
High-coordination Fe–N4SP single-atom catalysts via the multi-shell synergistic effect for the enhanced oxygen reduction reaction of rechargeable Zn–air battery cathodes†
Jiaqi
Liu‡
a,
Weibin
Chen‡
b,
Shuang
Yuan
 *ade,
Tie
Liu
c and
Qiang
Wang
*ac
*ade,
Tie
Liu
c and
Qiang
Wang
*ac
aSchool of Metallurgy, Northeastern University, Shenyang 110819, China. E-mail: yuans@smm.neu.edu.cn; wangq@mail.neu.edu.cn
bSchool of Materials Science and Engineering, Peking University, Beijing 100871, China
cKey Laboratory of Electromagnetic Processing of Materials (Ministry of Education), Northeastern University, Shenyang 110819, China
dKey Laboratory of Ecological Metallurgy of Multimetallic Mineral (Ministry of Education), Northeastern University, Shenyang 110819, China
eEngineering Research Center of Frontier Technologies for Low-carbon Steelmaking (Ministry of Education), Liaoning Low-carbon Steelmaking Technology Engineering Research Center, Shenyang 110819, China
First published on 16th November 2023
Abstract
Atomically dispersed iron–nitrogen–carbon (Fe–N–C) catalysts are immensely promising alternatives to noble metal catalysts for the oxygen reduction reaction (ORR), while their activity is bottlenecked by an unsatisfactory electronic structure. Accurately tuning the d-band centers and spin state of Fe sites is significant in enhancing intrinsic ORR activity but remains challenging. Herein, an innovative theoretical model of Fe–N4SP with a highly coordinated structure was rationally designed by decorating Fe–N4 sites with axial sulfur atoms in the first coordination shell and adjacent phosphorus atoms in the second coordination shell. The theoretical calculations and experimental validations confirmed that the proposed structure can break the symmetric structure of Fe–N4 sites to rearrange electrons and intensify spin polarization. The increased coordination number conferred enhanced d-orbital interactions in Fe atoms to depress their d-band centers, weakening the adsorption of oxygen intermediates. Fe–N4SP sites were successfully constructed in N, P, and S ternary co-doped hollow carbon nanocages to validate the proposed theoretical model. The obtained Fe–N4SP/NPS-HC catalysts exhibited impressive ORR activity with a decent half-wave potential of 0.912 V in alkaline media and 0.814 V in acidic media, coupled with ultra-long durability of up to 320 h in zinc–air battery systems. Overall, novel insights into the effect of the coordination environment on ORR catalytic activity were provided, useful for modulating the electronic structure of catalysts for efficient storage and energy production systems.
Broader contextSingle-atom catalysts represented by Fe-based materials are promising alternatives for the expensive conventional Pt-based catalysts in oxygen reduction reactions. Yet, the performances of Fe–N4 sites can further be improved owing to their suboptimal geometry and electronic structure. Herein, a nuanced strategy was developed to precisely tailor the coordination milieu and spin state of Fe sites. A highly coordinated Fe–N4SP model was designed to lower the d-band center and optimize the adsorption energy of oxygen intermediates. The intensified spin polarization also promoted the activation and reduction of oxygen. This work strengthens the understanding of the structure–activity relationship of ORR catalysts, laying the foundation for the development of new efficient energy conversion and storage systems. |
Introduction
Rechargeable zinc–air batteries (ZABs), as a new generation of green energy conversion devices, have been extensively explored due to the abundance of the zinc anode and high theoretical energy density (1086 W h kg−1).1–8 However, their large-scale application is still restricted by the sluggish kinetics of the oxygen reduction reaction (ORR), as well as the high-cost and scarce benchmark Pt/C catalysts.9,10 Alternatively, single-atom catalysts (SACs), prominently featuring M–Nx sites (where M refers to Fe, Co, Ni, etc.) on carbon substrates, have been validated to be the most promising contributors to the ORR owing to their maximized atom-utilization efficiency.11–14 Among these, Fe–N4–C catalysts exhibit the most exceptional ORR potential.15–18 Nevertheless, the symmetric D4h electron distribution in the Fe–N4 site endows oxygen intermediates with strong binding energy, resulting in unsatisfactory ORR catalytic activity.19–23 Therefore, the accurate manipulation of geometry and electronic structure of Fe–N–C catalysts to optimize the binding energy of key intermediates is significant but challenging.The adsorption strength of intermediates on Fe–N4 active sites is primarily determined by the d-band centers of Fe atoms, heavily depending on their coordination environments and 3d orbitals’ electronic configurations.19,24–28 To regulate the electronic distribution and d-band energy levels of the surrounding metal sites, tremendous efforts have so far been devoted to adjusting the electron-withdrawing/donating properties of carbon carriers in the second coordination shell (CS) or further outside. For instance, the introduction of heterogeneous heteroatoms (B, P, S, O, etc.) with different atomic radii and electronegativity can distinctly improve the charge distribution on the carbon matrix surface.16,29–32 In this view, Yin et al. noticed that long-distance P atoms could drive electron delocalization of edge Fe–N4 and enhance ORR activity in acidic media.33 However, such long-range electron modulation may be limited by the strength of absorbing/donor moieties and the spatial interaction distance, leading to a diminished effect of electron delocalization.34,35 Hence, efficient modulation of the first CS has been proposed and extensively studied.36–40 For instance, ligand atoms have been directly anchored to host atoms, aimed at modulating the microenvironment or the electronic spin state of metal active centers for tailored d-band centers and intrinsic activity of Fe–N–C catalysts.41–44 In this respect, Li et al. induced the electron rearrangement and spin state transition of Fe–N3S catalysts by incorporating S atom-coordination to accelerate the nitrogen reduction reaction.42 Nevertheless, most studies have focused on coordination modulation or heteroatom substitution in the Fe–N4 plane while there is only a handful of reports on the modulation of the coordination number and geometrical structure design of metal centers. Furthermore, the promotion mechanism of electrocatalysis by the synergistic coordination effect of the first and second shells is still not clear.
Herein, a nuanced strategy was developed to meticulously tailor the coordination milieu and spin state of Fe sites. An innovative Fe–N4SP configuration with a high coordination structure was specifically designed through a multi-shell coupling strategy by decorating with axial-bridged sulfur atoms in the first CS surrounded by phosphorus atoms in the second CS. Density functional theory (DFT) calculations predicted broken electron symmetry of Fe–N4 sites using a high-coordination Fe–N4SP model along with a depressed d-band center and elevated spin density, resulting in declined *OH adsorption energy. Guided by the theoretical model, a single atom catalyst with Fe–N4SP sites on the N, P, and S ternary co-doped hollow carbon nanocage (Fe–N4SP/NPS-HC) was experimentally and precisely synthesized. Detailed structural characterization confirmed the critical role of highly coordinated configurations in tailoring the d-band centers, spin polarizations, and electronic structures. Consequently, the obtained Fe–N4SP/NPS-HC catalyst exhibited superior ORR activity, improved reaction kinetics, and enhanced stability in both alkaline and acidic media. When used in ZABs, the Fe–N4SP/NPS-HC exhibited high power density and amazing extra-long durability, superior to most reported catalysts even noble metal catalysts, demonstrating the potential practical application value.
Results and discussion
Theoretical prediction of Fe–N4SP model
In this study, the restricted ORR activity of conventional Fe–N–C catalysts was explored by setting up a standard four-coordination model (Fe–N4) as a foundation for DFT computations (Fig. S1 of the ESI†). Initially, the Gibbs free energy profiles were calculated for the ORR's basic steps on the standard Fe–N4 model at variable pH levels. The step bearing the most significant thermodynamic hurdle was deemed the rate-determining step (RDS) of the overall ORR pathway. In Fig. S1a and b (ESI†), the *OH desorption step on the standard Fe–N4 model's surface emerged as the RDS in the ORR, pointing to the suboptimal activity of the Fe–N4 sites stemming from the overly robust binding between the *OH intermediate and Fe sites. Nevertheless, the Sabatier principle suggested that the catalyst's adsorption for reaction intermediates should neither be too strong nor too weak. Over-adsorption can lead to a detrimental buildup of reaction intermediates on the catalyst's surface, thereby obstructing the reaction. The binding energy of the intermediate on the Fe site would be contingent upon its d-band center position. Consequently, fine-tuning the d-band center to weaken the adsorption of *OH could significantly improve ORR catalysis in Fe–N–C materials. Meanwhile, the d-band center of the same element would be closely related to the width of its d-orbitals. As illustrated in Fig. 1(a), the broadening of the d-band resulted in an elongated and narrowed shape (state α to state β) since the change in the bandwidth did not affect the total number of energy levels. Under constant electron filling number, the entire band shifted downward after narrowing, accompanied by a drop in the d-band center (state β to state γ). Harnessing this principle, we aim to augment the coordination atoms (without replacements) of the Fe sites in Fe–N–C single-atom catalysts. In this case, higher coordination numbers would induce more intense d-orbital interactions of the single Fe atom, thereby widening the d-band and lowering the d-band center, conducive to weakening the adsorption of *OH.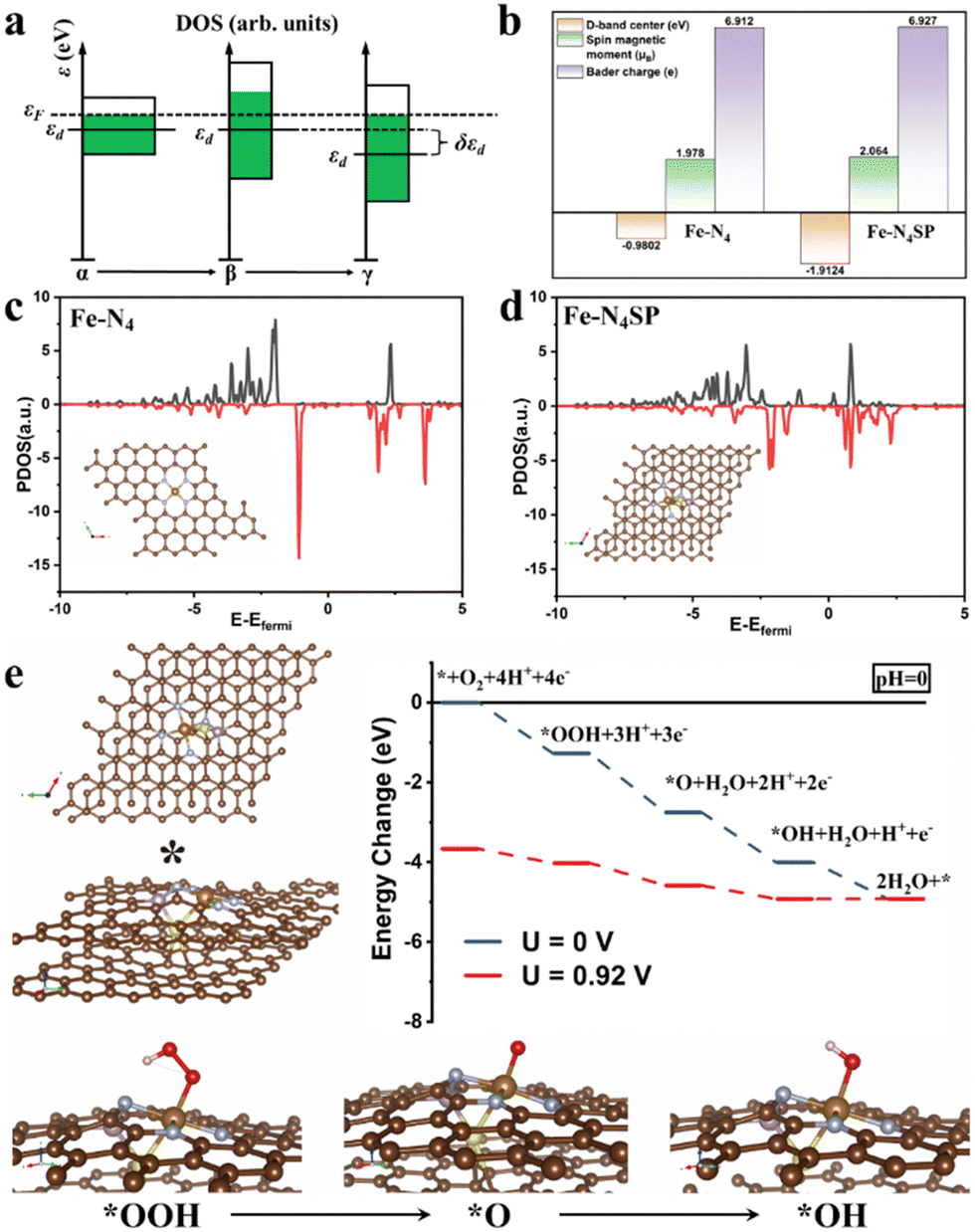 | ||
| Fig. 1 Theoretical prediction. (a) Effect of energy bandwidth on d-band center position (εd: d-band center). (b) D-band centers, spin magnetic moments, and Bader charges of Fe–N4 and Fe–N4SP. Partial density of states (PDOS) for Fe 3d of (c) Fe–N4 and (d) Fe–N4SP. (e) The optimized structure of ORR intermediates on the Fe–N4SP model and free energy diagrams of Fe–N4SP at different electrode potentials in an acidic electrolyte. | ||
Accordingly, the Fe–N4SP configuration was designed to increase the coordination number of Fe sites by introducing axially bridged S atoms in the first CS and P atoms in the second CS. As expected, the d-band center of Fe–N4SP shifted to −1.9124 eV, a value 0.9 eV lower than the d-band center of the standard Fe–N4 configuration (−0.9802 eV, Fig. 1(b)–(d)). Furthermore, the computed ORR free energy diagrams of the Fe–N4SP model are displayed in Fig. 1(e). At U = 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V (vs. RHE), every reaction step in the Fe–N4SP configuration moved downhill, suggesting spontaneous and thermodynamically exothermic ORR processes. Notably, the free energy displayed an identical trend under alkaline correction, suggesting no altered RDS of the ORR process by pH adjustment (Fig. S2, ESI†). The limiting potential of Fe–N4SP was identified as 0.92V (Fig. 1(e)), highlighting the enhanced ORR activity that can be credited to the adsorption energy refinement of *OH intermediates by the highly coordinated structures in both acidic and alkaline environments. Moreover, we also developed the Fe–N4S and the Fe–N4P models to illustrate the functionality of sole S or P on the Fe–N4 configuration. As shown in Fig. S3 and S4 (ESI†), the individual doping of S or P also optimized the adsorption of *OH to a certain extent. However, the overpotentials for both models did not exceed 0.76 V, which is significantly less efficient for the ORR compared to the Fe–N4SP model (0.92 V). This finding underscored the synergistic effect between the axially bridged S atoms and the second CS-doped P atoms, where sulfur increased the coordination number at the Fe center, and phosphorus disrupted the square planar symmetry of the Fe–N4 configuration. In particular, sulfur positioned axially and phosphorus situated at a near-neighbor location together contributed to a charge redistribution of the Fe–N4 sites (Fig. S5, ESI†). Consequently, this distinctive high-coordination Fe–N4SP configuration not only promoted the downward shift of the d-band center but also diminished the electronegativity and increased the spin density of the Fe center (Fig. 1(b)), conducive to facilitating the desorption of *OH intermediates.
V (vs. RHE), every reaction step in the Fe–N4SP configuration moved downhill, suggesting spontaneous and thermodynamically exothermic ORR processes. Notably, the free energy displayed an identical trend under alkaline correction, suggesting no altered RDS of the ORR process by pH adjustment (Fig. S2, ESI†). The limiting potential of Fe–N4SP was identified as 0.92V (Fig. 1(e)), highlighting the enhanced ORR activity that can be credited to the adsorption energy refinement of *OH intermediates by the highly coordinated structures in both acidic and alkaline environments. Moreover, we also developed the Fe–N4S and the Fe–N4P models to illustrate the functionality of sole S or P on the Fe–N4 configuration. As shown in Fig. S3 and S4 (ESI†), the individual doping of S or P also optimized the adsorption of *OH to a certain extent. However, the overpotentials for both models did not exceed 0.76 V, which is significantly less efficient for the ORR compared to the Fe–N4SP model (0.92 V). This finding underscored the synergistic effect between the axially bridged S atoms and the second CS-doped P atoms, where sulfur increased the coordination number at the Fe center, and phosphorus disrupted the square planar symmetry of the Fe–N4 configuration. In particular, sulfur positioned axially and phosphorus situated at a near-neighbor location together contributed to a charge redistribution of the Fe–N4 sites (Fig. S5, ESI†). Consequently, this distinctive high-coordination Fe–N4SP configuration not only promoted the downward shift of the d-band center but also diminished the electronegativity and increased the spin density of the Fe center (Fig. 1(b)), conducive to facilitating the desorption of *OH intermediates.
Preparation and morphological characterization of Fe–N4SP/NPS-HC
Based on the theoretical predictions, we precisely synthesized N, P, and S ternary co-doped hollow carbon nanocages with the Fe–N4SP configuration (Fe–N4SP/NPS-HC), followed by performance testing and mechanistic validation. The preparation procedure of Fe–N4SP/NPS-HC is schematically illustrated in Fig. 2(a). Initially, ZIF-8/FePc@PZS was synthesized by adding iron phthalocyanine (FePc) into the typical zeolite imidazole framework (ZIF-8) followed by coating with a uniform layer of phosphonitrile polymer (PZS) on its surface. This method allowed the encapsulation of FePc in the cavity of ZIF-8 without a change in the crystal structure (Fig. S6 and S7, ESI†) and the thickness of the PZS shell layer is about 20 nm (Fig. 2(b)). Afterward, Fe–N4SP/NPS-HC was fabricated by one-step pyrolysis and acid etching of ZIF-8/FePc@PZS, in which doped N served as the effective anchoring sites for the Fe atom, while abundant P and S elements in PZS were synchronously doped into the carbon shell. Additionally, the S element was also directly and axially bonded to the Fe atom to modulate the electronic structure of Fe–N4. The transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of Fe–N4SP/NPS-HC in Fig. 2(c), (d) and Fig. S8 (ESI†) displayed a hollow carbon nanocage without any lattice striations, revealing Fe moieties present as single atoms or tiny clusters. Such a unique hollow structure would maximize the accessibility of the active sites to the electrolyte, as well as facilitate ion transport and mass transfer during electrocatalysis.45–47 Furthermore, only a broad carbon peak (at 24.4°) was observed in the X-ray diffraction (XRD) pattern, proving the complete etching of FeP caused by pyrolysis (Fig. S8b, ESI†). The aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM) also confirmed uniformly dispersed Fe species as individual atoms throughout the hollow carbon shell (Fig. 2(e)). Additionally, energy-dispersive X-ray spectroscopy (EDX) mapping illustrated the presence of well-distributed C, N, P, S, and Fe elements throughout the entire carbon matrix (Fig. 2(f)). Among them, the elemental contents of Fe, P, and S were determined to be 1.33%, 3.90%, and 2.15%, respectively (Table S1, ESI†).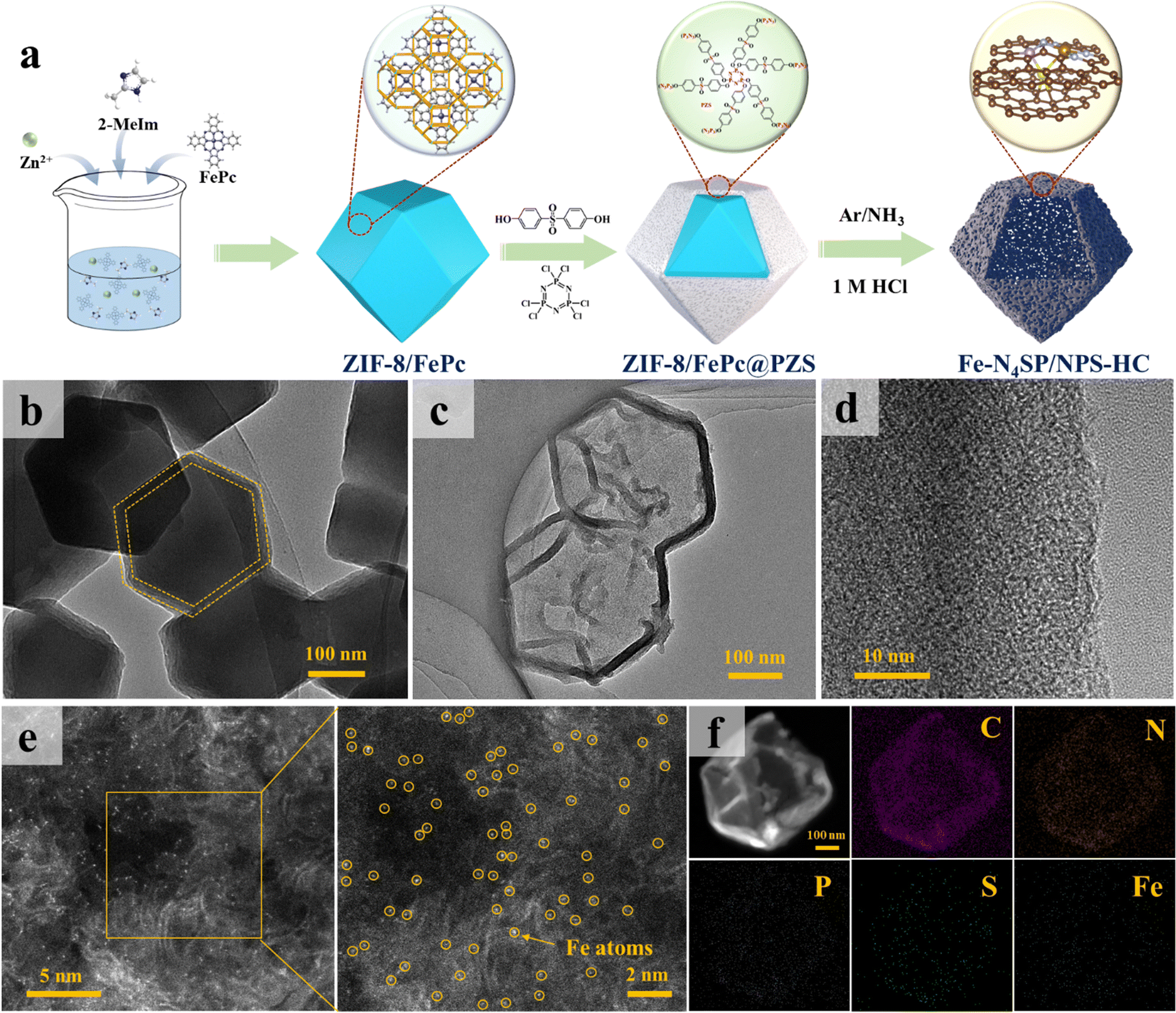 | ||
| Fig. 2 Synthesis and morphological characterizations. (a) Schematic illustration of the synthesis process of Fe–N4SP/NPS-HC. TEM images of (b) ZIF-8/FePc@PZS and (c) Fe–N4SP/NPS-HC. (d) HRTEM, (e) HAADF-STEM, and (f) corresponding EDX mapping images of Fe–N4SP/NPS-HC. | ||
Formation mechanism of the hollow structure
To gain insights into the formation of hollow structures and the influence of heteroatom doping, a series of controlled experiments were conducted and the corresponding morphologies are summarized in Fig. S9 (ESI†). Note that details of the experiments can be found in the ESI.† Obviously, Fe–N4/NC and Fe–N4P/NPC without S-doping depicted a similar porous solid polyhedron, with Fe–N4P/NPC accompanied by slight surface collapse, while Fe–N4S/NS-HC displayed a hollow nanocage (Fig. S10, ESI†). Thus, the S element played a key role in the formation of hollow structures. Further validation was implemented by thermogravimetric (TG) and XRD analyses. In Fig. S11a (ESI†), ZIF-8/FePc@PZS depicted a preferential decomposition at about 250 °C, while ZIF-8/FePc started to break down at 500 °C. Besides, the XRD pattern exhibited ZIF-8/FePc with the retained intact lattice structure of ZIF-8, while ZIF-8/FePc@PZS showed significant decomposition at 400 °C (Fig. S11b and c, ESI†). As the temperature rose to 600 °C, ZIF-8/FePc@PZS completely transformed into an amorphous carbon structure, while ZIF-8/FePc still retained the characteristic peak of ZnO. Therefore, the PZS shell contributed to the decomposition of ZIF-8/FePc. Note that the TG curve of ZIF-8/FePc@PZM highly coincided with ZIF-8/FePc, while ZIF-8/FePc@MZS also decomposed at 250 °C but at a lower decomposition rate than that of ZIF-8/FePc@PZS (Fig. S11a, ESI†). Thus, S played a vital role in the formation of hollow structures, while P accelerated the reaction to some extent.Based on this information, a formation mechanism of the hollow structure was proposed and the details are shown in Fig. S11d (ESI†). During the pyrolysis process, ZIF-8/FePc underwent decomposition induced by S2− ions from the PZS shell, releasing Zn2+ ions. The difference in radii between Zn2+ and S2− resulted in a lower inward transport rate of S2− than the outward diffusion of Zn2+, invoking the Kirkendall effect.48 The internal ZIF-8/FePc gradually decomposed and ultimately formed a hollow structure as a function of the occurrence of the Kirkendall effect. Simultaneously, the FePc molecules trapped in the ZIF-8 cavity were reduced in situ to form atomically dispersed Fe–N4 sites.
Coordination configurations and spin states of catalysts
The chemical valences and local coordination environments of Fe sites were clarified by X-ray absorption spectroscopy (XAS). The Fe K-edge absorption of Fe–N4SP/NPS-HC was situated between those of FePc and Fe2O3, elucidating an average valence of isolated Fe atoms of +2 to +3, as shown in the X-ray absorption near-edge structure (XANES, Fig. 3(a)). The coordination configurations of the Fe site were investigated by k3-weighted Fourier transform of Fe K-edge extended X-ray absorption fine structure (FT-EXAFS) spectroscopy in the R-space (Fig. 3(b)). Only the peak of Fe–N4SP/NPS-HC can be observed near 1.5 Å, consistent with FePc and corresponding to the predominant Fe–N scattering. The absence of the peak position at 2.2 Å demonstrated mainly anchored Fe atoms by N atoms instead of the Fe–Fe bond. It is noteworthy that a Fe–S peak can be detected in Fe–N4SP/NPS-HC via the quantitative least-squares curve-fitting, with coordination numbers of Fe–N and Fe–S calculated as 4.0 and 1.1 (Fig. 3(c), Fig. S12–S14 and Table S2, ESI†), respectively. In order to pinpoint the exact coordination structures of Fe–N4SP/NPS-HC, we considered all possible models for the position of coordination S atoms according to EXAFS fitting results, including four in-plane sites and the axial position. And the total energies of these models were calculated by the DFT (Fig. S15, ESI†), which indicates that the energy is the lowest and the structure is the most stable when sulfur is positioned axially. This convincingly demonstrated each Fe atom coordinated to four in-plane nitrogen atoms and one axially oriented sulfur atom, unambiguously fitting with the Fe–N4SP configuration, where the shortened Fe–S bond implied the effect of P atoms in the second CS (Table S2, ESI†). The unique axial S-coordination and adjacent P-coordination moved the central Fe slightly out of the Fe–N4 plane, thereby breaking the symmetry interactions of the original Fe–N4 sites.49,50 Compared to Fe–N4/NC, a negative shift of 0.3 eV accompanied by a decrease in the L3/L2 ratio of Fe–N4SP/NPS-HC were seen in Fe L2,3-edge XANES (Fig. 3(d)). Both indicated the declined valence state of the Fe center, consistent with DFT calculations (Fig. 1(b)). This revealed the strong electron transfer between Fe atoms and axial S atoms, as well as surrounding doped P atoms through d–p orbital hybridization. Additionally, the wavelet transform (WT) analysis shown in Fig. 3(e) illustrated only a maximum intensity at about 5 Å−1, corresponding to the Fe–N bond. The deviation in the contour toward ∼9 Å−1 can be attributed to the Fe–S contribution. Overall, the Fe–N4SP configuration was identified as the most probable structure of the Fe–N4SP/NPS-HC catalyst. Specifically, the Fe species were stabilized by the four N atoms as isolated atoms, while being modified by the S atom in the first CS combined with the P atom in the second CS.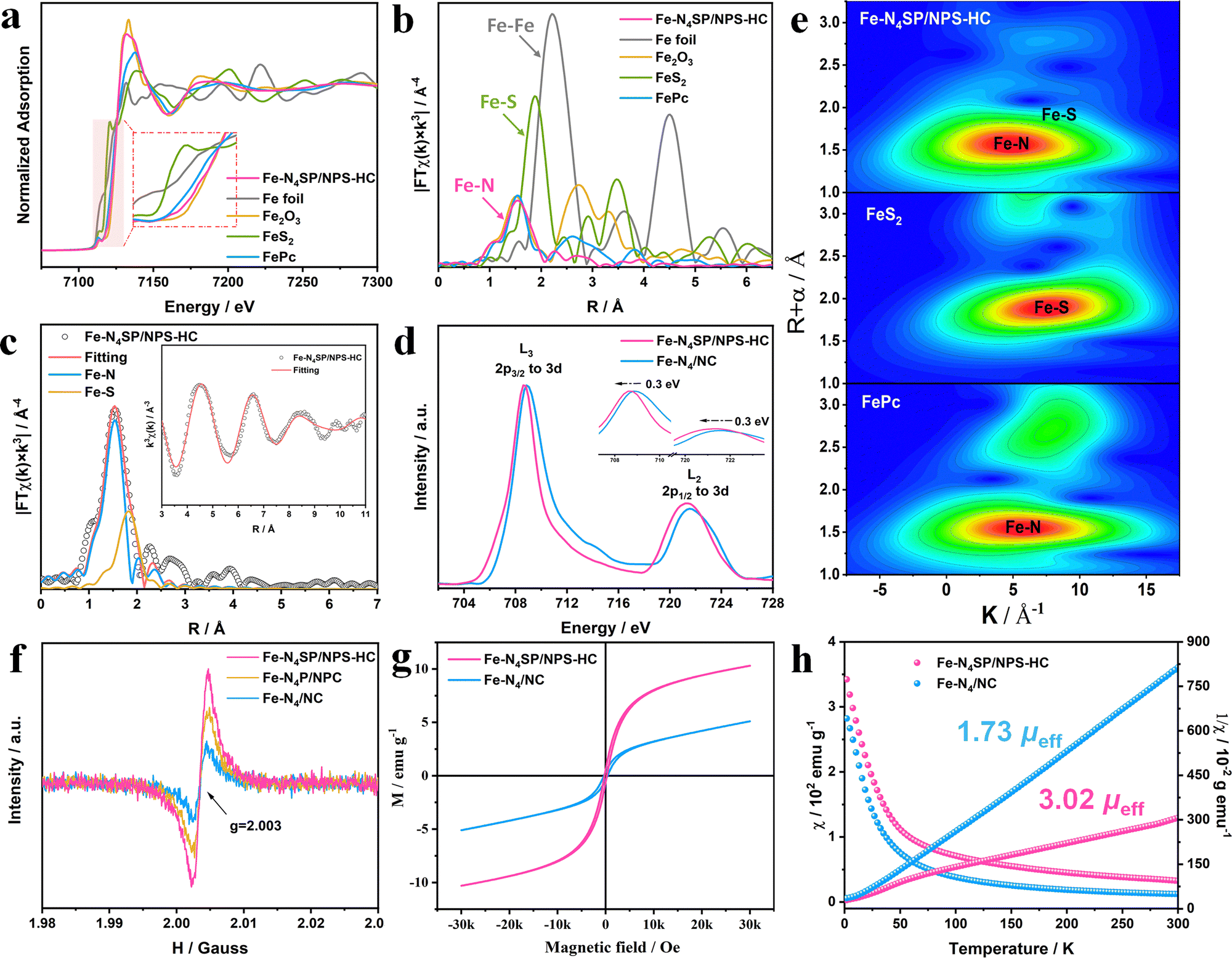 | ||
| Fig. 3 Atomic and electronic structure analysis. (a) Fe K-edge XANES, (b) FT-EXAFS spectra, and (e) WT contour plots of Fe–N4SP/NPS-HC and reference materials. (c) FT-EXAFS fitting curve of Fe–N4SP/NPS-HC. (d) Fe L-edge XANES and (f) EPR spectra of Fe–N4SP/NPS-HC and Fe–N4/NC. (g) The magnetic hysteresis loops of Fe–N4SP/NPS-HC and Fe–N4/NC at room temperature (300 K). (h) Temperature-dependent magnetic susceptibilities of Fe–N4SP/NPS-HC and Fe–N4/NC. | ||
Electron paramagnetic resonance (EPR) measurements were carried out to highlight the importance of 3d orbital electron arrangement in ORR activity. Note that in this technique, the g factor would directly be related to the paramagnetic absorption signal of the unpaired spin electron.51 As shown in Fig. 3(f), Fe–N4SP/NPS-HC exhibited the highest g value compared to Fe–N4P/NPC and Fe–N4/NC, indicating increased unpaired 3d electron numbers of the Fe center through doping of heteroatoms, especially axial S atoms. The higher saturation magnetization also meant more unpaired electrons in Fe–N4SP/NPS-HC (Fig. 3(g)). The zero-field cooling (ZFC) temperature-dependent magnetic susceptibility tests were then performed to further explore the d-electrons spin states of Fe sites. The effective magnetic moments of Fe–N4SP/NPS-HC and Fe–N4/NC calculated by fitting the curve of χ−1–T according to the Curie–Weiss law were 3.02 μeff and 1.73 μeff (Fig. 3(h)). Obviously, the highly coordinated configuration triggered by axial S atoms and proximal P atoms intensified the spin polarization in the 3d orbitals of Fe atoms, validating the DFT calculations (Fig. 1(b)). Thus, high-energy eg orbitals with more electron filling can be obtained in Fe–N4SP/NPS-HC for facilitated penetration of oxygen antibonding π-orbitals, thereby promoting O2 activation and reduction.52–55
Modulation of the Fe content and porosity
The influence of Fe sites was studied by preparing Fe–N4SP/NPS-HC with different Fe contents. As depicted in Fig. S16 (ESI†), all as-prepared Fe–N4SP/NPS-HC with different Fe contents exhibited similar hollow nanocage structures, further proving the unaffected structure of the catalysts by adding FePc. As the amount of added FePc increased, the metal phase gradually transformed from the FeP phase to the Fe2P phase, and all metal phosphides were removed after acid leaching (Fig. S17, ESI†). The Fe contents of Fe–N4SP/NPS-HC-0.5, Fe–N4SP/NPS-HC, Fe–N4SP/NPS-HC-1.5, and Fe–N4SP/NPS-HC-2 determined by inductively coupled plasma optical emission spectrometry (ICP-OES) were 0.63, 1.33, 0.39, and 0.21 wt.% (Table S3, ESI†), respectively. Notably, the addition of excess FePc led to a decline in Fe content in the catalyst due to the weakened spatial confinement effect of ZIF-8, insufficient in limiting the migration of excessive FePc during pyrolysis and consequently triggering agglomeration. Most Fe moieties were removed as phosphate iron during subsequent acid etching, resulting in a lower Fe content in the catalysts. Interestingly, FePc was involved in modulating not only the densities of metal sites but also the specific surface area and porosity of the catalysts. The Brunauer–Emmett–Teller (BET) measurements shown in Fig. S18 and Table S4 (ESI†) revealed an increment in the specific surface area as a function of the amount of added FePc. Moreover, pore diameters of Fe–N4SP/NPS-HC-0.5 and Fe–N4SP/NPS-HC were predominantly distributed between 2 and 4 nm, while larger mesopores (7–10 nm) started to form after the addition of 150 mg FePc, further confirming the aggregation of excess FePc. A large number of aggregated metal compounds were etched, leaving abundant hierarchical micro/mesopores in the carbon matrix. The large specific surface area and abundant pores could enhance the contact between the active site and electrolyte, as well as accelerate electron and mass transport.56,57Electrocatalytic ORR performance of catalysts
The unique coordination configuration and electronic environment of the as-prepared catalysts prompted the investigation of their ORR properties in alkaline and acidic electrolytes in a three-electrode system. The linear sweep voltammetry (LSV) curves of as-prepared catalysts evaluated in 0.1 M KOH are displayed in Fig. 4(a). The Fe–N4SP/NPS-HC presented a more positive half-wave potential (E1/2) of 0.912 V than Fe–N4P/NPC (0.858 V), Fe–N4/NC (0.828 V), Pt/C (0.889 V), and most published state-of-the-art Fe-based electrocatalysts (Table S5, ESI†), indicating the excellent ORR activity of Fe–N4SP/NPS-HC in alkaline media. Since a smaller Tafel slope would mean faster reaction kinetics, Fe–N4SP/NPS-HC with a much smaller Tafel slope of 39.18 mV dec−1 than others would indicate the highly coordinated configuration contributing to an accelerated ORR process (Fig. 4(b)). Undeniably, the kinetic current density (Jk) is also a key parameter to evaluate the ORR activity. Fe–N4SP/NPS-HC showed a record Jk value of 60 mA cm−2 at 0.85 V, exceeding those of Fe–N4P/NPC, Fe–N4/NC, and Pt/C by 8.5-, 20-, and 4-fold, respectively (Fig. 4(c)). Furthermore, the effect of the hollow structure was explored by preparing Fe–N4/NPSC with a solid structure. As expected, Fe–N4SP/NPS-HC exhibited better ORR performances, indicating the superiority of the hollow structure (Fig. S19, ESI†).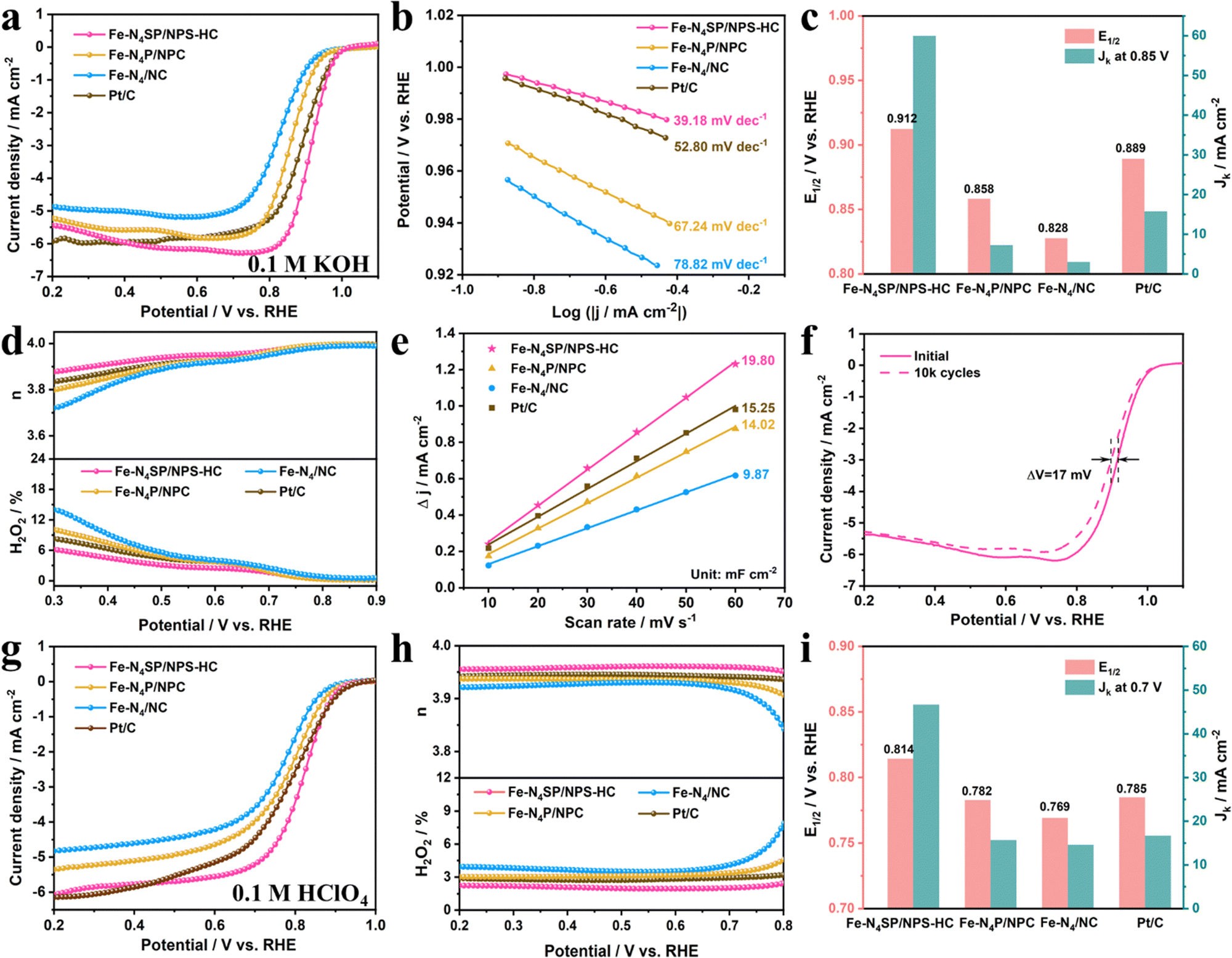 | ||
| Fig. 4 Electrocatalytic performance of the ORR. (a) ORR polarization curves at a rotation rate of 1600 rpm, (b) Tafel plots, (c) comparison of E1/2 and Jk at 0.85 V, (d) electron-transfer number and H2O2 yield, and (e) double-layer capacitance (Cdl) values of Fe–N4SP/NPS-HC, Fe–N4P/NPC, Fe–N4/NC, and Pt/C in 0.1 M KOH. (f) LSV curves of Fe–N4SP/NPS-HC before and after 10k cycles in 0.1 M KOH. (g) ORR polarization curves at a rotation rate of 1600 rpm, (h) electron-transfer number and H2O2 yield, and (i) comparison of E1/2 and Jk at 0.7 V of Fe–N4SP/NPS-HC, Fe–N4P/NPC, Fe–N4/NC, and Pt/C in 0.1 M HClO4. | ||
The in-depth ORR details of Fe–N4SP/NPS-HC are demonstrated in Fig. S20 (ESI†). Compared to the N2-saturated electrolyte, the pronounced reduction peak of cyclic voltammetry (CV) at 0.895 V illustrated good intrinsic reactivity in the O2-saturated electrolyte. The first-order reaction kinetics and a 4-electron transfer pathway were extrapolated by the parallel linear relationships in Koutecky–Levich (K–L) plots. Furthermore, rotating ring disk electrode (RRDE) tests confirmed a transfer electron number of Fe–N4SP/NPS-HC close to 4 accompanied by a lower H2O2 yield (below 6%) than other catalysts, indicating its higher selectivity toward the 4-electron mediated ORR process (Fig. 4(d)). Afterward, the electrochemical double-layer capacitance (Cdl) was used to estimate the electrochemical surface areas (ECSA). As shown in Fig. 4(e) and Fig. S21 (ESI†), the Cdl value of Fe–N4SP/NPS-HC was identified as 19.8 mF cm−2, superior to those of Fe–N4P/NPC (14.02 mF cm−2), Fe–N4/NC (9.87 mF cm−2), and Pt/C (15.25 mF cm−2). This result implied that the hollow structure endowed Fe–N4SP/NPS-HC with maximum exposure of active sites, thereby facilitating the ORR reaction. Besides, the catalyst stability was assessed by the LSV after cycling and chronoamperometric response (i–t) curves. As presented in Fig. 4(f), only 17 mV of decay in E1/2 was recorded after 10k cycles for Fe–N4SP/NPS-HC. Moreover, the current of Fe–N4SP/NPS-HC still remained above 95% after continuously operating for 70 h, while Pt/C decayed to 68% only after 37 h (Fig. S20d, ESI†). All of the above results demonstrated the excellent ORR activity, reaction kinetics, and stability of Fe–N4SP/NPS-HC in the alkaline media.
The broadened usefulness of Fe–N4SP/NPS-HC was further investigated by evaluating the ORR performance in acidic media. As shown in Fig. 4(g)–(i), Fe–N4SP/NPS-HC also exhibited a striking ORR activity in 0.1 M HClO4 with an E1/2 value of 0.814 V and a Jk value of 46.7 mA cm−2 at 0.7 V, values much higher than those of Fe–N4P/NPC (E1/2 = 0.782 V, Jk = 15.7 mA cm−2) and Fe–N4/NC (E1/2 = 0.769 V, Jk = 14.3 mA cm−2), even exceeding those of commercial Pt/C (E1/2 = 0.785 V, Jk = 16.5 mA cm−2). Compared to other catalysts, the smallest Tafel slope (49.72 mV dec−1) and the largest Cdl (36.72 mF cm−2) of Fe–N4SP/NPS-HC also implied faster reaction kinetics and a larger electrochemically active area (Fig. S22a, b and S23, ESI†). Meanwhile, RRDE experiments confirmed a high-efficiency 4e− ORR pathway with a transfer of 3.96 electrons per process accompanied by an H2O2 yield below 3% (Fig. 4(h)). Compared to Pt/C exhibiting 69% current retention attenuation after 52 h, Fe–N4SP/NPS-HC showed a high current retention of 97% after 76 h and only 12.2 mV attenuation after 10k cycles, indicating outstanding long-cycle durability in an acidic electrolyte (Fig. S22c and d, ESI†). In addition, unlike the Pt/C, the CV curves and the current density of Fe–N4SP/NPS-HC remained essentially unchanged after methanol crossover, inferring a favorable methanol tolerance in both acidic and alkaline environments (Fig. S24, ESI†). Significantly, the performances of the proposed Fe–N4SP/NPS-HC catalyst also compared well with previously reported single-atom catalysts (Table S6, ESI†). All these results corroborated the predominance of the designed theoretical model.
The effects of the metal site density on the catalytic ORR performance of Fe–N4SP/NPS-HC with different Fe contents were also studied. Both the half-wave potential, the Tafel slope, and kinetic current density were positively correlated with the Fe content and far exceeded those of HPS-HC without Fe species (Fig. S25, ESI†), confirming the critical role played by isolated Fe–N4 sites in the ORR activity. Therefore, modulating the electronic structure and spin state of the Fe–N4 sites is essential to enhance ORR activity. Besides, the impacts of the calcination atmosphere on ORR activity were also compared in Fig. S26a and b (ESI†). Unlike results under an Ar atmosphere, Fe–N4SP/NPS-HC obtained in Ar/NH3 exhibited a better E1/2 value and a smaller Tafel slope. Furthermore, X-ray photoelectron spectroscopy (XPS) spectra were recorded to reveal the differences in chemical composition between Fe–N4SP/NPS-HC obtained under an Ar/NH3 and an Ar atmosphere. The high-resolution N 1s spectra of Fe–N4SP/NPS-HC and Fe–N4SP/NPS-HC-Ar both displayed three peaks at 401.3, 400.4, and 398.5 eV, corresponding to graphitic-N, pyrrolic-N, and pyridinic-N, respectively (Fig. S26c, ESI†). Meaningfully, the results showed that a small amount of ammonia contributed to the enhancement of the N content in the catalyst, especially the percentage of pyridinic-N and pyrrolic-N (Fig. S26d, ESI†), which were also considered important ORR active sites in previous reports.58–60 Overall, the single Fe atoms coordinated with pyridinic-N and pyrrolic-N served as predominant catalytic active sites. Also, the enhanced ORR activity can be attributed to the near/long-range electronic regulation of the axial S-coordination and adjacent P-coordination, synergizing the unique hollow structure.
Performance in zinc–air battery applications
Inspired by the excellent ORR performance, a Zn–air battery (ZAB) was prepared with Fe–N4SP/NPS-HC as the catalyst and tested to verify its practical application value. The schematic illustration is shown in Fig. 5(a) and further details can be found in the ESI.† As expected, the ZAB based on Fe–N4SP/NPS-HC showed an open circuit voltage of 1.42 V and a specific capacity of 893 mA h gZn−1 at a current density of 5 mA cm−2, outperforming ZAB based on Pt/C (1.39 V and 752 mA h gZn−1, Fig. S27, ESI†). Moreover, Fe–N4SP/NPS-HC-based ZAB possessed a better current density than others at the same voltage (Fig. 5(b)), and the peak power densities (225 mW cm−2) were 1.2-, 1.5-, and 1.3-fold higher than those of Fe–N4P/NPC, Fe–N4/NC, and Pt/C, respectively. These data were also superior to recently reported ZABs (Fig. S28 and Table S7, ESI†). The galvanostatic discharge curves of Fe–N4SP/NPS-HC and Pt/C at different current densities revealed a decline in discharge potential plateau as the current density increased (Fig. 5(c)). However, the voltage of Fe–N4SP/NPS-HC was permanently higher than that of Pt/C. Fe–N4SP/NPS-HC still maintained a voltage plateau of 1.12 V even at a high current density of 100 mA cm−2. As the current density gradually decreased, the voltage plateau also returned to the initial state, indicating the remarkable rate performance of Fe–N4SP/NPS-HC. Significantly, unlike the obvious voltage drop of Pt/C-based ZAB at 130 h, Fe–N4SP/NPS-HC-based ZAB still maintained an ultra-stable voltage window after continuous operation for over 320 h (approximately 2880 cycles), revealing superb long-term durability (Fig. 5(d) and Fig. S29, ESI†). It is worth mentioning that the charging voltage of Fe–N4SP/NPS-HC-based ZAB is significantly lower than that of Pt/C-based ZAB, which can be attributed to the excellent oxygen evolution reaction (OER) activity of Fe–N4SP/NPS-HC (Fig. S30, ESI†). Besides, as shown in Fig. 5(e), the charge/discharge voltage gap was only 0.704 V in the initial 10 cycles and the round-trip efficiency reached as high as 62.9%. Note that the voltage gap only increased by 2% and 9% at the 1505th and 2875th cycles, respectively. Also, the cycling efficiency still remained above 60% even after the 2875th cycle (Fig. 5(f) and (g)). In sum, the extra-long stability would greatly be important for the practical use of Fe–N4SP/NPS-HC-based ZAB.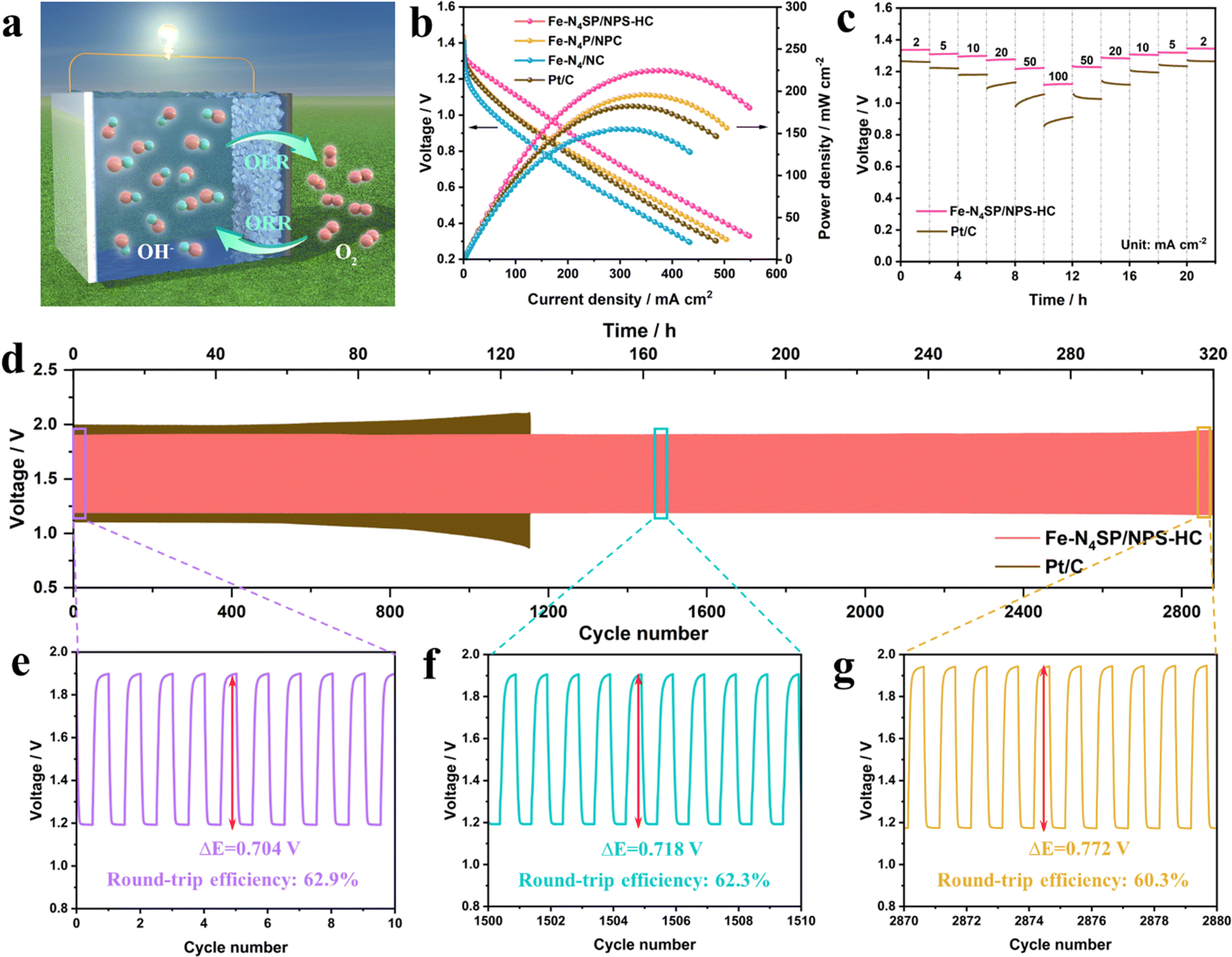 | ||
| Fig. 5 Zinc–air battery performance. (a) Schematic illustration of a Zn–air battery. (b) Discharge polarization and power density curves of Fe–N4SP/NPS-HC, Fe–N4P/NPC, Fe–N4/NC, and Pt/C. (c) Galvanostatic discharge curves of Fe–N4SP/NPS-HC and Pt/C at different current densities. (d) Charge/discharge cycling stabilities of Fe–N4SP/NPS-HC and Pt/C at a current density of 5 mA cm−2. (e)–(g) Charge/discharge cycle curves and round-trip efficiency of different cycles. | ||
Conclusions
In summary, a meticulous multi-shell synergistic coordination strategy was successfully used to construct Fe–N4SP configurations with an elevated coordination number. DFT calculations and detailed structural characterization demonstrated that the precisely tailored interaction of axial S-coordination and adjacent P-coordination effectively induced electronic delocalization and decreased the d-band center of the Fe–N4 site, accompanied by an increase in the spin state. This strategy contributed to the activation and reduction of oxygen, as well as the desorption of *OH intermediates in the RDS, endowing Fe–N4SP with an enhanced ORR intrinsic activity. The optimal Fe–N4SP/NPS-HC catalyst displayed an impressive high half-wave potential (E1/2, 0.912 V in alkaline media and 0.814 V in acidic media) coupled with excellent kinetic current density (Jk, 60 mA cm−2 at 0.85 V in alkaline media and 46 mA cm−2 at 0.7 V in acidic media), along with an outstanding maximum power density of 225 mW cm−2 and continuous stable operation up to 320 h when used as the ZAB catalyst. This work unveils the critical role of the coordination micro milieu and spin state on the ORR catalytic performance, promising to guide the design and optimization of high-efficiency single-atom catalysts from the electronic level.Author contributions
Conceptualization, S. Y. and J. Q. L.; methodology, J. Q. L.; theoretical calculations, W. B. C.; formal analysis, investigation and data curation, J. Q. L.; writing – original draft preparation, J. Q. L. and W. B. C.; writing – review & editing, S. Y., T. L. and Q. W.; supervision, S. Y.; project administration, S. Y. and Q. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant no. 52274294) and the Fundamental Research Funds for the Central Universities (grant no. N2124007-1). We would like to thank MogoEdit (https://www.mogoedit.com) for its English editing during the preparation of this manuscript.Notes and references
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS.
- X. Zhu, C. Hu, R. Amal, L. Dai and X. Lu, Energy Environ. Sci., 2020, 13, 4536–4563 RSC.
- C.-X. Zhao, J.-N. Liu, J. Wang, C. Wang, X. Guo, X.-Y. Li, X. Chen, L. Song, B.-Q. Li and Q. Zhang, Sci. Adv., 2022, 8, eabn5091 CrossRef CAS PubMed.
- X. W. Zhong, Z. Y. Zheng, J. H. Xu, X. Xiao, C. B. Sun, M. T. Zhang, J. B. Ma, B. M. Xu, K. Yu, X. Zhang, H.-M. Cheng and G. M. Zhou, Adv. Mater., 2023, 35, 2209980 CrossRef CAS.
- B. J. Hopkins, C. N. Chervin, J. W. Long, D. R. Rolison and J. F. Parker, ACS Energy Lett., 2020, 5, 3405–3408 CrossRef CAS.
- W. Li, L. Cheng, X. Chen, Y. Liu, Y. Liu, Q. Liu and Y. Huang, Nano Energy, 2023, 106, 108039 CrossRef CAS.
- W. Shao, R. Yan, M. Zhou, L. Ma, C. Roth, T. Ma, S. Cao, C. Cheng, B. Yin and S. Li, Electrochem. Energy Rev., 2023, 6, 11 CrossRef CAS.
- T. Cui, Y.-P. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CrossRef CAS PubMed.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- A. E. Thorarinsdottir, D. P. Erdosy, C. Costentin, J. A. Mason and D. G. Nocera, Nat. Catal., 2023, 6, 425–434 CrossRef CAS.
- M. L. Jiao, Q. Zhang, C. L. Ye, Z. B. Liu, X. W. Zhong, J. X. Wang, C. Li, L. X. Dai, G. M. Zhou and H.-M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202202119 CrossRef CAS.
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS.
- J.-B. Liu, H.-S. Gong, G.-L. Ye and H.-L. Fei, Rare Met., 2022, 41, 1703–1726 CrossRef CAS.
- Q. Yang, Y. Jiang, H. Zhuo, E. M. Mitchell and Q. Yu, Nano Energy, 2023, 111, 108404 CrossRef CAS.
- X. W. Zhong, Y. F. Shao, B. Chen, C. Li, J. Z. Sheng, X. Xiao, B. M. Xu, J. Li, H.-M. Cheng and G. Zhou, Adv. Mater., 2023, 35, 2301952 CrossRef CAS.
- Y. Li, Y. Ding, B. Zhang, Y. Huang, H. Qi, P. Das, L. Zhang, X. Wang, Z.-S. Wu and X. Bao, Energy Environ. Sci., 2023, 16, 2629–2636 RSC.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M.-T. Sougrati, M. Primbs, A. M. Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, Nat. Catal., 2022, 5, 311–323 CrossRef CAS.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- X.-F. Cheng, J.-H. He, H.-Q. Ji, H.-Y. Zhang, Q. Cao, W.-J. Sun, C.-L. Yan and J.-M. Lu, Adv. Mater., 2022, 34, 2205767 CrossRef CAS.
- S. Zha, D. Wang, C. Liu, W. Wang, N. Mitsuzaki and Z. Chen, Sustainable Energy Fuels, 2022, 6, 3895–3906 RSC.
- J. Cho, T. Lim, H. Kim, L. Meng, J. Kim, S. Lee, J. H. Lee, G. Y. Jung, K.-S. Lee, F. Viñes, F. Illas, K. S. Exner, S. H. Joo and C. H. Choi, Nat. Commun., 2023, 14, 3233 CrossRef CAS PubMed.
- Y. Wang, H. Li, F. Peng and F. Gao, Chem. Eng. J., 2022, 445, 136692 CrossRef CAS.
- J. Wang, Y.-C. Huang, Y. Wang, H. Deng, Y. Shi, D. Wei, M. Li, C.-L. Dong, H. Jin, S. S. Mao and S. Shen, ACS Catal., 2023, 13, 2374–2385 CrossRef CAS.
- S. Wang, B. Huang, Y. Dai and W. Wei, Small, 2023, 19, 2205111 CrossRef CAS PubMed.
- L. Li, Y. Wen, G. Han, F. Kong, L. Du, Y. Ma, P. Zuo, C. Du and G. Yin, Small, 2023, 19, 2300758 CrossRef CAS PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS.
- C. Zheng, X. Zhang, Z. Zhou and Z. Hu, eScience, 2022, 2, 219–226 CrossRef.
- L. Yin, S. Zhang, M. Sun, S. Wang, B. Huang and Y. Du, Adv. Mater., 2023, 35, 2302485 CrossRef CAS PubMed.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215136 CrossRef CAS.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS PubMed.
- M. Wang, W. Yang, X. Li, Y. Xu, L. Zheng, C. Su and B. Liu, ACS Energy Lett., 2021, 6, 379–386 CrossRef CAS.
- H. Yin, P. Yuan, B.-A. Lu, H. Xia, K. Guo, G. Yang, G. Qu, D. Xue, Y. Hu, J. Cheng, S. Mu and J.-N. Zhang, ACS Catal., 2021, 11, 12754–12762 CrossRef CAS.
- Y. Gao, B. Liu and D. Wang, Adv. Mater., 2023, 35, 2209654 CrossRef CAS.
- C. Hu, Y. Zhang, A. Hu, Y. Wang, X. Wei, K. Shen, L. Chen and Y. Li, Adv. Mater., 2023, 35, 2209298 CrossRef CAS.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS.
- Y. Pan, Y. Chen, K. Wu, Z. Chen, S. Liu, X. Cao, W.-C. Cheong, T. Meng, J. Luo, L. Zheng, C. Liu, D. Wang, Q. Peng, J. Li and C. Chen, Nat. Commun., 2019, 10, 4290 CrossRef PubMed.
- H. Shang, W. Sun, R. Sui, J. Pei, L. Zheng, J. Dong, Z. Jiang, D. Zhou, Z. Zhuang, W. Chen, J. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 5443–5450 CrossRef CAS PubMed.
- J. Wan, Z. Zhao, H. Shang, B. Peng, W. Chen, J. Pei, L. Zheng, J. Dong, R. Cao, R. Sarangi, Z. Jiang, D. Zhou, Z. Zhuang, J. Zhang, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 8431–8439 CrossRef CAS.
- T. Zhang, Z. Sun, S. Li, B. Wang, Y. Liu, R. Zhang and Z. Zhao, Nat. Commun., 2022, 13, 6996 CrossRef CAS.
- Y. Dai, B. Liu, Z. Zhang, P. Guo, C. Liu, Y. Zhang, L. Zhao and Z. Wang, Adv. Mater., 2023, 35, 2210757 CrossRef CAS.
- Y. Li, Y. Ji, Y. Zhao, J. Chen, S. Zheng, X. Sang, B. Yang, Z. Li, L. Lei, Z. Wen, X. Feng and Y. Hou, Adv. Mater., 2022, 34, 2202240 CrossRef CAS PubMed.
- Z. Chen, H. Niu, J. Ding, H. Liu, P.-H. Chen, Y.-H. Lu, Y.-R. Lu, W. Zuo, L. Han, Y. Guo, S.-F. Hung and Y. Zhai, Angew. Chem., Int. Ed., 2021, 60, 25404–25410 CrossRef CAS.
- R. Wang, L. Zhang, J. Shan, Y. Yang, J.-F. Lee, T.-Y. Chen, J. Mao, Y. Zhao, L. Yang, Z. Hu and T. Ling, Adv. Sci., 2022, 9, 2203917 CrossRef CAS.
- R. Ma, X. Cui, X. Xu, Y. Wang, G. Xiang, L. Gao, Z. Lin and Y. Yang, Nano Energy, 2023, 108, 108179 CrossRef CAS.
- Y. Zhao, X. F. Lu, Z.-P. Wu, Z. Pei, D. Luan and X. W. (David) Lou, Adv. Mater., 2023, 35, 2207888 CrossRef CAS.
- H. Yang, Y. Liu, X. Liu, X. Wang, H. Tian, G. I. N. Waterhouse, P. E. Kruger, S. G. Telfer and S. Ma, eScience, 2022, 2, 227–234 CrossRef.
- U. Sen, M. Erkartal, C.-W. Kung, V. Ramani, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2016, 8, 23015–23021 CrossRef CAS.
- G. Qu, K. Guo, J. Dong, H. Huang, P. Yuan, Y. Wang, H. Yuan, L. Zheng and J.-N. Zhang, Energy Storage Mater., 2023, 55, 490–497 CrossRef.
- J. Yu, X. Yong and S. Lu, Energy Environ. Mater., 2023, e12587 CrossRef.
- M. M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC.
- D. Xue, P. Yuan, S. Jiang, Y. Wei, Y. Zhou, C.-L. Dong, W. Yan, S. Mu and J.-N. Zhang, Nano Energy, 2023, 105, 108020 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS.
- S. Li, L. Xia, J. Li, Z. Chen, W. Zhang, J. Zhu, R. Yu, F. Liu, S. Lee, Y. Zhao, L. Zhou and L. Mai, Energy Environ. Mater., 2022, e12560 Search PubMed.
- B. Yang, X. Li, Q. Cheng, X. Jia, Y. Liu and Z. Xiang, Nano Energy, 2022, 101, 107565 CrossRef CAS.
- M. L. Jiao, L. X. Dai, H.-R. Ren, M. T. Zhang, X. Xiao, B. R. Wang, J. L. Yang, B. L. Liu, G. M. Zhou and H.-M. Cheng, Angew. Chem., Int. Ed., 2023, 135, e202301114 CrossRef.
- Q. Shao, J. Liu, Q. Wu, Q. Li, H. Wang, Y. Li and Q. Duan, Nano-Micro Lett., 2019, 11, 4 CrossRef CAS PubMed.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- J. Liu, J. Dang, M. Wang, X. Wang, X. Duan, S. Yuan, T. Liu and Q. Wang, J. Colloid Interfaces Sci., 2022, 606, 38–46 CrossRef CAS PubMed.
- Z. Zhu, Z. Li, J. Wang, R. Li, H. Chen, Y. Li, J. S. Chen, R. Wu and Z. Wei, eScience, 2022, 2, 445–452 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03183g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |