | 491 | 80.7% | 75.6% | 90.7% |
|
||||
| OPERA | ||||
| Test set | 411 | 79% | 81% | 77% |
2.1 Scientific work
Howard et al. (1992)10 built linear and nonlinear classification models using 264 compounds and 35 molecular substructures. The models achieved an accuracy (number of correct substances by number of total substances) of 81.5% (linear) and 88.8% (nonlinear) on the validation set, respectively.Boethling et al. (1994)1 continued the work of Howard et al. (1992)10 and built linear and nonlinear classification models using 295 compounds and 36 molecular substructures plus molecular weight. The modeling approach was the same as in Howard et al. (1992), just using slightly different molecular substructures. No validation set was created, therefore the performance of the two models was only given for the training set. The linear model achieved an accuracy of 89.5%, the nonlinear model an accuracy of 93.2% on the training set. The models of Boethling et al. (1994)1 were used later on for BIOWIN1 and BIOWIN2 (see Section 2.2).
Loonen et al. (1999)11 trained models on a dataset containing 894 compounds tested under the Ministry of International Trade and Industry of Japan (MITI) protocol. The chemicals were characterized by a set of 127 predefined structural fragments. Partial least squares (PLS) discriminant analysis was used for the model development. The authors pointed out that hydroxy, ester, and acid groups that were present were easily degraded, while aromatic rings and halogen substituents were not conducive to biodegradation. The average percentage of correct predictions from four external validation studies was 83%. However, no predictions were made for <10% of the substances because the calculated scores were in the borderline area between readily and not readily biodegradable. Model optimization by including fragment interactions improved the model predicting capabilities to 89%.
Tunkel et al. (2000)12 refitted the molecular substructures of Boethling et al. (1994)1 to 884 compounds tested under the MITI protocol. Two-third of the compounds were used for the training set, one-third for the validation set. Again, a linear and non-linear model were developed. The linear model had an accuracy on the validation set of 74.9%, the nonlinear model an accuracy of 73.6%, respectively. The models of Tunkel et al. (2000)12 were used later on for BIOWIN5 and BIOWIN6 (see Section 2.2).
Cheng et al. (2012)13 trained models on a dataset containing 1440 compounds tested under the MITI protocol. Different features and molecular fingerprints were used to construct Support Vector Machines (SVM), k-Nearest Neighbors (kNN), Naive Bayes (NB), and Decision Tree (DT). The best model (SVM with genetic algorithm – GASVM-kNN) achieved a balanced accuracy of 81.9% in 5-fold Cross-Validation (CV). The best seven combinations of models and features and a consensus model were also tested on 27 new chemicals, which were experimentally tested for their biodegradability under the Japanese MITI test protocol. The consensus model and two of the other models predicted the test results of all 27 substances 100% correctly.13 In contrast, the formerly best model (GASVM-kNN) only achieved a balanced accuracy of 53.8%.
Mansouri et al. (2013) trained kNN, Partial Least Squares Discriminant Analysis (PLSDA), and SVM models on a dataset of 1055 experimental biodegradation data points. The dataset originated from the National Institute of Technology and Evaluation of Japan (NITE) and underwent thorough data screening and improvement. Additionally, an external test set of 670 substances was created based on data from Cheng et al. (2012)13 and the Canadian DSL database. All three models (kNN, PLSDA, and SVM) performed similarly well. Mansouri et al. (2013) created two consensus models based on the three models. The first consensus model assigned each substance the most common label predicted from the three models. The second consensus model only assigned a class to a substance if the three models agreed on one label. The second consensus model performed best, achieving an accuracy of 91% and 87% on the test and external test set, respectively. However, the second consensus model only made predictions on 85% of the test set and on 87% of the external test set as a molecule was only assigned if the three models classified it in the same class; otherwise, it was not assigned. Overall, all models showed conservative behavior with a higher specificity than sensitivity.14
Cao and Leung (2014)15 used the data of Mansouri et al. (2013)14 and introduced the differential evolution (DE) algorithm into the SVM to optimize the parameters of the classifier in order to produce an improved classifier called DE-SVC. The DE-SVC had a slightly lower performance than the consensus II model of Mansouri et al. (2013)14 but was able to classify all substances, which was not the case for the consensus model II of Mansouri et al. (2013).14
Lombardo et al. (2014)16 built a decision tree with a seven rule-set based on 728 compounds that were split in a training set (80%) and an internal test set (20%). Additionally, a set of 874 compounds that originate from the study of Cheng et al. (2012)13 was used as external test set. The fragments for this model derive both from a statistical part (SARpy) and an expert-based part. The balanced accuracy was 82.1% on the internal test set and 78.4% on the external test set, respectively. The model of Lombardo et al. (2014) was used later on for VEGA (see Section 2.2).16
Blay et al. (2016)17 developed a ready-biodegradable prediction for fragrants using 130 compounds. They applied linear discriminant analysis (LDA) and artificial neural networks (ANNs) to build two classification models. To use external validation, a random set of molecules was held out before the training. This hold-out set contained a 20% of the original dataset of 130 molecules. Additionally, internal validation in LDA was applied as 5-fold cross validation. The LDA model had a balanced accuracy based on the 5-times cross validation of 86.5%. The ANN had a balanced accuracy based on the external validation set of 91.5%.
Zhan et al. (2017)18 developed a naïve Bayesian classifier (NBC) to classify the 1055 compounds from Mansouri et al. (2013).14 Three representative structure partitioning methods, including Murcko framework, Scaffold Tree and a scheme based on different complexities of ring combinations and side chains, were used to characterize the structural features of the studied molecules. About 284 RB and 553 NRB chemicals (80%) served as training set and the remaining chemicals as the test set I. In addition, the test set II collected by Mansouri et al. (2013)14 was also used. The best descriptors achieved a balanced accuracy of 85.6% on test set I and 83.8% on test set II, respectively.
Lunghini et al. (2020)19 created a new ready biodegradability dataset by curating and combining data from multiple data sources and additional industry data. This new dataset contained 3146 data points. Furthermore, an additional test set was created based on data from Cheng et al. (2012)13 and Mansouri et al. (2013).14 Lunghini et al. (2020) trained three models based on SVM with linear and Radial Basis Function Kernels (RBF kernels), Random Forest (RF) and NB. Finally, a consensus model was created, which makes a decision based on the majority vote of the three sub-models. The consensus model had balanced accuracies of 81 ± 1.4% on the test set and 75% on the external test set.
Yin et al. (2023)21 trained models on a dataset containing 1928 compounds of which 1424 were used in the training set and 504 in the test set. CORINA descriptors, MACCS fingerprints, and ECFP_4 fingerprints were utilized to characterize the molecules and were used as input features for models after filtering. Models were built using the SVM, DT, RF, and deep neural network (DNN) algorithms. In addition, models based on Graph- and Transformer-CNN models were constructed. The balanced accuracy of the best performing model (Transformer-CNN with 77 MACCS key fingerprints) achieved a balanced accuracy of 87.3%.
The models from the scientific work are summarized in Table 1.
2.2 Open-access applications
BIOWIN is a component of the Estimation Program Interface Suite (EPI Suite™) software, which is a collection of computational tools developed by the United States Environmental Protection Agency (U. S. EPA).28 BIOWIN is designed to predict the biodegradability of organic chemicals in water and wastewater treatment systems. It consists of seven sub-models that focus on different aspects of biodegradation.28The predictive models BIOWIN1 and BIOWIN2 were trained on a dataset of only 295 substances. The BIOWIN1 model is based on multiple linear regressors, while the BIOWIN2 model is based on logistic regression.1,10,28 Therefore, they are also called linear and non-linear models, respectively. BIOWIN1 and BIOWIN2 have a reported accuracy of 65% and 67% on an external test set containing 884 substances, and a reported accuracy of 54% and 67% on an external test set containing 305 substances, respectively.
The models BIOWIN5 and BIOWIN6, which are also part of EPI Suite™, were developed with a similar approach as BIOWIN1 and BIOWIN2 but were trained on a dataset of 884 discrete organic substances from the MITI ready biodegradation tests.12,28 Again, multiple linear regressions were performed to obtain a linear model, BIOWIN5, and a logistic regression was fitted to create a non-linear model, BIOWIN6. BIOWIN5 and BIOWIN6 have a reported accuracy of 83% on an external test set containing 305 substances.
VEGA, which stands for Virtual models for property Evaluation of chemicals within a Global Architecture, is a non-proprietary and openly available tool designed to predict the ready biodegradability of chemical compounds.29 The model behind VEGA is based on Lombardo et al. 2014 and a dataset of 728 mono-constituent organic substances tested according to the OECD 301C Modified MITI(I) Test. An external testing dataset was extracted from Cheng et al. (2012).13 VEGA was developed based on 78 substructures statistically related to ready biodegradability, which were extracted using expert knowledge and the SARpy software.16,29 VEGA's performance scores are similar to the performance of BIOWIN5 and BIOWIN6 on the test set and the external test set.16
OPERA is a freely accessible application that contains Quantitative Structure–Activity Relationship (QSAR) models to predict thirteen different physicochemical and environmental fate properties of organic chemicals.8 Among those thirteen models is a model for assessing the ready biodegradability of organic substances.30 The biodegradation model is based on data from the PHYSPROP database. To ensure data quality, a workflow was utilized for data curation, which involved standardizing chemical structures, correcting the identity of chemicals, and only selecting high-quality data.8 The data curation resulted in a dataset of 1609 substances. The ten most impactful molecular descriptors were calculated using PaDEL, an open-source software for calculating molecular descriptors and FPs.30 The model was trained using a weighted k-nearest neighbor approach and was validated using 5-fold CV.8 The OPERA model predicted the ready biodegradability of the substances in the test set with a balanced accuracy of 79%, a sensitivity of 81%, and a specificity of 77%.8,30
The open-access applications are summarized in Table 2.
3 Methods
The analysis and processing of the data in the current study included a data-centric and a model-centric approach. The data-centric approach had three main steps: analysis of the dataset of Huang and Zhang;20 creation of new datasets; and analysis of the new datasets. Each of these steps included several sub-steps which are shown in Fig. 1. In the model-centric approach, three additional sub-steps were performed which included selecting the best-performing model, testing different features, and hyperparameter tuning. All sub-steps are explained in the following subsections in detail. Section S1 in the ESI-1† contains additional information, including definitions of the chemical identifiers (CAS RN™, SMILES, International Chemical Identifier (InChI™)) and definitions of the terms ‘label’, ‘feature’, ‘data leakage’, ‘MACCS keys’, and ‘balanced accuracy’. | ||
| Fig. 1 Overview of the different steps and sub-steps that were performed in the data-centric and the model-centric approach. Key characteristics means here the molecular weight of the substances, the proportion of halogens present, and the distribution of the biodegradation labels. UMAP stands for Uniform Manifold Approximation and Projection. | ||
3.1 Analysis of the dataset of Huang and Zhang20
In the following, the classification dataset from Huang and Zhang20 will be referred to as Huang-Classification-Dataset, and the regression dataset will be referred to as Huang-Regression-Dataset. The Huang-Regression-Dataset contains inherent- and ready-biodegradation study results retrieved from the eChemPortal. It can contain multiple study results per substance, and the biodegradability is given in percent. The Huang-Classification-Dataset was created based on the Huang-Regression-Dataset and in theory should only contain one data point per substance, which is labeled as RB or NRB.3.2 Creation of new datasets
First, the CAS RN™ and their corresponding SMILES were split into two groups based on whether or not they were verified by Glüge et al.31 In cases where a CAS RN™ was included in the Gluege-Dataset, the verified and valid SMILES for this CAS RN™ from the Gluege-Dataset was used as the SMILES for this data point. Furthermore, for the substances in the Gluege-Dataset, it was also checked if the experimental study was based on read-across. Studies based on read-across were removed. For the substances not checked by Glüge et al.,31 valid SMILES had to be retrieved. For one-component substances, the SMILES were retrieved via an Application Programming Interface (API) based on the CAS RN™ from CAS Common Chemistry.32 For the remaining substances, a weight-of-evidence approach was taken. The SMILES had to be found from at least two independent sources. If this was not possible, the substance was removed from the dataset.
Once the SMILES were found by CAS RN™, further processing steps were performed. Mixtures and organometallic substances were removed, and all counterions were removed from the SMILES representations. For stereoisomers, the SMILES of one stereoisomer was randomly selected. Furthermore, for all ionizable substances, the retrieved SMILES was replaced with the SMILES of the substance's dominant species at pH 7.4 and 298 K. The dominant species was retrieved from the pKa plugin in MarvinSketch 22.18 by using the option “show distribution chart” in “macro” mode.33 Substances were removed when no dominant species existed under the specified conditions. Huang and Zhang20 did not adjust the SMILES of ionizable substances but rather introduced extra features (pKa and α-values) that represent the chemical specification of the substances. They reported a performance increase in the balanced accuracy from 84.9% to 87.6% when including pKa and α-values as extra features. However, using the same model, we could not reproduce this performance increase (see Table S6 in ESI-I†). Therefore, we did not include information on chemical specification directly as features. However, this information is reflected in the SMILES.
 | ||
| Fig. 2 Overview of the different data curation and label noise filtering steps and the resulting datasets. | ||
To overcome these problems, we applied two existing estimation models for biodegradability to filter the data for label noise. Specifically, BIOWIN5 and BIOWIN6 were used to identify and filter out data points with noisy labels (see also Section 2). If BIOWIN5 and/or BIOWIN6 disagreed with the experimental study result, the substance was removed from the CuratedSCS dataset. The subsequently obtained dataset was called CuratedBIOWIN. All substances that were removed in this step were grouped in the CuratedProblematic dataset (Fig. 2). The substances from the CuratedProblematic dataset were tested afterward with a third classifier which was a XGBClassifier trained on the CuratedBIOWIN dataset. In cases where the third classifier agreed with the experimental label of a substance in CuratedProblematic, that substance was readded to the CuratedBIOWIN dataset. Otherwise, the substance remained removed. This led to the creation of the CuratedFinal and the CuratedRemoved datasets. The workflow is also explained again in Table S2 in the ESI-1.†
3.3 Analysis of the new datasets
The new datasets, CuratedBIOWIN, and CuratedFinal, were thoroughly examined to ensure that the removal of data points with potentially noisy labels was not biased towards a specific data type or limited to challenging-to-predict data. The CuratedSCS dataset was used as a benchmark as its labels were not evaluated for noise. Further, it is the dataset from which the others were created.The analysis explored three key characteristics: the molecular weight of the substances, the proportion of halogens present, and the distribution of the biodegradation labels. Further, the Applicability Domain (AD) of the models trained on the three datasets was determined using the Tanimoto similarity. The Tanimoto similarity calculates similarities between two chemicals based on the number of common molecular fragments.20,38 The defined ADs were then used to evaluate how many of the substances in the Distributed Structure-Searchable Toxicity (DSSTox) database are in the ADs of the models. As Huang and Zhang20 did the same for their model, it was possible to compare the broadness of the ADs of the models. More information regarding the similarity threshold and how the AD was applied to the DSSTox database is given in Section S4 in ESI-I.†
Finally, the feature space of the Curated-Datasets was visualized and analyzed using Uniform Manifold Approximation and Projection (UMAP). UMAP is a dimensionality reduction technique used to visualize high-dimensional data in a lower-dimensional space that preserves the underlying structure of the data.39 UMAP was also used to evaluate the impact of using three different chemical representations as model input. More information on the three chemical representations tested can be found in ESI-1† Section S7.2.
3.4 Model training
3.5 Model testing
ML performance metrics reported based on a single test set are subject to biases introduced by the fixed size and distribution of the test dataset. Therefore, in this work, the models were trained and tested five times to average out the potential noise and biases (5-fold cross-validation).To compare the performance metrics of models trained on the different Curated-Datasets, the test sets were kept fixed for all models. Maintaining a consistent test set is typically recommended when a data-centric approach is used and the dataset is augmented. This ensures that any observed changes in model performance are genuinely attributed to data augmentation rather than variations in the test sets. Due to the limitations found and a lack of information regarding the original test set used by Huang and Zhang,20 the test sets were derived from the CuratedSCS dataset. However, a partial objective of the data augmentation was to eliminate data points with noisy labels. Therefore, the models were also tested on fixed test sets from the CuratedBIOWIN dataset.
All models trained on the different datasets were evaluated using these identical test sets. To do so, the CuratedSCS or the CuratedBIOWIN dataset was randomly split into five training (80%) and test (20%) sets using a random seed of 42. Stratified splitting was used to maintain an approximate class distribution across all training and test subsets (cf.44 Ch. 7.10). Further, this ensures that every sample from the dataset was in the test set once. The test sets were then employed as the test sets for all datasets. The training sets were constructed for each dataset by removing the substances of the test set from the dataset.
3.6 Model improvement
Throughout the data curation outlined above, the ML models trained for predicting ready biodegradability were kept constant. To evaluate the effects of the data curation, XGBClassifiers were trained utilizing the optimal hyperparameters reported by Huang and Zhang,20 which were the default hyperparameters of the XGBClassifier. However, upon completing the data curation, we shifted from a data-centric to a model-centric approach. Instead of keeping the models constant, the CuratedFinal dataset was selected to investigate the performance of different ML models. To find the best models for predicting ready biodegradability, the LazyPredict tool was used.45LazyPredict offers the capability to evaluate the performance of nearly all estimators from the SKLEARN library on a given dataset.45 SKLEARN is an open-source ML library containing diverse algorithms.46 Beyond the SKLEARN estimators, LazyPredict also assesses the performance of XGBoost. The outcome of LazyPredict is a table that ranks the most effective models, showing their performance metrics alongside the time taken in seconds for fitting the model on the provided dataset.45
3.7 Hyperparameter tuning
After identifying the most promising models for the CuratedFinal dataset, hyperparameter tuning was carried out for the top five models. As proposed by Huang and Zhang,20 Bayesian Optimization was employed to find the optimal hyperparameters.3.8 Importance of certain features for ready-biodegradation
The importance of certain chemical substructures on ready-biodegradation was analyzed by means of the SHapley Additive exPlanations (SHAP) method.47 Specifically, the SHAP method was used to analyze which of the 166 MACCS keys have the most influence on ready-biodegradation.4 Results
4.1 Analysis of the dataset of Huang and Zhang20
3721 of the substances in the Huang-Classification-Dataset were also in the Gluege-Dataset. It was found that for approximately 20% of these substances, the SMILES added by Huang and Zhang20 converted to InChI™ that did not match the InChI™ associated with the CAS RN™. 5.0% of the SMILES added by Huang and Zhang20 did not even convert into InChI™ with the same chemical formula as the InChI™ corresponding to the CAS RN™ and 8.5% did not have the same InChI™-main-layer. Examples of added SMILES that were not according to the CAS RN™ can be found in Table S3 in ESI-1.† Table S4 in ESI-1† shows examples of substances that appeared in the Huang-Classification-Dataset multiple times with different versions of the same SMILES. We concluded overall that the quality of the SMILES is not sufficient to continue working with them. Therefore, all SMILES that were added by Huang and Zhang20 were removed and new validated SMILES were added (see Section 3.2).
4.2 Creation of the new datasets
| Dataset | Data points | RB | NRB |
|---|---|---|---|
| Huang-Classification-Dataset | 6139 | 34.9% | 65.1% |
| CuratedSCS | 5185 | 34.6% | 65.4% |
| CuratedBIOWIN | 3864 | 31.9% | 68.1% |
| CuratedProblematic | 1321 | 42.6% | 57.4% |
| CuratedFinal | 4371 | 31.7% | 68.3% |
| CuratedRemoved | 814 | 50.1% | 49.9% |
A third classifier (an XGBClassifier trained on the CuratedBIOWIN dataset) was consulted for the substances in the CuratedProblematic dataset to make a decision. According to this third classifier, 507 substances were added back to the CuratedBIOWIN classification dataset. The CuratedFinal dataset thus contains 4371 substances, the CuratedRemoved dataset 814 (Table 3).
4.3 Model performance
Fig. 3a shows the balanced accuracies of the XGBClassifiers trained on the four different datasets. All trained classifiers were tested five times on fixed test sets from the CuratedSCS dataset. Actually, we should have used the Huang-Classification-Dataset to generate the test sets, as this was the initial dataset. However, as some substances were present multiple times (with different chemical identifiers) in the Huang-Classification-Dataset, this would have led to data leakage. For this reason, the test sets were generated from the CuratedSCS dataset. The Replicated-Huang-Classifier shows a balanced accuracy of 80.6 ± 1.5%, which is significantly lower than the reported balanced accuracies of 87.6% (ref. 20). The reason for this difference is not entirely clear, but is probably due to the different test sets. Huang and Zhang20 did not publish their test set and it seems that they only tested their model with one test set. For the Replicated-Huang-Classifier, on the other hand, we divided the dataset into 5 parts and tested each part once. As will be shown later on, the balanced accuracy depends very much on the test set, which is why the difference in the balanced accuracy might be explained by the test set. | ||
| Fig. 3 Model balanced accuracy reported by Huang and Zhang20 and balanced accuracies for the XGBClassifiers trained on the Huang-Classification-Dataset and the Curated-Datasets. The trained classifiers were tested five times on fixed test sets from (a) the CuratedSCS and (b) the CuratedBIOWIN dataset. The definition of “balanced accuracy” is given in ESI-1† Section S1.8. | ||
The model trained on the CuratedSCS dataset also has a balanced accuracy of 80.9 ± 1.7%. The XGBClassifiers trained on the CuratedBIOWIN and the CuratedFinal datasets show balanced accuracies of 78.3 ± 0.9% and 79.4 ± 0.8%, respectively.
Fig. 3a shows that despite correcting dataset limitations such as incorrect CAS RN™–SMILES pairings or removing read-across studies, no improvement was observed in the performance of the classifier that was trained on the Huang-Classification-Dataset and the CuratedSCS dataset. Furthermore, removing substances with potentially wrong labels also did not increase the performance of the model. This might be attributed to three different reasons: (1) a significant portion of the data points in the test sets may have noisy labels due to high variance in experimental studies, (2) the used features may not cover all required information to predict the ready biodegradability, and (3) the model may have been unable to learn and generalize well enough to make correct predictions for difficult-to-predict substances.
As a matter of fact, no dataset of substances that contain only accurate labels could be identified. For the majority of substances, only one experimental study result for ready biodegradation carried out for 28 days exists. The substances with multiple such test results could often not be labeled with certainty because conflicting study results exist.
However, to build robust models, label noise should be reduced as much as possible. Therefore, the model performance was also evaluated on test sets derived from the CuratedBIOWIN dataset as shown in Fig. 3b. The Replicated-Huang-Classifier had a balanced accuracy of 88.0 ± 1.3% and, therefore, performed similarly to the best-performing classifier reported by Huang and Zhang.20 The XGBClassifier trained on the CuratedSCS dataset performed similarly with a balanced accuracy of 88.4 ± 2.1%.
The model trained on the CuratedFinal dataset, the CuratedFinal-Classifier, was the best-performing model and achieved a balanced accuracy of 94.2 ± 1.2%, a sensitivity of 91.6 ± 2.8%, and a specificity of 96.9 ± 0.4%. Therefore, the CuratedFinal-Classifier showed a higher performance than any other previously published classifier (see also Section 2). The classifier trained on the CuratedBIOWIN dataset only had a slightly lower balanced accuracy of 93.7 ± 1.0%. The performance metrics of all classifiers are provided in Table S7 in ESI-1.†
However, it has to be kept in mind that the models were tested on test sets from the CuratedBIOWIN dataset, which underwent label noise filtering. This might have introduced bias, or the difficult-to-predict data points could have been removed. A thorough analysis of the new datasets was therefore carried out to understand if this is the case.
4.4 Analysis of the new datasets
| Characteristic | CuratedSCS | CuratedBIOWIN | CuratedFinal |
|---|---|---|---|
| All substances | 100% | 74.5% | 84.3% |
|
|||
| Molecular weight | |||
| 0 to 250 Da | 100% | 68.9% | 80.8% |
| 250 to 500 Da | 100% | 79.7% | 87.0% |
| 500 to 750 Da | 100% | 75.4% | 83.1% |
| 750 to 1000 Da | 100% | 87.0% | 88.0% |
| 1000 to 2000 Da | 100% | 62.9% | 65.7% |
|
|||
| Halogens | |||
| F | 100% | 86.1% | 96.6% |
| Br | 100% | 79.4% | 93.4% |
| Cl | 100% | 85.4% | 92.0% |
|
|||
| Biodegradation class | |||
| NRB | 100% | 75.9% | 86.8% |
| RB | 100% | 67.4% | 75.6% |
Table 4 shows that there are slight compositional differences between the CuratedSCS and the CuratedBIOWIN and CuratedFinal datasets. The largest difference was observed for the group “1000 to 2000 Da” with a difference of up to 19% compared to “all substances”. However, all other sub-groups in Table 4 are within −9% and +13% of the percentages of “all substances”, meaning that no characteristic or chemical group in these other groups could be identified that was disproportionally more or less present in the CuratedBIOWIN or the CuratedFinal datasets than in the CuratedSCS dataset. Therefore, based on the three characteristics analyzed, the CuratedProblematic and CuratedRemoved datasets can be considered to be very similar in composition to the CuratedBIOWIN and CuratedFinal datasets.
Fig. 4 shows the visual representations of the datasets after dimensionality reduction with the UMAP algorithm. Subplots (a–c) show the results for the CuratedBIOWIN and CuratedProblematic datasets. Subplots (d–f) show the results for the CuratedFinal and CuratedRemoved datasets.
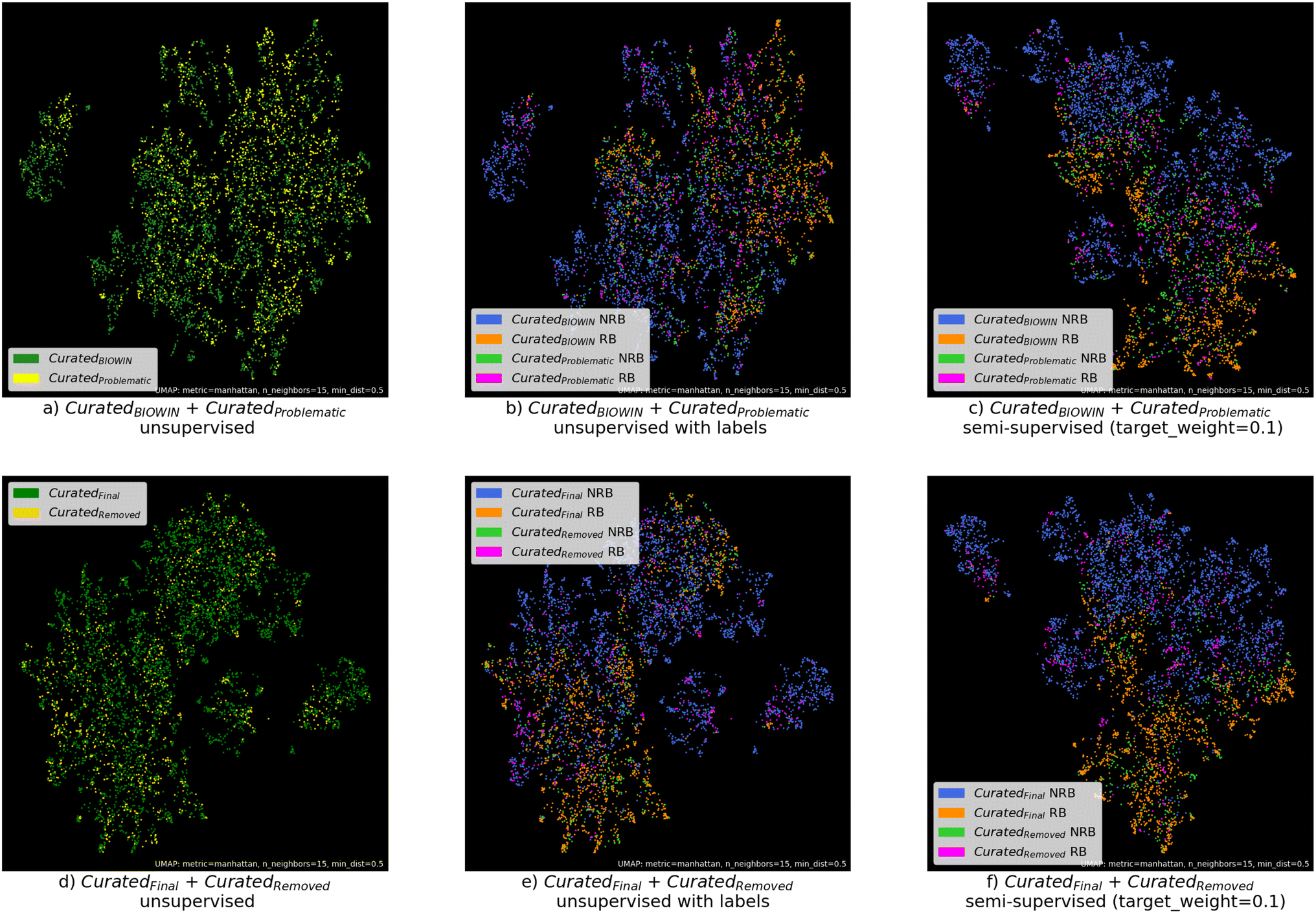 | ||
| Fig. 4 Visual representations of the CuratedBIOWIN, CuratedProblematic, CuratedFinal, and CuratedRemoved datasets after dimensionality reduction with the UMAP algorithm. Subplots (a–c) show the results for the CuratedBIOWIN and CuratedProblematic datasets. Subplots (d–f) show the results for the CuratedFinal and CuratedRemoved datasets. Subplots (a), (b), (d), and (e) are unsupervised, and subplots (c and f) are semi-supervised. | ||
4.4.2.1 Removed data are not different from remaining data. For the subplots (a and d), we applied unsupervised UMAP, i.e. without using the labels for biodegradability. As one can see, the substances of the CuratedProblematic and the CuratedRemoved datasets, both displayed in yellow, are distributed in the same way as the substances in the CuratedBIOWIN and CuratedFinal datasets (green). No clustering or grouping of the yellow data points can be observed. Hence, there is no meaningful similarity or association among those data points. However, it is also possible that the MACCS keys are just not suitable for representing the differences between the CuratedRemoved data and the remaining data.
4.4.2.2 Inherent complexity. Subplots (b and e) are also plots of the unsupervised UMAP. This time, the substances are colored according to their labels for ready- or not ready-biodegradation. As one can see, there are no clear clusters according to the labels. This suggests that predicting the ready biodegradability of organic chemicals based on their structure is a challenging task. Furthermore, we can state that the data have a significant amount of variability and that the features might only capture some of the characteristics that define the classes.
4.4.2.3 Using a third vote for label noise filtering reduced noise or class overlapping. For subplots (c and f), we applied semi-unsupervised UMAP, i.e. we provided some data with and some without labels. For subplot (c), the labels of the data points in the CuratedBIOWIN dataset were used, while the data of the CuratedProblematic dataset were arranged in the UMAP plot according to their feature characteristics without the label being known. For subplot (f), UMAP was provided with the labels of the data points in the CuratedFinal dataset but did not see the labels of the data in the CuratedRemoved dataset. From subplot (c), one can see that it is still difficult to separate the NRB from the RB substances. This separation of the NRB and RB substances is better in subplot (f), i.e. there is less overlap between the classes. This suggests that using the XGBClassifier trained on the CuratedBIOWIN dataset to cast a third vote on the data in the CuratedProblematic dataset reduced the class overlap, hence, led to a reduction in noise in the datasets.
4.4.2.4 CuratedFinal is more similar to data of opposing biodegradation label. The distance between points in a UMAP plot reflects their distance in the original high-dimensional feature space. Points that are close to each other in the UMAP plot are considered more similar, while those that are farther apart are less similar.39 Subplot (f) shows that the majority of the substances in the CuratedFinal dataset, which were given without label, were placed close to substances from the CuratedFinal dataset with opposing labels. This trend is actually also visible in subplots (b), (c), and (e). The majority of the substances from the CuratedProblematic and the CuratedRemove datasets that were labeled as RB are closer to substances labeled as NRB from the CuratedFinal and CuratedFinal datasets, respectively. The same is observed for substances from the CuratedProblematic or the CuratedRemoved datasets that were labeled as NRB. The closeness of the substances in the CuratedProblematic or the CuratedRemoved to the substances with opposing labels from the CuratedBIOWIN and CuratedFinal datasets suggests that the labels of the substances in the CuratedProblematic or the CuratedRemoved are difficult to learn – either because they are incorrect or because the features do not cover the required information.
Huang and Zhang20 had found that 98.4% of the substances in the DSSTox database would fall within the AD of the Huang-Classifier. For the XGBClassifier trained on the CuratedSCS, CuratedBIOWIN, and the CuratedFinal datasets, it was found that 97.9%, 97.3%, and 97.7% of the substances in the DSSTox database are in the AD, respectively. Therefore, reducing the dataset size due to curation based on the CAS RN™–SMILES pairings and removing data points with noisy labels did not significantly reduce the AD. This indicates that the substances in the curated datasets comprise a similarly broad chemical space as the substances in the Huang-Classification-Dataset. If the substances in the CuratedProblematic and CuratedRemoved datasets had different structural characteristics, the AD of the XGBClassifier trained on the CuratedBIOWIN and CuratedFinal datasets should have been much narrower than the reported AD of the Huang-Classifier. However, one has to note that the Tanimoto Index is based on molecular fragments. If these molecular fragments do not cover certain properties of substances (such as intramolecular hydrogen bonds), then the AD would also not reveal if substances with these properties were excluded from our test set.
4.5 Model selection with LazyPredict
Once the data analysis and data curation were completed, a model-centric approach was taken to improve the model performance further. The dataset used was CuratedFinal. Overall, model selection with LazyPredict and hyperparameter tuning did not lead to a significantly higher performance on both the CuratedSCS and the CuratedBIOWIN test sets (Tables S9–S12 in ESI-1†). For the models tested on data from the CuratedSCS dataset, feature creation using RDK FPs in combination with the LogisticRegression classifier leads to a slightly but insignificantly higher model performance. For the models tested on data from the CuratedBIOWIN dataset, feature creation using MACCS keys leads to the highest balanced accuracies. Importantly, no algorithm could be found that significantly outperformed the XGBClassifier that was trained with the default hyperparameters on the CuratedFinal dataset and tested with data from the CuratedBIOWIN dataset. Therefore, it was confirmed that XGBClassifier is among the most suitable algorithms for predicting ready biodegradability. The trained XGBClassifier that achieved a balanced accuracy of 94.2 ± 1.2% is now also available in a graphical user interface on https://biodegradability-prediction-app.streamlit.app/.4.6 Importance of certain features for ready-biodegradation
Fig. 5a shows the MACCS keys that contribute most to non ready-biodegradability while Fig. 5b shows the MACCS keys that contribute most to ready-biodegradability. The results reveal that halogens, 6-member rings, and aromatic rings that are bound to a nitrogen contribute most to non ready-biodegradable while carboxyl and hydroxyl groups can substantially improve the biodegradability. These findings are in line with previous literature, e.g., Loonen et al.11 | ||
| Fig. 5 Box plots showing the SHAP values for the highest negative and highest positive SHAP values. The SHAP values were only calculated for those substances that contained the feature. Atom symbols are: A – any valid periodic table element symbol; Q – hetero atoms (any non-C or non-H atom); X – halogens; Z – other than H, C, N, O, Si, P, S, F, Cl, Br, I. The bond types are: – single; = double; % an aromatic query bond; $ ring bond; ! chain or non-ring bond. | ||
4.7 Discussion and environmental implications
This study set out to train an improved ML model for predicting the aerobic ready-biodegradability of organic chemicals. First, a data-centric approach was taken based on the dataset from Huang and Zhang,20 the largest dataset of RBT published thus far. The initial findings reveal that, despite meticulous data curation which included correction of CAS RN™–SMILES pairings and handling noisy labels, the classification model's performance when using the test set from CuratedSCS could not be improved. This can be due to three reasons: inadequate features for capturing important information about the chemicals, limitations in the model's ability to learn and generalize, or noisy data labels in the test set.The first two point were addressed in the model-centric approach. On the first point, four different feature creation methods were tested. The resulting number of features for each data point ranged from 167 to 2048. However, none of the methods led to an improved model performance for the CuratedFinal dataset when the test set from CuratedSCS was used. To evaluate whether the lack of performance improvement was due to the model's inability to learn and generalize well enough (point 2), 31 ML models were screened. No ML algorithm could be identified that led to a significant performance increase for the CuratedFinal dataset. However, even though we could not find an improved feature creation method and ML algorithm, that does not mean they do not exist. Our findings do not indicate it, but the lack of performance increase might still be due to an inadequate model algorithm or features.
Another explanation is the presence of data with noisy labels in the test datasets. RBT results depend on various factors and have also been shown to depend on the used test protocol and the laboratory that carried out the test.19 However, test sets without label noise are necessary to build and evaluate robust ML models. Given the inherent noise in RBT results, no fully reliable test set could be identified, so label noise filtering was employed here in the second approach to the test set as well. All models tested on the CuratedBIOWIN dataset performed significantly better than when tested on the CuratedSCS dataset, which was not filtered for label noise. When the training set was also filtered for label noise, the balanced accuracy increased from 88.4 ± 2.1% to 94.2 ± 1.2%.
Overall, 184 substances could not be labeled due to contradicting experimental test results and 814 substances were identified as having potentially noisy labels (ESI-2†). Of these 814 substances, 408 were assigned to be RB. This is concerning because substances that have been found to be RB in RBT do not have to undergo further testing for their biodegradability.
Our findings indicate that label noise filtering can lead to a more robust and reliable classifier for predicting the aerobic ready-biodegradability of chemicals. We could not find any indications that the label noise filtering led to the removal of difficult-to-learn substances. However, we cannot completely exclude that instead of data points with noisy labels, data points with difficult-to-learn substances have been removed. Therefore, we recommend that those substances that prevent the model from being more accurate (substances in CuratedRemoved) should be tested further experimentally to investigate whether the labels were noisy or not.
Data availability
The data supporting this article are included as part of the ESI.† The entire python code including the XGBClassifier that was trained on the CuratedFinal dataset is provided in our GitHub repository (https://github.com/pkoerner6/Prediction-of-Aerobic-Biodegradability-of-Organic-Chemicals).Author contributions
PK: investigation, methodology, data curation, validation, writing – original draft; JG: conceptualization, methodology, supervision, writing – review & editing; SG: methodology, writing – review & editing MS: funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Paulina Körner and Juliane Glüge acknowledge funding from the Swiss Federal Office for the Environment. We thank Kuan Huang and Huichun Zhang for providing additional data and information on their publication at the beginning of the project.Notes and references
- R. S. Boethling, P. H. Howard, W. Meylan, W. Stiteler, J. Beauman and N. Tirado, Group contribution method for predicting probability and rate of aerobic biodegradation, Environ. Sci. Technol., 1994, 28, 459–465, DOI:10.1021/es00052a018.
- S. Solomon, D. Wuebbles, I. Isaksen, J. Kiehl, M. Lal, P. Simon and N.-D. Sze, Ozone Depletion Potentials, Global Warming Potentials, and Future Chlorine/Bromine Loading, in Scientific Assessment of Ozone Depletion, World Meteorological Organization, 1994, ch. 13 Search PubMed.
- C. J. van Leeuwen and T. G. Vermeire, Risk Assessment of Chemicals: an Introduction, Springer, 2007, vol. 94 Search PubMed.
- M. Scheringer, J. H. Johansson, M. E. Salter, B. Sha and I. T. Cousins, Stories of global chemical pollution: will we ever understand environmental persistence?, Environ. Sci. Technol., 2022, 56(24), 17498–17501, DOI:10.1021/acs.est.2c06611.
- European Chemicals Agency (ECHA), Guidance on Information Requirements and Chemical Safety Assessment – Chapter R.11: PBT/vPvB Assessment (Version 4.0), 2023, https://echa.europa.eu/documents/10162/17224/information_requirements_r11_en.pdf/a8cce23f-a65a-46d2-ac68-92fee1f9e54f Search PubMed.
- A. H. Neilson and A.-S. Allard, Environmental Degradation and Transformation of Organic Chemicals, CRC Press, 2007 Search PubMed.
- M. Pavan and A. P. Worth, Review of estimation models for biodegradation, QSAR Comb. Sci., 2008, 27(1), 32–40, DOI:10.1002/qsar.200710117.
- K. Mansouri, C. M. Grulke, R. S. Judson and A. J. Williams, OPERA models for predicting physicochemical properties and environmental fate endpoints, J. Cheminf., 2018, 10(10), 1–19, DOI:10.1186/s13321-018-0263-1.
- ECHA, QSAR models, https://echa.europa.eu/support/registration/how-to-avoid-unnecessary-testing-on-animals/qsar-models, 2023, Accessed: 2023-09-01.
- P. H. Howard, R. S. Boethling, W. M. Stiteler, W. M. Meylan, A. E. Hueber, J. A. Beauman and M. E. Larosche, Predictive model for aerobic biodegradability developed from a file of evaluated biodegradation data, Environ. Technol. Chem., 1992, 11, 593–603, DOI:10.1002/etc.5620110502.
- H. Loonen, F. Lindgren, B. Hansen, W. Karcher, J. Niemelä, K. Hiromatsu, M. Takatsuki, W. Peijnenburg, E. Rorije and J. Struijś, Prediction of biodegradability from chemical structure: modeling of ready biodegradation test data, Environ. Toxicol. Chem., 1999, 18, 1763–1768, DOI:10.1002/etc.5620180822.
- J. Tunkel, P. H. Howard, R. S. Boethling, W. Stiteler and H. Loonen, Predicting ready biodegradability in the Japanese Ministry of International Trade and Industry test, Environ. Toxicol. Chem., 2000, 19, 2478–2485, DOI:10.1002/etc.5620191013.
- F. Cheng, Y. Ikenaga, Y. Zhou, Y. Yu, W. Li, J. Shen, Z. Du, L. Chen, C. Xu, G. Liu, P. W. Lee and Y. Tang, In silico assessment of chemical biodegradability, J. Chem. Inf. Model., 2012, 52, 655–669, DOI:10.1021/ci200622d.
- K. Mansouri, T. Ringsted, D. Ballabio, R. Todeschini and V. Consonni, Quantitative structure–activity relationship models for ready biodegradability of chemicals, J. Chem. Inf. Model., 2013, 53, 867–878, DOI:10.1021/ci4000213.
- Q. Cao and K. M. Leung, Prediction of chemical biodegradability using support vector classifier optimized with differential evolution, J. Chem. Inf. Model., 2014, 54, 2515–2523, DOI:10.1021/ci500323t.
- A. Lombardo, F. Pizzo, E. Benfenati, A. Manganaro, T. Ferrari and G. Gini, A new in silico classification model for ready biodegradability, based on molecular fragments, Chemosphere, 2014, 108, 10–16, DOI:10.1016/j.chemosphere.2014.02.073.
- V. Blay, J. Gullón-Soleto, M. Gálvez-Llompart, J. Gálvez and R. García-Domenech, Biodegradability Prediction of Fragrant Molecules by Molecular Topology, ACS Sustain. Chem. Eng., 2016, 4, 4224–4231, DOI:10.1021/acssuschemeng.6b00717.
- Z. Zhan, L. Li, S. Tian, X. Zhen and Y. Li, Prediction of chemical biodegradability using computational methods, Mol. Simul., 2017, 43, 1277–1290, DOI:10.1080/08927022.2017.1328556.
- F. Lunghini, G. Marcou, P. Gantzer, P. Azam, D. Horvath, E. Van Miert and A. Varnek, Modelling of ready biodegradability based on combined public and industrial data sources, SAR QSAR Environ. Res., 2020, 31, 171–186, DOI:10.1080/1062936X.2019.1697360.
- K. Huang and H. Zhang, Classification and Regression Machine Learning Models for Predicting Aerobic Ready and Inherent Biodegradation of Organic Chemicals in Water, Environ. Sci. Technol., 2022, 56, 12755–12764, DOI:10.1021/acs.est.2c01764.
- H. Yin, C. Lin, Y. Tian and A. Yan, Prediction and Structure-Activity Relationship Analysis on Ready Biodegradability of Chemical Using Machine Learning Method, Chem. Res. Toxicol., 2023, 36, 617–629, DOI:10.1021/acs.chemrestox.2c00330.
- OECD, eChemPortal, https://www.echemportal.org/echemportal/property-search, 2023, Accessed: 2023-09-01.
- M. H. Jarrahi, A. Memariani and S. Guha, The Principles of Data-Centric AI, Commun. ACM, 2023, 66, 84–92, DOI:10.1145/3571724.
- D. Zha, Z. P. Bhat, K.-H. Lai, F. Yang and X. Hu, Data-centric ai: Perspectives and challenges, in Proceedings of the 2023 SIAM International Conference on Data Mining (SDM), 2023, 945–948, https://epubs.siam.org/doi/10.1137/1.9781611977653.ch106 Search PubMed.
- J. Jakubik, M. Vössing, N. Kühl, J. Walk and G. Satzger, Data-centric artificial intelligence, Bus. Inf. Syst. Eng., 2024, 66(4), 507–515, DOI:10.1007/s12599-024-00857-8.
- C. G. Northcutt, A. Athalye and J. Mueller, Pervasive label errors in test sets destabilize machine learning benchmarks, arXiv, 2021, preprint, arXiv:2103.14749, DOI:10.48550/arXiv.2103.14749.
- B. Frénay and M. Verleysen, Classification in the presence of label noise: a survey, IEEE Transact. Neural Networks Learn. Syst., 2013, 25, 845–869, DOI:10.1109/TNNLS.2013.2292894.
- United States Environmental Protection Agency (US EPA), Estimation Programs Interface Suite™ for Microsoft® Windows, V 4.11, 2012, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface Search PubMed.
- A. Lombardo, F. Pizzo, E. Benfenati, A. Manganaro and T. Ferrari, QMRF for VEGA Ready Biodegradation model, Joint Reserach Center, Technical Report, 2022, https://www.vegahub.eu/vegahub-dwn/qmrf/QMRF_RB_IRFMN.pdf Search PubMed.
- K. Mansouri and A. Williams, QMRF for OPERA-model for Readily Biodegradability, Joint Reserach Center, Technical Report, 2019, https://jeodpp.jrc.ec.europa.eu/ftp/jrc-opendata/EURL-ECVAM/datasets/QSARDB/LATEST/PDF/_qmrf_protocol_Q17-23a-0014_document.pdf Search PubMed.
- J. Glüge, K. McNeill and M. Scheringer, Getting the SMILES right: identifying inconsistent chemical identities in the ECHA database, PubChem and the CompTox Chemicals Dashboard, Environ. Sci.: Adv., 2023, 2, 612–621, 10.1039/D2VA00225F.
- CAS, CAS Common Chemistry, 2023, https://commonchemistry.cas.org/.
- Chemaxon, pKa plugin, 2022, https://docs.chemaxon.com/display/docs/pka-plugin.md.
- OECD, OECD Test No. 301: Ready Biodegradability, Organisation for Economic Cooperation and Development Technical Report, 1992, https://www.oecd-ilibrary.org/environment/test-no-301-ready-biodegradability_9789264070349-en Search PubMed.
- T. J. Martin, J. R. Snape, A. Bartram, A. Robson, K. Acharya and R. J. Davenport, Environmentally relevant inoculum concentrations improve the reliability of persistent assessments in biodegradation screening tests, Environ. Sci. Technol., 2017, 51, 3065–3073, DOI:10.1021/acs.est.6b05717.
- A. Kowalczyk, T. J. Martin, O. R. Price, J. R. Snape, R. A. van Egmond, C. J. Finnegan, H. Schäfer, R. J. Davenport and G. D. Bending, Refinement of biodegradation tests methodologies and the proposed utility of new microbial ecology techniques, Ecotoxicol. Environ. Saf., 2015, 111, 9–22, DOI:10.1016/j.ecoenv.2014.09.021.
- P. Li, X. Rao, J. Blase, Y. Zhang, X. Chu and C. Zhang, CleanML: A Study for Evaluating the Impact of Data Cleaning on ML Classification Tasks, 2021 IEEE 37th International Conference on Data Engineering (ICDE), 2021, pp. 13–24, https://www.research-collection.ethz.ch/bitstream/handle/20.500.11850/444041/CleanML__ICDE2021_Submission_.pdf?sequence=8&isAllowed=y Search PubMed.
- S. Kar, K. Roy and J. Leszczynski, Applicability Domain: A Step Toward Confident Predictions and Decidability for QSAR Modeling, Methods Mol. Biol., 2018, 141–169, DOI:10.1007/978-1-4939-7899-1_6.
- L. McInnes, J. Healy and J. Melville, Umap: Uniform manifold approximation and projection for dimension reduction, arXiv, 2018, preprint, arXiv:1802.03426, DOI:10.48550/arXiv.1802.03426.
- T. Chen and C. Guestrin, XGBoost: A Scalable Tree Boosting System, Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785–794, https://dl.acm.org/doi/10.1145/2939672.2939785 Search PubMed.
- A. Jadhav, S. M. Mostafa, H. Elmannai and F. K. Karim, An empirical assessment of performance of data balancing techniques in classification task, Appl. Sci., 2022, 12, 3928, DOI:10.3390/app12083928.
- H. He, Y. Bai, E. A. Garcia and S. Li, ADASYN: Adaptive synthetic sampling approach for imbalanced learning, 2008 IEEE International Joint Conference on Neural Networks, IEEE world congress on computational intelligence, 2008, pp. 1322–1328, https://ieeexplore.ieee.org/document/4633969 Search PubMed.
- xgboost developers, XGBoost Tutorial – Categorical Data, https://xgboost.readthedocs.io/en/stable/tutorials/categorical.html, 2023, Accessed: 2023-09-01.
- T. Hastie, R. Tibshirani and J. Friedman, The Elements of Statistical Learning, SpringerNew York, New York, 2009 Search PubMed.
- S. R. Pandala, Lazy Predict, https://github.com/shankarpandala/lazypredict, 2023, Accessed: 2023-09-01.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M Brucher, M Perrot and É. Duchesnay, Scikit-learn: Machine learning in Python, J. Mach. Learn. Res., 2011, 12, 2825–2830, DOI:10.5555/1953048.2078195.
- S. Lundberg, SHAP documentation, 2018, https://shap.readthedocs.io/en/latest/.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Large-scale chemical language representations capture molecular structure and properties, Nat. Mach. Intell., 2022, 4, 1256–1264, DOI:10.1038/s42256-022-00580-7.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-1 contains seven sections with additional information on inter alia some terms and definitions used in the article, the SMILES-retrieval pipeline, label noise filtering, applicability domain and model performance. ESI-2 is an MS Excel file that contains all datasets that were used and generated in this study. See DOI: https://doi.org/10.1039/d4em00431k. The entire python code including the XGBClassifier that was trained on the CuratedFinal dataset is provided in our GitHub repository under https://github.com/pkoerner6/Prediction-of-Aerobic-Biodegradability-of-Organic-Chemicals. The final classification model is also available in a graphical user interface under https://biodegradability-prediction-app.streamlit.app/ |
| This journal is © The Royal Society of Chemistry 2024 |