
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An oxide-promoted, self-supported Ni4Mo catalyst for high current density anion exchange membrane water electrolysis†
Ariana
Serban
 a,
Meng-Ting
Liu
b,
Nanjun
Chen
a,
Hao Ming
Chen
*b and
Xile
Hu
*a
a,
Meng-Ting
Liu
b,
Nanjun
Chen
a,
Hao Ming
Chen
*b and
Xile
Hu
*a
aLaboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne (EPFL) Lausanne CH-1015, Switzerland. E-mail: xile.hu@epfl.ch
bDepartment of Chemistry, National Taiwan University, Taipei, Taiwan
First published on 2nd December 2024
Abstract
Anionic exchange membrane (AEM) water electrolyzers are emerging as a cost-effective technology for green hydrogen production. However, state-of-the-art AEM electrolyzers rely on platinum group metal (PGM) catalysts for the hydrogen evolution reaction (HER). Currently, PGM-free HER catalysts exhibit inadequate activity and stability at high current densities in electrolyzer environments. Here, we report a simple electrodeposition method for a self-supported Ni4Mo–MoOx catalyst. This catalyst exhibits remarkable HER activity, as demonstrated both in three-electrode cells as well as in prototype AEM electrolyzers. In particular, the catalyst enables AEM electrolyzers to operate stably at current densities as high as 3 A cm−2, which had not been reported for a non-PGM HER catalyst. The performance (2 V@3 A cm−2) is comparable to the benchmark Pt/C, whereas the stability is even higher. Characterization and particularly operando X-ray diffraction and absorption spectroscopy reveal that the catalyst is an unconventional tetragonal Ni4Mo with a D1a superlattice whose surface contains in situ formed MoOx species. The cooperative action of MoOx and Ni4Mo enhances the volmer step of HER, attributing to the superior activity.
Broader contextThis research contributes to the ongoing efforts to develop efficient and cost-effective catalysts for hydrogen production, crucial for advancing the viability of green hydrogen technologies. It addresses key challenges associated with PGM-free catalysts, offering a promising alternative that could facilitate the commercialization and widespread adoption of AEM water electrolyzers in sustainable energy applications. Additionally, the insights gained from the characterization techniques provide a valuable understanding of the catalyst's structure–property relationships, informing future catalyst design strategies.This work discusses advancements in anionic exchange membrane (AEM) water electrolyzers, focusing on the development of a novel catalyst for the hydrogen evolution reaction (HER) without relying on platinum group metals (PGMs). It highlights the limitations of current PGM-free catalysts, particularly in maintaining activity and stability at high current densities typical of electrolyzer environments. The study introduces a new self-supported Ni4Mo–MoOx catalyst fabricated via a simple electrodeposition method. The main findings indicate that this catalyst demonstrates exceptional HER activity, validated in both three-electrode cells and prototype AEM electrolyzers. Notably, the catalyst allows AEM electrolyzers to operate stably at high current densities of up to 3 A cm−2. Moreover, its performance at 2 V@3 A cm−2 rivals that of the benchmark Pt/C catalyst, while exhibiting even higher stability. Characterization techniques, including operando X-ray diffraction and absorption spectroscopy, provide insights into the catalyst's structure and composition. Specifically, it identifies the catalyst as an unconventional tetragonal Ni4Mo with a D1a superlattice, with surface features containing in situ formed MoOx species. The superior activity of the catalyst is attributed to the cooperative action of MoOx and Ni4Mo, while its high stability is linked to the strong adhesion resulting from the unique electrodeposition conditions employed during fabrication. |
Introduction
The electrochemical splitting of water into green hydrogen fuels, driven by renewable electricity, offers a promising path toward a carbon-neutral energy society. Alkaline water electrolysis is a mature industrial hydrogen (H2) production technology. However, some of its limitations include a high ohmic resistance, a low partial load range, and a low operating pressure.1 Anion exchange membrane water electrolysis (AEMWE) combines alkaline and proton exchange membrane electrolysis benefits and offers a promising technology for green hydrogen production.2,3 Nevertheless, current state-of-the-art AEM electrolyzers primarily employ carbon-supported platinum (Pt/C) as the cathodic catalyst for the hydrogen evolution reaction (HER).4 Reducing Pt loading at the cathode or substituting it with platinum group metal (PGM)-free materials is essential to enable the wide adoption of AEM electrolyzers. Numerous PGM-free catalysts have been developed for alkaline HER.5–9 However, current studies focus on the electrochemical activity of these catalysts in model conditions (e.g., 3-electrode cells) and at low current densities (<100 mA cm−2). Catalysts that deliver high current densities (e.g., >1 A cm−2) at low overpotentials remain scarce.10 Moreover, few PGM-free catalysts have demonstrated operational stability devices in prototype devices at such high current densities. A possible reason for the poor stability is that most catalysts are sprayed onto GDLs, leading to weak adhesion and potential delamination under stress. The uneven coverage and the low structural integrity make sprayed layers prone to degradation, while poor electrical conductivity at the interface increases resistance. Over time, the binders may degrade, causing the catalyst to detach from the substrate. Here, we describe the development of a new electrochemical deposition method that leads to the direct growth of a novel NiMo catalyst on various industrially relevant gas diffusion layers (GDLs). The resulting catalytic electrodes exhibit remarkable activity in HER, especially at high current densities (>1 A cm−2). The electrodes can be integrated into a prototype, PGM-free AEMWE, demonstrating excellent performance at current densities as high as 3 A cm−2. The activity and stability of this catalyst are much superior to other PGM-free HER catalysts. Operando X-ray diffraction and absorption spectroscopy reveal the actual active form of the catalyst to be an unconventional Ni4Mo species whose surface is decorated by Mo oxides. The promotional effect of the oxide is likely the origin of the catalyst's surprisingly high activity.Results and discussion
Synthesis of catalysts
Self-supported catalysts, where catalysts are grown directly on GDLs, have been identified as a strategy to achieve high current densities at low overpotentials. The advantages of this approach include better adhesion on the substrate and less catalyst leaching compared to the conventional sprayed powders on the GDLs.11 However, achieving an optimal catalyst-support interface is challenging, so the resulting catalytic assembly can exhibit high activity and stability simultaneously. We chose to electrodeposit a NiMo catalyst on GDLs to achieve this goal. Although electrodeposited and chemically synthesized NiMo HER catalysts are known,12 they have not been deposited on high-surface-area GDLs for high-current density applications. While exploring conditions to electrodeposit self-supported NiMo catalysts, we discovered a new method of electrodeposition that led to a high-performance catalyst, described below. Our synthesis involved a one-step electrodeposition procedure at a high current density. Under the optimized conditions for a sample size of 1 cm2 (NiMo-AS1), the deposition involved reduction at −1.5 A cm2 for 1000 s. Voltage profiles during the electrodeposition process are shown in Fig. S1 and S2, ESI.†This method's novelty lies in the composition of the electrodeposition bath and the utilization of high-current density for deposition. Unlike the well-established NiMo electrodeposition baths containing boric acid as a pH buffer13 or citric acid as a complexing agent,14 or methods that employ low concentrations of NH4Cl with NH3 for pH adjustments; our approach features high-concentration of NH4Cl as the sole supporting electrolyte without any pH adjustment.15 The deposition contained NiCl2 and (NH4)6Mo7O24 as sources of Ni and Mo. If NH4Cl is replaced by the same concentration of KCl under identical electrodeposition current and time, no deposition occurs; if NH4Cl is replaced by (NH4)2(SO4) using the same parameters, the resulting sample has much lower activity and falls off the electrode after one measurement (Fig. S3 and S4, ESI†). These results indicate the critical role of NH4Cl in the deposition. We identified Cl2 as a by-product in the deposition, which was attributed to the chlorine evolution reaction at the anode. The dissolution of Cl2 in the deposition bath would lead to a decrease in pH, which was indeed observed experimentally (Fig. S5, ESI†). Our deposition current densities are at least seven times higher than the conventional ones (e.g., 50–250 mA cm−2). The higher deposition current density in our method is a deliberate choice to achieve a more compact and thicker electrode structure. This is feasible because the conductivity of the electrodeposition bath is sufficiently high: 26 mS cm−1 for the NiMo-AS1 bath, compared to 5.55 mS cm−1 for the NiMo-citric bath and 7.36 mS cm−1 for the NiMo-boric acid bath. We could deposit the catalysts on other conducting substrates such as Ni foam and Ni felt. As references, we also prepared “conventional” NiMo catalysts using standard Ni plating baths reported in the literature,12,13 In these cases, NiMo-citric represents the catalyst deposited from a citric acid-containing bath, whereas NiMo-boric represents the catalyst deposited from a boric acid-containing bath.
Electrocatalytic activity and water electrolysis performance
We first used linear sweep voltammetry (LSV) to screen for the catalytic activity of different samples in HER. Through these experiments, we observed the influences of deposition current density, deposition time, and Ni/Mo precursor ratio (Fig. S6, ESI†), and identified the optimal deposition conditions. The optimal catalyst is labeled as NiMo-AS1. We found the activity of the as-deposited catalyst improved with time, and it reached a steady-state under a 2 h in situ activation under HER conditions (Fig. S7, ESI†). A similar activation process was found for other electrodeposited NiMo catalysts: NiMo-citric and NiMo-boric, on C paper (Fig. S8, ESI†). Various catalysts' double-layer capacitances and surface roughness factors were measured (Fig. S9–S12 and Tables S1, S2, ESI†). The Ni mesh itself has a roughness factor of about 1.4, which is increased by 2–3.5 times upon deposition of NiMo catalysts. The roughness factor increases in the order of NiMo-boric < NiMo-AS1 < NiMo-citric. A similar result was found for catalyst deposited on C paper. Fig. 1A shows that on the same Ni mesh support, the NiMo-AC-activated catalyst exhibits a high activity for HER at high current densities. The overpotential for 1 A cm−2 (η@1 A cm−2) was merely 0.19 V (Table 1). Notably, this activity is the best for all electrodeposited catalysts (Fig. S13, ESI†). For comparison, the NiMo-citric catalyst has a η@1 A cm−2 of 0.34 V, and the NiMo-boric catalyst has a η@1 A cm−2 of 0.43 V (Table 1). Ni mesh alone has a low activity (η@1 A cm−2 of 0.45 V). A Pt-mesh sample also has a relatively low activity. The NiMo-AS1 catalysts supported by Ni foam and Ni felt were less active, having overpotentials of about 300 mV at 1 A cm−2 according to the LSV data (Fig. S14, ESI†). The capacitance/roughness-averaged LSV data of NiMo catalysts are shown in Fig. 1B. NiMo-AS1 has the highest specific activity, followed by NiMo-boric and then NiMo-citric. We measured the loading of the NiMo catalysts (Table S3, ESI†) and obtained the mass-averaged LSV data (Fig. 1C). Our NiMo-AS1 has a much higher mass-averaged activity than NiMo-citric and NiMo-boric, which have similar mass-activity. | ||
| Fig. 1 (A) Linear sweep voltammetry (LSV) data showing geometric current densities of NiMo-AS1, NoMo-citric, NiMo-boric on Ni mesh support, bare Ni mesh, and Pt mesh; (B) LSV data showing electrochemical active surface area (ESCA) averaged current densities of NiMo- AS1, NiMo-citric, NiMo-boric deposited on Ni mesh; (C) LSV data showing catalyst loading averaged current densities of NiMo-AS1, NiMo-citric, NiMo-boric on Ni mesh. | ||
| Sample name | η@ 0.1 A cm−2 (V) | η@ 1 A cm−2 (V) | Sample name | η@ 0.1 A cm−2 (V) | η@ 1 A cm−2 (V) |
|---|---|---|---|---|---|
| NiMo-AS1/Ni mesh | 0.04 | 0.19 | NiMo-AS1/C paper | 0.13 | n.d. |
| NiMo-citric/Ni mesh | 0.11 | 0.34 | NiMo-citric/C paper | 0.38 | n.d. |
| NiMo-boric/Ni mesh | 0.13 | 0.34 | NiMo-boric/C paper | 0.53 | n.d. |
| Ni mesh | 0.23 | 0.45 | Pt/C | 0.14 | n.d. |
| Pt mesh | 0.20 | 0.42 |
We also electrodeposited the catalysts on carbon paper because of applications in AEMWEs. In the measurements using a 3-electrode setup in 1 M KOH solutions, the C-supported catalysts exhibit generally lower activity than their Ni mesh-supported counterparts (compare Fig. 2A to Fig. 1A). For comparison among different catalysts, we used the overpotential at 0.1 A cm−2 (η@0.1 A cm−2). As shown in Fig. 2A, our NiMo-AS -activated catalyst has a η@0.1 A cm−2 of 0.13 V, which is much lower than those of NiMo-citric (0.38 V) and NiMo-boric (0.53 V). Compared to a Pt/C reference, the NiMo-AS1 is less active at low current densities (10–100 mA cm−2), but more active at high current densities, especially in the range of 150–400 mA cm−2 (Fig. 2A). We think this result might be understood by considering a high intrinsic activity of Pt/C over NiMo, which leads to a lower on-set overpotential. As the current densities go higher, mass transport becomes more important. It is possible that H2 release from the NiMo-AS1 is more facile than from Pt/C, leading to higher activity at the former. The roughness factor-averaged LSV data (Fig. 2B), and the mass-averaged LSV data (Fig. 2C) for the electrodeposited catalysts show that NiMo-AS1 is much more active than NiMo-citric and NiMo-boric catalysts.
 | ||
| Fig. 2 (A) Linear sweep voltammetry data showing the geometric current densities of NiMo-AS1, NiMo-citric, NiMo-boric on C paper support and Pt/C sprayed on C paper support; (B) LSV data showing electrochemical active surface area (ESCA) averaged current density of NiMo-AS1, NiMo-citric, NiMo-boric deposited on C paper support; (C) LSV data showing catalyst loading averaged current densities of NiMo-AS1, NiMo-citric, and NiMo-boric on C paper support. | ||
We then used the C-supported catalysts as the cathode for AEMWEs. The setup of our prototypes has been described previously.16,17 In brief, a Branion AEM from NovaMea was used as the membrane, and a NiFe catalyst supported on Ni foam was used as the anode. The electrolyte was 1 M KOH for the anode, and the cathode was “dry”. The transient IV curves are shown in Fig. 3A. The AEMWE, having our NiMo-AS1 HER catalyst, exhibits a high performance, with a cell voltage of 1.75 V at 1 A cm−2. An analogous cell with a Pt/C cathode performs similarly at low current densities (>0.5 A cm−2). But at higher current densities, this cell has slightly higher cell voltages (1.81 V at 1 A cm−2) than the cell with our NiMo-AS1 cathode. By comparison, the NiMo-citric-containing AEMWE performs worse, giving a cell voltage of 1.88 V at 1 A cm−2. The NiMo-boric-containing AEMWE performs poorly, with a cell voltage of 2.37 V at 1 A cm−2. The trend observed in the current–voltage profiles of these AEMWEs is consistent with the activity trend observed in the 3-electrode settings (Fig. 1A and 2A).
 | ||
| Fig. 3 (A) Polarization curves of AEMWEs with NiMo-AS1, NiMo-citric, and NiMo-boric on C paper as the cathode, respectively; (B) long-term electrolysis data at a constant current density of 1 A cm−2 and cell temperature of 80 °C; (C) electrolysis at a current density of 3 A cm−2 at 80 °C. | ||
We further tested these AEMWEs in long-term (potentiometric) electrolysis. A discrepancy was observed in the voltages of AEMWEs obtained from I–V (Fig. 3A) and potentiometric electrolysis experiments (Fig. 3B) as a result of the different time scales of the measurements. The AEMWE containing our NiMo-AS1 cathode operated stably at 1 A cm−2 for 150 hours, with a final cell voltage of 1.81 V. After an initial “activation” process of several hours, the voltage remains largely constant. When the benchmark precious catalyst Pt/C was used, the AEMWE could also be operated at 1 A cm−2 for 150 hours, with a final voltage of 1.77 V. The AEMWE containing the NiMo-citric catalyst had a final cell voltage of 2.1 V, 290 mV higher than that of our NiMo-AS1 catalyst. Furthermore, there is a noticeable degradation rate of 0.53 mV h−1. For the AEMWE with the NiMo-boric catalyst, the initial voltage spiked to 2.42 V before stabilizing at approximately 2.2 V at a current density of 1 A cm−2. However, the cell ultimately broke down after 100 hours of operation.
To further demonstrate the utility of our NiMo-AS1/C paper catalyst for even higher current density applications, we tested the AEMWEs at 3 A cm−2. The AEMWEs containing either the NiMo-AS1 catalyst or the Pt/C catalyst had very close performance: 1.96 V for Pt/C and 2.03 V for NiMo-AS1. Both cells can be stably operated for at least 100 hours at a cell voltage of about 2 V. After 100 h, the cell containing Pt/C showed voltage fluctuations compared to the NiMo-AS1-containing cell.
We also tested NiMo-AS1 catalysts deposited on Ni foam and Ni felt for AEMWEs. The former led to a cell voltage of around 1.8 V at 1 A cm−2, while the latter led to a cell voltage of about 1.76 V at 1 A cm−2 (Fig. S15, ESI†). These results indicate a similar performance of NiMo-AS1 catalysts in AEMWEs on different GDLs, although their activity appears to be different in LSV measurements.
Ex situ characterization of catalysts
Inductively coupled plasma mass spectrometry (ICP-MS) revealed a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ni/Mo atomic ratio for NiMo-AS1 after activation (Fig. S18 and Table S4, ESI†). Scanning electron microscopy (SEM) measurements reveal that the NiMo-AS1 catalyst, when deposited under optimal conditions, achieves homogeneous coverage on both Ni mesh and carbon paper supports (Fig. 4A–F) as well as on Ni foam and Ni felt (Fig. S19, ESI†). Shorter deposition times and lower current densities resulted in partial coverage, whereas longer deposition times and higher current densities caused more visible cracks, indicating reduced adhesion (ESI† Fig. S20 and S21). After electrolysis at 1 A cm−2 for 1.5 hours, NiMo-AS1 on Ni mesh remained intact with only minor cracks (Fig. 4C). For NiMo-AS1 on C, the carbon fibers were initially fully covered by the catalyst (Fig. 4E). After electrolysis, a small number of fibers became exposed (Fig. 4F), likely due to the evolved gas bubbles. Among different catalysts, NiMo-AS1 formed the most compact layer on carbon paper, while the NiMo-citric and NiMo-boric catalysts did not cover the fibers (ESI† Fig. S16). After 150 hours at 3 A cm−2, morphological changes occurred for all carbon-supported catalysts. NiMo-AS1 and NiMo-citric remained on the C support, whereas NiMo-boric was partially detached (ESI† Fig. S17). EDX mapping confirmed a homogeneous distribution of nickel and molybdenum in all samples before and after electrolysis, with no phase segregation (ESI,† Fig. S16 and S17). As a reference, Pt/C on carbon paper also showed a substantial morphological change and suffered from Pt loss after electrolysis (ESI† Fig. S16 and S17).
:1 Ni/Mo atomic ratio for NiMo-AS1 after activation (Fig. S18 and Table S4, ESI†). Scanning electron microscopy (SEM) measurements reveal that the NiMo-AS1 catalyst, when deposited under optimal conditions, achieves homogeneous coverage on both Ni mesh and carbon paper supports (Fig. 4A–F) as well as on Ni foam and Ni felt (Fig. S19, ESI†). Shorter deposition times and lower current densities resulted in partial coverage, whereas longer deposition times and higher current densities caused more visible cracks, indicating reduced adhesion (ESI† Fig. S20 and S21). After electrolysis at 1 A cm−2 for 1.5 hours, NiMo-AS1 on Ni mesh remained intact with only minor cracks (Fig. 4C). For NiMo-AS1 on C, the carbon fibers were initially fully covered by the catalyst (Fig. 4E). After electrolysis, a small number of fibers became exposed (Fig. 4F), likely due to the evolved gas bubbles. Among different catalysts, NiMo-AS1 formed the most compact layer on carbon paper, while the NiMo-citric and NiMo-boric catalysts did not cover the fibers (ESI† Fig. S16). After 150 hours at 3 A cm−2, morphological changes occurred for all carbon-supported catalysts. NiMo-AS1 and NiMo-citric remained on the C support, whereas NiMo-boric was partially detached (ESI† Fig. S17). EDX mapping confirmed a homogeneous distribution of nickel and molybdenum in all samples before and after electrolysis, with no phase segregation (ESI,† Fig. S16 and S17). As a reference, Pt/C on carbon paper also showed a substantial morphological change and suffered from Pt loss after electrolysis (ESI† Fig. S16 and S17).
 | ||
| Fig. 4 (A) SEM image of bare Ni mesh; (B) SEM image of the as-prepared NiMo-AS1 on Ni mesh; (C) SEM image of the NiMo-AS1 on Ni mesh after HER at 1 A cm−2; (D) SEM image of the bare C paper; (E) SEM image of the as-prepared NiMo-AS1 on C paper; (F) SEM image of the NiMo-AS1 on C paper after HER at 1 A cm−2. | ||
Transmission electron microscopy (TEM) analyses of catalyst layers of NiMo-AS1 detached from the Ni mesh support show a nanosheet structure of the catalyst (Fig. S22, ESI†), which was transformed into a more layered structure after HER at 1 A cm−2 for 1.5 h (Fig. S22, ESI†), selected area electron diffraction (SAED) show that the NiMo-AS1 catalyst (Fig. S22, ESI†) after activation is polycrystalline. The most intense diffraction spots, with a d-spacing of 0.2, 0.16,0.12 and 0.11 nm, were attributed to (121), (101), (132) and (431) facets of a tetragonal Ni4Mo (Fig. S23 and S24, ESI†). Energy dispersive X-ray spectroscopy (EDX) mapping confirmed the uniform distribution of Ni and Mo in the catalyst, both in the as-prepared and activated forms (Fig. S25, ESI†).
To probe the influence of the temperature on the morphology of the NiMo-AS1 catalyst, we performed electrolysis at 22 °C, 40 °C, and 80 °C for 100 hours and analyzed the surface morphology using SEM with a secondary electron (SE) detector (Fig. S26, ESI†). After electrolysis at 24 °C, the catalyst surface remained relatively heterogeneous, with a mix of rounded particles and undefined structures, suggesting a limited reorganization at this temperature. After operation at 40 °C, the surface underwent a noticeable reorganization and became more homogeneous with a smoother and more uniform morphology. After operation at 80 °C, the catalyst surface acquired distinct layered microstructures. These results indicate substantial surface reconstruction driven by heating.
The X-ray photoelectron spectroscopy (XPS) data of the as-prepared and activated NiMo catalysts (Fig. S27–S29, ESI†), including NiMo-AS1, NiMo-citric, and NiMo-boric, all indicate the presence of Ni and Mo in various oxidation states (Ni: 0, 2, 3; Mo: 4, 6, and more). These data suggest that the surfaces of the catalysts contain various Ni and Mo oxide species, which could be intrinsic to the catalyst or could be formed at the surface of the catalysts due to oxidation during the sample preparation and transfer stages. Because of this complication, a detailed analysis of the XPS data is difficult. Nevertheless, the XPS data clearly show the increase in the percentage of Mo on the surface of our NiMo-AS1 after activation (Fig. S27, ESI†). Such an increase is not observed for the NiMo-citric and NiMo-boric catalysts.
Operando characterization
To probe the composition and structure of the active species of the catalyst under HER, we conducted operando studies of these catalysts. These studies are particularly necessary because we expect the catalysts to be metallic species under the reductive conditions of HER, yet the metals are prone to oxidation outside of such a reducing environment (e.g., in the ex situ characterization as described above). We first performed operando X-ray diffraction (XRD) analysis. A “non-diffracting” substrate is needed for these measurements, so we chose catalysts deposited on carbon paper. The sharp diffraction peaks at 43.8° and 50.7°, assigned to the (121) phase and (310) phase of Ni4Mo, respectively, were observed using the synchrotron radiation instrument. The data indicates a tetragonal Ni4Mo species in the dry state and at the open circuit potential for NiMo-AS1 (Fig. 5A). Under HER, however, two new peaks at 2θ = 33.7° and 60° emerged. This result indicates substantial structure change and the formation of a D1a superlattice of tetragonal Ni4Mo under HER.18–20 Notably, these features are absent for the XRD data of NiMo-citric (Fig. 5B), which indicates only a conventional tetragonal Ni4Mo for the latter.21 As for NiMo-boric, the XRD data suggest an amorphous nature for the catalyst (Fig. 5C). | ||
| Fig. 5 (A) Operando XRD pattern of NiMo-AS1. (B) Operando XRD pattern of NiMo-citric. (C) Operando XRD pattern of NiMo-boric. | ||
Operando X-ray absorption spectroscopy (XAS) measurements were further used to probe the chemical state and structure of the active sites during activation and operation. The operando Ni K-edge X-ray absorption near-edge structure (XANES) spectra and extended X-ray absorption fine structure (EXAFS) of all three samples remained similar during both the activation process and the HER process (Fig. 6 and Fig. S30, ESI†), indicating minimal changes in the Ni sites. The Ni K-edge XANES spectra and EXAFS spectra (Fig. 6 and Fig. S30A–C, ESI†) indicate that the as-deposited NiMo-AS1 and NiMo-citric catalysts have Ni metallic bulk species, whereas NiMo-boric contains mostly NiO species with the presence of both Ni–O and Ni–Ni paths (Fig. 6F). The chemical state of the Ni sites was determined from the first derivatives of the Ni K-edge XANES spectra (Fig. S31 and S32, ESI†). Notably, for NiMo-AS1, the Ni sites remained almost unchanged at a “negative” oxidation state of −0.25 during both the activation and the HER processes (Fig. S33A and S34A, ESI†). This result might be understood by considering charge transfer from Mo. Conversely, for NiMo-citric and NiMo-boric, the chemical state of the Ni sites remained almost unchanged oxidation states at 0 and +1.8, respectively, during both the activation process and the HER process (Fig. S33B, C and S34B, C, ESI†).
 | ||
| Fig. 6 Operando XAS data: (A)–(C)-Ni K-edge XANES spectra of A-NiMo-AS1; B-NiMo-citric; C-NiMo-boric; (D)–(F)-Ni K-edge k3-weighted EXAFS spectra for D-NiMo-AS1; E-NiMo-citric; F-NiMo-boric during activation in 1.0 M KOH. | ||
The operando Mo K-edge XANES of NiMo-AS1 and NiMo-boric indicates dynamic changes in the Mo sites, while NiMo-citric remained similar during both the activation process and the HER process. (Fig. 7A–C and Fig. S35, ESI†) The first derivatives of the Mo K-edge XANES spectra of NiMo-AS1 indicate that Mo sites reduced from approximately +1.5 to around +0.5 during the activation process and remained at approximately +0.5 during the HER process (Fig. S36A, S37A, S38A and S39A, ESI†). The operando Mo K-edge EXAFS spectra of NiMo-AS1 indicate the emergence of Mo–O bonds during the activation process, while the local structure of Mo centers remained similar during HER (Fig. S35D and S36D, ESI†). In contrast, the chemical state of Mo centers in NiMo-citric remained almost unchanged at an oxidation state of −0.5 during both the activation and the HER processes (Fig. S35B, S36B, S37B and S38B, ESI†). Additionally, no Mo–O bonds were formed during the activation and the HER process, which may be induced by the protection of citric acid (Fig. S34B and S35E, ESI†). As for NiMo-boric, the Mo centers were slightly reduced and remained almost stable during the HER process (Fig. S36C, S37C, S38C and S39C, ESI†). Furthermore, there were no Mo–Mo/Ni bonds observed in the Mo K-edge EXAFS spectra, indicating that the Mo sites in NiMo-boric were atomically dispersed with the presence of Mo–N/O/C path only (Fig. S35F, ESI†). A quantitative EXAFS analysis was further performed to characterize the coordination environment of NiMo-AS1 and NiMo-citric (Fig. S40–S55 and Table S5–S12, ESI†). In NiMo-AS1, the coordination number (CN) for Ni–Mo was approximately 1, while that for Mo–Ni was around 3. This result suggests a significant heteroatomic interaction between Ni and Mo, leading to a notable deviation from the ideal model of random distribution. Additionally, the total CNs for Ni–(Ni + Mo) was significantly lower than that of Ni foil, indicating a poor crystalline nature on the catalytic surface through the total-fluorescence-yield mode (see materials and methods). Notably, one can see that the interatomic distance for Mo–Ni is similar to those of Ni–Mo and Ni–Ni cases, which further supports the suggested charge transfer between Ni and Mo.
 | ||
| Fig. 7 Operando XAS data: (A)–(C)-Mo K-edge XANES spectra of A-NiMo-AS1; B-NiMo-citric; C-NiMo-boric; (D)–(F)-Mo K-edge k3-weighted EXAFS spectra for D-NiMo- AS1; E-NiMo-citric; F-NiMo-boric during activation in 1.0 M KOH. | ||
Furthermore, the CN of Mo–O in NiMo-AS1 increased from 0.5 to around 1.5 during the activation process, and the structure remained stable during the HER. The CN of Mo–Mo in NiMo-citric was around 2, while there were no Mo–Mo bonds observed in NiMo-AS1. After activation, Mo is reduced towards metallic, and according to EXAFS (Fig. 7), the CN of Mo–Ni increases by about 0.6 (see Table S6, ESI†) during activation. Moreover, the CN of Mo–O increases substantially, and the corresponding bond distance decreases by 0.07 Å in this process. In contrast, the EXAFS fitting results indicate the Mo species in NiMo-citric is metallic in nature even in the as-prepared sample; during activation and HER, the Mo species remain similar, while no Mo–O path can be revealed in this catalyst (Table S9–S12, ESI†).
Discussion of the electrodeposition method
The common methods for electrodeposition of NiMo catalysts involve cathodic deposition in a chemical bath of Ni and Mo salts as well as an additive such as citric acid or boric acid.22 The widely accepted mechanism of the electrodeposition of NiMo involves the following eqn (1)–(4) | (1) |
 | (2) |
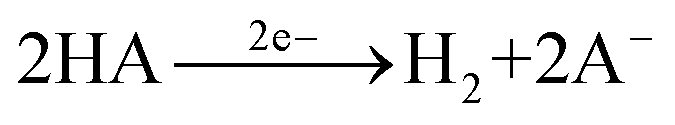 | (3) |
| 2H2 + NiMoO2 → NiMo + 2H2O | (4) |
As HER occurs, the local pH of the deposition bath increases so that precipitation of Ni and Mo oxides might occur, especially for a poorly buffered solution. Thus, the formation of NiMo oxides or hydroxides from the bath containing boric acid can be attributed to the latter’ poor buffering capacity. Such a problem can be circumvented by a complex agent such as citric acid that solubilizes the Ni and Mo precursors.23,24 For the deposition of our catalyst, NH4Cl plays a key role here in that the anodic Cl2 evolution and subsequent dissolution leads to a decrease, rather than an increase, in pH (Fig. S5, ESI†). This effect allows for a deposition current density to be an order of magnitude higher than previous methods. Moreover, NH4+ cation provides a better source of proton than water, which explains why KCl could not be used as the electrolyte.
The active species
Although NiMo catalysts have long been studied for HER, only very few reports studied the composition and structure of these catalysts.18,25–28 These studies were done ex situ on as-prepared catalysts, and on rare occasions, on post-catalytic samples. Here, by in situ and operando XRD and XAS, we revealed the compositions and structures of the three NiMo catalysts in the as-prepared state and during catalysis. We show that the particular deposition method described here results in an unconventional Ni4Mo catalyst with D1a superlattice distortions that might facilitate the Mo migration to the surface. Upon catalysis, this catalyst undergoes surface re-organization. Some Mo atoms in the bulk migrate to the surface and become MoOx species (Fig. 8). This process is similar to the re-organization of a thermally produced Ni4Mo catalyst during HER.25 We can hypothesize a mechanism of such re-organization: (1) the presence of absorbed H at the surface lowers the surface energy of Mo, making it migrate to the surface; (2) the OH− and air at the surface oxidizes some Mo atoms into Mo(OH)x clusters. Further studies such as DFT calculations are needed to probe this mechanism. The operando measurements further show that such an active species remains stable during catalysis. Additionally, our measurements identify the active forms of NiMo-citric and NiMo-boric, which were also previously unclear. | ||
| Fig. 8 Schematic diagram of the surface re-organization of the catalyst during hydrogen evolution reaction. | ||
Activity and origin
The geometric activity of our NiMo-AS1 catalyst on Ni mesh, when determined in a 3-electrode electrochemical cell, is among the highest in non-PGM HER catalysts (Fig. S13, ESI†). The geometric activity of a catalyst may originate from multiple factors, among which is a high surface area. The ECSA measurements show a small roughness factor for our catalyst, ruling out surface area as the main contribution to its high activity. Both the roughness factor- and mass-averaged activity of our NiMo-AS1 catalyst are much higher than those of reference NiMo catalysts, indicating a higher intrinsic activity of the former.A more important parameter to compare for catalyst applications is the performance of devices using such catalysts. Indeed, our catalysts were designed for applications with AEMWEs in mind. Gratifyingly, comparing the device performance with state-of-the-art PGM-free catalysts demonstrates the superiority of our catalyst16,27–33-(Fig. 9). For this comparison, we focus on electrolysis data from relatively long-term operations (>100 hours) rather than those from transient IV measurements, as the latter does not reflect the steady-state performance of the systems.
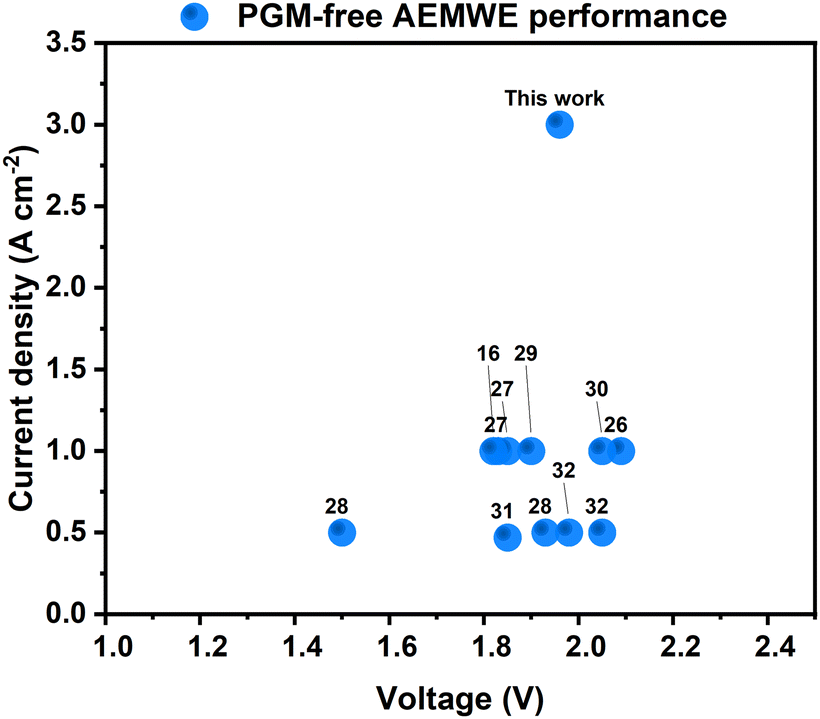 | ||
| Fig. 9 Comparison of the state-of-the-art PGM-free AEMWE cells. The numbers in the figure refer to the numbers of the references. | ||
The stable operation of our AEMWE at 3 A cm−2 is particularly noteworthy, as it had not been reported previously for any PGM-free AEMWEs. Our group recently reported that cathode ionomers can improve the performance of AEMWEs.16 Thus, appropriate ionomers might further improve the current AEMWEs using NiMo-AS1 as the HER catalyst. This objective is subject to future work.
We think the superior intrinsic activity of NiMo-AS1 might originate from its structure. Probably due to the D1a superlattice structure, our Ni4Mo pre-catalyst is subject to a re-organization in the activation process in which some bulk Mo atoms migrate to the surface to form MoOx clusters. Analogous Ni4Mo/MoOx assembly catalysts have been prepared by alternative methods34 such as growing Ni4Mo on MoOx nanosheet35 or by making NiMoO4 and subsequent annealing in a reductive atmosphere.26 Mechanistic studies, primarily based on DFT computations, suggest that the MoOx species facilitate the water dissociation to provide proton for the hydrogen adsorption on the Ni4Mo surface26,28,34 (the Volmer step, Fig. 10). In the absence of an oxide, the water dissociation in an alkaline medium is known to be difficult for metals. We expect a similar explanation for the superior activity of our catalyst described here.34
 | ||
| Fig. 10 Schematic diagram of synergetic catalysis of alloy–oxide interface structures, in which the molybdenum oxide promotes water dissociation, and the alloy accelerates hydrogen combination. | ||
Conclusions
We have developed a simple electrodeposition method for a NiMo catalyst that exhibits superior geometric and intrinsic activity at industrially relevant high current densities in an alkaline medium compared to other NiMo and, in general, other PGM-free HER catalysts. The catalyst can be applied in AEMWEs, delivering device performance comparable to Pt/C. Moreover, the applicability of this catalyst in AEMWE is demonstrated at a current density as high as 3 A cm−2. Operando characterization reveals the active form of the catalyst to be an unconventional Ni4Mo species whose surface is decorated by MoOx species. The active form remains stable during catalysis, and its high activity originates from the cooperative action of the Ni4Mo and MoOx components. Thanks to its simple and inexpensive fabrication process, as well as its demonstrated activity and stability in AEMWE, this catalyst has the potential to be used for large-scale green hydrogen production.Materials and methods
Materials synthesis
Nickel chloride (NiCl2), ammonium chloride (NH4Cl), and ammonium heptamolybdate tetrahydrate (NH4)6Mo7O24 4H2O) were of analytical grade and were purchased from Carl Roth Chemicals and used as received. Nickel and Pt meshes with an open area of 16% and a purity of 99.99% were purchased from Goodfellow, U.K. Toray TGP-H-060 carbon paper were from Fuelcellstore. The counter electrode used in electrodeposition was a graphite rod purchased from Redoxme. Electrodeposition of NiMo-AS1 was performed in a mixed solution of 0.1 M NiCl2, 5.25 × 10−4M (NH4)6Mo7O24 × 4H2O, and 2 M NH4Cl with an initial pH of 4.9. No buffering agent or complexing agent was used. The electrodeposition was carried out at a constant cathodic current (−1.5 A cm2 for 1000 s) and room temperature, using the nickel mesh, nickel paper; nickel foam, and C paper as a working electrode and a graphite rod as a counter electrode under N2 protection without stirring as depicted in Fig. S56, ESI.† The deposition conditions, such as electrolyte composition, current density, and deposition time, were optimized as shown in ESI.† NiMo-boric samples were electrodeposited from a sulfamate solution that consisted of 325 g L−1 Ni(II) sulfamate, 30 g L−1 H3BO3, and 5 g L−1 Na2MoO4 with an initial pH of 4.2. After adding the molybdate precursor, precipitates started to be observed in the solution. Therefore, the pH was decreased to 1.7 using 1 M HCl. Electrodeposition was carried out by application of a bias sufficient to produce a cathodic current density of 200 mA cm−2 for 500 s. Depositions were performed in air, in a one-compartment cell, using a large-area Ni counter electrode and an Ag/AgCl reference electrode. NiMo-citric samples were deposited from nickel chloride hexahydrate (NiCl2·6H2O), nickel sulfate hexahydrate (NiSO4·6H2O), citric acid (C6H8O7) sodium molybdate dihydrate (Na2MoO4·2H2O), ammonia (NH3·H2O). The initial pH of the electrodeposition bath was 4. The electrodeposition was carried out at a constant cathodic current of 1 A cm2 for 600 s at room temperature, using the nickel mesh/C paper as a working electrode and a graphite rod as a counter electrode. For the AEMWE measurements, the anode was a NiFe self supported catalyst previously reported and characterized by our lab.16Materials characterization
SEM, corresponding elemental mapping, and EDX analysis were carried out with a Merlin Column II (Carl Zeiss) system. HRTEM was performed using a JEM-F200 (Jeol) operating at an accelerating voltage of 200 kV. XPS experiments were carrthe ied out on Axis Supra (Kratos) system using Al Kα radiation. XRD patterns were recorded on a PANalytical X'Pert X-ray diffraction system (40 kV, 15 mA, Cu Kα radiation). A Gamry Reference 3000 electrochemical instrument was used to measure the electrochemical activity with an automatic iR compensation of 90% at ambient temperature and pressure. ICP-MS was performed on an ICP-MS NexIon 350 (PerkinElmer). X-ray absorption spectroscopy (XAS), which included XANES and EXAFS, was obtained to determine the oxidation state and local structure of samples. The operando XAS measurements at the Ni K-edge (8333 eV) and Mo K-edge (20000 eV) were conducted in total-fluorescence-yield (TFY) mode by the Lytle detector. The scan ranges for the Ni K-edge, and the Mo K-edge were 8133–8933 eV and 19800–20600 eV, respectively. The XAS data were calibrated using an E0 value of 8333.0 eV for Ni foil and 20000.0 eV for Mo foil, referencing their respective first inflection points in absorption K-edge. The XAS data were analyzed using standard procedures, including background subtraction and edge height normalization. EXAFS spectra were obtained using the Fourier transform on k3-weighted oscillations to determine the local environment of the Ni and Mo atoms. The operando XRD measurements were conducted using a wavelength of 0.6889 Å (18 keV). The above operando measurements were performed at the 12B2 Taiwan beamline (SPring-8, Japan) of the National Synchrotron Radiation Research Center (NSRRC), operated at an 8.0 GeV storage ring with a constant current of ∼99.5 mA. A customized Teflon cell, which utilized Kapton tapes as an X-ray window, was employed for conducting operando measurements at ambient air/temperature in a 1.0 M KOH electrolyte. The sample electrodeposited on the carbon paper was used as the working electrode, while a graphite rod and Ag/AgCl in a 3 M KCl solution were employed as the counter and reference electrodes, respectively.
Electrochemical measurements
In a three-electrode electrochemical setup, linear sweep voltammetry (LSV) with a scan rate of 1 mV s−1 was done to establish and characterize the electrochemical performance of each substrate. The activity of the catalysts was measured in a saturated 1 M KOH N2 solution using an Ag/AgCl (3 M KCl) reference electrode and graphite as the counter electrode. The RHE calibration of the Ag/AgCl electrode was performed in an H2-saturated 1 M KOH standard solution (pH = 14) at 23 °C as described elsewhere.36ERHE = EAg/AgCl + 1.016. The potentiostat automatically did the IR drop correction.AEMWE assembly procedure: 40 μm-thick NovaMea's Branion AEMs with 1 mol% of 1,3,5- triphenylbenzene monomer (b-PTP-1, IEC = 2.81 mmol g−1) were used in this study for performance tests. The cathode PGM catalyst was Pt (weight fraction of 40%) supported on Vulcan XC-72R (Alfa Aesar HiSPEC 4000). The ink dispersions were sprayed onto Toray TGP-H-060 PTLs with PTFE wetproofing (weight fraction of 5%). The catalyst loadings were kept at 0.6 mgPt cm−2. The PGM-free self-supported NiFe-anode and NiMo-AS1 cathode were directly used in the AEMWE without any further treatment.
The membrane was soaked in 1 M KOH for 12 h prior to use and then the wet membrane was placed with OER and HER electrodes to prepare the PGM-free MEA. Subsequently, the MEAs were loaded into a 5 cm2 Dioxide Materials AEMWE hardware between two single-pass serpentine flow nickel plates. The MEA was sealed and compressed with 10 mil (270 μm) and 6 mil (160 μm) PTFE for anode and cathode, respectively, at 2.5 N m torque, resulting in 20–25% pinch. RND lab DC power supply was used to control the operating current density. The IV curves were recorded by holding each current density for 5 seconds and measuring the potential, starting from 0 A and increasing in increments of 0.3 A up to 2.3 A. A home-assembled temperature controller and sensor-controlled cell temperature at 80 °C. A Golander peristaltic pump controlled the electrolyte flow rate at 1 mL min−1.
Author contributions
Ariana Serban – conceived and designed the catalyst; collected the data; performed the analysis; wrote the text. Meng-Ting Liu – performed and analyzed XAS experiments. Nanjun Chen – synthesized the anionic exchange membranes used in this work. Hao Ming Chen – analyzed XAS data and supervised XAS study; wrote the XAS section. Xile Hu – directed the research; analyzed data; wrote and revised the paper.Data availability
The data supporting this article have been included as part of the ESI.† The data can be also downloaded at Zenodo: https://doi.org/10.5281/zenodo.14276766.Conflicts of interest
The authors declare the following competing financial interest(s): A. S. and X. H. are inventors of a patent application that includes the HER catalyst used in this paper. The branched PAP AEM used in this work is commercialized under the brand name Branion by an EPFL Startup NovaMea SA, of which X. H. has a financial interest.Acknowledgements
The authors from EPFL gratefully acknowledge the financial support of the Swiss State Secretariat for Education, Research and Innovation (SERI) under contract number 22.00117. The SERI support is the Swiss contribution to the participation of Swiss partners in the EU project ANEMEL (the European Union's Horizon Europe research and innovation programme under Grant Agreement No. 101071111). We thank Dr Qiucheng Xu for the discussions.Notes and references
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef PubMed.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122(13), 11830–11895 CrossRef PubMed.
- C. Santoro, A. Lavacchi, P. Mustarelli, V. Di Noto, L. Elbaz, D. R. Dekel and F. Jaouen, ChemSusChem, 2022, 15(8), e202200027 CrossRef PubMed.
- N. Chen, S. Y. Paek, J. Y. Lee, J. H. Park, S. Y. Lee and Y. M. Lee, Energy Environ. Sci., 2021, 202114(12), 6338–6348 RSC.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef PubMed.
- Y. Xu, R. Wu, J. Zhang, Y. Shi and B. Zhang, Chem. Commun., 2013, 49, 6656–6658 RSC.
- L. A. King, M. K. A. Hubert, C. Capuano, J. Manco, N. Danilovic, E. Valle, T. R. Hellstern, K. Ayers and T. F. Jaramillo, Nat. Nanotechnol., 2019, 14, 1071–1074 CrossRef PubMed.
- C. G. Morales-Guio., L. A. Stern and X. Hu., Chem. Soc. Rev., 2014, 43(18), 6555–6569 RSC.
- Y. Yang, P. Li, X. Zheng, W. Sun, S. X. Dou, T. Ma and H. Pan, Chem. Soc. Rev., 2022, 51(23), 9620–9693 RSC.
- L. Zhang, Z. Shi, Y. Lin, F. Chong and Y. Qi, Front. Chem., 2022, 10, 866415 CrossRef.
- M. Jin, X. Zhang, S. Niu, Q. Wang, R. Huang, R. Ling, J. Huang, R. Shi, A. Amini and C. Cheng, ACS Nano, 2022, 16(8), 11577–11597 CrossRef.
- S. H. Park, D. T. To and N. V. Myung, Appl. Catal., A, 2023, 651, 119013 CrossRef.
- Y. Tsuru, M. Nomura and F. R. Foulkes, J. Appl. Electrochem., 2002, 32, 629–634 CrossRef.
- H. Liu, D. Liu, X. Cheng, Z. Hua and S. He, Mater. Des., 2022, 224, 111427 CrossRef.
- A. Serban and X. Hu, Nickel Molybdenum Catalysts and Their Methods of Preparation, EP23172741.3, 2024.
- Y. Zheng, A. Serban, H. Zhang, N. Chen, F. Song and X. Hu, ACS Energy Lett., 2023, 8(12), 5018–5024 CrossRef.
- N. Chen, Q. Jiang, F. Song and X. Hu, ACS Energy Lett., 2023, 8(10), 4043–4051 CrossRef.
- H. M. Tawancy, Metallogr., Microstruct., Anal., 2019, 8, 591–606 CrossRef.
- H. M. Tawancy, Metallogr., Microstruct., Anal., 2014, 3, 126–130 CrossRef.
- R. H. Banerjee, A. Arya and H. Donthula, Trans. Indian Inst. Met., 2022, 75, 949–957 CrossRef.
- Y. Duan, Z. Y. Yu, L. Yang, L. R. Zheng, C. T. Zhang, X. T. Yang, F. Y. Gao, X. L. Zhang, X. Yu, R. Liu and H. H. Ding, Nat. Commun., 2020, 11, 4789 CrossRef.
- J. R. McKone, E. L. Warren, M. J. Bierman, S. W. Boettcher, B. S. Brunschwig, N. S. Lewis and H. B. Gray, Energy Environ. Sci., 2011, 4(9), 3573–3583 RSC.
- E. Beltowska-Lehman and P. Indyka, Thin Solid Films, 2012, 520(6), 2046–2051 CrossRef.
- E. Chassaing, K. Vu Quang and R. Wiart, J. Appl. Electrochem., 1989, 19, 839–844 CrossRef.
- W. Du, Y. Shi, W. Zhou, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60(13), 7051–7055 CrossRef.
- Y. Y. Chen, Y. Zhang, X. Zhang, T. Tang, H. Luo, S. Niu, Z. H. Dai, L. J. Wan and J. S. Hu, Adv. Mater., 2017, 29(39), 1703311 CrossRef PubMed.
- J. Hou, Y. Wu, S. Cao, Y. Sun and L. Sun, Small, 2017, 13(46), 1702018 CrossRef PubMed.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8(1), 15437 CrossRef PubMed.
- I. Vincent, E.-C. Lee and H. Kim, RSC Adv., 2020, 10(61), 37429–37438 RSC.
- B. Motealleh, Z. Liu, R. I. Masel, J. P. Sculley, Z. R. Ni and L. Meroueh, Energies, 2021, 46(5), 3379–3386 Search PubMed.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10(40), 9165–9181 RSC.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Front. Chem., 2018, 6, 263 CrossRef.
- L. Wang, T. Weissbach, R. Reissner, A. Ansar, A. S. Gago, S. Holdcroft and K. A. Friedrich, ACS Appl. Energy Mater., 2019, 2(11), 7903–7912 CrossRef CAS.
- M. Luo, J. Yang, X. Li, M. Eguchi, Y. Yamauchi and Z. L. Wang, Chem. Sci., 2023, 14(13), 3400–3414 RSC.
- Y. An, X. Long, M. Ma, J. Hu, H. Lin, D. Zhou, Z. Xing, B. Huang and S. Yang, Adv. Energy Mater., 2019, 9(41), 1901454 CrossRef CAS.
- J. A. Z. Zeledón, A. Jackson, M. B. Stevens, G. A. Kamat and T. F. Jaramillo, J. Electrochem. Soc., 2022, 169(6), 066505 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04528a |
| This journal is © The Royal Society of Chemistry 2025 |