
Achieving superior oxygen evolution of perovskite via phase transition and electrochemical reconstruction strategy†

Abstract
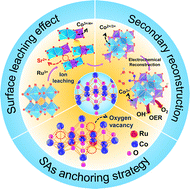
Surface reconstruction is an effective strategy to improve the OER performance of perovskites. However, understanding the reconstruction kinetics of perovskites and revealing real active sites for the OER remain elusive. Herein, we propose a secondary reconstruction strategy including phase transformation and electrochemical reconstruction to promote the electrocatalytic performance of perovskite. The as-prepared perovskite SrCoO2.52 is first reconstructed into Ru atomically dispersed spinel Co3O4 (RuSA-Co3O4) by selective leaching, whose surface is further reconfigured into CoOOH through electrochemical reconstruction. The RuSA-Co3O4/CoOOH catalyst with massive oxygen vacancies exhibits an ultralow overpotential of 175 mV at 10 mA cm−2 and maintains stability for up to 1000 h, which is the best perovskite-derived electrocatalyst to date. In situ XAS analysis and DFT calculations indicate that the valence states of Co species and isolated Ru single atoms are optimized and combined with –O ligands to form Co–O–O and Ru–O–O active sites. The secondary reconstruction can effectively regulate the electronic structure of the active metals thus providing the optimal adsorption energy for the reaction intermediates to promote the charge transfer in the OER process.
 Please wait while we load your content...
Please wait while we load your content...

