
Achieving a high loading of cathode in PVDF-based solid-state battery†
Yang
Liu‡
a,
Xufei
An‡
a,
Ke
Yang
a,
Jiabin
Ma
a,
Jinshuo
Mi
a,
Danfeng
Zhang
 a,
Xing
Cheng
a,
Yuhang
Li
a,
Yuetao
Ma
a,
Ming
Liu
*a,
Feiyu
Kang
ab and
Yan-Bing
He
*a
a,
Xing
Cheng
a,
Yuhang
Li
a,
Yuetao
Ma
a,
Ming
Liu
*a,
Feiyu
Kang
ab and
Yan-Bing
He
*a
aShenzhen All-Solid-State Lithium Battery Electrolyte Engineering Research Center, Institute of Materials Research (IMR), Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, P. R. China. E-mail: he.yanbing@sz.tsinghua.edu.cn; liuming@sz.tsinghua.edu.cn
bLaboratory of Advanced Materials, School of Materials Science and Engineering, Tsinghua University, Beijing, 100084, P. R. China
First published on 22nd November 2023
Abstract
The lack of fundamental understanding of ion transport in the cathode of polyvinylidene fluoride (PVDF)-based solid-state lithium metal batteries restricts their rate performance and cycle stability, especially under high cathode mass loadings. Herein, we reveal that the lithium ion (Li+) solvated with N,N-dimethylformamide ([Li(DMF)x]+) in PVDF electrolyte spontaneously diffuses into the cathode, but its diffusion depth is limited, and a continuous Li+ transport network can only be built in cathodes with low loadings. We further find that carbon-coated Li1.4Al0.4Ti1.6(PO4)3 nanowires (C@LATP NW) as a cathode filler not only conduct Li+, but also exhibit strong adsorption of the [Li(DMF)x]+ complex, which promotes the uniform diffusion of [Li(DMF)x]+ in a thick cathode to construct a highly efficient Li+ transport network and achieve full reaction of the thick cathode. The carbon layer on C@LATP NW greatly suppresses the side decomposition reactions of DMF and LiFSI to improve the stability of the conductive network and structure of the cathode materials. The cathode with 3 wt% C@LATP NW enables excellent rate performance and cycle stability of solid-state batteries with high mass loadings of up to 15 mg cm−2, which opens a way for practical cathode design of solid-state batteries.
Broader contextPolyvinylidene fluoride (PVDF)-based solid-state polymer electrolytes are particularly appealing for high-performance solid-state Li metal batteries (SSLMBs) due to their merits of high ionic conductivity, great thermal stability and excellent electrochemical performance at room temperature. However, PVDF-based SSLMBs are still difficult to pair with high-mass-loading cathodes to achieve high-energy-density SSLMBs towards practical application. This is mainly due to the unclear mechanism of ion transport through the cathode and the sluggish ion transport caused by the limited contact between the solid electrolytes and the active materials. In this work, we reveal that the ion transport medium [Li(DMF)x]+ spontaneously diffuses through the cathode, and its diffusion depth and side reactions are the bottlenecks to achieving high cathode loading. On this basis, we construct a composite cathode with a highly efficient Li+ transport network via carbon-coated Li1.4Al0.4Ti1.6(PO4)3 nanowires, which also exhibit strong adsorption for [Li(DMF)x]+ complex to promote its uniform diffusion in the cathode. As a result, the utilization of the active materials is significantly improved in the thick cathode. This work provides a basic understanding of the Li+ diffusion pathways in the cathode and a new solution strategy for stable cycling of PVDF-based SSLMBs with high energy density. |
Introduction
Solid-state lithium metal batteries (SSLMBs) with a lithium (Li) metal anode and high-voltage cathodes (i.e., LiNi0.8Co0.1Mn0.1O2, NCM811) are considered to be a promising combination to improve both the safety and energy density of secondary batteries.1–8 Specifically, solid-state polymer electrolytes (SPEs) have favorable plasticity, flexibility and scalable properties, which are compatible with commercial lithium-ion battery manufacturing processes, and they are thus regarded as the most promising solid-state electrolytes (SSEs) for lithium metal batteries.9,10 In comparison with other SPEs, polyvinylidene fluoride (PVDF) solid polymer electrolytes have excellent flexibility and mechanical properties, along with high electrochemical and thermal stability, giving them great application potential in SSLMBs.11 It is generally recognized that the [Li(DMF)x]+ solution structure is formed by complexing trace amounts of residual N,N-dimethylformamide (DMF) molecules in the PVDF electrolyte with lithium ions (Li+), and the Li+ is transported through the interaction between [Li(DMF)x]+ and PVDF chains.12,13After years of effort, the ionic conductivity, mechanical strength and electrochemical stability window of PVDF-based electrolytes have been improved to a considerable level11 through inorganic filler modification,14–18 organic polymer grafting,19–21 liquid additives22,23 and electrode/electrolyte interface design.24–26 For instance, our group prepared lattice-matched BaTiO3–Li0.33La0.56TiO3−x (BTO–LLTO) nanowires with a side-by-side heterojunction structure as a filler for PVDF-based electrolytes.17 A high concentration of free Li+ was generated by Li salt dissociation stimulated by BTO and spontaneously migrated to LLTO for efficient transport due to the decreased energy barrier of Li+ transport across the lattice-matched interface. However, PVDF-based SSLMBs are still difficult to pair with high-mass-loading cathodes to achieve high-energy-density SSLMBs towards practical application. One issue is that the ion transport mechanism through the cathode when using PVDF-based SSEs is not clear yet, which restricts the rational design of a high-loading cathode. This is because the internal components of the cathode, including the active materials, binder, solid electrolyte and Li salt, are quite complex, and can present poor interactions with each other, creating great challenges for constructing a stable ion transport network. Additionally, the much smaller contact area and relatively large interspace between the SSEs and active materials limit the efficient transport of Li+.27–30 As a result, SSLMBs with high-loading cathodes present quite poor electrochemical performance.
To solve these problems, various SSEs, such as a lithiated polyvinyl formal-based Li+ conductor,31 poly(ethylene oxide),32 Li5.5PS4.5Cl1.5,33 and Li7La3Zr2O12,34 have been introduced into the cathode to construct an efficient and stable ion transport network and enhance the cathode performance. Unfortunately, high temperature is still required for them to achieve high performance. PVDF-based SSEs with trace [Li(DMF)x]+ can activate the performance of SSLMBs with low-mass-loading cathodes at room temperature (RT), but it is still difficult to promote high-performance SSLMBs with high-mass-loading cathodes towards practical application.27 Therefore, it is quite important to obtain a fundamental understanding of the ion transport mechanism inside the cathode of PVDF-based SSLMBs and subsequently design a robust, highly efficient ion transport network to improve the performance of the PVDF-based SSLMBs with high cathode loading.
Herein, we reveal that the [Li(DMF)x]+ in the PVDF electrolyte can spontaneously diffuse into the cathode to act as a mediator for the transport of Li+, but the diffusion depth is limited, and a continuous Li+ transport network across the cathode can only be built at low mass loading. Moreover, DMF is easily consumed by the side reactions in the high-voltage cathode during the desolvation processes, which not only hinders the transport path of Li+, but also seriously damages the structure of the cathode materials. As a further step, we design carbon-coated Li1.4Al0.4Ti1.6(PO4)3 nanowires (C@LATP NW) as a filler to construct an efficient and stable Li+ transport network in the cathode with a higher loading. The LATP bulk phase in C@LATP NW can provide an efficient and stable solid Li+ transport path. In addition, the C@LATP NW presents high adsorption for DMF, which promotes the uniform diffusion of [Li(DMF)x]+ inside the cathode with a higher loading along C@LATP NW. Moreover, the C@LATP NW can anchor [Li(DMF)x]+ to block its diffusion to the surface of the active material particles and significantly improve its stability. As a result, the solid-state NCM811/PVDF-based electrolyte/Li cell with 3 wt% C@LATP NW can stably cycle for 1200 cycles at 3C, and shows excellent cycle stability and rate performance even when the cathode mass loading is increased to 15 mg cm−2. This work provides a fundamental understanding of Li+ diffusion pathways in the cathode and a new solution strategy for stable cycling of PVDF-based SSLMBs with high cathode loading.
Results and discussion
It is recognized that the Li+ in PVDF-based SSLMBs is transported through the interaction between [Li(DMF)x]+ and the PVDF chains.13 However, it is unclear whether the [Li(DMF)x]+ diffuses from the electrolyte to the cathode for Li+ transport. A PVDF-based SPE consisting of PVDF and 25 wt% Li6.4La3Zr1.4Ta0.6O12 (LLZTO) particles (PVDF-LLZTO) was prepared by a solution casting method to assemble the NCM811/Li SSLMBs (Fig. S1, ESI†). The PVDF-LLZTO electrolyte exhibits a dense structure with a thickness of 70 μm and an ionic conductivity of 1.97 × 10−4 S cm−1 to tolerate a high cathode loading and achieve high utilization. Fourier transform infrared (FTIR) measurements at different cross-sectional depths were performed for an NCM811 cathode (P-NCM) with a loading of 15 mg cm−2 from NCM811/PVDF-LLZTO/Li solid-state batteries after standing for 12 h and before cycling to identify the diffusion of [Li(DMF)x]+. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak of DMF at 1650.31 cm−1 is obvious at P-NCM cathode depths from 0 to 20 μm, i.e., close to the PVDF-LLZTO electrolyte, but it slowly disappears from 30 to 60 μm, i.e., close to the current collector (Fig. 1(a)). Moreover, there is no N–CO peak corresponding to free DMF at 658 cm−1, only one at 672.08 cm−1 for the solvated [Li(DMF)x]+ (Fig. S2, ESI†), indicating that all the DMF molecules are bound to Li+.13,35 This suggests that the [Li(DMF)x]+ in the PVDF-LLZTO electrolyte spontaneously diffuses into the P-NCM cathode before electrochemical cycling of PVDF-based SSLMBs. The cross-sectional energy dispersive spectroscopy (EDS) mapping images of the cathode can further verify the limited diffusion depth of [Li(DMF)x]+ (Fig. S3, ESI†). Thus, it can be summarized that [Li(DMF)x]+ can spontaneously diffuse into the P-NCM cathode to a depth of 20 μm and act as a transport mediator for Li+. As a result, the PVDF-based SSLMB with a low mass loading of NCM811 (1.5 mg cm−2) shows a much higher discharge specific capacity of 182.5 mA h g−1 compared to that with a high mass loading of 15 mg cm−2 (97.4 mA h g−1) (Fig. S4, ESI†). In addition, the PVDF-based SSLMBs with a high cathode loading of 15 mg cm−2 present sudden capacity loss during cycling, and the impedance after the cycle is greatly increased from 108 Ω to 212 Ω (Fig. S5, ESI†), indicating the depletion of DMF during the cycle, which damages the cycle stability of P-NCM.
O peak of DMF at 1650.31 cm−1 is obvious at P-NCM cathode depths from 0 to 20 μm, i.e., close to the PVDF-LLZTO electrolyte, but it slowly disappears from 30 to 60 μm, i.e., close to the current collector (Fig. 1(a)). Moreover, there is no N–CO peak corresponding to free DMF at 658 cm−1, only one at 672.08 cm−1 for the solvated [Li(DMF)x]+ (Fig. S2, ESI†), indicating that all the DMF molecules are bound to Li+.13,35 This suggests that the [Li(DMF)x]+ in the PVDF-LLZTO electrolyte spontaneously diffuses into the P-NCM cathode before electrochemical cycling of PVDF-based SSLMBs. The cross-sectional energy dispersive spectroscopy (EDS) mapping images of the cathode can further verify the limited diffusion depth of [Li(DMF)x]+ (Fig. S3, ESI†). Thus, it can be summarized that [Li(DMF)x]+ can spontaneously diffuse into the P-NCM cathode to a depth of 20 μm and act as a transport mediator for Li+. As a result, the PVDF-based SSLMB with a low mass loading of NCM811 (1.5 mg cm−2) shows a much higher discharge specific capacity of 182.5 mA h g−1 compared to that with a high mass loading of 15 mg cm−2 (97.4 mA h g−1) (Fig. S4, ESI†). In addition, the PVDF-based SSLMBs with a high cathode loading of 15 mg cm−2 present sudden capacity loss during cycling, and the impedance after the cycle is greatly increased from 108 Ω to 212 Ω (Fig. S5, ESI†), indicating the depletion of DMF during the cycle, which damages the cycle stability of P-NCM.
 | ||
| Fig. 1 Li+ transport mechanism of PVDF-based battery using a P-NCM cathode with a high loading of 15 mg cm−2. (a) FTIR spectra of P-NCM cathode at different depths before cycling. (b) O 1s and (c) N 1s XPS spectra of cycled P-NCM cathode. (d) TOF-SIMS analysis of pristine and cycled P-NCM cathode. (e) FTIR spectra of cycled P-NCM cathode at different depths. (f) HOMO and LUMO energy levels of DMF and [Li(DMF)3]+. Schematics of (g) Li+ transport paths in the traditional cathode and (h) the limited diffusion of [Li (DMF)x]+ in the traditional P-NCM cathode. | ||
To further explore the capacity decay of PVDF-based SSLMBs with high cathode loading, the surface composition of the P-NCM cathode from NCM811/PVDF-LLZTO/Li cells after 25 cycles was studied using X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (TOF-SIMS). As shown in Fig. 1(b), the intensity of the N–CO peak of DMF (533.37 eV) decreases significantly after cycling, accompanied by the appearance of LixSOy (531.97 eV) and Li2O (527.84 eV) peaks and an increase in the intensity of the Li2CO3 (530.59 eV) peak.13,27 The LixSOy and Li2CO3 are mainly derived from the decomposition of DMF and Li salt (LiFSI). This side reaction can be further confirmed in the N 1s spectrum (Fig. 1(c)), which presents an increase in the intensity of the LixN (398.59 eV) peak and a decrease in the N–CO peak of DMF (400.34 eV) after cycling.13 From the TOF-SIMS data of the cycled P-NCM cathode (Fig. 1(d)), it can be confirmed that the species with CN−, NCO− and NCHO− appear at m/z = 26, m/z = 42 and m/z = 43, respectively, and their intensity increases significantly after cycling, in accordance with the oxidization of DMF on the surface of the P-NCM cathode.36
To further examine the state of DMF present in the P-NCM cathode after cycling, FTIR tests were performed at different cross-sectional positions of the P-NCM cathode (Fig. 1(e)). The intensity of the CO peak of DMF in the cathode near the electrolyte side was significantly weakened to almost disappear after cycling, suggesting that most of the DMF is consumed by the severe side reactions with the NCM811 active materials. Density functional theory (DFT) calculations indicated that the highest occupied molecular orbital (HOMO) level of DMF is −6.96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV, which is higher than that of [Li(DMF)x]+ (−10.01 eV, Fig. 1(f)). The above results indicate that the remaining free DMF molecules after the desolvation of [Li(DMF)x]+ are easily oxidized, which damages the transport of Li+ and the stability of the cathode structure. Therefore, the diffusion depth of [Li(DMF)x]+ in the P-NCM cathode is responsible for the utilization of the NCM811 active materials, and it is easily decomposed because of the higher HOMO level of DMF (Fig. 1(g) and (h)).
eV, which is higher than that of [Li(DMF)x]+ (−10.01 eV, Fig. 1(f)). The above results indicate that the remaining free DMF molecules after the desolvation of [Li(DMF)x]+ are easily oxidized, which damages the transport of Li+ and the stability of the cathode structure. Therefore, the diffusion depth of [Li(DMF)x]+ in the P-NCM cathode is responsible for the utilization of the NCM811 active materials, and it is easily decomposed because of the higher HOMO level of DMF (Fig. 1(g) and (h)).
In order to improve the Li+ diffusion process, it is important to construct an efficient and stable Li+ transport network inside the cathode for PVDF-based SSLMBs. LATP has high Li+ conductivity, excellent compatibility with cathodes and strong adsorption of DMF,37–39 which may promote the uniform distribution of DMF in the cathode. As a further step, carbon-coating can achieve electron conduction; thus, carbon-coated LATP nanowires (C@LATP NW) were introduced into a thick P-NCM cathode as a filler (PCL-NCM) (Fig. 2(a) and (b)). The C@LATP NW were prepared by an electrospinning method and annealed in argon atmosphere. The X-ray diffraction (XRD) patterns indicate that the C@LATP NW with a stable tetragonal phase were successfully synthesized at an annealing temperature of 800 °C, and match well with the standard pattern of LATP electrolyte (PDF#35-0754) (Fig. 2(c)). The prepared C@LATP NW precursor presents an average diameter of 200 nm (Fig. S7a, ESI†). After the calcination, the structure is well maintained, and there are many bumps on the surface of C@LATP NW, which can increase the specific surface area (Fig. 2(d) and Fig. S7b, ESI†). The high-resolution transmission electron microscopy (HRTEM) image presents a d-spacing of 0.39 nm inside the C@LATP NW (Fig. 2(e)), which is consistent with the (113) facet of LATP. The Raman spectrum of the C@LATP NW exhibits D and G bands at 1358 and 1596 cm−1, respectively40 (Fig. 2(f)), indicating that an amorphous carbon layer with a thickness of about 5 nm was successfully coated on the LATP surface (Fig. 2(e)), and the carbon content in C@LATP NW is 24.2% (Fig. S8, ESI†), which is beneficial to enhancing the electronic conductivity inside the cathode. In addition, the C@LATP NW have a specific surface area of 289.62 m2g−1, which is much higher than that of LATP NW (7.98 m2g−1, Fig. 2(g) and Table S1, ESI†). The composite PCL-NCM was prepared by the conventional slurry-coating process, and the PL-NCM cathode with LATP NW and the P-NCM cathode without filler were used for equal comparison. As shown in the SEM images and energy dispersive spectroscopy (EDS) mapping images of PCL-NCM, the C@LATP NW are uniformly dispersed in the PCL-NCM cathode (Fig. 2(h) and Fig. S9, ESI†), which can connect the NCM811 particles for highly efficient ion and electron transport (Fig. 2(i)).
 | ||
| Fig. 2 Physical properties of the C@LATP NW filler and PCL-NCM cathode. Schematic of the (a) multiple Li+ transport channels and (b) diffusion of [Li (DMF)x]+ in the composite cathode with C@LATP NW. (c) XRD patterns of C@LATP NW and LATP NW. (d) SEM image of C@LATP NW. (e) HRTEM image of C@LATP NW; the inset is the corresponding FFT pattern. (f) Raman spectra of the C@LATP NW and LATP NW. (g) N2 adsorption and desorption isotherms of the C@LATP NW and LATP NW. (h) SEM images of PCL-NCM cathode with 3 wt% C@LATP NW and (i) corresponding EDS mapping of the elements C, Ni and P. | ||
To verify the interaction between C@LATP NW and DMF, FTIR tests were performed on the cathode of uncycled PCL-NCM/PVDF-LLZTO/Li and P-NCM/PVDF-LLZTO/Li SSLMBs after standing for 12 h. It can be clearly found that the CO peak of DMF shifts from 1650.31 cm−1 to 1663.27 cm−1 as a result of the addition of C@LATP NW, indicating that there is a strong interaction between C@LATP NW and DMF (Fig. S10, ESI†).37,41 The distribution of DMF in the PCL-NCM cathode with 3 wt% C@LATP NW was further detected using atomic force microscope-nano infrared spectroscopy (AFM-nano-IR) (Fig. 3(a) and (b)). The intensity of the CO group is much stronger at the C@LATP NW, suggesting that most of the DMF is anchored on the surface of the high-surface-area C@LATP NW due to their higher adsorption energy (Fig. S11, ESI†). To characterize the diffusion depth of DMF in the PCL-NCM cathode, FTIR spectra at different cross-sectional positions were obtained. The CO peak of DMF is clearly found in PCL-NCM even at a thickness of 60 μm (Fig. 3(c) and Fig. S12, S13, ESI†), which is sharply different from the diffusion depth of only 20 μm in P-NCM (Fig. 1(a)). Additionally, EDS mapping images of the cross-sections of the PCL-NCM cathode show that the DMF diffuses more deeply and more evenly (Fig. S14, ESI†). These results indicate that the strong adsorption of C@LATP NW on DMF changes the distribution of DMF in the PCL-NCM cathode, which provides a driving force for the uniform diffusion of DMF in the cathode to build a continuous and highly efficient Li+ transport network.
 | ||
| Fig. 3 Li+ transport mechanism of the PCL-NCM cathode. (a) AFM image of the PCL-NCM cathode and (b) nano-IR overlap of the CO vibration of DMF solvent at 1663.27 cm−1. (c) FTIR spectra of PCL-NCM at different depths before cycling. (d) GITT plots of the PCL-NCM and P-NCM cathode in the charge process. Cyclic voltammogram measurements of the (e) PCL-NCM/PVDF-LLZTO/Li and (f) P-NCM/PVDF-LLZTO/Li solid-state batteries. Raman spectra of the NCM811 particles at the top and bottom of the original (g), PCL-NCM charged to 4.3 V (h), and P-NCM charged to 4.3 V (i). (j) Ratio of Eg/A1g intensities at the top and bottom of the original, PCL-NCM charged to 4.3 V, and P-NCM charged to 4.3 V. | ||
The galvanostatic intermittent titration technique (GITT) results indicate that the voltage polarization of PCL-NCM is significantly weaker than that of P-NCM due to the addition of the C@LATP NW filler (Fig. 3(d)). In addition, the Li+ diffusion coefficient of PCL-NCM is also much larger than that of P-NCM (Fig. S15, ESI†), demonstrating that a Li+ transport network is successfully constructed by C@LATP NW for the rapid diffusion of Li+ in the composite cathode. Cyclic voltammetry (CV) experiments at various sweep rates between 2.8 and 4.3 V (Fig. 3(e) and (f)) also suggest that PCL-NCM has lower voltage polarization and a higher diffusion coefficient (2.22 × 10−14 cm2 s−1) than P-NCM (6.84 × 10−15 cm2 s−1) (Fig. S16, ESI†). It can be concluded that the C@LATP NW promote the rapid diffusion of Li+ in the composite cathode, which alleviates polarization and enhances transportation kinetics.42 From Kelvin probe force microscopy analysis of the PCL-NCM composite cathode (Fig. S17, ESI†), the average interfacial potential between C@LATP NW and NCM811 is 81.01 mV, which is much lower than that between PVDF binder and NCM811 (166.46 mV). The results further prove a lower diffusion barrier for Li+ through the interface of the NCM811 active material, which is favorable to the electrochemical reaction and interfacial kinetics of the PCL-NCM composite cathode.43
To understand the usage of the active material participating in the electrochemical reaction in the NCM811 cathode, Raman spectroscopy was carried at both the top (near the electrolyte) and the bottom (near the current collector) (Fig. S18, ESI†). The local structural transformation of NCM811 from the monoclinic phase to the hexagonal phase can be characterized using the A1g mode and Eg mode of the Raman spectrum, which can be directly related to the state of charge (SOC) of the NCM811 particles.44,45 The Eg/A1g intensity ratio can qualitatively characterize the degree of delithiation in NCM811 particles during the charging process. A larger Eg/A1g suggests more delithiation of the NCM811 particles and a higher SOC. In the initial state of the NCM811 particles, the intensity of Eg is significantly lower than that of A1g, and the Eg/A1g intensity ratio at the top and bottom of the cathode are nearly the same (0.46, Fig. 3(g) and (j)). After charging to 4.3 V, the Eg/A1g ratio at the top and bottom of PCL-NCM significantly increases to 1.36 and 1.26, respectively (Fig. 3(h) and (i)). In sharp contrast, while the Eg/A1g ratio at the top of P-NCM is 1.35, that at the bottom of P-NCM is only 0.63, which is very similar to the initial state. These results indicate that after charging to 4.3 V, the NCM811 particles at the top and bottom of PCL-NCM undergo a similar degree of delithiation process, whereas the NCM811 particles at the bottom of P-NCM show almost no participation in the delithiation process. Therefore, the C@LATP NW constructs a continuous and highly efficient Li+ transport network, which promotes the diffusion of bulk and interfacial Li+ in the cathode, leading to the full delithiation reaction of the thick NCM811 cathode.
PCL-NCM/PVDF-LLZTO/Li SSLMBs with different weight ratios of C@LATP NW (0%, 1%, 3%, 5% and 7%) were examined, and their discharge specific capacities at 5C are 58.1, 80.3, 122.8, 118.5 and 65.8 mA h g−1, respectively. The battery with 3 wt% C@LATP NW with the smallest resistance presents the best rate performance (Fig. S19 and S20, ESI†). When the amount of C@LATP NW added exceeds 3%, they agglomerate inside the electrode and cannot construct a highly efficient ion transport network, which reduces the reactivity of NCM and limits the rate performance of the batteries (Fig. S21, ESI†). Therefore, in subsequent electrochemical performance tests, the amount of C@LATP NW was set at 3 wt%. The discharge specific capacities of PCL-NCM/PVDF-LLZTO/Li cells at 0.1C, 0.3C, 0.5C, 1C, 3C and 5C are 198.1, 186.2, 178.8, 167.2, 139.6 and 122.8 mA h g−1, respectively (Fig. 4(a)). Its discharge specific capacity at 5C is much higher than that of the PL-NCM/PVDF-LLZTO/Li cells (93.76mA hg−1) and P-NCM/PVDF-LLZTO/Li cells (58.02mA hg−1). It should be noted that the high capacity is not attributed to the 3 wt% C@LATP NW (Fig. S23, ESI†). In addition, the capacity retention of the PCL-NCM/PVDF-LLZTO/Li cell after 1200 cycles at 3C is 70.5% (Fig. 4(b)). In sharp contrast, the discharge capacity of the PL-NCM/PVDF-LLZTO/Li cell decays rapidly after 400 cycles and the capacity retention after 1200 cycles is only 5.6%, while the P-NCM/PVDF-LLZTO/Li cells have almost no capacity after 200 cycles. The greatly enhanced electrochemical performance of the PCL-NCM/PVDF-LLZTO/Li cells is mainly due to the efficient and stable Li+ transport network constructed by C@LATP NW in the NCM811 cathode, which greatly improves both the stability of DMF in the NCM811 cathode and the structural stability of NCM811.
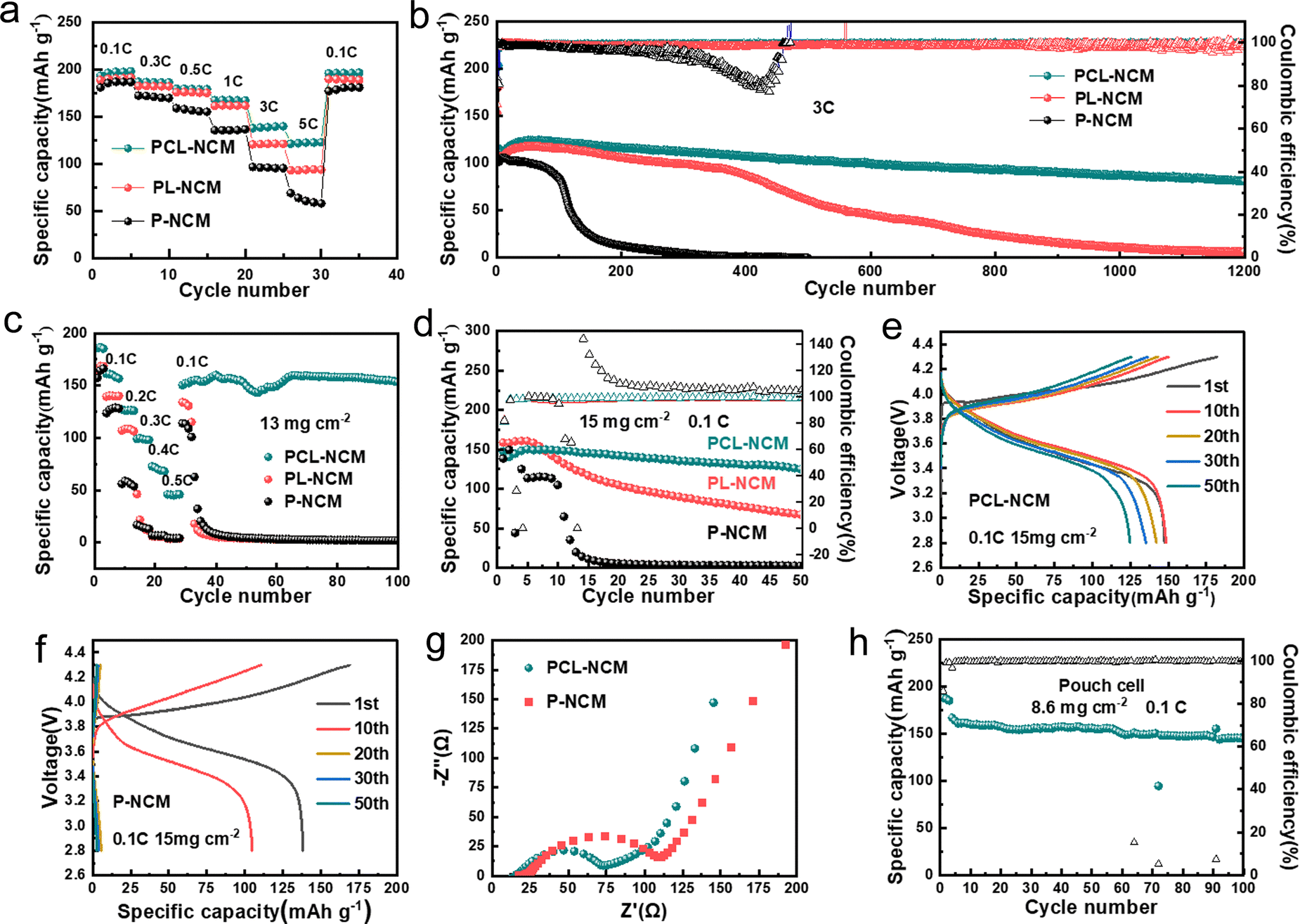 | ||
| Fig. 4 Electrochemical performances of the NCM811/Li solid-state batteries. (a) Rate performance of the NCM811/Li solid-state cells. (b) Cycling stability of the NCM811/Li solid-state cells at 3C. (c) Rate performance of the NCM811/Li solid-state cells with NCM811 loading of 13 mg cm−2. (d) Cycling stability of the NCM811/Li solid-state cells with NCM811 loadings of 15 mg cm−2 at 0.1C. Charge/discharge curves of (e) PCL-NCM/PVDF-LLZTO/Li and (f) P-NCM/PVDF-LLZTO/Li full cells at 15 mg cm−2 at 0.1C. (g) Electrochemical impedance spectroscopy of P-NCM/PVDF-LLZTO/Li and PCL-NCM/PVDF-LLZTO/Li full cells at 50% charge state. (h) Cycling performance of PCL-NCM/PVDF-LLZTO/Li pouch cell with an NCM811 loading of 8.6 mg cm−2 at 0.1C. | ||
Further, PCL-NCM/PVDF-LLZTO/Li cells with a high NCM811 loading of 13 mg cm−2 could deliver capacities of 161.2, 126.9, 98.7, 72.6 and 46.08 mA h g−1 at 0.1C, 0.2C, 0.3C, 0.4C and 0.5C, respectively (Fig. 4(c)), while the PL-NCM/PVDF-LLZTO/Li and P-NCM/PVDF-LLZTO/Li cells could not discharge any capacity at 0.4C. As shown by the charge and discharge curves under different currents, the PCL-NCM/PVDF-LLZTO/Li cells have a smaller voltage polarization (Fig. S22, ESI†). This means that the C@LATP NW can significantly improve the rate performance of the cathodes, especially at high loading. In addition, the PCL-NCM/PVDF-LLZTO/Li cell with an NCM811 loading of 11.5 mg cm−2 achieves a discharge specific capacity of 139.6 mA hg−1 and a coulombic efficiency of 99.6% at 0.2C, and maintains 98% capacity after 100 cycles (Fig. S24, ESI†). However, the NCM811/PVDF-LLZTO/Li cells using P-NCM and PL-NCM cathodes suffer from rapid capacity decay. Even when the cathode loading is increased to 15 mg cm−2 (Fig. 4(d)), the PCL-NCM/PVDF-LLZTO/Li cell shows excellent cycle stability, with 85% capacity retention after 50 cycles at 0.1C. In sharp contrast, the capacity retention of the PL-NCM/PVDF-LLZTO/Li and P-NCM/PVDF-LLZTO/Li cells after 50 cycles are only 42% and 2%, respectively. Moreover, the PCL-NCM/PVDF-LLZTO/Li cell shows a much smaller charge/discharge voltage polarization than the P-NCM/PVDF-LLZTO/Li cells due to its much smaller charge transfer resistance (Rct, 74 Ω) compared to that of the P-NCM/PVDF-LLZTO/Li cell (106 Ω) (Fig. 4(e)–(g)). Furthermore, we assembled a flexible PCL-NCM/PVDF-LLZTO/Li pouch cell using a PCL-NCM cathode with an NCM811 loading of 8.6 mg cm−2 (Fig. S25 and S26, ESI†). Its initial discharge capacity reaches 166.9 mA h g−1 at 0.1C with capacity retention of 87% after 100 cycles (Fig. 4(h) and Fig. S27, ESI†), showing excellent cycle stability. The above electrochemical performances of PCL-NCM/PVDF-LLZTO/Li cells with high NCM811 loading are much better than those of other recently reported PVDF-based SSLMBs (Fig. S28, ESI†).
In situ XRD measurements were carried out to investigate the structural evolution of the PCL-NCM and P-NCM cathode during the charge/discharge process (Fig. 5(a) and (b)). The (003) and (101) peak intensities and the Bragg angle of the P-NCM cathode change significantly during the phase transition from H1 to M, which means that a large structural change may lead to phase collapse and cracking of the layered oxide cathode material.46,47 After the transition to the H2 phase, the (003) peak begins to shift to the right, which corresponds to the transition from the H2 phase to the H3 phase with reduced interlayer spacing. The P-NCM cathode clearly presents a quite incomplete phase transition from H2 to H3 due to inadequate delithiation process, which is ascribed to the high voltage polarization in the P-NCM cathode. During the second charging cycle, the P-NCM can hardly change from the H2 to the H3 phase, and irreversible phase transition occurs after two cycles, which is harmful to the crystal structural stability of NCM811. In obvious contrast, the PCL-NCM cathode underwent sufficient and mild H1–M–H2–H3 phase transition, and there was no obvious irreversible phase transition.46,47 Therefore, the high-efficiency Li+ transmission network constructed by C@LATP NW significantly improves the kinetics of the PCL-NCM cathode and facilitates the thorough phase transformation of NCM811, which is beneficial not only to the full utilization of the active materials in the thick PCL-NCM cathode, but also to the crystal structure stability of the cathode materials.
 | ||
| Fig. 5 Structure characterizations of the PCL-NCM and P-NCM cathodes. Operando XRD characterization for (a) P-NCM and (b) PCL-NCM cathode during the initial two charge–discharge cycles at 0.1C. (c) C 1s, (d) F 1s, and (e) S 2p XPS spectra of cycled PCL-NCM and P-NCM cathodes. TEM and FFT images of cycled (f) P-NCM and (g) PCL-NCM cathodes. | ||
To confirm that DMF remains stable in the PCL-NCM cathode during long cycling, we assembled PCL-NCM/PVDF-LLZTO/Li cells with a high cathode loading to conduct EIS and FTIR tests. The PCL-NCM/PVDF-LLZTO/Li cells show excellent cyclic stability (Fig. S29a, ESI†), and the EIS results show that Rct remains stable before and after cycling (Fig. S29b, ESI†). From the FTIR test results of the cathode, DMF also can remain stable in the cathode after cycling (Fig. S29c, ESI†), suggesting that the C@LATP NW can prevent the decomposition of DMF in the PCL-NCM cathode to significantly enhance the battery performance. The structure and surface composition of the cathode from the NCM811/PVDF-LLZTO/Li cells after 60 cycles were also studied using HRTEM and XPS. The peak intensities in the C 1s spectra of the PCL-NCM cathode are much lower than those of P-NCM cathode (Fig. 5(c)), indicating that the decomposition of DMF at the cathode is greatly suppressed. The cathode electrolyte interface (CEI) of the PCL-NCM cathode contains more LiF (685 eV) (Fig. 5(d)), which is beneficial to the stabilization of CEI.48 In addition, the content of SO3− (166 eV) derived from LiFSI decomposition in PCL-NCM was significantly lower than that of P-NCM (Fig. 5(e)).49 The carbon layer on the C@LATP NW surface might reduce electron accumulation in PCL-NCM to greatly suppress the decomposition of DMF and LiFSI.50,51 Furthermore, as observed by HRTEM (Fig. S30, ESI†), the CEI on the cathode surface of PCL-NCM (∼2 nm) is much thinner and more uniform than that of P-NCM (∼11 nm). The nanostructures of the cycled NCM811 particles consists of a rock-salt phase (site A and D), a mixed phase (site B and E) and a layered structure (site C and F) (Fig. 5(f) and (g)). The structure of the NCM811 after 60 cycles shows that the thickness of the rock-salt phase and the mixed phase is only approximately 9nm in the cycled NCM811 of PCL-NCM (Fig. 5(g)), which is much smaller than that of P-NCM (∼37nm, Fig. 5(f)), indicating that the NCM811 of PCL-NCM suffers from less phase transformation during cycling. Moreover, to further verify the stability of the Li+ transport network, XRD and XPS analysis of the C@LATP NW after cycling tests was carried out. The XRD peaks of C@LATP NW do not change after cycling, indicating that the C@LATP NW maintains a stable tetragonal phase to provide highly efficient ion transport pathways (Fig. S31, ESI†). In addition, the XPS analysis demonstrates that the Ti4+ of C@LATP does not exhibit any change after cycling, suggesting that no side reaction occurred on C@LATP NW (Fig. S32, ESI†). In summary, C@LATP NW redistribute DMF in the thick cathode, reducing the side reactions with the NCM811 cathode, thus maintaining the stability of ion transport and the structure of the active materials.
Conclusions
In summary, we revealed that the solvation structure of [Li(DMF)x]+ formed by DMF and Li+ diffuses spontaneously into the cathode and acts as a transport medium for Li+. The diffusion depth of [Li(DMF)x]+ is limited and unstable in the cathode, and a stable Li+ transport network cannot be constructed throughout the cathode at high loading. We constructed a composite cathode with a highly efficient Li+ transport network via C@LATP NW. The uniform distribution of C@LATP NW in the cathode has a strong adsorption effect on DMF, which greatly promotes the uniform diffusion and stability of DMF in the cathode. The C@LATP NW in the composite cathode greatly improve the utilization of the charge/discharge reaction and the structural stability of cathode materials. The solid-state PCL-NCM811/PVDF-LLZTO/Li battery using C@LATP NW operates stably for 1200 cycles at 3C, and the performance of the solid-state battery with a high NCM811 loading of 15 mg cm−2 is also significantly improved. This work reveals the mechanism of Li+ transport in the cathode of PVDF-based SSLMBs and establishes an efficient Li+ transport network for full utilization of active materials in a thick cathode for high-energy-density SSLMBs.Author contributions
Y.-B. H. and M. L. conceived and supervised the project. Y.-B. H. and Y. L. designed the experiments. Y. L. and F. X. A. performed the experiments with help from K. Yang, J. B. Ma, J. S. Mi, D. F. Zhang, X. Cheng, Y. H. Li, Y. T. Ma., Y. L., F. X. A., K. Yang, X. Cheng., M. L., F. Y. K., and Y. -B. H. discussed the results. Y. L., F. X. A., M. L. and Y.-B. H. wrote the initial manuscript which was approved by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFF0500600), National Natural Science Foundation of China (no. U2001220 and 52203298), Local Innovative Research Teams Project of Guangdong Pearl River Talents Program (no. 2017BT01N111), Shenzhen All-Solid-State Lithium Battery Electrolyte Engineering Research Center (XMHT20200203006), Shenzhen Technical Plan Project (no. RCJC20200714114436091, JCYJ20220530143012027, JCYJ20220818101003008 and JCYJ20220818101003007) and Shenzhen Pengrui Young Faculty Program Research Plan (SZPR2023006).Notes and references
- Z. Zou, Y. Li, Z. Lu, D. Wang, Y. Cui, B. Guo, Y. Li, X. Liang, J. Feng, H. Li, C.-W. Nan, M. Armand, L. Chen, K. Xu and S. Shi, Chem. Rev., 2020, 120, 4169–4221 CrossRef CAS PubMed.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- X. Wang, R. Kerr, F. Chen, N. Goujon, J. M. Pringle, D. Mecerreyes, M. Forsyth and P. C. Howlett, Adv. Mater., 2020, 32, e1905219 CrossRef PubMed.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- L.-Z. Fan, H. He and C.-W. Nan, Nat. Rev. Mater., 2021, 6, 1003–1019 CrossRef CAS.
- Y. Guo, S. Wu, Y.-B. He, F. Kang, L. Chen, H. Li and Q.-H. Yang, eScience, 2022, 2, 138–163 CrossRef.
- S. Yang, F. Ai, Z. Li, G. Zhao and Y. Bi, Chem. Res. Chin. Univ., 2021, 38, 603–608 CrossRef.
- S. Wang, J. Zuo, Y. Li, Y. Zhong, X. Ren, P. Zhang and L. Sun, Chem. Res. Chin. Univ., 2022, 39, 240–245 CrossRef.
- Q. Wang, Z. Cui, Q. Zhou, X. Shangguan, X. Du, S. Dong, L. Qiao, S. Huang, X. Liu, K. Tang, X. Zhou and G. Cui, Energy Storage Mater., 2020, 25, 756–763 CrossRef.
- Q. Yu, K. Jiang, C. Yu, X. Chen, C. Zhang, Y. Yao, B. Jiang and H. Long, Chin. Chem. Lett., 2021, 32, 2659–2678 CrossRef CAS.
- S. Zhou, S. Zhong, Y. Dong, Z. Liu, L. Dong, B. Yuan, H. Xie, Y. Liu, L. Qiao, J. Han and W. He, Adv. Funct. Mater., 2023, 33, 2214432 CrossRef CAS.
- X. Zhang, S. Wang, C. Xue, C. Xin, Y. Lin, Y. Shen, L. Li and C. W. Nan, Adv. Mater., 2020, 32, e2000026 CrossRef PubMed.
- X. Zhang, J. Han, X. Niu, C. Xin, C. Xue, S. Wang, Y. Shen, L. Zhang, L. Li and C.-W. Nan, Batteries Supercaps, 2020, 3, 876–883 CrossRef CAS.
- L. Li, Y. Shan and X. Yang, Mater. Today Commun., 2021, 26, 101910 CrossRef CAS.
- S. Zhang, Z. Li, Y. Guo, L. Cai, P. Manikandan, K. Zhao, Y. Li and V. G. Pol, Chem. Eng. J., 2020, 400, 125996 CrossRef CAS.
- T. Zhang, J. Li, X. Li, R. Wang, C. Wang, Z. Zhang and L. Yin, Adv. Mater., 2022, 34, 2205575 CrossRef CAS PubMed.
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y.-B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS PubMed.
- Y. Yuan, L. Chen, Y. Li, X. An, J. Lv, S. Guo, X. Cheng, Y. Zhao, M. Liu, Y.-B. He and F. Kang, Energy Mater. Devices, 2023, 1, 9370004 CrossRef.
- J.-P. Zeng, J.-F. Liu, H.-D. Huang, S.-C. Shi, B.-H. Kang, C. Dai, L.-W. Zhang, Z.-C. Yan, F. J. Stadler, Y.-B. He and Y.-F. Huang, J. Mater. Chem. A, 2022, 10, 18061–18069 RSC.
- T. K. L. Nguyen, G. Lopez, C. Iojoiu, R. Bouchet and B. Ameduri, J. Power Sources, 2021, 498, 229920 CrossRef CAS.
- J. Mi, J. Ma, L. Chen, C. Lai, K. Yang, J. Biao, H. Xia, X. Song, W. Lv, G. Zhong and Y.-B. He, Energy Storage Mater., 2022, 48, 375–383 CrossRef.
- G. Chen, F. Zhang, Z. Zhou, J. Li and Y. Tang, Adv. Energy Mater., 2018, 8, 1801219 CrossRef.
- K. Deng, Z. Xu, S. Zhou, Z. Zhao, K. Zeng, M. Xiao, Y. Meng and Y. Xu, J. Power Sources, 2021, 510, 230411 CrossRef CAS.
- F. Liu, Y. Cheng, X. Zuo, R. Chen, J. Zhang, L. Mai and L. Xu, Chem. Eng. J., 2022, 441, 136077 CrossRef CAS.
- B. Li, Q. Su, L. Yu, S. Dong, M. Zhang, S. Ding, G. Du and B. Xu, J. Membr. Sci., 2021, 618, 118734 CrossRef CAS.
- Z. Fang, M. Zhao, Y. Peng and S. Guan, ACS Appl. Mater. Interfaces, 2022, 14, 18922–18934 CrossRef CAS PubMed.
- J. Ma, G. Zhong, P. Shi, Y. Wei, K. Li, L. Chen, X. Hao, Q. Li, K. Yang, C. Wang, W. Lv, Q.-H. Yang, Y.-B. He and F. Kang, Energy Environ. Sci., 2022, 15, 1503–1511 RSC.
- S. Huo, L. Sheng, W. Xue, L. Wang, H. Xu, H. Zhang, B. Su, M. Lyu and X. He, Adv. Energy Mater., 2023, 13, 2204343 CrossRef CAS.
- H. Zhang, L. Huang, H. Xu, X. Zhang, Z. Chen, C. Gao, C. Lu, Z. Liu, M. Jiang and G. Cui, eScience, 2022, 2, 201–208 CrossRef.
- S. Yang, B. Wang, Q. Lv, N. Zhang, Z. Zhang, Y. Jing, J. Li, R. Chen, B. Wu, P. Xu and D. Wang, Chin. Chem. Lett., 2023, 34, 107783 CrossRef CAS.
- H. Li, F. Lian, N. Meng, C. Xiong, N. Wu, B. Xu and Y. Li, Adv. Funct. Mater., 2021, 31, 2008487 CrossRef CAS.
- X. Tao, Y. Liu, W. Liu, G. Zhou, J. Zhao, D. Lin, C. Zu, O. Sheng, W. Zhang, H.-W. Lee and Y. Cui, Nano Lett., 2017, 17, 2967–2972 CrossRef CAS PubMed.
- L. Ye and X. Li, Nature, 2021, 593, 218–222 CrossRef CAS PubMed.
- Z. Wan, D. Lei, W. Yang, C. Liu, K. Shi, X. Hao, L. Shen, W. Lv, B. Li, Q.-H. Yang, F. Kang and Y.-B. He, Adv. Funct. Mater., 2019, 29, 1805301 CrossRef.
- M. M. E. Jacob and A. K. Arof, Electrochim. Acta, 2000, 45, 1701–1706 CrossRef CAS.
- D. Zhang, M. Liu, J. Ma, K. Yang, Z. Chen, K. Li, C. Zhang, Y. Wei, M. Zhou, P. Wang, Y. He, W. Lv, Q.-H. Yang, F. Kang and Y.-B. He, Nat. Commun., 2022, 13, 6966 CrossRef CAS PubMed.
- K. Yang, L. Chen, J. Ma, C. Lai, Y. Huang, J. Mi, J. Biao, D. Zhang, P. Shi, H. Xia, G. Zhong, F. Kang and Y.-B. He, Angew. Chem., Int. Ed., 2021, 60, 24668–24675 CrossRef CAS PubMed.
- W. Hou, Y. Ou and K. Liu, Chem. Res. Chin. Univ., 2022, 38, 735–743 CrossRef CAS.
- X. Zhao, M. Yang, J. Wang and D. Wang, Chem. Res. Chin. Univ., 2023, 39, 630–635 CrossRef CAS.
- J. Wei, B. Jiang, X. Zhang, H. Zhu and D. Wu, Chem. Phys. Lett., 2003, 376, 753–757 CrossRef CAS.
- Y. Zeng, L. Zhao, J. Zhang, Q. Li, D. Sun, Y. Ren, Y. Tang, G. Jin and H. Wang, Small Sci., 2023, 3, 2300017 CrossRef CAS.
- J. Y. Liang, X. X. Zeng, X. D. Zhang, P. F. Wang, J. Y. Ma, Y. X. Yin, X. W. Wu, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2018, 140, 6767–6770 CrossRef CAS PubMed.
- J. Haruyama, K. Sodeyama, L. Han, K. Takada and Y. Tateyama, Chem. Mater., 2014, 26, 4248–4255 CrossRef CAS.
- J. Nanda, J. Remillard, A. O'Neill, D. Bernardi, T. Ro, K. E. Nietering, J.-Y. Go and T. J. Miller, Adv. Funct. Mater., 2011, 21, 3282–3290 CrossRef CAS.
- J. Lei, F. McLarnon and R. Kostecki, J. Phys. Chem. B, 2005, 109, 952–957 CrossRef CAS PubMed.
- Y.-S. Hong, X. Huang, C. Wei, J. Wang, J.-N. Zhang, H. Yan, Y. S. Chu, P. Pianetta, R. Xiao, X. Yu, Y. Liu and H. Li, Chem, 2020, 6, 2759–2769 CAS.
- S. Kalluri, M. Yoon, M. Jo, S. Park, S. Myeong, J. Kim, S. X. Dou, Z. Guo and J. Cho, Adv. Energy Mater., 2017, 7, 1601507 CrossRef.
- P. Bai, X. Ji, J. Zhang, W. Zhang, S. Hou, H. Su, M. Li, T. Deng, L. Cao, S. Liu, X. He, Y. Xu and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202202731 CrossRef CAS PubMed.
- A. Tornheim, J. C. Garcia, R. Sahore, H. Iddir, I. Bloom and Z. Zhang, J. Electrochem. Soc., 2019, 166, A440–A447 CrossRef CAS.
- X. Chen, L. Qin, J. Sun, S. Zhang, D. Xiao and Y. Wu, Angew. Chem., Int. Ed., 2022, 61, e202207018 CrossRef CAS PubMed.
- Z. Chang, H. Yang, Y. Qiao, X. Zhu, P. He and H. Zhou, Adv. Mater., 2022, 34, 2201339 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03108j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |