
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
HCO3−-mediated highly efficient photoelectrochemical dioxygenation of arylalkenes: triple roles of HCO3−-derived radicals†
Jie
Yang
ab,
Yukun
Zhao
ab,
Mengyu
Duan
ab,
Chaoyuan
Deng
 ab,
Yufan
Zhang
ab,
Yu
Lei
ab,
Jikun
Li
ab,
Wenjing
Song
ab,
Chuncheng
Chen
*ab and
Jincai
Zhao
ab
ab,
Yufan
Zhang
ab,
Yu
Lei
ab,
Jikun
Li
ab,
Wenjing
Song
ab,
Chuncheng
Chen
*ab and
Jincai
Zhao
ab
aKey Laboratory of Photochemistry, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: ccchen@iccas.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 13th November 2023
Abstract
Alkene dioxygenation reactions are significant organic transformation process, but the direct oxidation of alkenes on photoanodes exhibits quite poor yields for dioxygenation products. Here, we report that the presence of bicarbonate in a heterogenous photoelectrochemical (PEC) cell can achieve efficient dioxygenation of alkenes under mild conditions. A broad range of alkene substrates with a variety of substitute groups can be effectively oxygenated to diols or α-hydroxy ketones, with a product yield of up to 89%. Furthermore, we identified that the diol product is formed via a peroxydiol intermediate. Spin-trap electron paramagnetic resonance (EPR) experiments show that the HCO3−-derived radicals are important active species. Accordingly, we propose a triple role for formed HCO3−-derived radicals in the dioxygenation of alkenes: attacking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the alkene to initiate the reaction, producing ˙CO4− active species for the formation of a peroxydiol intermediate, and reducing the peroxydiol to a diol. The present work provides a promising strategy for the transformation of alkenes with green radicals and paves the way for the application of HCO3− in PEC organic synthesis.
C bond of the alkene to initiate the reaction, producing ˙CO4− active species for the formation of a peroxydiol intermediate, and reducing the peroxydiol to a diol. The present work provides a promising strategy for the transformation of alkenes with green radicals and paves the way for the application of HCO3− in PEC organic synthesis.
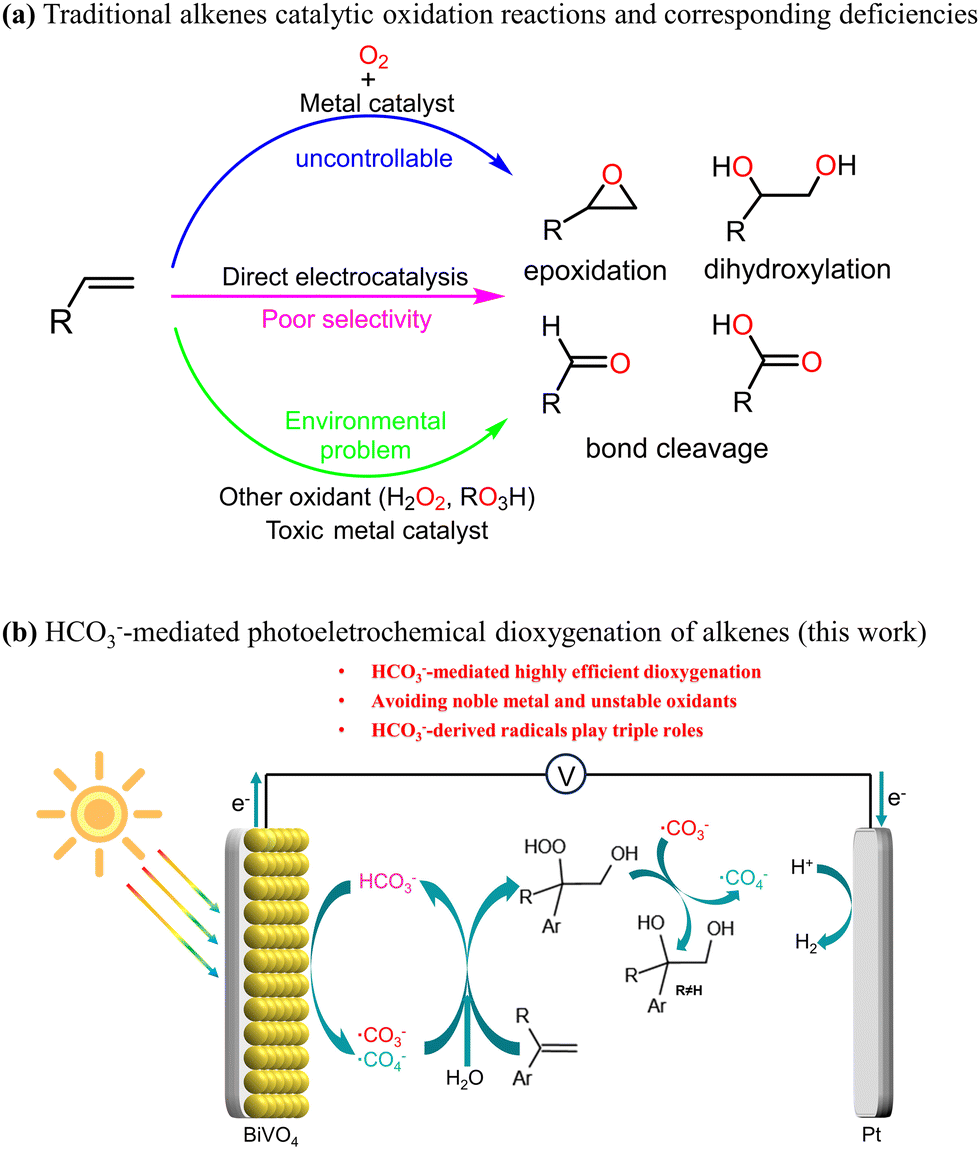
Broader contextSolar-driven photoelectrochemical (PEC) processes, including the splitting of water, CO2 reduction and pollutant degradation, represent sustainable methods for harnessing solar energy. Among these, water splitting for hydrogen production is the most extensively investigated. However, the kinetics of the oxygen evolution reaction are inherently unfavorable, and the resulting O2 holds minimal economic value. Thus, it is imperative to explore alternative reactions that yield high-value fine chemicals at the photoanode. The oxidation of alkenes has garnered significant attention, given the critical role of their products, such as diols and epoxides, as essential intermediates in numerous valuable chemical processes. PEC alkene oxidation has emerged as a sustainable and secure pathway to achieve this objective, while concurrently enhancing the rate of hydrogen production at the cathode. Nevertheless, the direct selective oxidation of alkenes on the photoanode surface poses formidable challenges. This study presents an innovative HCO3−-mediated PEC system, leveraging diverse reaction pathways facilitated by HCO3−-derived radicals, to achieve the highly selective dioxygenation of olefin coupled with the hydrogen evolution reaction. The triple roles of HCO3−-derived radicals were identified experimentally for selective dioxygenation. We believe that our research provides a promising strategy for the transformation of alkenes with “green” radicals, and paves the way for the application of HCO3− in PEC synthesis. |
Introduction
Photoelectrochemistry1 represents a sustainable method for solar energy conversion by driving reactions such as water splitting,2,3 CO2 reduction4 and pollutant degradation.5,6 PEC water splitting for hydrogen production is the most widely studied PEC process. In this reaction, the water molecule is oxidized to generate O2 (oxygen evolution reaction, OER) at the photoanode, while the released proton is reduced to produce hydrogen (hydrogen evolution reaction, HER) on the cathode. The OER is kinetically unfavourable, and the formed dioxygen has little economic value. It is also feasible to explore alternative reactions that produce high-value fine chemicals instead of dioxygen on the photoanode. For example, the oxidation of organic substrates or the mediated oxidation of organic substrates not only increases the rate of cathode hydrogen evolution,7,8 but also produces high value-added chemicals, exhibiting a higher economic benefit than that of water oxidation.The oxidation of olefins is one of the most important methods for converting mineral oil into high value-added chemicals9,10 (Scheme 1). In recent years, there has been significant attention on the electrochemical (EC) or PEC oxidation of olefins coupled with the HER, in which the requirement for external harsh oxidants can be avoided. However, achieving the direct selective oxidation of olefins at photoanodes towards selectivity of a given product is challenging.11 This difficulty is likely attributed to the large steric hindrance and low polarity of CC double bonds, making it unfavourable for the olefins to interact efficiently with the surface of the photoanode.12 Utilizing redox mediators, indirect EC or PEC oxidations have been reported to markedly promote the selective performance of high-value products.13,14 For instance, Sargent's group reported the chloride-mediated EC epoxidation of ethylene and propylene with 97% selectivity and 71% Faraday efficiency (FE).13 Recent research conducted by our team and Li's team revealed the bromine-mediated PEC epoxidation of olefins on α-Fe2O312 and BiVO4,8 respectively, which both demonstrated good selectivity, while they exhibited extremely poor PEC epoxidation performance in the absence of bromine.
 | ||
| Scheme 1 Comparison of the different pathways for the oxidation of alkenes. | ||
In recent years, HCO3− has been utilized as an EC or PEC mediator for producing hydrogen peroxide15–18 and degrading contaminants.19,20 For example, it has been reported that carbonate-mediated EC water oxidation shows high selectivity for H2O2 production through the formation of carbonate radical and percarbonate intermediates.18 The PEC degradation rate of rhodamine B (a pollutant model) with HCO3− as an electrolyte was found to be 30% higher than that using SO42− as a electrolyte,19 which is attributed to the mediating effect of the carbonate radical. It is worth noting that the redox potential of ˙CO3−/HCO3− is relatively low (E0 = 1.78 V, pH = 7).21 In various hydroxyl radical-based advanced oxidation processes, HCO3− has been found to transform the photoinduced hole or hydroxyl free radical to the less active carbonate radical, and thus significantly inhibit the degradation of organic pollutants.22 Although the mild oxidation capacity of the carbonate radical is unfavourable for the degradation of organic pollutants, it provides a significant advantage in selectively oxidizing substrates with certain functionality by avoiding overoxidation or mineralization. Therefore, theoretically, HCO3− should be ideal for selective oxidation in EC or PEC reactions. However, the application of HCO3−-mediated oxidation reactions in organic synthesis has rarely been reported to date.
In this study, we present HCO3− as an excellent redox mediator for the highly-selective PEC dioxygenation of alkenes, in which these dioxygenation products have been applied in organic synthesis23–25 and pharmaceuticals.26,27 HCO3−-mediated dioxygenation reactions can effectively address these challenges suffered in other systems, including toxic noble metal catalysts, the requirement for external oxidants and the incompatibility between the hydrophilicity of the photoanode surface and the hydrophobicity of the CC bond, which arises mainly because the hydrophilic HCO3− is easily oxidized to ˙CO3− on the BiVO4 surface, and the ˙CO3− can diffuse into the bulk solution and react with the organic substrate.28,29 Moreover, the protocol demonstrates excellent functional group tolerance. Mechanistic studies further verify that HCO3−-derived radicals play triple roles in the alkene dioxygenation reaction.
Results and discussion
HCO3−-mediated arylalkene dioxygenation reactions
All PEC reactions were conducted in an undivided cell with a three-electrode configuration (unless otherwise stated). BiVO4 was first employed as the photoanode, with Pt wire and Ag/AgCl serving as the counter electrode and reference electrode, respectively. The PEC performance of synthetic BiVO4 was tested in 0.5 M potassium borate buffer (KBi) solution with a pH of 9.5, and the addition of CH3CN was found to not affect the photocurrent of BiVO4 under the same applied potential (Fig. S2, ESI†). 4-Chloro-alpha-methylstryene (4aa) was chosen as the model substrate to explore the reaction conditions (Table 1). The reaction was conducted in a solution mixture of H2O and CH3CN (volumetric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with KHCO3 as the electrolyte. Under these conditions, after 1.5 h of reaction at an applied potential of 0.8 V (potentials given in this study are referenced to the Ag/AgCl electrode unless otherwise stated), approximately 95% of 4aa was converted, and the selectivity of the diol product (4a) reached around 89%, with a yield of 85% (Table 1, entry 1) and a FE of 15%. The low FE may be caused by the formation of by-products (Fig. S3a, ESI†), and the hydrolysis of HCO4− to H2O2 (Fig. S3b, ESI†) and further to O2. As the reaction progresses, the substrate is gradually consumed, resulting in a corresponding increase in the diol product (Fig. S4a, ESI†), and the selectivity of the product remains almost unchanged (Fig. 1(a)). In addition, the FE of the product gradually decreases with an increase in the reaction time. This phenomenon may be attributed to the substrate concentration also decreasing with an increase in the reaction time, so the probability of the collision between the substrate and the carbonate active species also decreases, resulting in a decrease in the FE of the product. However, no product was detected in the absence of light or applied potential (Table 1, entries 2 and 3). When the applied potential exceeds 0.8 V, the selectivity of 4a is well maintained (Table S1, ESI† entry 16 and Table 1, entry 4), while the conversion significantly decreases over the same photoelectrolysis time due to the competition of the OER at a higher potential. At a lower applied potential (Table 1, entry 5 and Table S1, ESI† entries 14 and 15), a decrease in selectivity is observed, which may be assigned to the poor separation efficiency of photogenerated carriers, leading to inadequate charge accumulation. The FE of H2 produced at the cathode reaches approximately 95% (Fig. S5, ESI†), indicating that the cathodic reaction predominantly involves a water reduction reaction.
:1) with KHCO3 as the electrolyte. Under these conditions, after 1.5 h of reaction at an applied potential of 0.8 V (potentials given in this study are referenced to the Ag/AgCl electrode unless otherwise stated), approximately 95% of 4aa was converted, and the selectivity of the diol product (4a) reached around 89%, with a yield of 85% (Table 1, entry 1) and a FE of 15%. The low FE may be caused by the formation of by-products (Fig. S3a, ESI†), and the hydrolysis of HCO4− to H2O2 (Fig. S3b, ESI†) and further to O2. As the reaction progresses, the substrate is gradually consumed, resulting in a corresponding increase in the diol product (Fig. S4a, ESI†), and the selectivity of the product remains almost unchanged (Fig. 1(a)). In addition, the FE of the product gradually decreases with an increase in the reaction time. This phenomenon may be attributed to the substrate concentration also decreasing with an increase in the reaction time, so the probability of the collision between the substrate and the carbonate active species also decreases, resulting in a decrease in the FE of the product. However, no product was detected in the absence of light or applied potential (Table 1, entries 2 and 3). When the applied potential exceeds 0.8 V, the selectivity of 4a is well maintained (Table S1, ESI† entry 16 and Table 1, entry 4), while the conversion significantly decreases over the same photoelectrolysis time due to the competition of the OER at a higher potential. At a lower applied potential (Table 1, entry 5 and Table S1, ESI† entries 14 and 15), a decrease in selectivity is observed, which may be assigned to the poor separation efficiency of photogenerated carriers, leading to inadequate charge accumulation. The FE of H2 produced at the cathode reaches approximately 95% (Fig. S5, ESI†), indicating that the cathodic reaction predominantly involves a water reduction reaction.
| Entry | Reaction conditionsa | Selectivity (%) | Yieldb (%) |
|---|---|---|---|
|
a Reaction conditions: BiVO4 photoanode (2.5 cm2), Pt cathode, an Ag/AgCl electrode (3.5 M KCl leak-free and 2.0 mm diameter) was used as the reference electrode, CH3CN/H2O (v:v = 1:1, 5 mL), under AM 1.5G illumination, 0.1 M KHCO3 was used as the electrolyte. The initial concentration of the substrate was 4 mM, the applied potential was 0.8 V versus Ag/AgCl, under an air atmosphere, room temperature, and the time of the reaction was 1.5 h, undivided cell.
b The quantity of the products determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard and substrates were analyzed and quantified using an Agilent HPLC1260 system.
c Isolated yield.
d Not detected.
e The reaction time was 3 h.
f Trace product was detected.
|
|||
| 1 | 50% H2O | 89 ± 2 | 85 ± 3 (80)c |
| 2 | Without light | n.d.d | n.d.d |
| 3 | Without applied potential | n.d.d | n.d.d |
| 4 | 1.4 V | 91 ± 1 | 64 ± 2 |
| 5 | 0.2 Ve | 72 ± 2 | 71 ± 2 |
| 6 | NaHCO3 | 82 ± 3 | 75 ± 3 |
| 7 | NH4HCO3 | 88 ± 1 | 79 ± 3 |
| 8 | Ar atmosphere | 87 ± 1 | 81 ± 1 |
| 9 | TiO2 | 81 ± 2 | 78 ± 1 |
| 10 | α-Fe2O3 | Tracef | Trace |
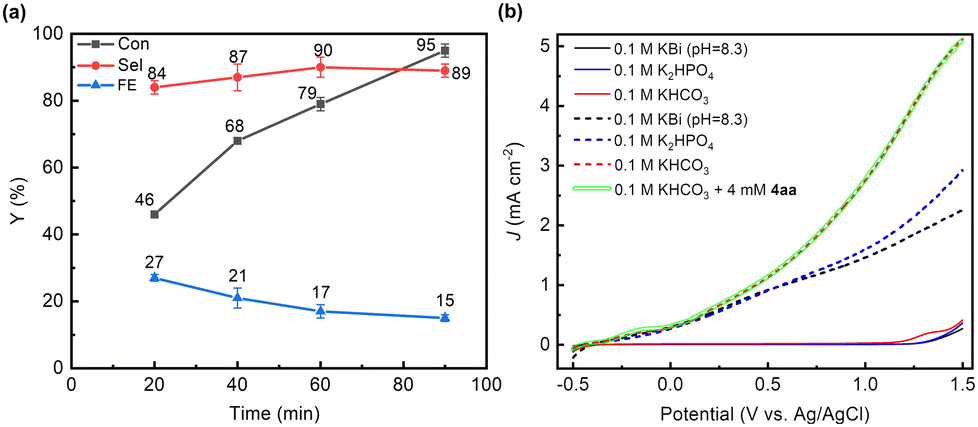 | ||
| Fig. 1 (a) Time-dependent conversion (Con) of 4.0 mM 4aa, selectivity (Sel) and Faraday efficiency (FE) of the product (4a) with 0.1 M KHCO3 solution (CH3CN/H2O = 1:1) under an air atmosphere at 0.8 V vs. Ag/AgCl on BiVO4 under AM 1.5G illumination. (b) LSV curves of BiVO4 under AM 1.5G illumination (dashed and green lines) and in the dark (solid lines) measured in 0.1 M KHCO3, 0.1 M KBi (potassium borate buffer (pH = 8.3)) or 0.1 M K2HPO4 solution (CH3CN/H2O = 1:1) on BiVO4. | ||
Replacing KHCO3 with equivalent amounts of K2HPO4, KBi (potassium borate buffer (pH = 8.3)), KAc (potassium acetate) and KNO3 completely suppresses the alkene dioxygenation process, in which the substrate is barely consumed (Fig. S6, ESI†). When Cl− that is able to be oxidized is used, alkenes can be converted but there is little dioxide product. These experimental results indicate that HCO3− plays a key role in mediating the dioxygenation process of alkenes. Moreover, when the cation K+ of KHCO3 is replaced by Na+ or NH4+ (Table 1, entries 6 and 7), high yields of the diol product are still observed. These experimental results strongly indicate that HCO3− plays a pivotal role in alkene dioxygenation reactions. We investigated the influence of electrolyte acidity/basicity by adjusting the pH of the water with KOH or CO2 gas. As shown in Table S1 (ESI†), the yields of the diol product remain almost unchanged (about 85%) when water with a pH range of 6.8–9.3 is used (Table S1, ESI† entries 1–3). A further increase in pH reduces the yield of diol (Table S1, ESI† entries 4 and 5). Considering the pKa1 (6.37) and pKa2 (10.32) of H2CO3, the high yield of diol in the pH range of 6.8–9.3 suggests that HCO3− is the dominant active species in mediating the alkene dioxygenation reaction. Furthermore, we carried out the reaction in an H-type cell with a proton exchange membrane (Fig. S7a, ESI†) and found that the HCO3−-mediated PEC alkene dioxygenation reaction only occurs at the photoanode (Fig. S7b, ESI†). When the reaction is carried out under an Ar atmosphere (Table 1, entry 8), similar selectivity and yield for the dioxygenation are observed compared to those in air, indicating that O2 is not involved in the alkene dioxygenation pathway.
The dioxygenation reaction can also occur on TiO2 photoanodes, although the yield is somewhat lower than that on BiVO4 (Table 1, entry 9), which is due to the strong oxidation capacity of TiO2 leading to an increase in by-products (Fig. S8b, ESI†). When the α-Fe2O3 is employed as the photoanode, negligible substrate reactivity is observed (Fig. S8c, ESI†). Moreover, the photocurrent of α-Fe2O3 photoanode shows little change when HCO3− is added to the solution (Fig. S9, ESI†). This phenomenon is attributed to the insufficient oxidation capacity of α-Fe2O3, making it difficult to oxidize HCO3− to ˙CO3− and consequently hinders the conversion of alkenes.
Scope of substrates
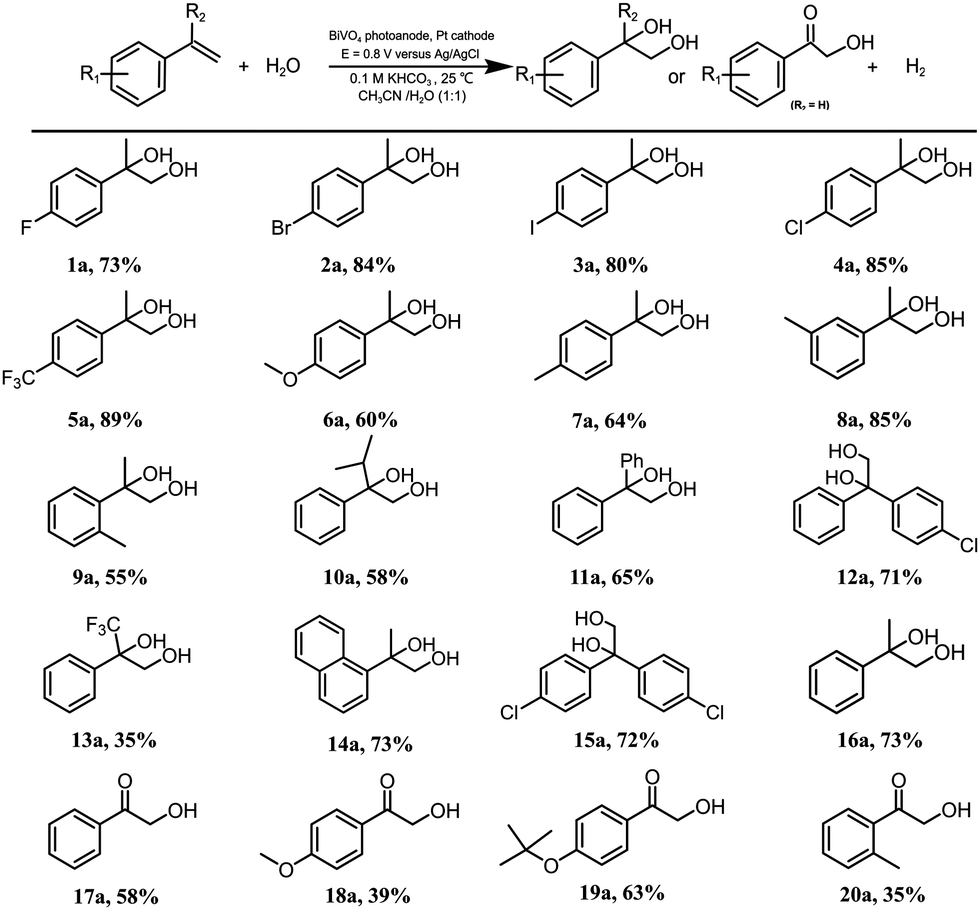
With the optimized protocol for alkene dioxygenation reactions in hand, the scope of the bicarbonate-mediated photoelectrochemical dioxygenation of alkenes was explored (Table 2). Initially, various monofunctionalized α-methyl-styrene substrates with para-, meta- or ortho-substituents were dioxygenated to the corresponding products in good yields. Notably, the substrates with electron-withdrawing groups can be efficiently converted to the corresponding diols smoothly (1a–5a), while the strong electron-donating groups (such as –OCH3 (6a), -CH3 (7a)) show a slight decrease in the yields of their products. The diol yield of the meta-methyl-substituted α-methyl-styrene (8a) is significantly higher than those of the para- and ortho-substituted substrates (7a, 9a), confirming that the substituent effect is important to the dioxygenation. In addition, when the α position of styrene is substituted by isopropyl (10a), phenyl (11a), 4-chlorobenzene (12a) or trifluoromethyl (13a), the corresponding products are obtained in good yields. Moreover, substrates with polycyclic rings, such as naphthalene (14a), also yield the dioxygenation product with good efficiency.| Reaction conditions: BiVO4 photoanode (2.5 cm2), Pt cathode, an Ag/AgCl electrode (3.5 M KCl leak-free and 2.0 mm diameter) was used as the reference electrode, CH3CN/H2O (2.5/2.5 mL), under AM 1.5G illumination, 0.1 M KHCO3 was used as the electrolyte and the initial concentration of the substrate was 4.0 mM, in air, room temperature, photoelectrolysis reaction time of 1.5-3 h. Quantity of products determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard and substrates were analyzed and quantified by using an Agilent HPLC1260 system. |
|---|
|
Various substituents of styrene derivatives without an α-substitutent could be oxidized to the corresponding α-hydroxy ketones under the aforementioned reaction conditions (17a–20a). The formation of α-hydroxy ketones may originate from the highly active hydrogen atom in the α-position, which undergoes further oxidation. In contrast, α-methyl styrene, which has a methyl group occupied at the α-position, could not be further oxidized, so the diol product remained. Generally, the selectivity and yield for α-hydroxy ketone formation during styrene oxidation are significantly lower than those of α-methyl-styrenes, which may be caused by further oxidation involving C–C bond breakage during the process of forming α-hydroxy ketone products. To assess the stability of the substrates under light exposure, several characteristic substrates were selected for stability testing experiments. As shown in Fig. S11 (ESI†), the concentration of these substrates remained unchanged over 2 h of light exposure, affirming that all organic substrates were stable under illumination.
For gram-scale synthesis, 4-chloro-alpha-methyl-stryene was chosen as a substrate to conduct the dioxygenation reaction. We chose a substrate concentration of 20 mM for gram-scale synthesis, which has a higher FE (Fig. S12, ESI†). The reaction solution volume was increased by thirty times (Fig. S13, ESI†) compared to that of the standard condition. 0.44 gram of diol product 4a with an 80% yield was obtained and the selectivity and FE were 90% and 52%, respectively. This higher FE may be caused by the increase in the substrate concentration, which improves the probability of interaction between the substrate and active species. For the stability of the BiVO4, PEC tests and characterization were conducted. As depicted in Fig. S14a and b (ESI†), the PEC behavior remained almost unchanged after 6 h of photoelectrolysis. XRD patterns (Fig. S14c, ESI†), UV-vis adsorption spectra (Fig. S14d, ESI†), surface components (XPS) (Fig. S15, ESI†) and SEM (Fig. S16, ESI†) of the BiVO4 photoanode before and after photoelectrolysis also revealed that the properties and morphology of the photoanodes showed no obvious changes. These results demonstrate that BiVO4 exhibits superior stability under standard reaction conditions.
Alkene dioxygenation mechanism
In order to gain more insight into the alkene dioxygenation reaction, linear sweep voltammetry (LSV) tests were performed under different conditions. Compared to using K2HPO4 and KBi as the electrolyte, a noticeable enhancement in photocurrent was observed in the KHCO3 system when the applied potential exceeded 0.2 V (Fig. 1(b)), indicating that the oxidation of HCO3− occurs prior to water oxidation. Although the oxidation of water to oxygen (EO2/H2O = 1.23 V vs. RHE) is more thermodynamically advantageous than the oxidation of HCO3−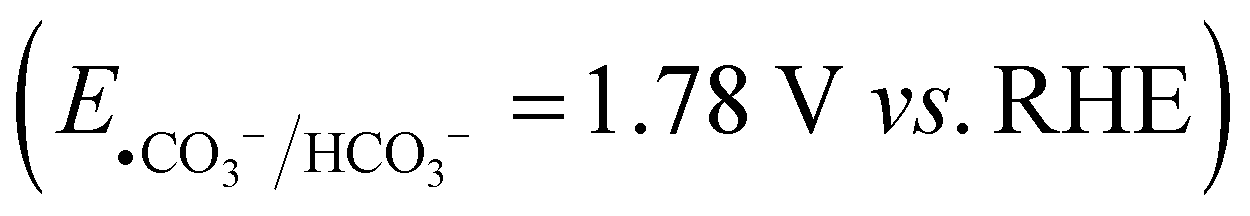 , HCO3− is oxidized to ˙CO3−via a single-electron-transfer pathway and water is oxidized to oxygen via a four-electron process, which is characterized by unfavorable kinetics. Therefore, the oxidation of HCO3− is superior to that of water, which has been reported in previous reports.17,18 Furthermore, the photocurrent exhibits negligible variation upon the addition of 4aa in the HCO3− system, suggesting that the direct oxidation of alkenes on the photoanode occurs at a neglectable rate. The redox potential of ˙CO3−/HCO3−via a single electron oxidation (eqn (1)) is 1.78 V vs. NHE.21 Notably, the valence band potential of BiVO4 registers at 2.4 V,30 surpassing the redox potential of ˙CO3−/HCO3−. Consequently, BiVO4 effectively promotes the oxidation of HCO3−, leading to formation of ˙CO3−. These experimental and theoretical data indicate that the alkene dioxygenation reaction may be mediated by generated carbonated active species. To validate this proposition, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 3 equiv.) or 2,6-di-tert-butyl-4-methylphenol (BHT, 3 equiv.), which are typical radical scavengers for trapping reactive radicals,31 were added into the reaction system. Strikingly, the presence of radical scavengers resulted in nearly complete inhibition of alkene dioxygenation (Table 3, entries 2 and 3), implying the involvement of active radical species in the dioxygenation process.
, HCO3− is oxidized to ˙CO3−via a single-electron-transfer pathway and water is oxidized to oxygen via a four-electron process, which is characterized by unfavorable kinetics. Therefore, the oxidation of HCO3− is superior to that of water, which has been reported in previous reports.17,18 Furthermore, the photocurrent exhibits negligible variation upon the addition of 4aa in the HCO3− system, suggesting that the direct oxidation of alkenes on the photoanode occurs at a neglectable rate. The redox potential of ˙CO3−/HCO3−via a single electron oxidation (eqn (1)) is 1.78 V vs. NHE.21 Notably, the valence band potential of BiVO4 registers at 2.4 V,30 surpassing the redox potential of ˙CO3−/HCO3−. Consequently, BiVO4 effectively promotes the oxidation of HCO3−, leading to formation of ˙CO3−. These experimental and theoretical data indicate that the alkene dioxygenation reaction may be mediated by generated carbonated active species. To validate this proposition, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 3 equiv.) or 2,6-di-tert-butyl-4-methylphenol (BHT, 3 equiv.), which are typical radical scavengers for trapping reactive radicals,31 were added into the reaction system. Strikingly, the presence of radical scavengers resulted in nearly complete inhibition of alkene dioxygenation (Table 3, entries 2 and 3), implying the involvement of active radical species in the dioxygenation process.| HCO3− → ˙CO3− + H+ + e− | (1) |
| HCO3− + H2O → HCO4−+ 2H+ + 2e− | (2) |
| HCO4− + H2O → HCO3−+ H2O2 | (3) |
| Entry | Reaction conditionsa | Selectivity (%) | Yield (%) |
|---|---|---|---|
| a Standard reaction conditions: BiVO4 photoanode (2.5 cm2), Pt cathode, CH3CN/H2O (2.5/2.5 mL), under AM 1.5G illumination, 0.1 M KHCO3 was used as the electrolyte and the initial concentration of the substrate was 4.0 mM, under air, room temperature, reaction time of 1.5 h, undivided cell. b Trace alkene dioxygenation product was detected. c 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO). d 2,6-Di-tert-butyl-4-methylphenol (BHT). e Not detected. f tert-butanol (t-BuOH). | |||
| 1 | 0.1 M KHCO3 | 89 ± 2 | 85 ± 3 |
| 2 | 3 equiv. TEMPOc | Traceb | Traceb |
| 3 | 3 equiv. BHTd | Traceb | Traceb |
| 4 | Add 1 mM H2O2 | 80 ± 1 | 76 ± 1 |
| 5 | Add 1 M H2O2 | n.d.e | n.d.e |
| 6 | Add 1 M H2O2, no PEC | n.d.e | n.d.e |
| 7 | 3 equiv. t-BuOHf | 86 ± 3 | 82 ± 2 |
It has been reported that HCO3− can be oxidized to ˙CO3− (eqn (1)) through single electron oxidation,18 or to HCO4−via a two-electron process (eqn (2))17,32 and HCO4− can be hydrolyzed to hydrogen peroxide (eqn (3))33 in the EC or PEC processes. Based on previous research, there are potential active species, ˙CO3−, H2O2 and HCO4−, which are capable of initiating the reaction. To elucidate the contributions of H2O2 and HCO4−, controlled experiments were conducted. Firstly, the addition of H2O2 triggered a slight decrease of dioxygenation activity (Table 3, entry 4), excluding the possibility that H2O2 (if formed) acts as the active species of dioxygenation. With an increase in the H2O2 concentration, the dioxygenation reaction is even suppressed (Table 3, entry 5), which might be attributed to the competitive oxidation caused by H2O2. Subsequently, to assess the role of HCO4−, the HCO4− species were in situ produced by a mixing reaction of abundant H2O2 and HCO3− (without an applied potential).34 No reaction was detected under these conditions (Table 3, entry 6), ruling out the participation of HCO4- species. Moreover, the addition of tert-butanol, a typical ˙OH scavenger,35 showed negligible effects on the selectivity and yield of the diol product (Table 3, entry 7), indicating that ˙OH is not the active species of dioxygenation. Consequently, it is reasonable to infer that ˙CO3− serves as the primary active species in the dioxygenation process.
Considering the difference in products obtained from styrene and alpha-methyl-styrene, it is plausible that the formation of α-hydroxyacetophenone through the oxidation of styrene proceeds via a diol-intermediate pathway, where a diol is initially formed and subsequently dehydrogenated to produce the α-hydroxy ketone. However, when the diol was directly employed as the substrate, the selectivity and yield of the α-hydroxyacetophenone product were only 64% and 35%, respectively. These values were lower than the selectivity (78%) and yield (57%) achieved with styrene as the substrate under the same reaction conditions (Table S2, ESI† entries 1 and 2). Thus, it is evident that the diol does not act as the intermediate for the formation of α-hydroxy ketone.
In addition, acetophenone and styrene epoxide were employed as substrates and a slight reaction was observed (Table S2, ESI† entries 3 and 4), suggesting that ketone and epoxide are also not the intermediates. Interestingly, when peroxydiol (4ab) was used as the substrate, excellent diol product selectivity (96%) and yield (95%) were obtained within 1 h of the PEC reaction (Fig. 2(b) and (d)), indicative that peroxydiol may be a true reaction intermediate in the dioxygenation process. This peroxydiol intermediate was also detected during the PEC oxidation of 4aa on BiVO4, and the concentration of the intermediate 4ab showed an initial increase followed by a subsequent decrease with an increase in the reaction time (Fig. 2(c)). It has been reported that the transformation from peroxydiol to diol is a two-electron reduction process, typically requiring a reductant such as triphenylphosphine (PPh3).36,37 To make clear whether the reduction of peroxydiol occurs on the anode or cathode, the reaction was carried out in an H-type cell. The results demonstrate that the reduction reaction from peroxydiol to diol occurs in the photoanode compartment (Fig. S17, ESI†). However, no reduction reaction was observed in the absence of HCO3−, irradiation or applied potential (Table S3, ESI†), highlighting the crucial role of ˙CO3− in facilitating the reductive reaction. These observations further imply that the formation of diol originates from the interaction between ˙CO3− and peroxydiol. Stoichiometrically, the attack of the peroxy group of the peroxydiol by ˙CO3− led to the formation of a ˙CO4− radical and diol product, which may be analogous to the reaction between HCO3− and H2O2 to form HCO4− and H2O, as reported in the previous literature.38
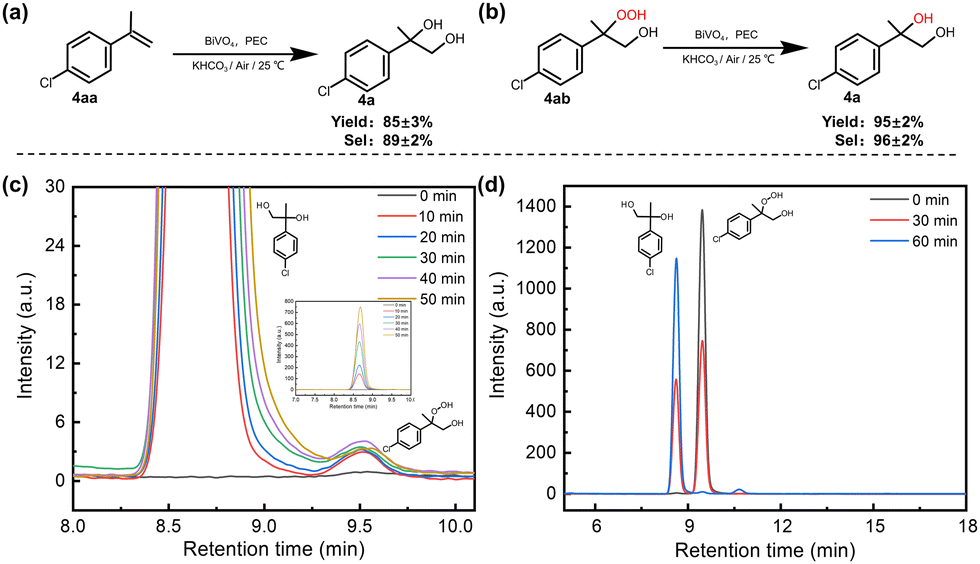 | ||
| Fig. 2 (a) and (b) Experiments of different substrate reactions under standard conditions. (c) The HPLC spectra obtained different photoelectrolysis time of 4.0 mM 4aa with 0.1 M KHCO3 at 0.8 V vs. Ag/AgCl on BiVO4 (illustration is the corresponding full-size image). (d) The HPLC spectra obtained different photoelectrolysis time of 4.0 mM peroxydiol (4ab) with 0.1 mM KHCO3 at 0.8 V vs. Ag/AgCl on BiVO4. Quantity of products determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard and substrates were analyzed and quantified using an Agilent HPLC1260 system. | ||
To examine the formation of radical species, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was used as a spin-trapping reagent for conducting EPR experiments. As shown in Fig. 3(a), a signal corresponding to peroxyradical species (DMPO-OOH) was detected in the presence of HCO3−, whereas no signal was observed when HCO3− was replaced by K2HPO4. The results suggest that HCO3− is involved in the formation of the superoxide adduct of DMPO. This DMPO-OOH signal remains unaltered upon adding superoxide dismutase (SOD) (0.2 mg ml−1) (Fig. 3(a)), meaning that this adduct is not formed from the trapping of the superoxide radical by DMPO. Therefore, a distinct radical species, rather than superoxide radicals, is responsible for the formation of the DMPO-OOH adduct. It has been reported that the interaction of ˙CO3− and HCO4− can yield ˙CO4−.39 Therefore, it is reasonable to propose that DMPO may trap ˙CO4− to form a DMPO-CO4− adduct, which is rapidly hydrolyzed to DMPO-OOH and HCO3− (Fig. S18b, ESI†). This is why the dioxygenation activity is well maintained upon the addition of SOD (Fig. 3(b)). Thus, it is highly plausible that ˙CO4− acts as the key active species to further trigger dioxygenation reaction. In addition, by conducting EPR experiments under different applied potentials (Fig. S19, ESI†), it is obvious that the signal of DMPO-OOH is significantly decreased when the applied potential is reduced. This indicates that the generation of ˙CO4− is indeed reduced at low potential, leading to a decrease in the product selectivity.
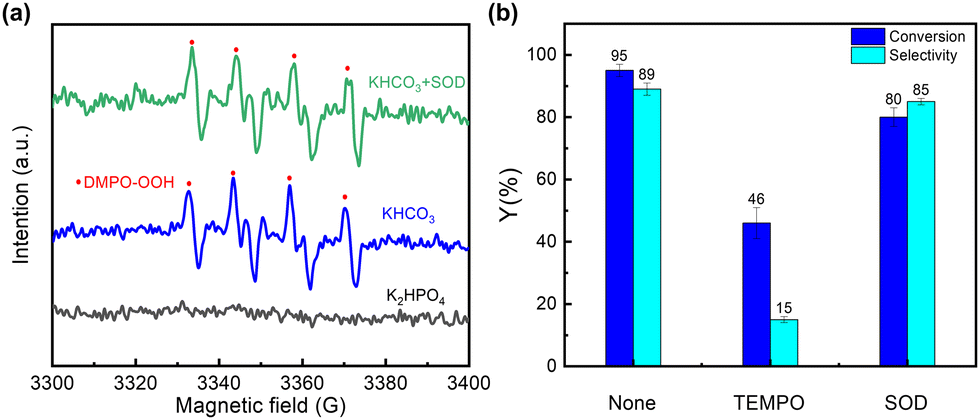 | ||
| Fig. 3 (a) EPR spectra of DMPO adducts in different systems. (b) Effects of different scavengers on the reaction activity. | ||
As shown in Scheme 2, the first step for PEC alkene dioxygenation is the oxidation of HCO3− to generate ˙CO3− and ˙CO4− species, which could be obtained by direct oxidation or a ˙CO3−-mediated process on the photoanode (step 1). Then, ˙CO3− further attacks the terminal C atom of the CC bond of the alkene (a1), leading to the formation of a carbonated radical (a2). This carbonated radical undergoes rapid hydrolysis to generate a hydroxylated carbon radical (a3). Subsequently, the generated carbon radical combines with ˙CO4− to form a peroxycarbonated intermediate, which ultimately undergoes hydrolysis to a peroxydiol (a4). Finally, the peroxydiol is reduced to a diol (a6) by ˙CO3−, with the simultaneous regeneration of ˙CO4− (step 5). Conversely, if styrene is employed as the substrate, the peroxydiol rapidly dehydrates to form an α-hydroxy ketone product (a5). According to this mechanism, the alkene dioxygenation is primarily determined by the reaction between the alkene and ˙CO3− (step 2). As shown in Fig. S20a (ESI†), the conversion rate of various para-substituent α-methyl-styrenes exhibit well linear relationships with the Hammett constants of the substituents, with a negative slope (ρ = −0.56). The negative linear free energy relationship verifies that the rate-determining step for the oxidation of alkenes involves electrophilic attack by ˙CO3−. However, as shown in Table 2, relatively lower yields were obtained for α-methyl-styrene with electron-donating groups (such as –OCH3 (6a), –CH3 (7a)). In contrast to the negative slope observed for the oxidation rate, there is a positive strong correlation between the selectivity for the diol and the Hammett constants of the substituents (Fig. S20b, ESI†), implying that the selectivity for the diol is determined by a nucleophilic process. According to our proposed mechanism, the reduction of peroxydiol to diol by ˙CO3− involves the nucleophilic attack of the peroxy moiety of peroxydiol by ˙CO3−. The substitution of an electron-withdrawing group would decrease the electron density on the peroxy bond, thereby facilitating its cleavage during the nucleophilic attack of ˙CO3−. The positive correlation further confirms that the reduction of peroxydiol to diol by ˙CO3− is the selectivity-determining step in the dioxygenation reaction.
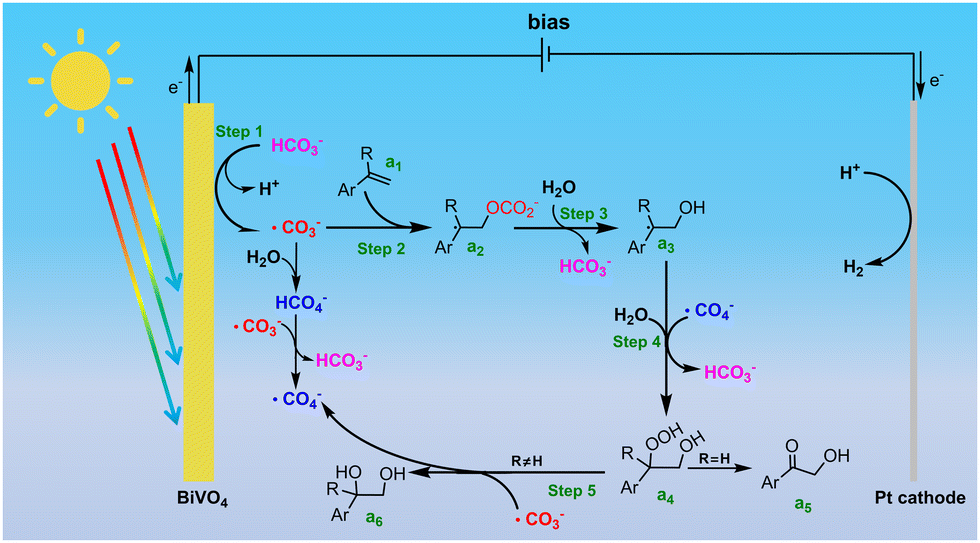 | ||
| Scheme 2 The proposed HCO3−-mediated dioxygenation mechanism on BiVO4. | ||
Conclusions
We developed a novel method for PEC bicarbonate-mediated alkene dioxygenation under mild conditions, coupled with the HER at the cathode, in which no noble metals or external oxidants are used. The PEC dioxygenation shows satisfying product selectivity and functional group tolerance, rendering the reaction straightforward, safe and amenable to diverse applications. Mechanistic studies verified the triple role played by HCO3−-derived radicals: firstly, as initiators of alkene reactions through ˙CO3−; secondly, ˙CO4− reacts with carbon radicals and undergoes further hydrolysis to form peroxyl alcohol intermediates; and thirdly, ˙CO3− serves as a reductant to facilitate the transformation from peroxyl alcohols to a diol product. As a result, this innovative strategy provides a scalable, highly efficient and economically feasible route for synthesizing a diverse range of high added-value diols or α-hydroxy ketones derivatives.Author contributions
J. Y. and C. C. conceived the idea. J. Y. designed and performed the experiments. J. Y., Y. K. Z. and M. D. analyzed the experiments data. J. Y., Y. F. Z. and J. L. analyzed the spin-trapped electron paramagnetic resonance (EPR) experiments. J. Y., C. C. and Y. L. analyzed the reaction mechanism. J. Y. and Y. K. Z. wrote the manuscript. Y. L. and C. D. helped with the writing. C. C., J. Z. and W. S. reviewed, edited the manuscript, and supervised the projects and processes. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22136005, 22188102, 22106164) and the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB36000000).References
- H. G. Cha and K. S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS.
- Y. Zhang, H. Zhang, A. Liu, C. Chen, W. Song and J. Zhao, J. Am. Chem. Soc., 2018, 140, 3264–3269 CrossRef CAS PubMed.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS.
- S. Chae, J. Choi, Y. Kim, D. L. T. Nguyen and O. Joo, Angew. Chem., Int. Ed., 2019, 58, 16395–16399 CrossRef CAS.
- Q. Zeng, J. Li, L. Li, J. Bai, L. Xia and B. Zhou, Appl. Catal., B, 2017, 217, 21–29 CrossRef CAS.
- R. Song, H. Chi, Q. Ma, D. Li, X. Wang, W. Gao, H. Wang, X. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2021, 143, 13664–13674 CrossRef CAS.
- Z. Q. Wang, Y. H. Guo, M. Liu, X. L. Liu, H. P. Zhang, W. Y. Jiang, P. Wang, Z. K. Zheng, Y. N. Liu, H. F. Cheng, Y. Dai, Z. Y. Wang and B. B. Huang, Adv. Mater., 2022, 34, 27 Search PubMed.
- X. Liu, Z. Chen, S. X. Xu, G. Q. Liu, Y. Zhu, X. Q. Yu, L. C. Sun and F. Li, J. Am. Chem. Soc., 2022, 144, 19770–19777 CrossRef CAS.
- P. Rajeshwaran, J. Trouve, K. Youssef and R. Gramage-Doria, Angew. Chem., Int. Ed., 2022, 61, 50 CrossRef PubMed.
- P. Xie, C. Xue, J. F. Luo, S. S. Shi and D. D. Du, Green Chem., 2021, 23, 5936–5943 RSC.
- B. de Bruin, P. H. M. Budzelaar and A. W. Gal, Angew. Chem., Int. Ed., 2004, 43, 4142–4157 CrossRef CAS.
- Y. Zhao, M. Duan, C. Deng, J. Yang, S. Yang, Y. Zhang, H. Sheng, Y. Li, C. Chen and J. Zhao, Nat. Commun., 2023, 14, 1943 CrossRef CAS PubMed.
- W. R. Leow, Y. Lum, A. Ozden, Y. H. Wang, D. H. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. W. Li, D. Sinton and E. H. Sargent, Science, 2020, 368, 1228 CrossRef CAS PubMed.
- M. J. Chung, K. Jin, J. S. Zeng and K. Manthiram, ACS Catal., 2020, 10, 14015–14023 CrossRef CAS.
- K. Fuku and K. Sayama, Chem. Commun., 2016, 52, 5406–5409 RSC.
- X. J. Shi, S. Siahrostami, G. L. Li, Y. R. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. L. Zheng and J. K. Norskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- T. M. Gill, L. Vallez and X. Zheng, ACS Energy Lett., 2021, 6, 2854–2862 CrossRef CAS.
- L. Fan, X. W. Bai, C. Xia, X. Zhang, X. H. Zhao, Y. Xia, Z. Y. Wu, Y. Y. Lu, Y. Y. Liu and H. T. Wang, Nat. Commun., 2022, 13, 2668 CrossRef CAS.
- F. Y. Chen, L. G. Xia, Y. Zhang, J. Bai, J. C. Wang, J. H. Li, M. Rahim, Q. J. Xu, X. Y. Zhu and B. X. Zhou, Appl. Catal., B, 2019, 259, 118071 CrossRef CAS.
- L. G. Xia, F. Y. Chen, J. H. Li, S. Chen, J. Bai, T. S. Zhou, L. S. Li, Q. J. Xu and B. X. Zhou, J. Hazard. Mater., 2020, 389, 122140 CrossRef CAS.
- F. Boccini, A. S. Domazou and S. Herold, J. Phys. Chem. A, 2004, 108, 5800–5805 CrossRef CAS.
- J. E. Grebel, J. J. Pignatello and W. A. Mitch, Environ. Sci. Technol., 2010, 44, 6822–6828 CrossRef CAS PubMed.
- C. P. Park, J. H. Lee, K. S. Yoo and K. W. Jung, Org. Lett., 2010, 12, 2450–2452 CrossRef CAS.
- C. M. Ho, W. Y. Yu and C. M. Che, Angew. Chem., Int. Ed., 2004, 43, 3303–3307 CrossRef CAS.
- X. H. Huo, R. He, X. Zhang and W. B. Zhang, J. Am. Chem. Soc., 2016, 138, 11093–11096 CrossRef CAS.
- B. M. Trost and L. R. Terrell, J. Am. Chem. Soc., 2003, 125, 338–339 CrossRef CAS PubMed.
- E. N. Jacobsen, I. Marko, W. S. Mungall, G. Schroder and K. B. Sharpless, J. Am. Chem. Soc., 1988, 110, 1968–1970 CrossRef CAS.
- S.-N. Chen, M. Z. Hoffman and G. H. Parsons, Jr, J. Phys. Chem. C, 1975, 79, 1911–1912 CrossRef CAS.
- T. Zeng and W. A. Arnold, Environ. Sci. Technol., 2013, 47, 6735–6745 CrossRef CAS PubMed.
- P. Lianos, Appl. Catal., B, 2017, 210, 235–254 CrossRef CAS.
- C.-Y. Huang, J. Li and C.-J. Li, Nat. Commun., 2021, 12, 4010 CrossRef CAS PubMed.
- A. K. Seitz, P. J. Kohlpaintner, T. Lingen, M. Dyga, F. Sprang, M. Zirbes, S. R. Waldvogel and L. J. Goossen, Angew. Chem., Int. Ed., 2022, 61, 25 CrossRef.
- J. Liu, Y. Zou, B. Jin, K. Zhang and J. H. Park, ACS Energy Lett., 2019, 4, 3018–3027 CrossRef CAS.
- X. J. Yang, Y. H. Duan, J. L. Wang, H. L. Wang, H. L. Liu and D. L. Sedlak, Environ. Sci. Technol. Lett., 2019, 6, 781–786 CrossRef CAS PubMed.
- L. Chen, S. Wang, Z. Yang, J. Qian and B. Pan, Appl. Catal., B, 2021, 292, 120193 CrossRef CAS.
- X. Sun, X. Li, S. Song, Y. Zhu, Y. F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 46, 6059–6066 CrossRef.
- B. Yang and Z. Lu, Chem. Commun., 2017, 53, 12634–12637 RSC.
- D. E. Richardson, H. R. Yao, K. M. Frank and D. A. Bennett, J. Am. Chem. Soc., 2000, 122, 1729–1739 CrossRef CAS.
- K. S. Haygarth, T. W. Marin, I. Janik, K. Kanjana, C. M. Stanisky and D. M. Bartels, J. Phys. Chem. A, 2010, 114, 2142–2150 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee02948d |
| This journal is © The Royal Society of Chemistry 2024 |