
Solvating lithium and tethering aluminium using di-coordination-strength anions for low-temperature lithium metal batteries†
Long
Kong  *a
*a
*a
Abstract
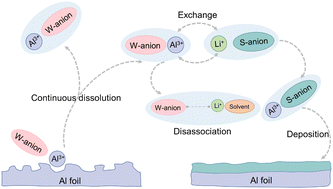
Lithium bis(fluorosulfonyl)imide (LiFSI) corrodes aluminium (Al) current collector, but this corrosion can be impeded using di-coordination-strength anions. This concept adopts the weakly coordinated anion (FSI−) to exert high ionic transport kinetics, and the strongly coordinated anion (nitride, NO3−) to stabilize the Al foil surface. Such simultaneous Li solvation and Al tethering, highlight the importance of solvation chemistry to mediating Li transport and Al corrosion.
 Please wait while we load your content...
Please wait while we load your content...

