
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the inherent stability of perovskite solar cells through chalcogenide-halide combinations
Cheng
Wang
a,
Riming
Nie
 *ab,
Yiming
Dai
a,
Huanyu
Tai
a,
Bingjian
Zhu
a,
Luyao
Zhao
a,
Yong
Wu
a,
Wanlin
Guo
*a and
Sang Il
Seok
*b
*ab,
Yiming
Dai
a,
Huanyu
Tai
a,
Bingjian
Zhu
a,
Luyao
Zhao
a,
Yong
Wu
a,
Wanlin
Guo
*a and
Sang Il
Seok
*b
aState Key Laboratory of Mechanics and Control of Mechanical Structures, Key Laboratory for Intelligent Nano Materials and Devices of the Ministry of Education, Institute for Frontier Science, College of Mechanical and Electrical Engineering, Nanjing University of Aeronautics and Astronautics, Nanjing 210016, P. R. China. E-mail: rmnie@nuaa.edu.cn; wlguo@nuaa.edu.cn
bDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology, 50 UNIST-gil, Eonyang-eup, Ulju-gun, Ulsan 44919, Republic of Korea. E-mail: seoksi@unist.ac.kr
First published on 15th January 2024
Abstract
Perovskite solar cells have attracted much attention due to their rapidly increasing power conversion efficiency, however, their poor inherent long-term stability limits their commercialization. One of the reasons for the low stabilities of halide perovskites can be attributed to the weak ionic binding energy between the monovalent halide and divalent lead ions. Here, we present an overview of several strategies for combining chalcogenides and halides to enhance the inherent stability of perovskite materials. Firstly, incorporating chalcogenides into halide perovskite materials can improve their ionic binding energies. However, it is important to note that this method can only improve their stability somewhat. Secondly, the exploitation of chalcogenide perovskite materials has the potential to significantly improve the inherent stability of perovskite materials. Nevertheless, their efficiencies remain relatively low, and the device fabrication needs high-temperature annealing and expensive experiments. Thirdly, the concept of combining chalcogenides and halides to create new perovskite materials shows promise, however, there is still limited research in this direction. Finally, we offer a perspective on the future development direction and trends of these three research areas.
Broader contextAs the world increasingly looks towards sustainable energy sources, perovskite solar cells have emerged as a promising alternative to traditional silicon-based photovoltaics, primarily due to their remarkable rise in power conversion efficiency. Nevertheless, their journey towards widespread commercialization faces a critical hurdle - the long-term stability of these materials. The precarious nature of halide perovskites, exemplified by their susceptibility to environmental factors, is a significant roadblock. It stems from the weak ionic binding energy between the monovalent halide and divalent lead ions within their crystal lattice. Within this context, this perspective provides an insightful exploration of strategies aimed at fortifying the stability of perovskite materials. The multidimensional landscape of potential solutions includes the introduction of chalcogenides into halide perovskites, a method that demonstrates notable improvements in ionic binding energies, although it does not entirely resolve the stability issues. On the other hand, delving into the unique properties of chalcogenide perovskite materials holds promise for significantly bolstering stability. Yet, the inherent trade-offs between stability and efficiency, along with the resource-intensive fabrication processes, must be carefully weighed. Additionally, the innovative approach of blending chalcogenides and halides to forge novel perovskite materials is a burgeoning frontier. However, it still remains relatively uncharted, necessitating further exploration. Consequently, the potential for a more stable, efficient, and cost-effective photovoltaic future is a tantalizing prospect that resonates with the broader goals of environmental sustainability and reduced dependence on fossil fuels. This broader context underscores the significance of ongoing research and the potential impact of the directions outlined in this overview. |
1. Introduction
In 2009, Miyasaka et al. introduced perovskite-sensitized solar cells as a noble alternative to traditional dye-sensitized solar cells (DSSCs), offering the potential for more efficient light absorption.1 Initially, these cells demonstrated a low efficiency of only 3.81%. The chemical composition of halide perovskite materials is represented by the formula ABX3. In this structure, the A site accommodates a positive monovalent metal cation, the B site hosts a divalent cation, and the BX6 arrangement exhibits classical octahedral connectivity.2 Typically, monovalent cations at the A site include MA+, FA+, or Cs+, while divalent cations at the B site are represented by Pb2−, Sn2−, or Ge2−, and halides at the X site are Cl−, Br−, or I−. The perovskite crystal structure comprised an A+ ion at the center of the BX6 octahedron, with the B2− ion exhibiting six-way coordination. Surrounding these ions are octahedra formed by halide anions, creating a unique perovskite crystal structure.3 Furthermore, these structures allow for the accommodation of various other elements, significantly expanding the range of perovskite composition. However, a challenge arises as most perovskite materials are susceptible to dissolving in the liquid electrolyte used in DSSCs, leading to rapid degradation in the device performance. To address this issue, researchers replaced the liquid electrolyte of DSSCs with a solid-state hole-transporting material (HTM), resulting in the fabrication of solid-state perovskite-sensitized solar cells.3 In 2014, Seok et al.4 proposed an anti-solvent method to deposit high-quality perovskite films, leading to significant advancements in real perovskite solar cells (PSCs). Subsequently, the anti-solvent method became a prominent technique for fabricating high-efficiency and stable PSCs. Notably, planar PSCs have recently made substantial progress, achieving a certified efficiency of 26.1%.5The advancement in PSCs increased their efficiency to around 26%5,24–33 through structural optimization. These improvements involve mesoporous titanium dioxide, the design of organic and inorganic carrier transport layers, adjustment of doping and composition with various additives, and enhancement in device structures, including the development of laminated solar cells. All these efforts aim to design and synthesize high-efficiency PSCs with long-term stability. However, there remains a considerable gap in stability compared to monocrystalline silicon inorganic solar cells. Therefore, how to efficiently design and synthesize long-term stable perovskite materials is the main research direction of researchers.
First, this review summarizes the work on improving stability for halide PSCs with high power conversion efficiency. Secondly, we discuss chalcogenide-based solar cells' long-term stability and high hydrophobicity. Thirdly, it is envisaged that an effective strategy to improve the stability of perovskite materials is to mix chalcogenide and halide perovskites. Finally, we propose the future research direction for mixed chalcogenide and halide PSCs.
2. Halide perovskite solar cells
Undoubtedly, in recent years, with the continuous improvement of the efficiency of halide PSCs, their poor chemical and environmental stability has also been exposed. After exposure for a long time, the crystal structure of halide perovskites will undergo phase change and decomposition, resulting in degraded performance. In addition, these solar cells are very sensitive to humid environments and oxygen, which further limits their stability in practical applications. This is a major factor limiting the commercialization of perovskites. Therefore, there is an urgent need to find stable alternatives in the natural environment, and researchers have adopted various strategies. Among them, the introduction of more stable sulfur elements has attracted much attention. Considering the atomic structure of chalcogenides and the stability of lone pair electrons, the interaction of metals with sulfur atoms in chalcogenides is more covalent, which makes the overall covalent bond of the material stronger, thereby improving stability.44 The introduction of chalcogenides into PSCs can significantly improve the device efficiency and stability.2.1. Chalcogenides in hole transport layers
One of the primary areas of research involves the incorporation of sulfur into the hole transport layer of PSCs. Considering the dipole moment interaction of the lone pair electrons of chalcogenides (such as sulfur S and oxygen O) with the unpaired Pb2+ of perovskites, the long-term stability of PSCs can be significantly improved. A typical application is methoxy interactions in trainline structures such as spiro-OMeTAD, which has been widely used as various doped/undoped hole transport layers, where derivatives substituted by ortho-methoxy groups exhibit higher LUMO (Lowest Unoccupied Molecular Orbital) levels and can effectively block electrons. At the same time, it has good solubility, weak absorption in the visible light range, good film-forming ability, and high hole mobility. However, it has the disadvantages of high processing cost, difficult purification, and low conductivity. It is necessary to improve its electrical performance by doping ion salts, which will cause problems with device stability.45 Pentaerythritol tetra (3-mercaptopropionate) (PETMP), an aliphatic crosslinker containing four mercaptan groups, has been employed to form anti-Markovnikov sulfide through mercaptan-alkene reaction. This process results in the production of cross-linked HTLs, significantly enhancing their photothermal stability even without additional packaging.46–48 Additionally, various approaches exist to bolster perovskite stability by establishing molecular interactions with chalcogenide elements. These strategies find extensive application in diverse types of hole transport materials.49 In 2021, Wong et al. deposited bifunctional aluminum-doped CuS films in p–i–n structure PSCs as the HTL, which not only played the role of an HTL but also improved the crystallization of perovskites at the interface. Characterization of Urbach energy, light intensity, and open circuit voltage showed that a better-quality interface was formed in the sulfide HTL–perovskite heterojunction. The degradation behaviour of sulphide-HTL-based perovskite devices was studied. It has been observed that the equipment remains at close to 95% of its initial efficiency after 2 weeks in a controlled environment50 (Fig. 1c).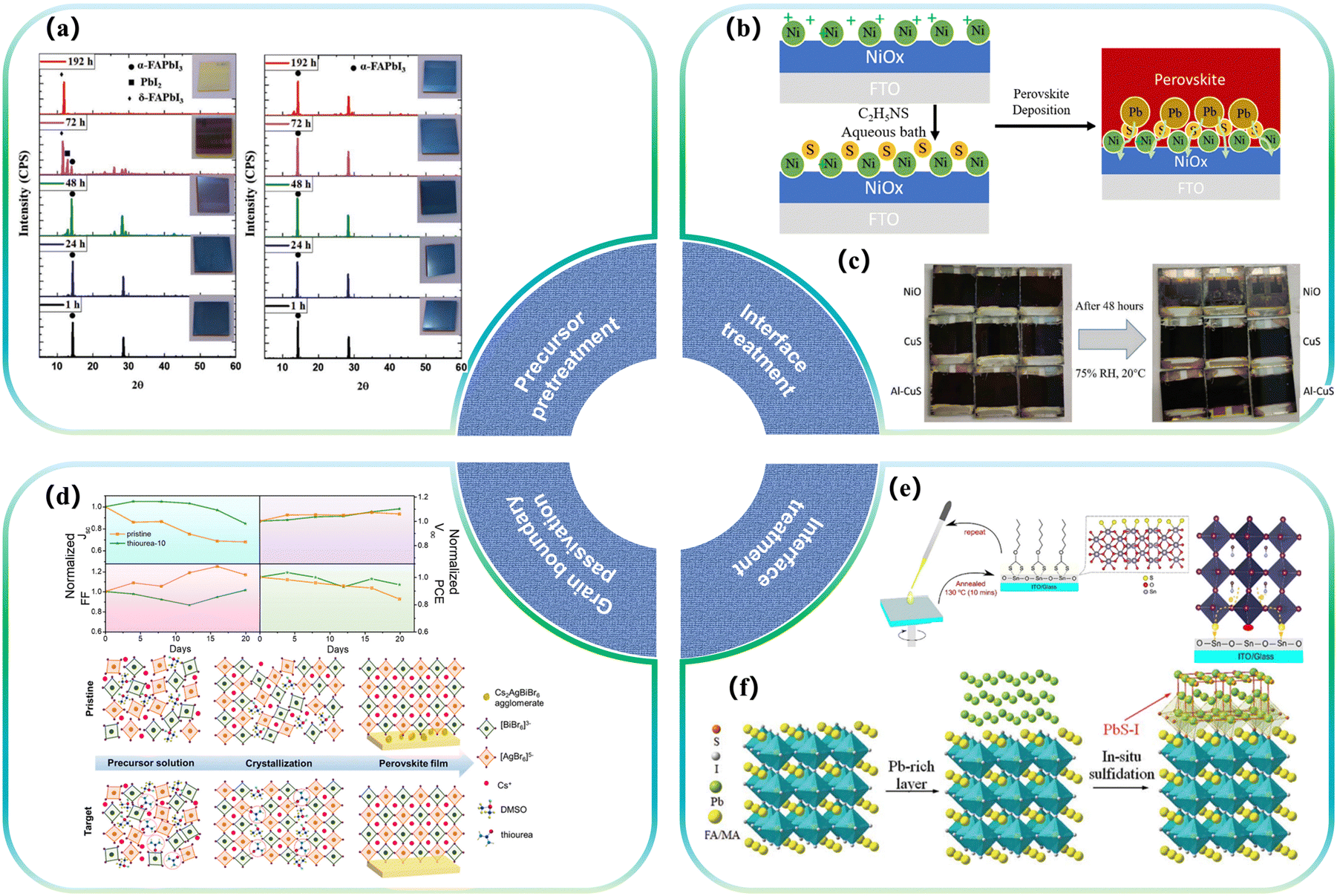 | ||
| Fig. 1 Effect of partial sulfides on the modification of perovskites. (a) XRD patterns of films deposited from a precursor solution aged for times ranging from 1 to 192 h without (left) and with (right) sulfur additives.51 Copyright 2019, Advanced Energy Materials. (b) Schematic illustration of the S doping process on the NiO surface.52 Copyright 2020, Solar RRL. (c) The visible degradation of the devices after storing them in 75% RH and 20 °C humidity for 48 h; the left-hand side shows the initial images of the devices, and the right-hand side shows the device images after 48 h. The top row of the devices is NiO, the middle row is CuS, and the bottom row is Al-CuS.50 Copyright 2020, Advanced Functional Materials. (d) Normalized performance parameters for the pristine Cs2AgBiBr6, thiourea-10 cells in the stability test under ambient conditions without any encapsulation, and the proposed mechanism of Cs2AgBiBr6 film formation assisted by a thiourea additive.53 Copyright 2022, Journal of Colloid and Interface Science. (e) Sulfur-functionalized electron transport layer formed by xanthate annealing and device interfacial structure.54 Copyright 2018, ChemSusChem. (f) Schematic diagram of SST vulcanization.55 Copyright 2022, Science. | ||
2.2. Chalcogenides in perovskite layers
Of course, there are many improvements and modifications to the use of chalcogenide compounds in the preparation of PSCs. These sulfide elements can effectively promote the crystallization performance and stability of halide PSCs. The most widespread impact was the introduction of sulfur (S8) into the halide perovskite precursor discovered by Seok et al. in 2019, which greatly stabilized the precursor solution due to amine-sulfur coordination without affecting the PCE of the derived PSC. At the same time, the introduction of sulfur to stabilize the precursor solution can improve the stability of PSCs (Fig. 1a).51 In 2020, Chen et al. prepared perovskite films to reduce defects by using a lead indicator (dithizone) as an additive to the perovskite layer. Dithizone can slow down the crystallization rate of perovskite films, passivate defects, and enhance the structural stability of perovskite films by coordinating with lead atoms. As a result, dithizone-doped devices have excellent PCE and stability.56 In 2022, Zhou et al. studied the effects of sulfur cations on the evolution of two-dimensional Ruddlesden–Popper (2DRP) perovskite intermediates and photovoltaic performance. The introduction of sulfur cations leads to preferential intermediate conversion of perovskites and improvement of film quality. Due to reduced recombination and enhanced charge transfer, the resulting device provides a championship efficiency of 19.08% at room temperature and an optimal efficiency of 20.52% at 180 K. What's more, the unpackaged device maintains an 84% initial efficiency over 1000 h with a maximum power point (MPP) track of 40 °C.57Shao et al. delayed the crystallization of the Cs2AgBiBr6 perovskite by introducing Lewis's base additive into the precursor solution. The addition of the strongly coordinated thiourea additive and sulfur donor led to the formation of Lewis acid–base adducts, delayed the crystallization process of Cs2AgBiBr6 crystals, improved the quality of Cs2AgBiBr6 films, reduced defect density, inhibited charge carrier recombination, and showed excellent stability. After 20 days of storage in air, the PCE retention was 95% (Fig. 1d).53 In 2023, Finkenauer et al. modified the crystal growth process of halide PSCs by introducing phenylmercaptan/selenol. In the second step, the combination of phenylselenol and amine ligands in the precursor solution dramatically changes the chemistry of the solution, increasing the conversion rate of PbI2 to films with high crystallization and orientation of α-FAPbI3.58 In conclusion, this method plays an important role in improving the stability and efficiency of the device.
2.3. Chalcogenides in interface layers
In 2018, Hayase et al. reported a strategy to effectively passivate charge traps and reduce interfacial recombination by functionalizing the surface of SnO2 with sulfur. The results show that the sulfur-functionalized atoms can coordinate with the undercoordinated Pb2+ near the interface, and the interfacial sulfur functionalization accelerates the interfacial charge transfer kinetics and reduces the traps in the perovskite block. The non-sealed interface of sulfur-functionalized solar cells also showed a considerable delay in solar cell degradation, degrading only 10% after 70 days of air storage54 (Fig. 1e). In 2020, Yang et al. introduced a sulfur doping strategy, which modifies the NiO surface through ion exchange reaction through simple and scalable chemical bath deposition technology, thereby greatly improving the photovoltaic performance of derived devices. Systematic studies were performed in which it was demonstrated that sulfur doping can generate favorable interfacial energy and reduce VOC loss. Pb–S interaction also improves the quality of perovskite films, reduces the number of grain boundaries, reduces the formation of structural defects, and improves the conductivity and stability of directly adjacent perovskite layers52 (Fig. 1b).Next, Jiang et al. introduced lead sulfide (PbS) into the perovskite film surface by performing an in situ reaction with thioacetamide (TAA) in solution. The PbS layer significantly reduces the trap density of the perovskite film and improves the charge extraction efficiency of the perovskite to the external charge transfer layer. In addition, it was also found that the water-insoluble PbS layer can effectively protect the perovskite film from water penetration. As a result, PSCs showed a stable output efficiency of 22.04% and high stability.59 In 2022, Fang et al. constructed a stable perovskite heterojunction for inverted solar cells by sulfidation of Pb-rich perovskite thin films. The lead–sulfur (Pb–S) bond formed increases the Fermi level at the perovskite interface and creates an additional backfield for electron extraction. Strong Pb–S bonds can stabilize the perovskite heterostructure and enhance the potential perovskite structure with a similar lattice. Surface vulcanization devices retain more than 90% of the initial PCE after 2200 h of aging at 85 °C or 1000 h of operation at the maximum power point under continuous illumination at 55 ± 5 °C (Fig. 1f).55 In addition, Wang et al. used 2-mercaptopyridine (2-MP) to surface-treat halide PSCs, because 2-MP contains nitrogen and sulfur heteroatoms, which have lone pair electrons and can act as Lewis bases. The optimized device shows impressive operational stability. After a maximum power point tracking experiment for 1000 h at 40 °C, the efficiency was reduced by 16%. However, since 2-MP cannot enter the perovskite lattice, only passivation and external treatment can be performed.60 At the same time, the environmental storage stability of the unencapsulated device is significantly improved, maintaining an initial efficiency of more than 80% after 600 h.
2.4. Chalcogenide quantum dots
As early as 2018, Han et al. added PbS quantum dots into CH3NH3PbI3 (MAPbI3) precursors to form a QD-in-perovskite structure, which is beneficial to the crystallization of perovskites, resulting in an increase in particle size, a decrease in fragmentation particles, an enhancement of characteristic peak intensity, a large proportion of (220) planes in X-ray diffraction patterns, and a highly regular and large crystal structure.61 Gaulding et al. carefully investigated the effects of surface chemistry and antisolvent on the emission and morphological properties of perovskite films embedded in metal halide PSCs. Compared with pure MAPbI3 thin films, the apparent grain size of MAPbI3 with a PbS quantum dot structure is smaller, and of course, it will also be taken into account that the choice of antisolvent will affect the growth mode of PbS/MAPbI3 perovskite crystals.62 Lee et al. synthesized quantum dots (QDs) composed of copper (Cu), zinc (Zn), indium (In), sulfur (S), and selenium (Se) in 2020 and doped them into CsPbBr3 inorganic perovskite films. To disperse CsPbBr3-encapsulated CZISSe-QDs in PbBr2 solution, the inorganic CsPbBr3 shell was replaced by ligand exchange. CZISSe-QDs act as the seeds of crystal nucleus in PbBr2 films, promoting the crystallization of CsPbBr3. In addition, they are present in the mesoporous TiO2-CsPbBr3 perovskite layer, which facilitates electron extraction and transport from the CsPbBr3 photo-absorbing layer to TiO2.63 These works on quantum dots achieve a finer and more regular lattice structure through the interaction of sulfur elements, which can significantly improve its stability to the environment and extend the lifetime of the device.2.5. Other ways to introduce chalcogenides
In the process of introducing sulfur into PSCs, it can be incorporated not only within PSCs but also through other ways. Introducing anions of organic structures, such as thiocyanate ions, is a common method for improving their crystallization ability and surface morphology in PSCs.64–70 As early as 2015, Jiang et al. replaced the two iodines in CH3NH3PbI3 with amphoteric halides of thiocyanate (SCN−), and fabricated PSCs related to CH3NH3Pb(SCN)2I by a simple spin-coating process, which generally maintained a lattice structure similar to halogen perovskites due to thiocyanate ions.71 In addition, in 2016, Tai et al. used a simple precursor solution treatment method to show that PSCs are prepared under environmental conditions using the lead(II) thiocyanate (Pb(SCN)2) precursor, and high-quality CH3NH3PbI3−X(SCN)X perovskite films can be easily prepared even if the relative humidity exceeds 70%.72 In the same year, Liu et al. also introduced thiocyanate into PSCs containing lead thiocyanate at room temperature. Due to the properties of thiocyanate, PSCs with (MA)2Pb(SCN)2I2 structures can be fabricated at a low temperature of 70 °C.73All in all, PSCs have gradually reached people's expectations in terms of performance, with a PCE of up to 26%, making them a leading technology in solar cells. However, their stability and sensitivity to water and oxygen present significant challenges that need to be solved. The former severely limits the environmental conditions for the use of these devices. The latter places further specific requirements on the processing conditions. Therefore, it is imperative to focus on designing and enhancing the stability of the active layer in PSCs.
3. Chalcogenide inorganic thin film solar cells
Compared with high-efficiency halide PSCs, chalcogenide inorganic thin film solar cells have attracted much attention due to their excellent long-term water and oxygen stability.74–76 As mentioned earlier, the perovskite-type crystal structure under the ABX3 structure is usually cubic and orthorhombic. In this traditional case, we generally use the X structure as a halide anion, of course, it is also modified by the chalcogenide anion. For halide PSCs, the stability of PSCs can be significantly improved by introducing sulfur into anions, enabling lighter adaptability to the environment and demand.77,78 To form perovskites with chalcogenide structures (i.e., X = O2−, S2−, and Se2−), based on these two points, the direct selection of the A site falls on the alkaline earth elements, and many other metal elements have a bivalent structure for reference.3.1. Theoretical calculations
Under these limitations, there are theoretically 3744 possible element groups forming the chalcogenide perovskite.79 However, only a limited number of these compositions are thermodynamically stable and will crystallize in the desired cubic or pseudo-cubic perovskite structure. Based on purely geometric considerations, the ion radius of the atoms in the composition can be used to calculate the Goldschmidt tolerance factor (t) and octahedral factor (μ) to predict the stability and formability of a specific perovskite form.80,81 It is expressed as: | (1) |
 | (2) |
In a strict sense, the main structures of perovskites include the GdFeO3 type and TaAgS3 type. In the latter type, B cations move slightly from the center of their BX6 octahedron. While other structures do not possess the properties of traditional perovskite crystal structures, they are still categorized as perovskite structures due to their elemental composition and inherent properties. Fig. 2 outlines the perovskite structure of the chalcogenide ABX3. The primary relevant structure is the GdFeO3 structure, often referred to as the β-phase structure, which typically forms at higher temperatures during processing.
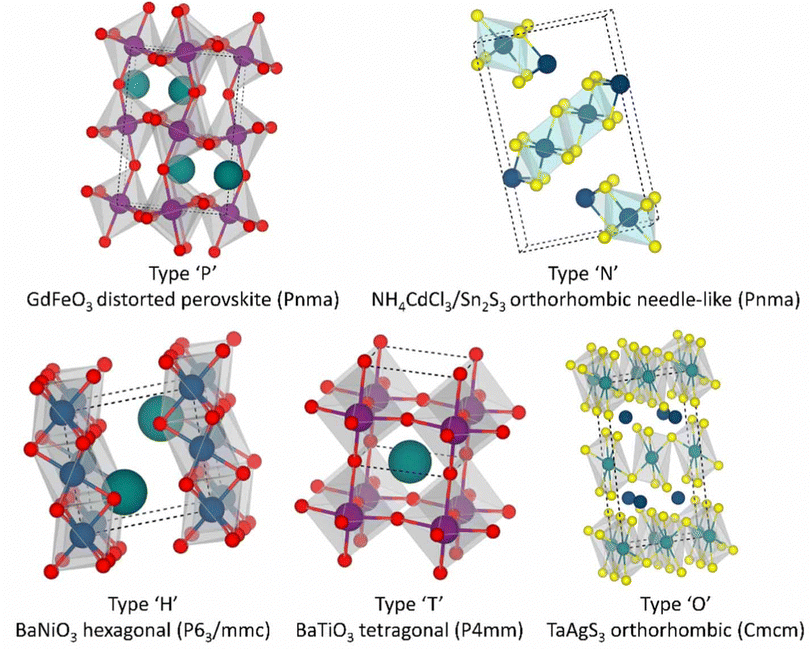 | ||
| Fig. 2 Perovskite structure of the chalcogenide ABX3. The cell diagram of the crystal structure of chalcogenide perovskites, as well as their geometric shape, space group, and prototype composition.82 Copyright 2021 Devendra Tiwari et al. J. Phys. Energy. | ||
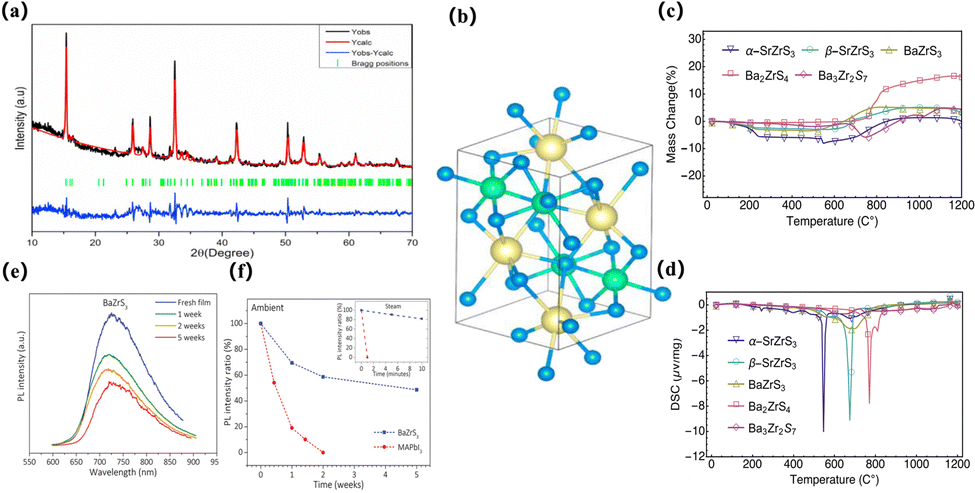
Compared to traditional halide PSCs, chalcogenides are challenging to synthesize, particularly due to the bonding requirements for the selenium-based structures. Thus, the current research on pure chalcogenide thin-film solar cells is more important. The developments and improvements lie in the field of theoretical simulation computing, and the computational methods envisaged by the researchers can better guide the development of synthesis work. After the introduction of metal sulfides, there will be 168 chalcogenide ABX3 compounds (A = Mg, Ca, Sr, Ba, Zn, Cd, Sn, Pb; B = Ti, Zr, Hf, Si, Ge, Sn, Pb; X = O, S, Se) with four different crystal structures (group symmetry Pm3m, Pnma, P63/mmc and Pnma (needle)). The band gap, light absorption, current-carrying effective mass, phase stability, thermodynamic stability, and dynamic stability were analysed, and these materials were screened out for photovoltaic applications. The potential of orthorhombic perovskites BaZrS3, BaHfSe3, SrZrSe3, SrHfSe3, and BaZrSe3 has been demonstrated for solar cell applications83,84 (Fig. 3b).
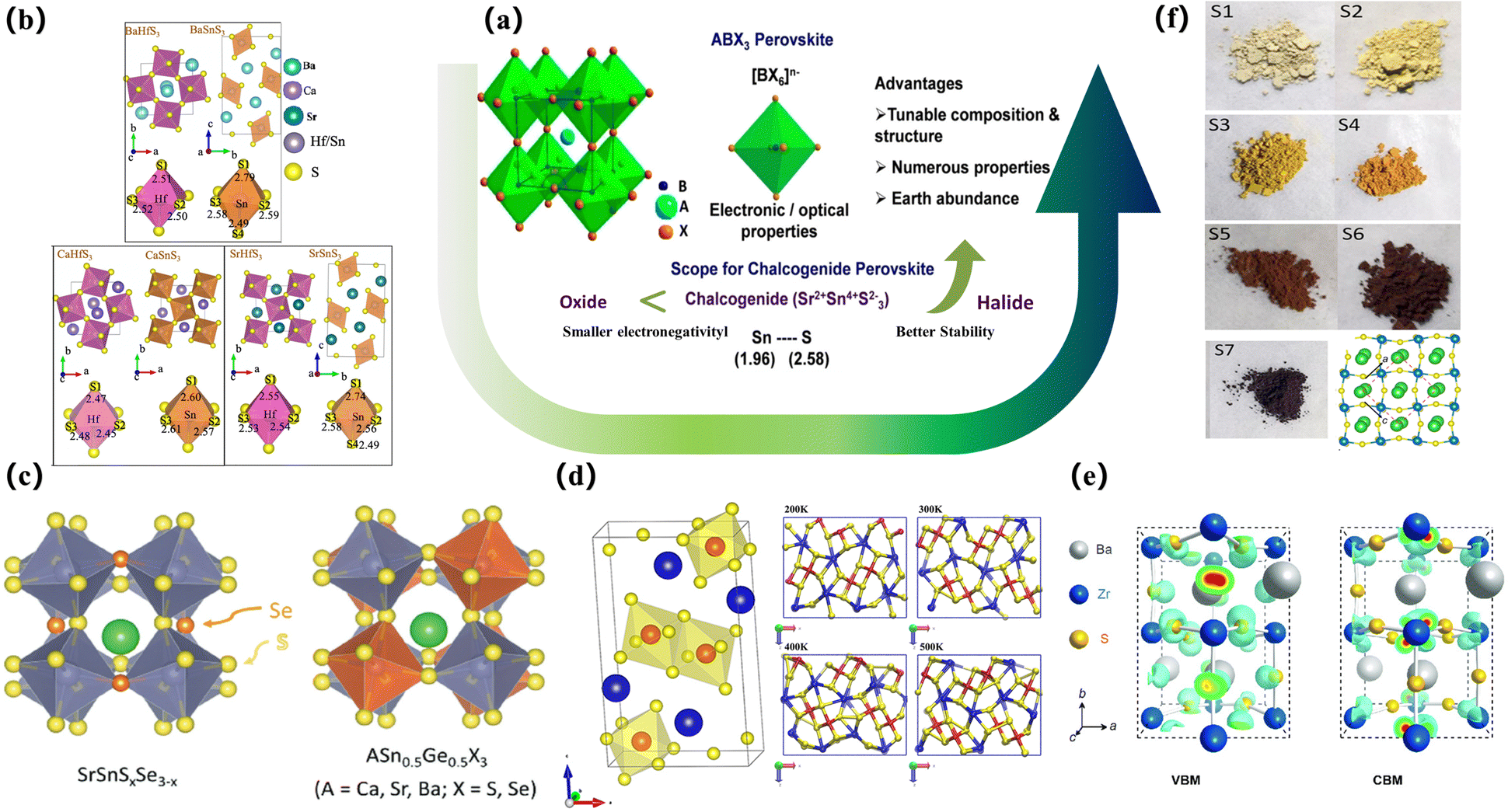 | ||
| Fig. 3 The simulated lattice structure of the sulfide perovskite. (a) Intuitive benefits of chalcogenide perovskites.85 Copyright 2019, Chem. Mater. (b) Crystal structures. BaHfS3 in orthorhombic distorted phase andBaSnS3 in needle-like phase; CaHfS3 andCaSnS3 in orthorhombic distorted phase; SrHfS3 in orthorhombic distorted phase and SrSnS3 in needle-like phase. Sulfur atom projected density of states.83 Copyright 2022, J. Phys. Chem. Lett. (c) Crystal structures of ABX3 perovskite chalcogenides with either a perfect perovskite structure or a distorted perovskite structure.86 Copyright 2017, Advanced Energy Materials (d) Snapshots of the BaZrSe3 crystal at different temperatures ranging from 200 K to 500 K. The projection of the structure is along the y-axis. Blue spheres represent Ba atoms, red spheres represent Zr atoms, and yellow spheres represent Se atoms.87 Copyright 2019, Journal of Applied Physics. (e) Charge density profiles of BaZrS3 for the bands of the VBM and CBM;7 Copyright 2020, Solar RRL. (f) The optical images of the BaZr-oxysulfides with different degrees of sulfurization and the top view of BaZrS3 in the distorted perovskite structure (space group Pnma). Red dashed frames and black arrows mark the unit cell and the crystallographic orientations, respectively.88 Copyright 2016, Nano Energy. | ||
SrSnSe3 and SrSnS3 are expected to be direct bandgap semiconductors with bandgap values in the optimal range of 0.9–1.6 eV. SrSnSe3 and SrSnS3 perovskites not only have good light absorption properties and carrier mobility, but also have flexible band gaps that can be continuously adjusted in the range of 0.9–1.6 eV by element mixing strategies, and these desirable properties give SrSnSe3 the potential to be a high-efficiency solar absorbing material, opening up possibilities for the design of chalcogenide solar cells86 (Fig. 3c). In 2019, Nag and his colleagues reviewed a series of chalcogenide perovskites with an appropriate band gap, strong absorption coefficient, and low effective carrier mass obtained by calculation. The theoretical maximum PCE of these materials is close to 30%. Under the guidance of these theoretical predictions, compounds such as BaZrS3, SrZrS3, and layered perovskite Ba3Zr2S7 were successfully synthesized. The experimentally obtained direct bandgap of BaZrS3 (∼1.8 eV) and SrZrS3 (∼2.1 eV) is slightly higher, but the direct bandgap of Ba3Zr2S7 (∼1.3 eV) is close to the ideal bandgap for single-junction solar cell applications. These materials exhibit strong absorption coefficients and near-band-edge emission characteristics. Zr-chalcogenides are promising solar harvesters, but achieving their efficacy in solar cell performance requires fabricating high-quality thin films and optimizing the charge transport process.85 At present, the synthesis and processing of chalcogenide perovskites pose greater challenges compared to halide perovskites. Both computational and experimental material design challenges need to be addressed in the near future. In particular, numerous chalcogenide perovskites have the advantages of earth-abundant resources, environment friendly nature, and robust thermal and humidity stability. Concurrently, Ong et al. studied the kinetic stability of BaZrSe3 formed by replacing S in BaZrS3 with selenium at non-zero temperature. They thermalized the predicted zero-temperature crystal structure of BaZrSe3 at different temperatures from 200 to 500 K by phonon frequency calculations and ab initio molecular dynamics simulations. The results show that the dynamic changes of the system have no obvious changes on the shells of BaSe and ZnSe at different temperatures, and the same structural units remain unchanged. Therefore, BaZrSe3 is predicted to be stable under typical conditions87 (Fig. 3d).
Machine learning methods have also been used to screen dichalcogenide perovskites for photovoltaic applications. Agiorgousis et al. developed a method based on a machine learning algorithm that uses atomic data from the periodic table to select potential absorber materials.89 By considering the thermodynamic stability, kinetic stability, and light absorption properties of the materials, they found that Ba2AlNbS6, Ba2GaNbS6, Ca2GaNbS6, Sa2GaNbS6, Sr2InNbS6, and Ba2SnHfS6 are the most promising new photovoltaic absorbers to replace CH3NH3PbI3.
3.2. Experimental research
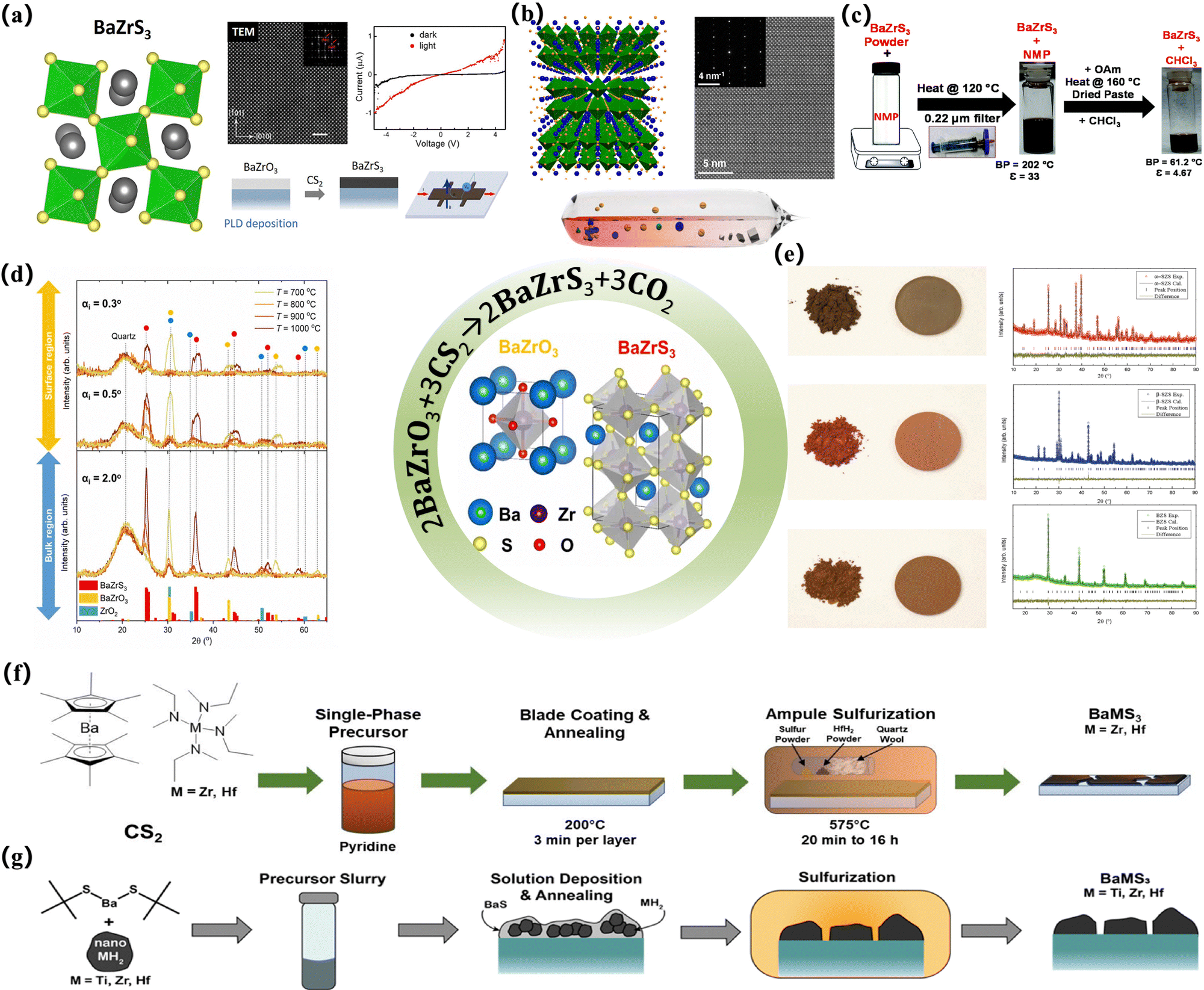 | ||
| Fig. 4 Preparation of partially vulcanized thin film devices. (a) Atomically resolved HAADF image of a BaZrS3 thin film sulfurized at 900 °C. The top right inset is the Fast Fourier Transformation (FFT) of the image. The I–V curves of the photodetector devices measured in the dark (black curves) and under illumination (red curves) for the BaZrS3 films sulfurized at 1050 °C for 2 h;91 Copyright 2020, Nano Energy. (b) A perspective view of the natural superlattice nanostructure in Ba3Zr2S7. The blue, yellow, and green spheres represent Ba, S, and Zr atoms, respectively. The ZrS6 octahedra are highlighted in green, the STEM image of a Ba3Zr2S7 crystal is viewed along the a-axis, and the inset shows the corresponding SAED pattern.16 Copyright 2018, Chem Mater. Phase identification of Ba–Zr–S–O thin films sulfurized at various temperatures: (c) schematic to make BaZrS3 NCs colloidally stable in N-methyl-2-pyrrolidinone (NMP) followed by treatment with oleylamine (OAm) to make a dispersion in chloroform (CHCl3).92 Copyright 2021, Nanoscale. (d) Representative GIWAXS patterns of Ba–Zr–S–O films synthesized at different temperatures. The measurements were performed with grazing incidence angles (αi) of 0.3°, 0.5° and 2°, allowing microstructural assessment of the surface (αi ⩽ 0.5°) and bulk (αi = 2.0°) regions of the thin films. Reference patterns of these phases are reported on the x-axis of the GIWAXS scans.93 Copyright 2023, Santhanu Panikar Ramanandan et al. J. Phys. Energy. (e) Optical images of synthesized powders and pelletized samples and plots of powder XRD patterns with Rietveld analysis for α-SrZrS3 (red), β-SrZrS3 (blue), and BaZrS3 (green).6 Copyright 2016, Advanced Materials. (f) The synthesis procedure for the formation of BaZrS3 and BaHfS3. Cp*2Ba, M(NMeEt)4 (M = Zr and Hf), and CS2 were combined and allowed to react. Excess CS2 was removed in-vacuo, and the resulting material was redissolved in pyridine to create a single-phase precursor ink. The ink was blade coated onto EXG glass and sulfurized at 575 °C in the presence of HfH2 to create the ternary chalcogenide perovskites.23 Copyright 2023, Angewandte Chemie International Edition. (g) Schematic outlining the synthesis method beginning with the use of Ba (SCMe3)2 and MH2 (M = Ti, Zr, and Hf) nanopowders to make a slurry that is deposited on glass and annealed to make a mixture of BaS and MH2. This material is then sulfurized to produce ternary BaMS3 materials.94 Copyright 2022, J. Am. Chem. Soc. | ||
It can be seen that most of the chalcogenide solar cells prepared in these experiments are synthesized by solid-state reaction.10 A common processing method is to seal the element or binary chalcogenide precursors in ampoules and heat them at high temperatures for hours or days. This classical processing and growth method often requires a temperature of more than 1000 °C and a reaction cycle of several hours or even days, so that the reaction material can be completely diffused on a micron basis, to obtain a uniform crystalline material (Table 1). For example, Meng et al. converted the material into a quartz tube, sealed the quartz tube in a dynamic vacuum, and maintained the reaction mixture at 800 °C for nearly 15 h to achieve a good crystal structure.11 As mentioned earlier, two twisted perovskite (BaZrS3,7 SrZrS3,7 BaHfS3,12 SrHfS3,12 BaZr(S1−xSeX)37) and non-perovskite materials (PbHfS3,95 PbTaS3,96 PbNbS397) were prepared in this way. Other variants of this technology, such as solid-state synthesis at high pressure, have also been developed and applied (for the synthesis of PbZrS3,98 BaSnS3,99 SrSnS3,99 and PbSnS397). However, this often puts forward different requirements for the preparation of PSCs. First of all, these processes introduce various issues. Due to the temperature requirements of this processing method, it is often impossible to achieve such high temperatures when preparing suitable thin-film devices, which means that if this method is blindly used to process a sulfur-containing perovskite structure, the non-perovskite structure of NH4CdCl3/Sn2S3 may emerge, unless subjected to some other externalities to stabilize it.9,13
| Material | Synthesis | Reaction conditions | Band gap(eV) | Ref. |
|---|---|---|---|---|
| BaZrS3 | Catalytic solid-state reaction | BaS+ Zr +I2 + S,600 °C/60 h | 1.81 | 6 |
| α-SrZrS3 | SrS+ Zr +I2 + S,850 °C/60 h | 1.53 | 6 | |
| β-SrZrS3 | SrS+ Zr +I2 + S,1000 °C/60 h | 2.13 | 6 | |
| BaZrS3 | Solid-state reaction | BaS + ZrSe2,1000 °C/48 h | 1.94 | 7 |
| SrZrS3 | SrS + ZrS2,1000 °C/48 h | 2.14 | 7 | |
| SrHfS3 | SrS + HfS2,1100 °C/48 h | 2.41 | 7 | |
| BaHfS3 | BaS + HfS2,1200 °C/48 h | 2.17 | 7 | |
| β-LaYbS3 | Solid-state reaction | M + Yb + Se + KI,1000 °C/96 h | 8 | |
| LaYbSe3 | 8 | |||
| CeYbSe3 | 8 | |||
| PrYbSe3 | 8 | |||
| NdYbSe3 | 8 | |||
| SmYbSe3 | 8 | |||
| BaZrS3 | Two-step Solid phase vulcanization | BaZrO3 + CS2,1050 °C/6 h | 1.75 | 9 |
| BaZrS3 | Solid-state reaction | BaS +ZrS2,900 °C/5 d | 10 | |
| BaZr1−xTixS3 | Solid-state reaction | BaS +ZrS2 + TiS2,800 °C/15 h | 1.47–1.7 | 11 |
| SrHfS3 | Solid-state reaction | SrS +La2S3+ HfS2,1100 °C/48 h | 2.3 | 12 |
| EuZrS3 | Solid-state reaction | Eu2O3 + ZrO2 + KI + S,1000 °C/10 d | 1.09 | 13 |
| BaZr−xTixS3 | Two-step Solid phase vulcanization | BaCO3, ZrO2 + TiO2,1000 °C/8 h; + CS2, + 1050 °C/4 h | 1.51–1.78 | 14 |
| BaZrS3 | Sputtering | BaS +ZrS2,3 h | 1.8 | 15 |
| Ba3Zr2S7 | Salt flux method | BaS+ Zr +S + BaCl2,1050 °C/40 h | 1.28 | 16 |
| Ba4Zr3S10 | 17 | |||
| Sr1+XTiS3 | Chemical vapor transport | SrS+ TiS2 +I2 + S,1000 °C/60 h | 18 | |
| NaTaO3 | Ultrasonic method | NaAcO + AcOH rt 2 h + Ta(EtO)5,6 h | 3.8 | 19 |
| NaTaO3 | Sol–gel synthesis, | CH3COONa + TaCl5 + citric acid,500 °C/5 h | 4 | 20 |
| CaSnS3 | Two-step ultrasonic spray | Ca + Sn + O2 800 °C + S 500 °C/2 h | 1.72 | 21 |
| LaYS3 | Sputter-deposited precursors + vulcanization | La + Y + O2; +S 900 °C | 2 | 22 |
| BaZrS3 | Solution deposition | Barium dithiocarboxylate species,575 °C | 23 |
Secondly, the process of high-temperature treatment may be accompanied by changes in the structure of some materials and the degradation of their physical and chemical properties. A relatively common phenomenon is the lack of anionic sulfur. This may be due to the low vacancy formation energy of the S element itself, which often leads to the degradation of the perovskite structure.88,100–102
One of the widely used solid-state syntheses of chalcogenide perovskites is through the solid-state synthesis of oxide perovskites, followed by CS2 vulcanization (for the synthesis of CaZrS3).7,88 However, it should be emphasized that the synthesis conditions play a fundamental role in the formability and phase determination of the required material. Vulcanization is a well-established method for synthesizing chalcogenides. Perera and colleagues observed that BaZrO3 (precursor) has a grain size of about 102 to 2 × 102 and does not show light absorption in the visible spectrum. However, after the vulcanization process of the sample, the particles become larger (μm), showing a wide range of visible spectra (light) absorption. They also observed that the bandgap (Eg) of these materials can be easily and systematically controlled by changing the chemical concentration ratio of chalcogenide samples to prepare oxygen sulfides.88 In fact, the sulfide structure of Ba(ZrXTi1−X)S3 can only be achieved by vulcanization of oxide perovskites with CS2,7,14 and direct synthesis by the solid-state reaction of sulfide precursors is not feasible. In contrast, SrZrS3 vulcanized with CS2 yields a non-perovskite structure,79 while a standard solid-state reaction yields a perovskite structure.7
| 2BaZrO3 + 3CS2 → 2BaZrS3 + 3CO2 | (3) |
However, when comparing various synthesis methods for preparing BaZrS3, a common issue with the direct synthesis of chalcogenide perovskites is the formation of unwanted products, such as binary sulfides or binary oxides.7,15 The BaZrS3 produced by the direct reaction of disulfide precursors or element precursors is more susceptible to oxidation,92 when contrasted with the structure produced by the conversion of oxide perovskites (BaZrO3) by carbon disulfide (Fig. 4c). This susceptibility to oxidation is not affected by the synthetic environment (such as a nitrogen atmosphere), but rather by the stoichiometry of elements. For chalcogenides, this can usually be achieved by providing a molten sulfur (S) phase. Wang and colleagues synthesized BaZrS3 by adjusting the ratio of S in the solid-state route and achieved higher BaZrS3 yield at growth temperatures as low as 600 °C.103,104 It was mentioned that if too little excess S is added, the BaZrS3 product is absent or poorly crystalline, and the main products are BaS, BaS2, and ZrS2. On the other hand, if too much S is added, the main products are S-rich binary compounds BaS3 and ZrS3. As also mentioned by Comparotto et al., unwanted phase formation is attributed to the S-rich factor.15 However, due to the lack of information about the formation process and kinetics, it is difficult to speculate on the optimal synthesis procedure. The synthesis temperature of BaZrS3 and other chalcogenides is very high, which indicates that there is a high energy barrier in the formation and nucleation of this compound, which may be responsible for its high stability. Therefore, understanding the thermodynamics and kinetics of the formation reaction is a fundamental step to optimize this material for energy applications. Recently, Kayastha and his colleagues have tried to improve the thermodynamic interpretation of the transition between 3D sulfur perovskites and 2D structures at high temperatures through a combination of experiments and calculations. Due to their similarity in Gibbs free energy, the overall S-element ratio in the synthesis process and the requirements of the synthesis environment are considered. The results suggest that chalcogenide perovskite compounds may need to master the equilibrium of the two-phase structure at low temperatures, and finally be deposited and integrated into optoelectronic devices.10
Ramanandan et al. further investigated the thermodynamic distribution and study of BaZrO3 (precursor) vulcanized by H2S. As the temperature rises, the overall structure is completely vulcanized at 1000 °C. The sensitivity of BaZrS to temperature was discussed. In practice, the formation mechanism of BaZrS is based on a two-step reaction involving (a) the intermediate amorphous phase of BaZrO, and (b) subsequently crystallized into the cBaZrS3 intermediate amorphous Ba–Zr–S–O phase93 (Fig. 4d). Ruddlesden–Popper perovskite chalcogenide Ba3Zr2S7 was successfully synthesized by Niu et al. Ba3Zr2S7 has a suitable band gap, high absorption coefficient near the absorption edge, a photoluminescence peak located at 1.28 eV, an external luminous efficiency of 0.15%, and an effective minority carrier recombination time of 65 ns, indicating that Ba3Zr2S7 is a promising photoabsorbent for solar cells16 (Fig. 4b). They also prepared high-quality BaZrS3 and its Ruddlesden–Popper, and Ba3Zr2S7 phase crystals by the flux growth method.17 Subsequently, they synthesized the hexagonal perovskite chalcogenide compound Sr1+XTiS3 with a quasi-one-dimensional structure. The material shows a strong dichroic window and strong dichroism peak from 2.5 mm to 12 mm, with a dichroic ratio in the infrared (MWIR) region exceeding 20.18
 | ||
| Fig. 5 Related optoelectronic performance of AMS3 (a) Rietveld refinement of the CaSnS3 X-ray diffraction pattern. (b) Side view of the structural presentation of CaSnS3 lattice shaping in a distorted perovskite structure (space group Pnma). Color code: Ca ¼ yellow, Sn ¼ green, and S¼ Blue.21 Copyright 2021, Journal of Alloys and Compounds. (c) TGA mass change of five sulphide samples and (d) DSC change with temperature.106 Copyright 2018, Journal of Materials Research. (e) PL spectra of BaZrS3 thin films kept under ambient conditions for 5 weeks. (f) PL intensity ratios of BaZrS3 and MAPbI3 thin films under ambient conditions. The inset shows PL intensity ratios for steam exposure.90 Copyright 2020, Advanced Functional Materials. | ||
Of course, for optoelectronic devices, a gentle approach to structuring solar cells is the ideal solution. Ravi et al. prepared solution-treated films using colloidal dispersions of solid synthesized BaZrS3. Chalcogenide perovskite thin-film transistors were prepared by low-temperature annealing.92 The results show that BaZrS3 nanocrystals exhibit bipolar behaviour in which hole mobility is three times higher than electron mobility, but the electrical conductivity is still very low (0.059 and 0.017 cm2 V s−1). This may be due to the small size of the nanoparticles processed by low-temperature treatment. This is the only report of working thin-film phototransistor devices based on chalcogenide perovskites. Of course, there have been reports of photovoltaic devices based on evaporating LaYS3 thin films, but the photocurrent has not yet been detected.22 In the latest study, Pradhan et al. synthesized the fourth group of metal dithiocarbamates by inserting carbon disulfide into a single-phase solution and then vulcanized BaZrS3 and BaHfS3 after treatment at 575 °C23 (Fig. 4f). In 2022, Jonathan and his partners used a combination of soluble barium mercaptan and nanoparticle zirconium hydride. After solution-based precursor deposition and subsequent vulcanization, BaZrS3 can be obtained at temperatures as low as 500 °C94 (Fig. 4g). However, compared to traditional halide PSCs, these solution deposition methods often require high-temperature post-treatment or long-term annealing, which may be related to the difficulty of the crystallization structure of sulfur atoms.
But in fact, one of the main advantages of halide PSCs over monocrystalline silicon cells is the improvement of solution processing and thin film growth. Halide perovskites are usually coated with solutions of metal halides (such as PbI2), organic cations (such as MA+), and various additives, followed by mild heating (<250 °C), in which the solvent evaporates, and the perovskite film crystallizes. Solvents are selected for good solubility and relatively low boiling point of precursor materials. Dimethylformamide and dimethylsulfoxide are common examples. Mild heating or the addition of anti-solvent can lead to perovskite saturation and precipitation. The cationic and anionic components of halide perovskites can be provided in a single solution without the need for additional counterions. Solution growth of halide perovskites is usually carried out in a glove box to prevent degradation of perovskite products from contact with air or moisture.
In general, for chalcogenides, it is more common to grow thin films by physical vapor deposition (PVD) methods, including thermal evaporation, electron beam evaporation, and sputtering. These methods can deposit high-purity and additive-free films. Various deposition processes exist in which the elements are deposited simultaneously or sequentially, and the thermal energy required to react with them can be provided during the deposition process or subsequently in a separate heating step. Chalcogenides can be included in the deposition step, added entirely during heating, or some combination of both. Regardless of the growth order, the temperature required to form a highly crystalline material is almost always in the range of 450–600 °C. Relatively speaking, the higher synthesis temperature and relatively more difficult processing methods explain the stability of the structural bonds of the chalcogenides themselves. In general, materials with stricter crystal structure processing tend to exhibit better external stability.
In this case, some of the processes used to obtain crystalline and phase-pure materials are summarized, but these very high temperatures are another important obstacle to be overcome in perovskite optoelectronic devices. After all, a high-temperature process is not compatible with most thin-film device processes. So far, among the perovskite components we have discussed, only BaZrS3 has been prepared in the form of thin films.
It can be observed that the chalcogenide material itself is highly stable in the water and oxygen environment, can operate in higher humidity environments, and has a promising alternative structure as a perovskite solar cell. However, theoretical calculation can only simulate the properties of materials to a certain extent, and whether chalcogenide compounds are highly stable or not remains to be verified by experimental results. In 2018, Niu et al. studied the thermal stability of five compounds: α-SrZrS3, β-SrZrS3, BaZrS3, Ba2ZrS4, and Ba3Zr2S7. They performed TGA and DSC measurements while heating the materials in air to 1200 °C. It was found that the needle-like phase of CSrZrS3 first degraded at 550 °C, while the other phases appeared to be fairly stable in air, and their oxidative decomposition temperature was much higher than 600 °C. Mass changes and post-XRD analysis confirmed this. It has also been reported that long-term ambient air storage and repeated water washing have no effect on the properties observed by XRD or UV-vis spectrophotometry of BaZrS3106 (Fig. 5c and d). Similarly, the stability of lead-free chalcogenide perovskite BaZrS3 films was also demonstrated by Gupta et al., who compared the properties of BaZrS3 and MAPbI3 in terms of moisture retention and light-induced degradation. BaZrS3 is more stable than methylammonium lead iodide (MAPbI3) in humid environments due to its weaker interaction with water and very low ion mobility. Under identical environmental conditions, the light response of the BaZrS3 device remained at 60% of its initial value after 4 weeks, while the light response of the MAPbI3 device decreased by 95% after a mere 4 days90 (Fig. 5e and f). In addition, in 2023, Zhang et al. discovered that LaScSe3 is a thermodynamically stable selenide perovskite through the first principles calculation and accurate prediction of phase stability standard, and successfully verified this in subsequent experimental synthesis.107
In general, the currently commonly used methods for pure sulfur-structured sulfides are often achieved by using alkaline earth metals and group IV transition metals or thiourea salt. Additionally, coating or heat treatment with thiourea is often employed, requiring constant temperature and humidity in the controlled environment.108 Of course, this is only required during preparation, and films tend to show ultra-long stability after preparation. Another way to circumvent the instability of precursor materials is to deliberately deposit an oxide film and then convert to chalcogenides by heat treatment with an S(e) source. Nevertheless, the exceptional stability of sulfides and their potential as light harvesters remain the primary advantages of thin-film solar cells. Challenges such as the requirement for high growth temperatures, slow crystallization, and compatibility issues with conductive substrates all arise from the same root issue. The use of oxide precursors presents inherent difficulties in removing oxygen, and attempts to crystallize directly from amorphous precursor membranes fail due to (possibly) slow diffusion kinetics. Unfortunately, owing to the current process requirements and limitations of processing methods, researchers have not been able to fabricate devices with an ultra-high PCE of more than 26% in this way. Therefore, the question of how to improve the performance of the device or enhance its ability to generate photogenerated carriers as a solar light harvester is a very important area of research. The improvement of its device manufacturing process is also a major research direction, especially the current relatively mild improvement method also requires an environment of about 200 °C. Finally, in order to improve its performance, the researchers adopted another method to improve its performance, that is, halides are introduced into the chalcogenide structure to prepare a light-absorbing layer with a “perovskite-like” structure.
4. Solar cells with a chalcogenide-halide blend structure
Considering the high efficiency of halide PSCs and the long-term water-oxygen stability of chalcogenide perovskite structures, the researchers tried to combine halides and chalcogenides, and gradually introduced a large number of halogen anions into the synthesis of chalcogenide compounds. Many chalcohalide crystal materials with a “perovskite-like” structure were synthesized. This type of solar cell structure combines the advantages of halide-soluble processing with long-term stability requirements (Table 2).| Material | Reaction conditions | Band gap(eV) | Device structure/measurement conditions | Performance | Ref. |
|---|---|---|---|---|---|
| CuBiSCl2 | CuCl + BiCl3 + Bi2S3 ground and pressed into tablets. The quartz tube was put into a box furnace, which was ramped to 430 °C within 10 h, dwelled for 10 h, cooled down to 200 °C within 60 h. | 1.37 | FTO/BL-TiO2/mp-TiO2/CuBiSCl2/Sn | J SC = 1.38 mA cm−2VOC = 1.09 V FF = 0.66 PCE = 1.00% | 34 |
| BiSI | Bi(NO3)3 + thiourea + NH4I in 2-methoxyethanol: acetylacetone (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v); heating at 200 °C in the air for 5 min. :1 v/v); heating at 200 °C in the air for 5 min. |
1.57 | FTO/SnO2/BiSI/F8/Au | J SC = 8.44 mA cm−2VOC = 0.445 V FF = 0.3514 PCE = 1.32% | 35 |
| Sb2S3-containing SbSI | Spin-coating method using metal ethylxanthate as the metal and sulfur source for the preparation of MSX and SAP thin films. | — | FTO/compact-TiO2/mp-TiO2/SbSI/PCPDTBT/PEDOT: PSS/Au | J SC = 12 mA cm−2VOC = 0.47 V FF = 0.52 PCE = 2.91% | 36 |
| (CH3NH3)3Bi2I9 | Bi(xt)3 (xt = ethyl xanthate) precursor solution was spin-coated on the substrate and then the samples were put in a covered Petri dish at 10 kPa with MAI powder scattered around uniformly. The sulfur incorporated MBI film was prepared at 150 °C with the different reaction times. | 2.1 | FTO/BL-TiO2/mp-TiO2/MA3Bi2I9/spiro-OMeTAD/Au | J SC = 0.58 mA cm−2VOC = 0.54 V FF = 0.472 PCE = 0.152% | 37 |
| Sb2S3-SbSI | In the solution process, SbSI is prepared by multiple cycles of spin-coating of the SbI3 solution onto the FTO/BL/mp-TiO2/Sb2S3 sample and thermal annealing. In the vapor process, SbSI is formed by simply annealing the FTO/BL/mp-TiO2/Sb2S3 sample and SbI3 powder (which vaporizes under the annealing condition) together. | — | FTO/TiO2-BL/mp-TiO2/Sb2S3-SbSI/PEDOT: PSS/Au | J SC = 14.31 mA cm−2VOC = 0.55 V FF = 0.661 PCE = 5.38% | 38 |
| SbSI | 1. Sb2S3: sodium thiosulfate in water + SbCl3 in acetone +water: reaction at 10 °C, followed by annealing in Ar at 300 °C for 5 min. 2. SbSI: Sbl3 in DMF spin coated on Sb2S3, followed by annealing at 150 °C for 5 min in an inert atmosphere. | 2.15 | FTO/BL-TiO2/mp-TiO2/SbSI/PCPDTBT/Au | J SC = 9.11 mA cm−2VOC = 0.58 V FF = 0.577 PCE = 3.05% | 39 |
| Pb2SbS2I3 | 1. Sb2S3: sodium thiosulfate in water + SbCl3 in acetone + water: reaction at 10 °C, + annealing in Ar at 300 °C for 5 min. 2. SbSI: Sbl3 in DMF spin coated at 500–2000 rpm for 60 s on Sb2S3, annealing in Ar at 300 °C for 2 min. | 2.19 | FTO/BL-TiO2/mp-TiO2/Pb2SbS2I3/PCPDTBT/Au | J SC = 8.79 mA cm−2VOC = 0.61 V FF = 0.582 PCE = 3.12% | 40 |
| SbSI | 1. Sb2S3: SbCl3, + thiourea in DMF spin-coated, followed by heat treatment at 150 °C for 5 min inside a glovebox. 2. SbSI: Spin coating of Sbl3 in NMP/DMSO solution on Sb2S3, followed by annealing at ∼200 °C for 1 h. | 1.96 | FTO/TiO2/SbSI/P3HT/Au | J SC = 5.45 mA cm−2VOC = 0.548 V FF = 0.31 PCE = 0.93% | 41 |
| Sb0.67Bi0.33SI | 1.Sb2S3: sodium thiosulfate in water + SbCl3 in acetone + water, reaction at 10 °C, followed by annealing in Ar at 300 °C for 5 min. 2. Sb0.67Bi0.33SI: Bil3 in DMF spin coated on Sb2S3, followed by annealing at 200 °C for 2 min in an inert atmosphere. | 1.62 | FTO/BL-TiO2/mp-TiO2/SbSI/PCPDTBT/Au | J SC = 14.54 mA cm−2VOC = 0.53 V FF = 0.528 PCE = 4.07% | 42 |
| Pb3S2X2 (X = Cl, Br, I) | PbX2 (X = Cl, Br, I) and Pb (SCN)2 in ODE + OLA + OA, reaction at 130 to 170 °C for 1 h. | 1.76 (Pb3S2I2) 1.98 (Pb3S2Br2) 2.02 (Pb3S2Cl2) | ITO/AZO/Pb3S2Br2/MoOX/Au | J SC = 1.2 mA cm−2VOC = 0.57 V PCE = 0.21% | 43 |
4.1. One-stage method
By replacing the ions in the perovskite with some sulfur-containing structural ions, this method aims to improve the crystallization properties and surface morphology of the perovskite structure and achieve the effect of improving the structural stability. In 2017, Kaltzoglou et al. prepared halide PSCs at room temperature by introducing trimethylthiotriiodide, which exhibits high chemical stability in the environment. Compared to protonated amine cations, (CH3)3S has no hydrolysis reaction and shows no signs of degradation in air up to 85 °C. But in contrast, methylamine in PSCs is replaced by trimethylsulfide molecules, and the feasibility of replacing methylamine with trimethylsulfide ions is proposed. This is also the first report on the lead sulfide perovskite compound109 (Fig. 6e). In 2022, Ming and his colleagues explored stable, lead-free, and defect-resistant photovoltaic materials. They combined computational screening with experimental synthesis, utilizing large cations with a single pair of electrons and mixed anions of sulfur and halogens as the marker structure, while achieving defect tolerance and high stability. Combining the criteria with direct bandgap and optimal bandgap values, the screening of the inorganic materials database identified CuBiSCl2 with an ideal bandgap of 1.37 eV in the perovskite structure. Experiments have found that the material remains stable under thermal decomposition at 300 °C and can be stored for 25 days under environmental conditions with a relative humidity of 60%. A prototype of the solar cell was made, proving that the open-circuit voltage is 1.09 V and the PCE is 1.00%34 (Fig. 6d).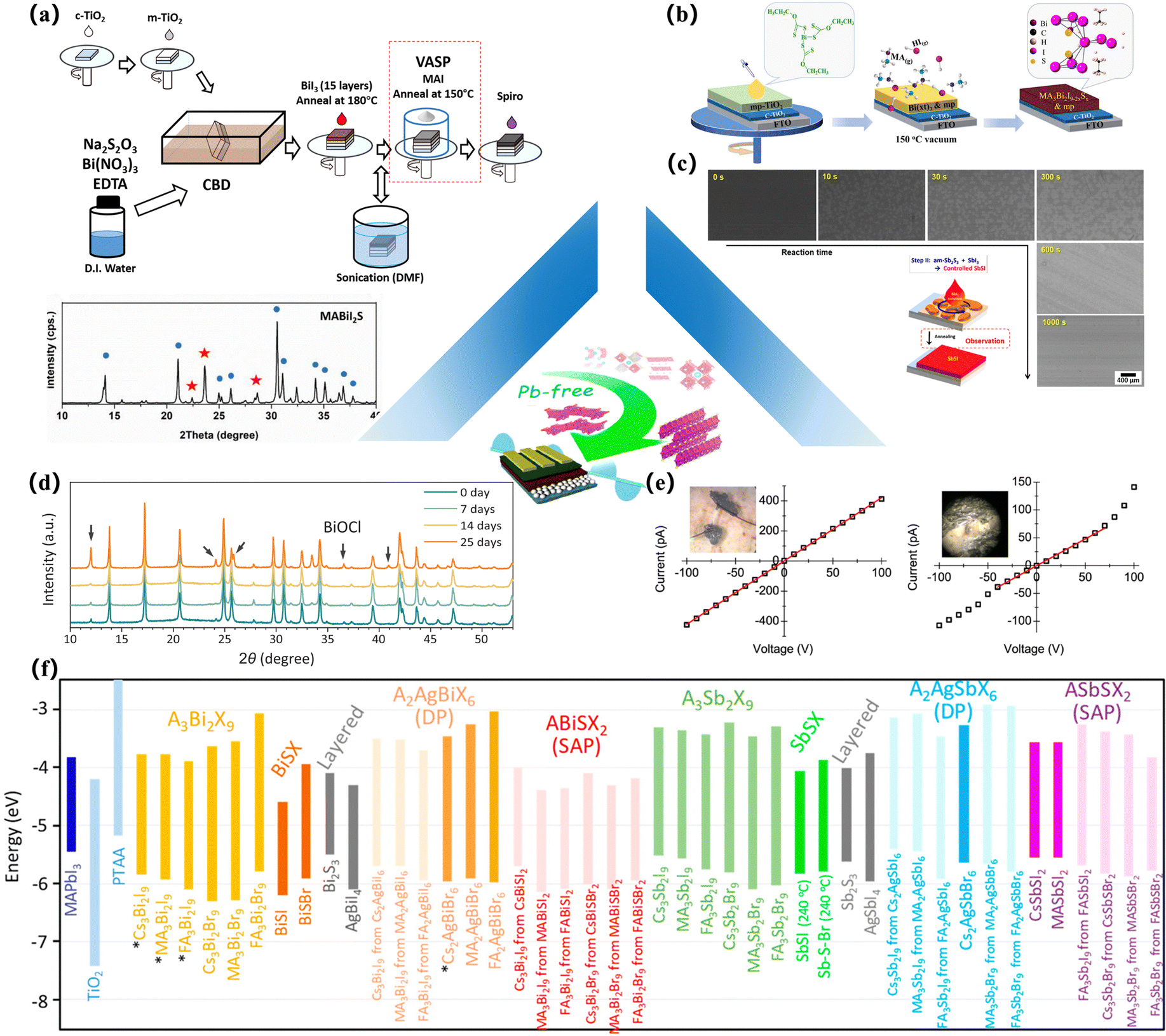 | ||
| Fig. 6 Preparation of some chalcogenide perovskite-like thin film devices. (a) The solid-state synthesis procedure of the MBIS perovskite for the device fabrication process.110 (b) A schematic illustration of the fabrication process of S-incorporated bismuth-based perovskites solar cells via low-pressure vapor-assisted solution process (LP-VASP).37 Copyright 2019, Solar RRL. (c) FESEM images of the samples obtained at different reaction times; Typical FESEM surface image around a dot.41 Copyright 2018, APL Materials. (d) Evolution of XRD patterns for the CuBiSCl2 samples stored at room temperature in air with 60% relative humidity. The black arrows mark the peaks attributed to the compound BiOCl.34 Copyright 2022, Advanced Functional Materials. (e) I–V plot for single crystals of (CH3)3SPbI3 at room temperature. Needle-shaped crystal from a DMF solution with dimensions (inset) 0.2 mm × 0.2 mm × 1.3 mm, and plate-like crystal from an HI solution with dimensions (inset) 0.1 mm × 0.1 mm × 0.03 mm. The former crystal exhibits a regular ohmic behavior in the ±100 V range, whereas the latter shows signs of polarization above ±50 V.109 Copyright 2017, Inorganic Chemistry. (f) Energy levels of the VBM and CBM for the direct transitions of the Bi- and Sb-based films.36 Copyright 2020, Chem Mater. | ||
Tiwari et al. applied spin coating technology to the one-step preparation of BiSI thin films.35 They spin-coated with a molecular solution synthesized by dissolving Bi(NO3)2·5H2O, thiourea, and NH4I in a mixture of 2-methoxyethanol and acetylacetone. Using this method, flake BiSI films are produced. Similarly, Nishikudo et al. reported that the Sb(EtX)3 single crystal was used for spin coating based on the one-step method. To make SbSI solar cells, the solution of the Sb(EtI)3 single crystal and SbI3 dissolved in dimethyl sulfoxide was spun onto the substrate of the mesoporous TiO2 (mp-TiO2)/TiO2 barrier (TiO2-BL)/FTO and annealed at 240 °C. Then, HTL and Au are deposited36 (Fig. 6f). In 2019, Yao and colleagues used the developed low-pressure vapor-assisted solution process (LP-VASP) method to obtain sulfur-doped bismuth-based perovskite films. Due to the presence of sulfur, the crystal quality and band properties were effectively improved in the prepared lead-free perovskite thin films. Through systematic study of the effect of reaction time on device performance, it is found that the optimized reaction time is 30 min, at which time the bandgap of the sulfur-doped MA3Bi2I9−2xSx perovskite film decreased to 1.67 eV, and the morphology is compact. The corresponding optimal PCE reaches 0.152%. This study provides a new method for incorporating sulfur into lead-free bismuth PSCs37 (Fig. 6b). In addition to ternary MChX, the one-step spin coating method can also be used to prepare quaternary sulfur halide (MII2MIIICh2X3).
4.2. Two-stage method
In the two-step deposition method, sulfide (M2CH3) is prepared and then converted to chalcohalides (MChX) by the reaction of M2CH3 and MX3 (step 2). This inverse application eqn (4) says.| M2Ch3 + MX3 → 3MChX | (4) |
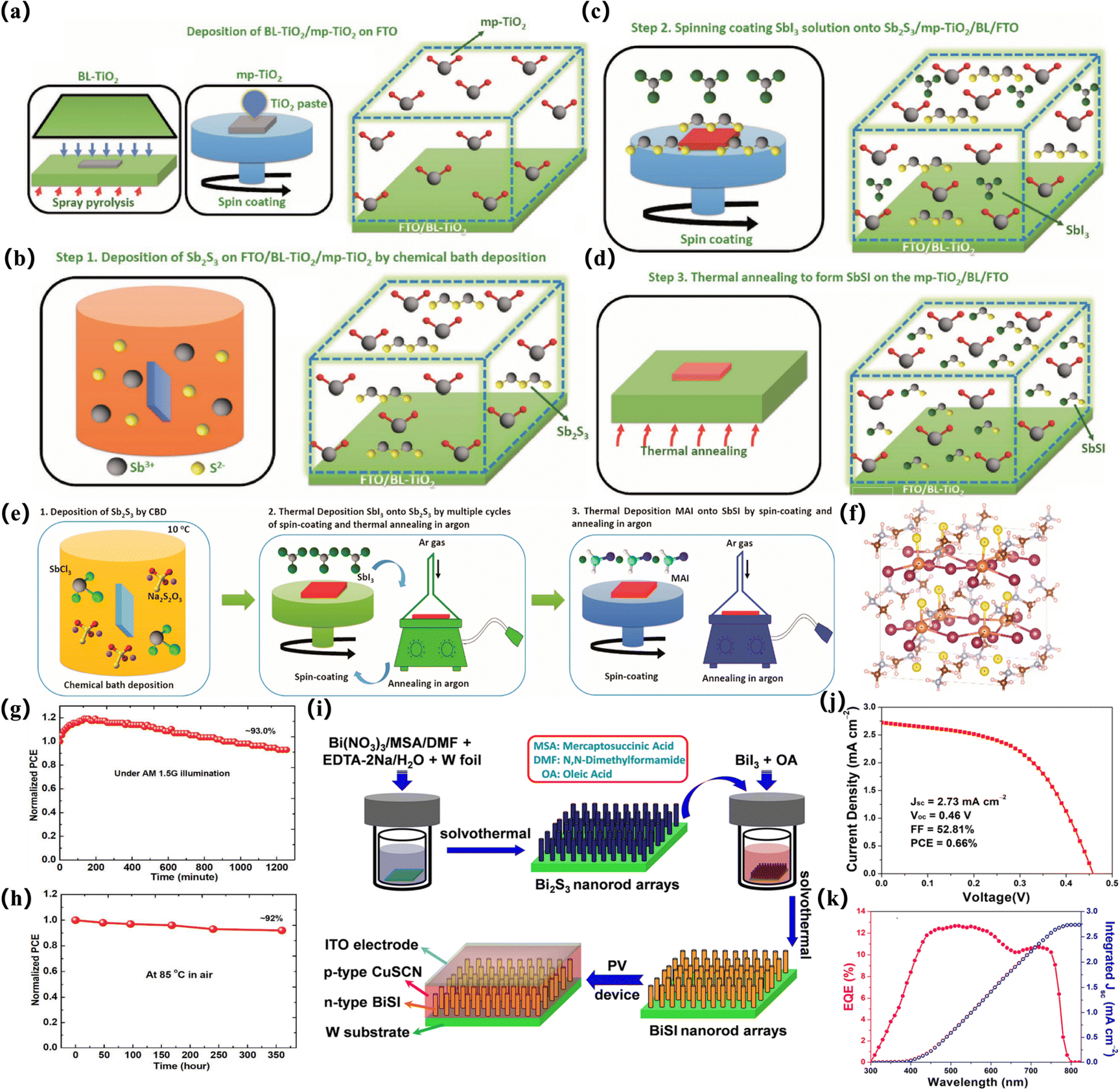
The most systematic and impactful work in this area is the related research of Nie and colleagues. The Pb2SbS2I3 material was synthesized based on an early report from Starosta et al. The stoichiometric elements Sb, Pb, S were weighed in quartz glass ampoules and reacted with sublimed iodine in a furnace at 650 °C for 36 h. The synthesized Pb2SbS2I3 material shows a bandgap of 2.0 eV.111 In 2018, Nie et al. prepared films by chemical bath deposition (CBD) of Sb2S3 films and SbI3 solution in N-dimethylformamide (DMF) in argon or nitrogen.38 A combination of solution treatment and solid state reaction prepares SbSI thin films. SbSI-based solar cells were fabricated by fluorine-doped SnO2 (FTO)/TiO2 resistance tomography (BL-TiO2)/mesoporous TiO2 (mp-TiO2)/SbSI/hole transport material (HTM)/Au cell structure39 (Fig. 7a–d). This protocol is also applied to prepare solar cells with an MASbSI2 structure and Pb2SbS2I3 as a light harvester, respectively. Pb2SbS2I3 solar cells without encapsulated nanostructures show good humidity stability within 30 days, and the effect of PbI2 solution concentration on device performance was studied. The role of trap states is studied to elucidate the potential mechanism of different reorganization processes. Based on their previous optimization work, such as spin-coating conditions, m-TiO2 thickness, and Sb2S3 deposition time, the PCE is approximately 3.12%, JSC is 8.79 mA cm−2, VOC is 0.61 V, and FF is 58.2%. An optimized crystal structure of the synthesized MASbSI2 perovskite was prepared, with sulfur atoms distributed along the Z axis of the octahedral unit. The Sb–S bond is divided into a long bond (long bond) and a short bond (short bond). The length of the Sb–I bond is 3.16 Å. As a whole, it shows a strict cubic structure. This explains the impressive stability of sulfur halide materials compared to lead halide perovskite materials40,78 (Fig. 7e–f). An optimized crystal structure of the synthesized MASbSI2 perovskite was prepared, with sulfur atoms distributed along the Z-axis of the octahedral unit. The Sb–S bond is divided into a long bond (∼3.6 Å) and a short bond (∼2.27 Å). This series of processing methods for preparing chalcogenide solar cells is also applied to selenium-based structures, and the PCE(PCE) is 4.1% under standard air mass. Even if illuminated at AM1.5G (100 mW cm−2) for the 2321 min, the device retains 90.0% of the initial PCE.112 In addition, Zhang et al. first prepared MABiI2S perovskite structure crystals by a two-step solid-state synthesis method, showing high crystallinity overall110 (Fig. 6a). Choi et al. reported a simple two-step solution treatment method based on the formation of amorphous Sb2S3 (am-Sb2S3) using the SbCl3-TU solution in step I and the application of SbI3 to am-Sb2S3 in step II to obtain the final SbSI film. In addition, the structure and morphology of the final SbSI film can be controlled by high-concentration treatment, and a dense SbSI film with a pure SbSI phase can be obtained at a low temperature of 200 °C. The successful application of the SbSI thin film to planar SbSI solar cells demonstrates the controlled growth of the SbSI thin film and its application in planar solar cells for the first time41 (Fig. 6c).
 | ||
| Fig. 7 Preparation of some sulfur “perovskite-like” solar cell devices. (a)–(d) Schematic illustration for preparing SbSI layers through the reaction between Sb2S3 deposited by the CBD process and SbI3 onto mp-TiO2 electrode.39 Copyright 2017, Advanced Energy Materials. (e) Process for preparing the MASbSI2 perovskite layer, (f) MASbSI2 configuration simulated by DFT calculation is presented by the ball and stick model with the orange ball for Sb, purple for I, yellow for S, gray for C, brown for N, and white for H.78 Copyright 2018, J Am Chem Soc. (g) under standard AM 1.5G illumination with a xenon lamp including UV radiation at room temperature and (h) measured at 85 °C in air, under 40% average relative humidity in the dark.42 Copyright 2019, Advanced Materials. (i) Schematic illustration of the growth procedures of BiSI nanorod arrays and their application for solar cells (j) Typical J–V curve of the ITO/p-CuSCN/n-BiSI/W solar-cell device based on BiSI nanorod arrays. (k) EQE spectrum and the corresponding integrated JSC of the fabricated solar cell device.113 Copyright 2020, ACS Sustain Chem Eng. | ||
However, in practice, it is found that the Sb2S3 structural lattice will cause lattice distortion, which further affects the trap density and photovoltaic performance, insufficient light absorption and charge transfer, or the device efficiency will be reduced if the concentration of the SbCl3 precursor solution is too low or too high.
The poor efficiency of the device (in high concentrations of SbCl3 precursor solutions) is mainly due to the high trap density and deep-level trap state. It is worth noting that the unpackaged device exhibits excellent thermal stability, light stability, and long-term stability when stored for 504 h, which indicates that Sn2SbS2I3 materials have higher stability than Sn-based halide perovskites. In 2019, Nie et al. deposited Sb2S3 on the FTO/TiO2-BL/mp-TiO2 substrate, and Sb2S3 was replaced and modified by Bi and I ions. After deposition of amorphous Sb2S3 and annealing, the solution of BiI3 in DMF is spin-coated on FTO/TiO2-BL/mp-TiO2/Sb2S3. The sample was annealed in argon or nitrogen at 250 °C, and the process was repeated several times until Sb2S3 was fully converted to Sb0.67Bi0.33SI42 (Fig. 7g and h).
However, these methods are time-consuming because they require multiple cycles in step 2 to obtain complete thioiodide. In addition, the film obtained is not entirely homogeneous. To overcome these limitations, we introduced the SbI3 vapor process instead of the SbI3 solution process in step 2, in order to produce higher SbSI uniformity without repeating step 2 and provide a better 3.62% PCE for SbSI solar cells. However, the inherent limitations of the CBD process, such as the formation of impurities and difficulties in controlling the proportions, may limit the controlled growth of sulfur halides.114 In addition, since the structure of mesoporous devices is optimized, factors such as morphology and thickness, which are important for planar devices, are not considered. Therefore, developing a two-step method for planar device architectures that allows for controlled growth of sulfur halide remains a challenge.
To apply a two-step approach to the planar device architecture, a thin film covering the entire surface is required. This is because incomplete surface coverage reduces the ability to absorb light and produces defects, which reduces the performance of the device.115 Recently, Xiong et al. presented their own views and reported a method for preparing BiSI nanorod arrays based on the two-step method. However, their method involves solvothermal synthesis rather than spin coating at each step. Bi2S3 deposited tungsten (W) foil is immersed in an autoclave containing BiI3 solution, and BiSI nanorods are prepared by heating. Compared with the two-step method, the prepared nanorods have an Eg value similar to 1.57 eV but show a preferred [010] orientation113 (Fig. 7i–k).
4.3 Other methods
In addition to the methods mentioned above for preparing solar cells, there are other methods for the processing and modification of solar cells. Manna et al. have developed a colloid synthesis method for the synthesis of Pb4S3X2 (X = Cl, Br, and I) NC, which has made further progress in the synthesis of Pb-based sulfur halides. A heating method has been developed in which hot thiocyanate (SCN−) ions (as a source of S2−) decompose at lower temperatures (below 190 °C) in the presence of corresponding PbBr2, PbI2, PbCl2 precursors and OLA and OA solvents.43 The energy arrangement on Pb4S3Br2 NC after ligand exchange was studied to test its applicability in the fabrication of solar cells. Unencapsulated solar cells configured with ITO/AZO/Pb4S3Br2/MoOX/Au have a PCE of 0.21 ± 0.02% under standard AM 1.5G illumination, a JSC of 1.09 mA cm−2, and a VOC of 0.58 V. Kunioku et al. heated BiOI particles in H2S or H2Se gas for a short time <1 h. By replacing O2− with S2−, BiSBr1−XIX was also synthesized by this low-temperature anion substitution, and the composition ratio between Br and I is retained, which is attributed to the successful inhibition of halogen volatilization at such low temperatures.116 Li fabricated a new type of ternary Bi chalcohalide, the tetragonal Bi13S18I2, in addition to BiSI, with both controlled by adjusting the mole ratio of CH4N2S/BiI3/CH3NH3I(CH3NH3I = MAI) in the solution used in the solvothermal process. They found that a pure Bi13S18I2 structure can be obtained from the conversion reaction of BiSI over 6 h at a CH4N2S/BiI3/MAI ratio of 4:2:3.117 Kanatzidis et al. re-examined Sn-based sulfur iodide and demonstrated the synthesis of novel quaternary Sn2BiS2I3 and Sn2BiSI5 materials using isothermal heating and chemical vapor transport. Interestingly, these compounds are stable in water and air with a band gap of 1.2–1.3 eV, which makes them promising for solar energy conversion applications. However, practical applications have not yet been proven.118 The specific process and results of Nie et al. are described above, and the excellent performance of these solar cells in terms of humidity, thermal, and light stability is shown. This study is of great significance for developing and applying solar cells.
5. Summary and perspective
In summary, we have proposed various strategies for augmenting the inherent stability of perovskite materials by combining chalcogenides and halides. Introducing chalcogenides into halide PSCs has shown considerable enhancement in device efficiency and stability. A crucial aspect of research involves the incorporation of sulfur into the hole transport layer of PSCs, significantly improving long-term stability through dipole moment interactions between the lone pair electrons in chalcogenides (e.g., sulfur S, oxygen O) and unpaired Pb2+ ions in perovskites. Additionally, integrating chalcogenides into the perovskite layer promotes crystallization properties and stability, notably through the inclusion of additives such as S8, dithizone, and Lewis bases into perovskites. Furthermore, interface modifications employing chalcogenides have been explored in halide PSCs, including surface treatments on materials like SnO2, NiO, and perovskites. Various methods such as solid-state reaction, vulcanization processes, and high-temperature, high-pressure synthesis are commonly used in the synthesis of chalcogenide perovskites. Catalytic synthesis, lowering reaction temperatures, has shown promise in creating pure sulfur perovskite structures. Additionally, techniques such as sputtering and pulsed laser deposition have been proposed for synthesizing chalcogenide perovskites. Despite theoretical possibilities for chalcogenide perovskites in inorganic thin film solar cells, only a limited number of stable compositions crystallize into the desired perovskite structure. Therefore, experimental research has focused on high-temperature synthesis methods and alternative catalyst-based approaches. Regarding solar cells utilizing a chalcogenide-halide blend structure, combining the high efficiency of halide perovskite solar cells with the long-term stability of chalcogenide perovskites has been explored. One-stage and two-stage methods have been employed for the preparation of mixed chalcogenide and halide perovskites. While the one-stage method offers convenience, temperature control becomes crucial due to the differing requirements of chalcogenides and halides. The two-stage method involves depositing chalcogenide at high temperatures followed by halide deposition at lower temperatures, albeit increasing preparation complexity. Moreover, various synthesis methods such as colloid synthesis, low-temperature anion substitution, solvothermal processes, isothermal heating, and chemical vapor transport have been utilized for mixed chalcogenide and halide perovskite synthesis. Despite advancements in utilizing chalcogenides to enhance solar cell stability, certain limitations persist, necessitating further research. To address these limitations, suggestions for future research can be categorized as follows:For halide PSCs:
(a) Further optimization of perovskite structures through lead ion substitution, anion modification, and the use of non-halide anions.
(b) Incorporation of quantum dot perovskites into antisolvents for enhanced crystallization during cell preparation.
(c) Exploration of perovskite-silicon tandem solar cell structures to bolster overall stability.
(d) Utilizing virtual modelling to understand degradation mechanisms at the atomic scale, particularly regarding material properties.
For more stable chalcogenide thin film materials:
(a) Detailed theoretical calculations to identify materials suitable for solar cells.
(b) Reduction of chalcogenide perovskite synthesis temperatures to suit substrate materials.
(c) Optimization of preparation processes, including device structure and crystallization orientation regulation.
For chalcogenide-halide compounds:
(a) Discovery and development of new chalcogenide-halide materials with improved optoelectronic performance.
(b) Investigation into the intrinsic properties of chalcogenide-halide compounds and their impact on solar cell efficiency.
(c) Exploration of growth regulation techniques for enhanced performance.
(d) In-depth study of crystallization mechanisms and electronic properties of chalcogenide-halide compounds.
In conclusion, leveraging chalcogenide-halide combinations to enhance the intrinsic stability of PSCs presents a promising avenue for future research. This approach could substantially contribute to the commercialization of perovskite-based devices by broadening the scope of stability-related studies.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by financial support from the National Natural Science Foundation of China (52203359), the National Key Research and Development Program of China (2019YFA0705400), the Natural Science Foundation of China (52073119, 21774040, and 22002162), the Natural Science Foundation of Jiangsu Province (BK20212008), the Research Fund of State Key Laboratory of Mechanics and Control of Mechanical Structures (MCMS-I-0421K01), the Fundamental Research Funds for the Central Universities (NJ2020003, NZ2020001), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. R.N. and S.I.S. acknowledge financial support from the Basic Science Research Program (NRF-2018R1A3B1052820) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (MSIP). The research work was also supported by the supporting funds for talents of Nanjing University of Aeronautics and Astronautics.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- J. A. Brehm, J. W. Bennett, M. R. Schoenberg, I. Grinberg and A. M. Rappe, J. Chem. Phys., 2014, 140, 224703 CrossRef PubMed.
- M. S. G. Hamed and G. T. Mola, Crit. Rev. Solid State, 2020, 45, 85–112 CrossRef CAS.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.pdf.
- S. Niu, H. Huyan, Y. Liu, M. Yeung, K. Ye, L. Blankemeier, T. Orvis, D. Sarkar, D. J. Singh, R. Kapadia and J. Ravichandran, Adv. Mater., 2017, 29, 1604733 CrossRef PubMed.
- Y. Nishigaki, T. Nagai, M. Nishiwaki, T. Aizawa, M. Kozawa, K. Hanzawa, Y. Kato, H. Sai, H. Hiramatsu, H. Hosono and H. Fujiwara, Sol. RRL, 2020, 4, 1900555 CrossRef CAS.
- K. Mitchell, R. C. Somers, F. Q. Huang and J. A. Ibers, J. Solid State Chem., 2004, 177, 709–713 CrossRef CAS.
- C.-S. Lee, K. M. Kleinke and H. Kleinke, Solid State Sci., 2005, 7, 1049–1054 CrossRef CAS.
- P. Kayastha, D. Tiwari, A. Holland, O. S. Hutter, K. Durose, L. D. Whalley and G. Longo, Sol. RRL, 2023, 7, 2201078 CrossRef CAS.
- W. Meng, B. Saparov, F. Hong, J. Wang, D. B. Mitzi and Y. Yan, Chem. Mater., 2016, 28, 821–829 CrossRef CAS.
- K. Hanzawa, S. Iimura, H. Hiramatsu and H. Hosono, J. Am. Chem. Soc., 2019, 141, 5343–5349 CrossRef CAS PubMed.
- S.-P. Guo, Y. Chi, J.-P. Zou and H.-G. Xue, New J. Chem., 2016, 40, 10219–10226 RSC.
- X. Wei, H. Hui, S. Perera, A. Sheng, D. F. Watson, Y.-Y. Sun, Q. Jia, S. Zhang and H. Zeng, ACS Omega, 2020, 5, 18579–18583 CrossRef CAS PubMed.
- C. Comparotto, A. Davydova, T. Ericson, L. Riekehr, M. V. Moro, T. Kubart and J. Scragg, ACS Appl. Energy Mater., 2020, 3, 2762–2770 CrossRef CAS.
- S. Niu, D. Sarkar, K. Williams, Y. Zhou, Y. Li, E. Bianco, H. Huyan, S. B. Cronin, M. E. McConney, R. Haiges, R. Jaramillo, D. J. Singh, W. A. Tisdale, R. Kapadia and J. Ravichandran, Chem. Mater., 2018, 30, 4882–4886 CrossRef CAS.
- S. Niu, B. Zhao, K. Ye, E. Bianco, J. Zhou, M. E. McConney, C. Settens, R. Haiges, R. Jaramillo and J. Ravichandran, J. Mater. Res., 2019, 34, 3819–3826 CrossRef CAS.
- S. Niu, H. Zhao, Y. Zhou, H. Huyan, B. Zhao, J. Wu, S. B. Cronin, H. Wang and J. Ravichandran, Chem. Mater., 2018, 30, 4897–4901 CrossRef CAS.
- O. Vázquez-Cuchillo, A. Manzo-Robledo, R. Zanella, N. Elizondo-Villareal and A. Cruz-López, Ultrason. Sonochem., 2013, 20, 498–501 CrossRef PubMed.
- W.-H. Lin, C. Cheng, C.-C. Hu and H. Teng, Appl. Phys. Lett., 2006, 89, 211904 CrossRef.
- H. Shaili, M. Beraich, A. Elhat, M. Ouafi, E. Salmani, R. Essajai, W. Battal, M. Rouchdi, M. Taibi, N. Hassanain and A. Mzerd, J. Alloys Compd., 2021, 851, 156790 CrossRef CAS.
- A. Crovetto, R. Nielsen, M. Pandey, L. Watts, J. G. Labram, M. Geisler, N. Stenger, K. W. Jacobsen, O. Hansen, B. Seger, I. Chorkendorff and P. C. K. Vesborg, Chem. Mater., 2019, 31, 3359–3369 CrossRef CAS.
- A. A. Pradhan, M. C. Uible, S. Agarwal, J. W. Turnley, S. Khandelwal, J. M. Peterson, D. D. Blach, R. N. Swope, L. Huang, S. C. Bart and R. Agrawal, Angew. Chem., Int. Ed., 2023, 62, e202301049 CrossRef CAS PubMed.
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- Q. Jiang, J. Tong, Y. Xian, R. A. Kerner, S. P. Dunfield, C. Xiao, R. A. Scheidt, D. Kuciauskas, X. Wang, M. P. Hautzinger, R. Tirawat, M. C. Beard, D. P. Fenning, J. J. Berry, B. W. Larson, Y. Yan and K. Zhu, Nature, 2022, 611, 278–283 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H.-X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531–534 CrossRef CAS PubMed.
- E. A. Duijnstee, B. M. Gallant, P. Holzhey, D. J. Kubicki, S. Collavini, B. K. Sturdza, H. C. Sansom, J. Smith, M. J. Gutmann, S. Saha, M. Gedda, M. I. Nugraha, M. Kober-Czerny, C. Xia, A. D. Wright, Y.-H. Lin, A. J. Ramadan, A. Matzen, E. Y.-H. Hung, S. Seo, S. Zhou, J. Lim, T. D. Anthopoulos, M. R. Filip, M. B. Johnston, R. J. Nicholas, J. L. Delgado and H. J. Snaith, J. Am. Chem. Soc., 2023, 145, 10275–10284 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef CAS PubMed.
- J.-P. Correa-Baena, M. Saliba, T. Buonassisi, M. Grätzel, A. Abate, W. Tress and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- H. J. Snaith, Nat. Mater., 2018, 17, 372–376 CrossRef CAS PubMed.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- C. Ming, Z. Chen, F. Zhang, S. Gong, X. Wu, J. Jiang, T. Ye, Q. Xu, K. Yang, L. Wang, X. Cao, S. Yang, S. Zhang, Y. Zhang, J. Shi and Y.-Y. Sun, Adv. Funct. Mater., 2022, 32, 2112682 CrossRef CAS.
- D. Tiwari, F. Cardoso-Delgado, D. Alibhai, M. Mombrú and D. J. Fermín, ACS Appl. Energy Mater., 2019, 2, 3878–3885 CrossRef CAS.
- R. Nishikubo, H. Kanda, I. García-Benito, A. Molina-Ontoria, G. Pozzi, A. M. Asiri, M. K. Nazeeruddin and A. Saeki, Chem. Mater., 2020, 32, 6416–6424 CrossRef CAS.
- J. Li, X. Liu, J. Xu, J. Chen, C. Zhao, M. Salma Maneno, B. Zhang and J. Yao, Sol. RRL, 2019, 3, 1900218 CrossRef.
- R. Nie and S. I. Seok, Small Methods, 2020, 4, 1900698 CrossRef CAS.
- R. Nie, H. Yun, M.-J. Paik, A. Mehta, B. Park, Y. C. Choi and S. I. Seok, Adv. Energy Mater., 2018, 8, 1701901 CrossRef.
- R. Nie, B. Kim, S.-T. Hong and S. I. Seok, ACS Energy Lett., 2018, 3, 2376–2382 CrossRef CAS.
- Y. C. Choi, E. Hwang and D.-H. Kim, APL Mater., 2018, 6, 121108 CrossRef.
- R. Nie, J. Im and S. I. Seok, Adv. Mater., 2019, 31, 1808344 CrossRef PubMed.
- S. Toso, Q. A. Akkerman, B. Martín-García, M. Prato, J. Zito, I. Infante, Z. Dang, A. Moliterni, C. Giannini, E. Bladt, I. Lobato, J. Ramade, S. Bals, J. Buha, D. Spirito, E. Mugnaioli, M. Gemmi and L. Manna, J. Am. Chem. Soc., 2020, 142, 10198–10211 CrossRef CAS PubMed.
- U. V. Ghorpade, M. P. Suryawanshi, M. A. Green, T. Wu, X. Hao and K. M. Ryan, Chem. Rev., 2023, 123, 327–378 CrossRef CAS PubMed.
- E. J. Juarez-Perez, M. R. Leyden, S. Wang, L. K. Ono, Z. Hawash and Y. Qi, Chem. Mater., 2016, 28, 5702–5709 CrossRef CAS.
- Z. Li, Z. Zhu, C.-C. Chueh, J. Luo and A. K.-Y. Jen, Adv. Energy Mater., 2016, 6, 1601165 CrossRef.
- X. Ji, T. Zhou, X. Ke, W. Wang, S. Wu, M. Zhang, D. Lu, X. Zhang and Y. Liu, J. Mater. Chem. A, 2020, 8, 5163–5170 RSC.
- R. Li, J. Zhang, M. Liu, S. K. Matta, J. Tian, Z. Deng, S. P. Russo, P. Vivo, Z. Zhou and H. Zhang, Sol. RRL, 2023, 7, 2300031 CrossRef CAS.
- S. Collavini, A. Cabrera-Espinoza and J. L. Delgado, Macromolecules, 2021, 54, 5451–5463 CrossRef CAS.
- A. Sadhu, M. Rai, T. Salim, X. Jin, J. M. R. Tan, S. W. Leow, M. G. Ahmed, S. Magdassi, S. G. Mhaisalkar and L. H. Wong, Adv. Funct. Mater., 2021, 31, 2103807 CrossRef CAS.
- H. Min, G. Kim, M. J. Paik, S. Lee, W. S. Yang, M. Jung and S. I. Seok, Adv. Energy Mater., 2019, 9, 1803476 CrossRef.
- C. Hu, Y. Bai, S. Xiao, K. Tao, W. K. Ng, K. S. Wong, S. H. Cheung, S. K. So, Q. Chen and S. Yang, Sol. RRL, 2020, 4, 2000270 CrossRef CAS.
- X. Yang, H. Xiang, J. Huang, C. Zhou, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Colloid Interface Sci., 2022, 628, 476–485 CrossRef CAS PubMed.
- Z. Wang, M. A. Kamarudin, N. C. Huey, F. Yang, M. Pandey, G. Kapil, T. Ma and S. Hayase, ChemSusChem, 2018, 11, 3941–3948 CrossRef CAS PubMed.
- X. Li, W. Zhang, X. Guo, C. Lu, J. Wei and J. Fang, Science, 2022, 375, 434–437 CrossRef CAS PubMed.
- W. Hou, G. Han, T. Ou, Y. Xiao and Q. Chen, Angew. Chem., Int. Ed., 2020, 59, 21409–21413 CrossRef CAS PubMed.
- B. Wang, Q. Cheng, G. Huang, Y. Yue, W. Zhang, X. Li, Y. Li, W. Du, X. Liu, H. Zhang, Y. Zhang and H. Zhou, Adv. Mater., 2023, 35, 2207345 CrossRef CAS PubMed.
- B. P. Finkenauer, Y. Zhang, K. Ma, J. W. Turnley, J. Schulz, M. Gómez, A. H. Coffey, D. Sun, J. Sun, R. Agrawal, L. Huang and L. Dou, Amine-Thiol/Selenol Chemistry for Efficient and Stable Perovskite Solar Cells, J. Phys. Chem. C, 2023, 127(2), 930–938 CrossRef CAS.
- Y. Zhang, Y. Han, Y. Xu, G. Yu, Y. Miao, C. Gao, X. Zhou, Y. Song and K.-J. Jiang, Sustainable Energy Fuels, 2021, 5, 3700–3704 RSC.
- C. Wang, Z. Zhu, G. Wang, X. Ding, Y. Chen, S. Xiao, X. Liu, X. Zhang and S. Wen, ACS Sustainable Chem. Eng., 2022, 10, 17318–17326 CrossRef CAS.
- J. Han, S. Luo, X. Yin, Y. Zhou, H. Nan, J. Li, X. Li, D. Oron, H. Shen and H. Lin, Small, 2018, 14, 1801016 CrossRef PubMed.
- E. A. Gaulding, X. Chen, Y. Yang, S. P. Harvey, B. To, Y.-H. Kim, M. C. Beard, P. C. Sercel and J. M. Luther, ACS Mater. Lett., 2020, 2, 1464–1472 CrossRef CAS.
- E. J. Lee, D.-H. Kim, R. P. H. Chang and D.-K. Hwang, ACS Appl. Energy Mater., 2020, 3, 10376–10383 CrossRef CAS.
- D. Li, T. Xia, W. Liu, G. Zheng, N. Tian, D. Yao, Y. Yang, H. Wang and F. Long, Appl. Surf. Sci., 2022, 592, 153206 CrossRef CAS.
- Q. Chen, J. Wu, J. T. Matondo, L. Bai, D. M. Maurice and M. Guli, Sol. RRL, 2020, 4, 2000584 CrossRef CAS.
- N. Cheng, W. Li, M. Zhang, H. Wu, S. Sun, Z. Zhao, Z. Xiao, Z. Sun, W. Zi and L. Fang, Curr. Appl. Phys., 2019, 19, 25–30 CrossRef.
- X. Wang, H. Huang, S. Du, P. Cui, Z. Lan, Y. Yang, L. Yan, J. Ji, B. Liu, S. Qu, Q. Zhang, X. Yue, X. Zhao and M. Li, Sol. RRL, 2022, 6, 2200717 CrossRef CAS.
- S. Zhong, Z. Li, C. Zheng, X. Luo, J. Gao, X. Lu, X. Gao, L. Shui, S. Wu and J.-M. Liu, Sol. RRL, 2022, 6, 2200088 CrossRef CAS.
- M.-S. Bae, Y. H. Chang, W. Lee, C.-S. Moon, S.-W. Kim, H.-S. Kim, J. Lim and T.-Y. Yang, Energy Fuels, 2023, 37, 4608–4615 CrossRef CAS.
- Y. Yu, C. Wang, C. R. Grice, N. Shrestha, D. Zhao, W. Liao, L. Guan, R. A. Awni, W. Meng, A. J. Cimaroli, K. Zhu, R. J. Ellingson and Y. Yan, ACS Energy Lett., 2017, 2, 1177–1182 CrossRef CAS.
- Q. Jiang, D. Rebollar, J. Gong, E. L. Piacentino, C. Zheng and T. Xu, Angew. Chem., Int. Ed., 2015, 54, 7617–7620 CrossRef CAS PubMed.
- Q. Tai, P. You, H. Sang, Z. Liu, C. Hu, H. L. W. Chan and F. Yan, Nat. Commun., 2016, 7, 11105 CrossRef CAS PubMed.
- J. Liu, J. Shi, D. Li, F. Zhang, X. Li, Y. Xiao and S. Wang, Synth. Met., 2016, 215, 56–63 CrossRef CAS.
- G. Harbeke, J. Phys. Chem. Solids, 1963, 24, 957–963 CrossRef CAS.
- A. Starczewska, M. Nowak, P. Szperlich, I. Bednarczyk, K. Mistewicz, M. Kepinska and P. Duka, Frontiers in Optics 2014 (2014), paper JW3A.28, Optica Publishing Group, 2014, p. JW3A.28 Search PubMed.
- T. Li, X. Wang, Y. Yan and D. B. Mitzi, J. Phys. Chem. Lett., 2018, 9, 3829–3833 CrossRef CAS PubMed.
- Y.-Y. Sun, J. Shi, J. Lian, W. Gao, M. L. Agiorgousis, P. Zhang and S. Zhang, Nanoscale, 2016, 8, 6284–6289 RSC.
- R. Nie, A. Mehta, B. Park, H.-W. Kwon, J. Im and S. I. Seok, J. Am. Chem. Soc., 2018, 140, 872–875 CrossRef CAS PubMed.
- Q. Sun, W.-J. Yin and S.-H. Wei, J. Mater. Chem. C, 2020, 8, 12012–12035 RSC.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 702–707 CrossRef CAS PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- D. Tiwari, O. S. Hutter and G. Longo, J. Phys.: Energy, 2021, 3, 034010 CAS.
- P. Basera and S. Bhattacharya, J. Phys. Chem. Lett., 2022, 13, 6439–6446 CrossRef CAS PubMed.
- Z. Huo, S.-H. Wei and W.-J. Yin, J. Phys. D: Appl. Phys., 2018, 51, 474003 CrossRef.
- A. Swarnkar, W. J. Mir, R. Chakraborty, M. Jagadeeswararao, T. Sheikh and A. Nag, Chem. Mater., 2019, 31, 565–575 CrossRef CAS.
- M.-G. Ju, J. Dai, L. Ma and X. C. Zeng, Adv. Energy Mater., 2017, 7, 1700216 CrossRef.
- M. Ong, D. M. Guzman, Q. Campbell, I. Dabo and R. A. Jishi, J. Appl. Phys., 2019, 125, 235702 CrossRef.
- S. Perera, H. Hui, C. Zhao, H. Xue, F. Sun, C. Deng, N. Gross, C. Milleville, X. Xu, D. F. Watson, B. Weinstein, Y.-Y. Sun, S. Zhang and H. Zeng, Nano Energy, 2016, 22, 129–135 CrossRef CAS.
- M. L. Agiorgousis, Y.-Y. Sun, D.-H. Choe, D. West and S. Zhang, Adv. Theory Simul., 2019, 2, 1800173 CrossRef.
- T. Gupta, D. Ghoshal, A. Yoshimura, S. Basu, P. K. Chow, A. S. Lakhnot, J. Pandey, J. M. Warrender, H. Efstathiadis, A. Soni, E. Osei-Agyemang, G. Balasubramanian, S. Zhang, S.-F. Shi, T.-M. Lu, V. Meunier and N. Koratkar, Adv. Funct. Mater., 2020, 30, 2001387 CrossRef CAS.
- X. Wei, H. Hui, C. Zhao, C. Deng, M. Han, Z. Yu, A. Sheng, P. Roy, A. Chen, J. Lin, D. F. Watson, Y.-Y. Sun, T. Thomay, S. Yang, Q. Jia, S. Zhang and H. Zeng, Nano Energy, 2020, 68, 104317 CrossRef CAS.
- V. K. Ravi, S. H. Yu, P. K. Rajput, C. Nayak, D. Bhattacharyya, D. S. Chung and A. Nag, Nanoscale, 2021, 13, 1616–1623 RSC.
- S. P. Ramanandan, A. Giunto, E. Z. Stutz, B. Reynier, I. T. F. M. Lefevre, M. Rusu, S. Schorr, T. Unold, A. F. I. Morral, J. A. Márquez and M. Dimitrievska, J. Phys.: Energy, 2023, 5, 014013 Search PubMed.
- J. W. Turnley, K. C. Vincent, A. A. Pradhan, I. Panicker, R. Swope, M. C. Uible, S. C. Bart and R. Agrawal, J. Am. Chem. Soc., 2022, 144, 18234–18239 CrossRef CAS PubMed.
- G. A. Wiegers, A. Meetsma, R. J. Haange and J. L. de Boer, Acta Crystallogr., 1989, C45, 847–849 CAS.
- T. Schmidt, K. Lischka and W. Zulehner, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 8989 CrossRef PubMed.
- L. Schmidt, Phys. Lett. A, 1970, 31, 551–552 CrossRef CAS.
- S. Yamaoka, J. Am. Ceram. Soc., 1972, 55, 111 CrossRef CAS.
- S. Yamaoka and B. Okai, Mater. Res. Bull., 1970, 5, 789–794 CrossRef CAS.
- Y. Wang, N. Sato, K. Yamada and T. Fujino, Shigen-to-Sozai, 2000, 116, 211–216 CrossRef CAS.
- N. Gross, Y.-Y. Sun, S. Perera, H. Hui, X. Wei, S. Zhang, H. Zeng and B. A. Weinstein, Phys. Rev. Appl., 2017, 8, 044014 CrossRef.
- J. Yan, M. Greenblatt, A. Sahiner, D. Sills and M. Croft, J. Alloys Compd., 1995, 229, 216–222 CrossRef CAS.
- Y. Wang, N. Sato, K. Yamada and T. Fujino, J. Alloys Compd., 2000, 311, 214–223 CrossRef CAS.
- Y. Wang, N. Sato and T. Fujino, J. Alloys Compd., 2001, 327, 104–112 CrossRef CAS.
- K. V. Sopiha, C. Comparotto, J. A. Márquez and J. J. S. Scragg, Adv. Opt. Mater., 2022, 10, 2101704 CrossRef CAS.
- S. Niu, J. Milam-Guerrero, Y. Zhou, K. Ye, B. Zhao, B. C. Melot and J. Ravichandran, J. Mater. Res., 2018, 33, 4135–4143 CrossRef CAS.
- H. Zhang, X. Wu, K. Ding, L. Xie, K. Yang, C. Ming, S. Bai, H. Zeng, S. Zhang and Y.-Y. Sun, Chem. Mater., 2023, 35, 4128–4135 CrossRef CAS.
- R. Nie, K. S. Lee, M. Hu, M. J. Paik and S. I. Seok, Matter, 2020, 3, 1701–1713 CrossRef.
- A. Kaltzoglou, C. C. Stoumpos, A. G. Kontos, G. K. Manolis, K. Papadopoulos, K. G. Papadokostaki, V. Psycharis, C. C. Tang, Y.-K. Jung, A. Walsh, M. G. Kanatzidis and P. Falaras, Inorg. Chem., 2017, 56, 6302–6309 CrossRef CAS PubMed.
- C. Zhang, S. Teo, Z. Guo, L. Gao, Y. Kamata, Z. Xu and T. Ma, Chem. Lett., 2019, 48, 249–252 CrossRef CAS.
- V. I. Starosta, J. Kroutil and L. Beneš, Cryst. Res. Technol., 1990, 25, 1439–1442 CrossRef CAS.
- R. Nie, M. Hu, A. M. Risqi, Z. Li and S. I. Seok, Adv. Sci., 2021, 8, 2003172 CrossRef CAS PubMed.
- J. Xiong, Z. You, S. Lei, K. Zhao, Q. Bian, Y. Xiao and B. Cheng, ACS Sustainable Chem. Eng., 2020, 8, 13488–13496 CrossRef CAS.
- Y. C. Choi, D. U. Lee, J. H. Noh, E. K. Kim and S. I. Seok, Adv. Funct. Mater., 2014, 24, 3587–3592 CrossRef CAS.
- L. Zheng, D. Zhang, Y. Ma, Z. Lu, Z. Chen, S. Wang, L. Xiao and Q. Gong, Dalton Trans., 2015, 44, 10582–10593 RSC.
- H. Kunioku, M. Higashi and R. Abe, Sci. Rep., 2016, 6, 32664 CrossRef CAS PubMed.
- S. Li, L. Xu, X. Kong, T. Kusunose, N. Tsurumachi and Q. Feng, J. Mater. Chem. C, 2020, 8, 3821–3829 RSC.
- S. M. Islam, C. D. Malliakas, D. Sarma, D. C. Maloney, C. C. Stoumpos, O. Y. Kontsevoi, A. J. Freeman and M. G. Kanatzidis, Chem. Mater., 2016, 28, 7332–7343 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |