
Unraveling and suppressing the voltage decay of high-capacity cathode materials for sodium-ion batteries†
Luoran
Sun‡
a,
Zhonghan
Wu‡
a,
Machuan
Hou
a,
Youxuan
Ni
a,
Haoxiang
Sun
a,
Peixin
Jiao
a,
Haixia
Li
ab,
Wei
Zhang
ab,
Liang
Zhang
 cd,
Kai
Zhang
*ab,
Fangyi
Cheng
ab and
Jun
Chen
ab
cd,
Kai
Zhang
*ab,
Fangyi
Cheng
ab and
Jun
Chen
ab
aState Key Laboratory of Advanced Chemical Power Sources, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Renewable Energy Conversion and Storage Center (RECAST), College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: zhangkai_nk@nankai.edu.cn
bHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, P. R. China
cInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou 215123, China
dJiangsu Key Laboratory of Advanced Negative Carbon Technologies, Soochow University, Suzhou 215123, China
First published on 10th November 2023
Abstract
Oxygen-redox-active (ORA) layered oxide cathodes for sodium-ion batteries have received considerable attention due to their ultrahigh capacity. However, the voltage decay during electrochemical cycling in such materials is still elusive and unsolved, which seriously limits their practical implementation. Herein, we unveil the intrinsic origin of voltage decay in sodium-based ORA cathodes by coupling spatially local electron energy loss spectroscopy with bulk-sensitive X-ray absorption spectroscopy. It is demonstrated that the steric heterogeneity of Mn redox derived from the surface formation of oxygen vacancies is responsible for the voltage deterioration upon cycling. Moreover, we propose an ORA cathode (Na0.8Li0.24Al0.03Mn0.73O2) with negligible voltage decay. Its oxygen redox reversibility is significantly strengthened because the strong Al–O bonds weaken the covalency of Mn–O bonds to promote the electron localization on oxygen. These findings suggest a new insight into the electronic structure design of high-energy-density cathode materials for advanced rechargeable batteries.
Broader contextNon-resource limited electrochemical energy storage technologies play a vitally important role in addressing the volatility and randomness of renewable energy power. Compared with lithium-ion batteries, sodium-ion batteries (SIBs) are more likely to be used in large-scale stationary energy storage due to their abundant resources and low cost. For the SIB technology, the cathodes primarily dominate the battery performance. With high capacity and two-dimensional Na-diffusion channels, oxygen-redox-active (ORA) sodium-based layered oxides are a promising class of cathode materials, but they suffer from substantial voltage decay during electrochemical cycling. Here, we unveil that the voltage decay originates from the steric redox heterogeneity, which is proved by a combination of X-ray absorption spectroscopy and electron energy loss spectroscopy. Furthermore, we propose an electron localization strategy to tackle this challenge and build an electronic-structure-tuned Na0.8Li0.24Al0.03Mn0.73O2 cathode without voltage decay. Because of the introduction of strong Al–O bonds, the covalency of Mn–O bonds is significantly weakened, which enhances the oxygen electron localization to eliminate the O2 release and the surface oxygen vacancy formation. Our work sheds light on the electrochemical nature of voltage decay and also offers a feasible route to develop high-capacity cathode materials for energy-storage batteries. |
Introduction
Sodium-ion batteries (SIBs), because of the natural abundance and wide availability of sodium resources, are regarded as the most promising complements to lithium-ion batteries (LIBs), especially in the application of large-scale electrochemical energy storage.1–3 As cathodes primarily dominate the battery performance and cost, formidable efforts have been devoted to seeking ideal cathode materials for SIBs.4–7 Among various candidates, the oxygen-redox-active (ORA) layered oxides, represented by P2-type Nax[AyM1−y]O2 (0.5 < x < 1, 0 < y ≤ 1/3), where M stands for transition metal ions and A represents Li+,8 Mg2+,9 Zn2+,10 or a vacancy,11 have garnered intense interest due to their high capacity based on the accumulated cationic and anionic oxygen redox. The oxygen redox activity is typically triggered via the Na–O–A electronic configurations associated with the newly-generated non-bonding oxygen 2p states.12–15 Therefore, both transition metal cations and oxygen anions can participate in the charge compensation reaction during Na+ (de)intercalation, achieving exceptionally high energy density.Unfortunately, the ORA cathode materials generally suffer from irreversible electrochemical performances, in which voltage decay is an intractable issue. The gradual drop in average operation voltage during battery cycling inevitably results in a substantial energy loss, impeding the practical utilization of the oxygen redox reaction. Though voltage decay is known to be linked with oxygen redox, its intrinsic inducement in sodium-based ORA materials is still unrevealed because of the intricate electrochemical behaviors involving cation migration,16 lattice oxygen loss,17 and structure degradation.18
In LIB chemistry, it is widely recognized that the undesirable voltage decay in Li-rich layered oxides (Li[LizM1−z]O2, 0 < z ≤ 1/3) correlates with the surface layered-to-spinel phase transition and the activation of the lower-potential redox couples.19–21 Nevertheless, this principle is inapplicable in sodium-deficient layered oxide cathodes owing to their discrepancy in the crystal structure (P2 phase for SIBs vs. O3 phase for LIBs) (Fig. S1, ESI†). Moreover, transition metal migration from MO2 layers to alkali metal layers is prohibited in P2 structures because the trigonal prismatic Na sites are typically unaccommodated for M cations. Therefore, understanding the electrochemical nature of voltage decay and further finding a targeted way to suppress it remain challenging for SIB cathode materials.
Herein, we unravel the root of voltage decay in sodium-based high-capacity cathodes through comprehensively investigating an archetypical ORA cathode, Na0.8Li0.24Mn0.76O2 (NLMO). The systematic research has been accomplished by using an arsenal of advanced analytical techniques and theoretical modelling, such as synchrotron-based hard and soft X-ray absorption spectroscopy (XAS), scanning transmission electron microscopy (STEM), electron energy loss spectroscopy (EELS) and density functional theory (DFT) calculations. These data reveal that the surface formation of oxygen vacancies caused by the lattice oxygen loss leads to the spatial heterogeneity of Mn redox during cycling, thus inducing the voltage fading. Then, we propose an electron localization strategy to address this issue. The electronic-structure-tuned Na0.8Li0.24Al0.03Mn0.73O2 (NLAMO) exhibits effectively restrained oxygen release and outputs highly stable voltage within 200 cycles, which is due to the weakened Mn–O bond covalency and the enhanced oxygen electron localization by the strong Al–O bonds. This study sheds light on the oxygen redox chemistry and paves a way toward the design of high-performance SIB cathode materials.
Results and discussion
Structure and electrochemistry analysis
A series of Na0.8Li0.24AlxMn0.76−xO2 (x = 0, 0.01, 0.03, 0.05) have been synthesized via a traditional solid-state method (see ESI† for details). In consideration of phase purity and electrochemical performance, the optimal Al substitution amount is determined to be 0.03 (Fig. S2 and S3, ESI†). The actual compositions of NLMO and NLAMO are consistent with the designed chemical formula, as confirmed by inductively coupled plasma optical emission spectrometry (ICP-OES) (Table S1, ESI†). The Rietveld refined X-ray diffraction (XRD) patterns of both materials are shown in Fig. 1a and Fig. S4–S6 (ESI†), and the detailed lattice parameters are listed in Tables S2–S5 (ESI†). All the diffraction peaks can be well indexed to a hexagonal P2-type structure with the P63 or P63/mmc space group. The weak diffraction peak at ∼22° is attributed to the Li/Mn honeycomb ordering in the MO2 layers.22 The high-angle annular dark-field (HAADF) image of NLAMO shows that the visible MO2 layers alternate with the dark Na layers, and the d-spacing is measured to be ∼5.51 Å, which is in line with the interslab distance of the P2-layered structure (Fig. 1b). Energy-filtered transmission electron microscopy (EFTEM) and 7Li solid-state nuclear magnetic resonance (NMR) techniques are employed to illustrate the sites of cations, and it is demonstrated that all the Al, Mn and Li reside in the MO2 layers (inset of Fig. 1b and Fig. S7, S8, ESI†). The results of relative formation energy by first-principles calculations further reveal that Al atoms prefer to occupy the Mn site rather than the Li site (Fig. 1c). In addition, the NLAMO particles exhibit a plate-like morphology as shown in the scanning electron microscopy (SEM) image (Fig. S9, ESI†). Furthermore, as verified by transmission electron microscopy (TEM) with energy-dispersive spectroscopy (EDS), the elements (O, Na, Al and Mn) are uniformly distributed throughout the NLAMO particles (Fig. S10, ESI†).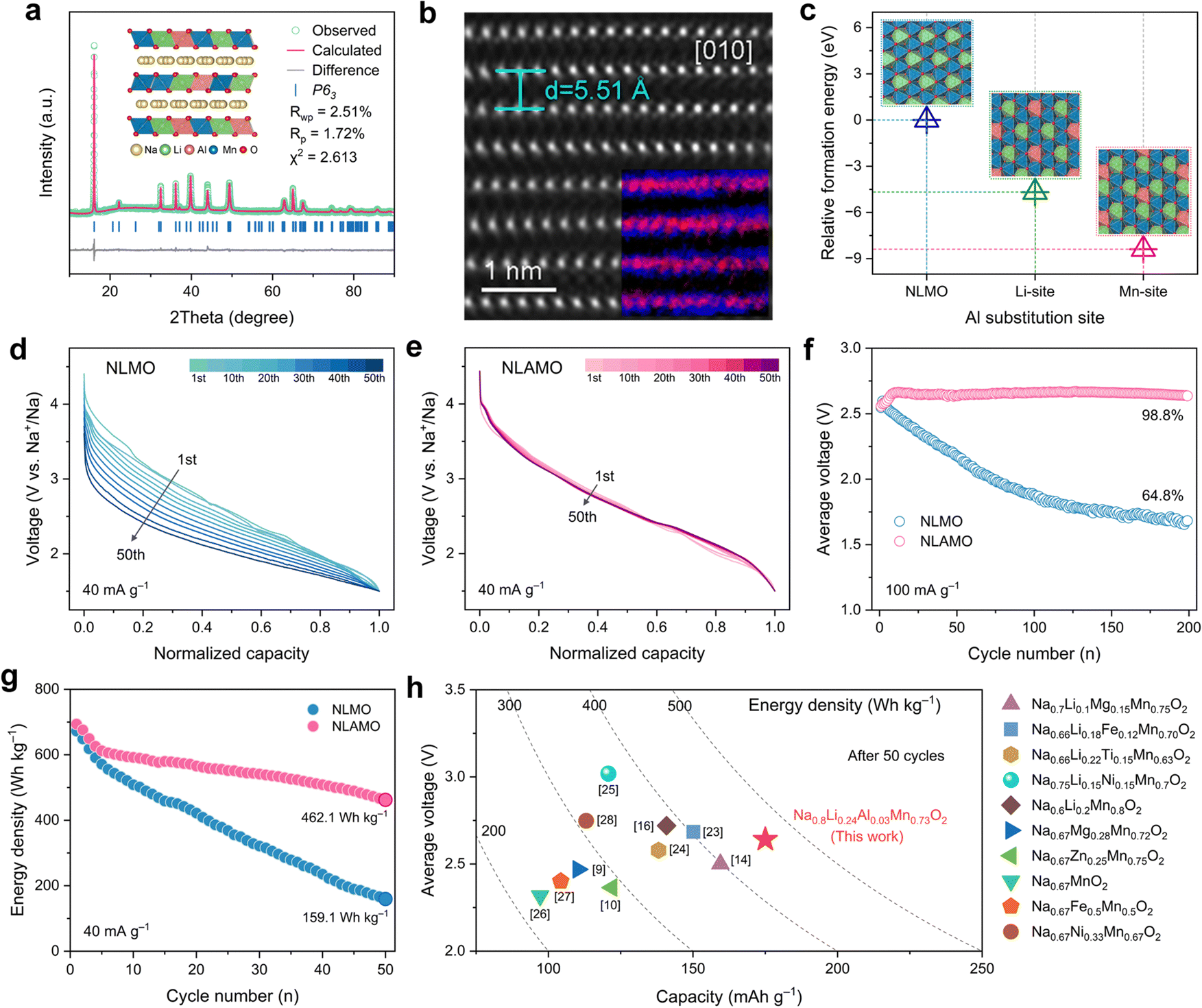 | ||
| Fig. 1 Crystal structure and electrochemical performance. (a) Rietveld refined XRD pattern of NLAMO. (b) HAADF-STEM image of NLAMO. Inset presents the corresponding EFTEM image. The blue-colored spots stand for Mn atoms and the red-colored spots represent Al atoms. (c) Relative formation energies of the crystal structure models for NLAMO with different Al substitution sites. (d) and (e) Normalized discharge curves of NLMO (d) and NLAMO (e). (f) Comparison of average voltage retention between NLMO and NLAMO over 200 cycles. (g) Comparison of energy density between NLMO and NLAMO over 50 cycles. (h) Statistics of discharge capacity, average voltage and energy density for the representative cathodes over 50 cycles. | ||
The electrochemical properties of NLMO and NLAMO are gauged within a voltage range of 1.5–4.5 V at 40 mA g−1 (0.2 C). In the galvanostatic charge–discharge profiles of both cathodes, a typical long plateau above 4.0 V appears during the first charge, indicating the oxidation process of oxygen anions (Fig. S11, ESI†).9 Although NLMO and NLAMO can deliver high initial discharge capacities of 257.1 mAh g−1 and 264.3 mAh g−1, respectively, their Na-storage performances differ in the subsequent cycles. NLMO exposes the distinct voltage fading behavior with an average discharge voltage retention of 79.3%, while NLAMO exhibits negligible voltage decay with an excellent voltage retention of 99.5% upon 50 cycles at 40 mA g−1 (Fig. 1d, e and Fig. S12, ESI†). Moreover, the average voltage of NLAMO can be well maintained even after 200 cycles at 100 mA g−1 (Fig. 1f and Fig. S13, ESI†). Note that a voltage rise appears in the early cycles due to the evolution of the charge compensation mechanism that correlates with the high-voltage side reactions between the electrolytes and oxygen species (Fig. S14, ESI†). The improved electrochemical performance of NLAMO is also well recognized in differential capacity (dQ dV−1) curves (Fig. S15, ESI†) and cyclic voltammetry plots (Fig. S16 and S17, ESI†).
Because of the stable output voltage during cycling, NLAMO retains a high specific energy of 462.1 Wh kg−1 (based on the mass of the active material) after 50 cycles, which is far superior to that of NLMO (159.1 Wh kg−1) (Fig. 1g). The outstanding energy density of NLAMO outperforms the other reported sodium-based layered cathode materials (Fig. 1h and Table S6, ESI†).9,10,14,16,23–28 Furthermore, the electrochemical performance of NLMO and NLAMO has been evaluated under different charge cutoff voltages, and the results show that the primary cause of voltage attenuation is basically relative to the oxygen-related anionic redox in the high-voltage range (Fig. S18 and S19, ESI†). Thus, it is critical to fundamentally investigate this unique behavior for such oxygen-redox cathode materials.
Investigation of the oxygen redox behavior
To study the stability of lattice oxygen, the oxygen vacancy formation energies of both cathodes are firstly calculated. As presented in Fig. 2a, the formation energy of NLAMO (−1.736 eV) is much higher than that of NLMO (−3.299 eV), verifying the enhanced lattice oxygen steadiness by Al incorporation. Next, the oxygen-related redox behavior is experimentally investigated by in situ differential electrochemical mass spectrometry (DEMS) (Fig. 2b). For NLMO, O2 evolution can be clearly observed at the end of the initial charge. Meanwhile, the lattice oxygen release has been effectively restrained in NLAMO, which proves its oxygen anions are stable, in agreement with the calculation results. It should be noted that the oxygen escape typically leads to the formation of oxygen vacancies from the surface to the bulk. And these oxygen vacancies will further induce a massive number of structural defects during cycling, resulting in phase boundaries and cracks of the cathode particles.29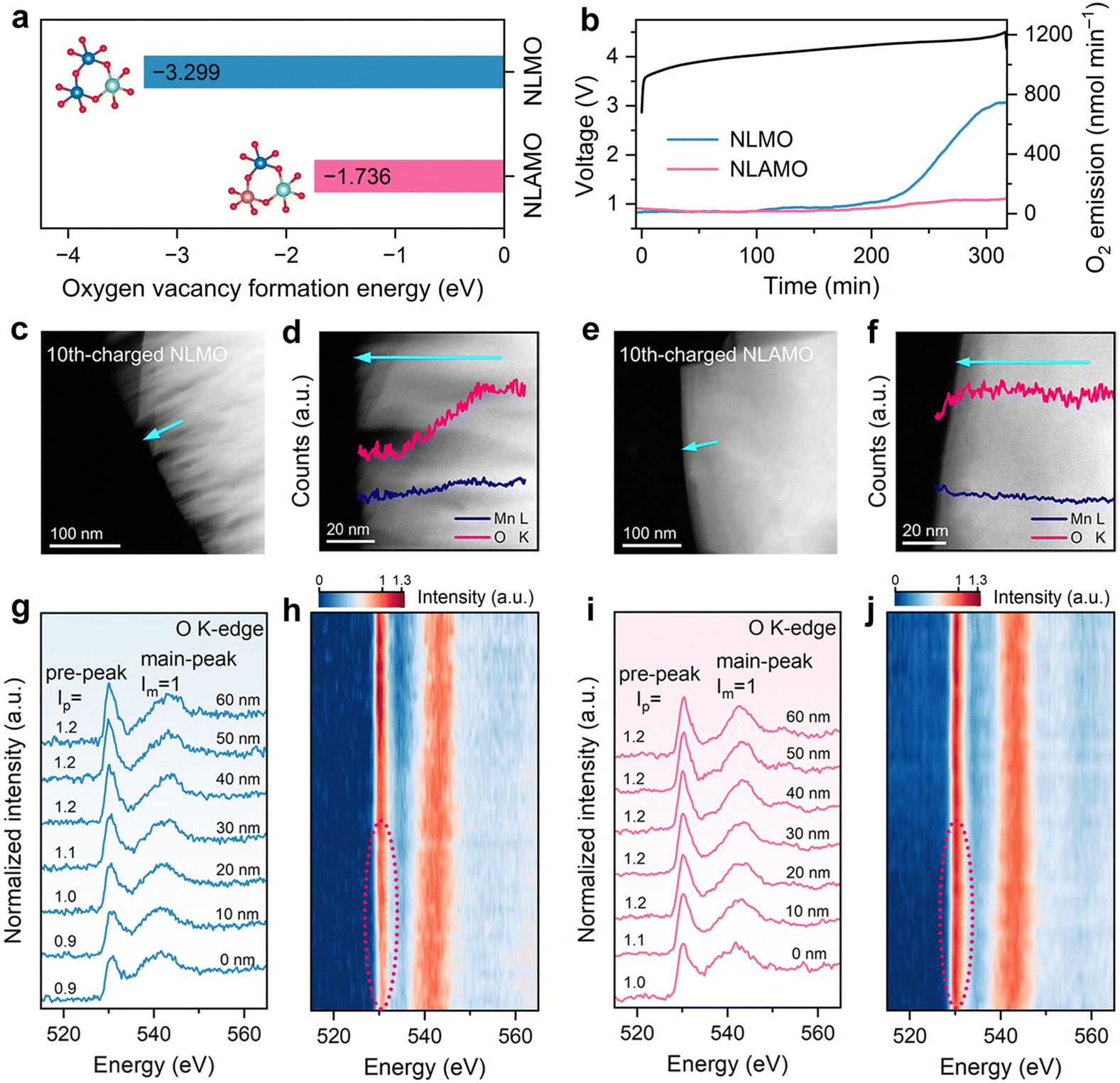 | ||
| Fig. 2 Oxygen redox behaviors of NLMO and NLAMO. (a) The oxygen vacancy formation energy. (b) In situ DEMS measurements from open circuit voltage to 4.5 V. (c) and (d) STEM image (c) and EELS quantification (d) of the O and Mn for the 10th-charged NLMO. (e) and (f) STEM image (e) and EELS quantification (f) of the O and Mn for the 10th-charged NLAMO. The arrows in (c)–(f) illustrate the direction of the EELS line scan. (g)–(j) O K-edge EELS spectra of the 10th-charged NLMO (g) and (h) and NLAMO (i) and (j). | ||
The STEM coupled with the EELS technique is employed to investigate the evolutions in micro morphology and oxygen states. Both NLMO and NLAMO particles are intact in the pristine state (Fig. S20 and S21, ESI†). After 10 cycles, the NLMO particle displays plenty of cracks at the surface, as a consequence of the irreversible oxygen loss and the resultant structural collapse (Fig. 2c). Comparatively, the 10th-charged NLAMO particle remains unchanged from the surface to the bulk (Fig. 2e). Quantitative EELS analysis shows that the oxygen content of the NLMO particle decreases progressively from the interior towards the surface, whereas it is almost invariable for the NLAMO particle, which confirms its improved lattice oxygen stability and structural integrity during electrochemical cycling (Fig. 2d and f).
To further explore the steric distribution of the oxygen states, the O K-edge EELS spectra of those 10th-charged particles are collected at different positions from the bulk to the surface. The first peak at ∼530 eV (pre-peak) is attributed to the electron transition from the 1s core state to the unoccupied 2p states hybridized with 3d metal states, while the other peak at ∼542 eV (main-peak) is associated with the electron transition from the 1s state to the hybridized states of O 2p and metal 4sp states.30 Each spectrum is normalized using the main-peak signal intensity. For NLMO, the intensity of the pre-peak reduces from the 40 nm-deep regions to the surface due to the formation of oxygen vacancies (Fig. 2g and h), which indicates the release of lattice oxygen from the surface, in good agreement with the DEMS result.31–33 By comparison, no obvious pre-peak intensity change is observed for NLAMO, except for a slight decrease near the surface region (approximately 10 nm) that might arise from the inevitable surface reaction with electrolyte (Fig. 2i and j).34 This proves that the lattice oxygen in NLAMO is well preserved during cycling.
Soft XAS characterization gives more evidence for oxygen redox.35 The XAS spectra show two sharp pre-edge peaks located at threshold energies of ∼530 and ∼533 eV, which are attributed to the transition of the O 1s electron to the unoccupied hybridized state of the TM 3d and O 2p orbitals with different energies of t2g and eg symmetries, respectively (Fig. S22, ESI†).8 For both NLMO and NLAMO at the 1st charged state, an increase in peak intensity appears at ∼532 eV in the pre-edge peak area, indicating the formation of O–O covalent bonding. Meanwhile, the noticeable change recovers to the initial state after discharging. Such changes in pre-edge range can be ascribed to electron loss and gain on the O 2p band, which confirms the redox reaction of oxygen anions. For the cycled NLAMO (at 30th charged state), the O K-edge spectrum still retains the change in the pre-edge feature, while the spectrum of the cycled NLMO (at 30th charged state) presents less change and is very similar to that of the pristine one. These results show that the oxygen redox reversibility has been significantly strengthened for NLAMO.
Investigation of the Mn redox behavior
The evolution of the feature and energy position of Mn L-edge in EELS spectra provides information on the change of the Mn valence state. The L3 and L2 signals denote the electronic transition from the split Mn 2p3/2 and 2p1/2 to the unoccupied 3d states, respectively.36 The Mn L-edge spectra of the pristine cathodes demonstrate that the Mn oxidation state from the bulk to the surface is +4, in which a shoulder at a lower energy (∼642.0 eV) represents Mn4+ species with octahedral coordination (Fig. S23, ESI†). For the 10th-charged NLMO, this shoulder peak gradually turns larger from the interior to the surface, and finally becomes the main peak, suggesting that almost all of Mn4+ are reduced to Mn3+ at the outmost surface (Fig. 3a, b and Fig. S24, S25, ESI†).37 By contrast, the significant variations in the shape and energy position of Mn L3-edge for the 10th-charged NLAMO are inhibited, which confirms that Mn4+ can be maintained at different particle positions during cycling (Fig. 3c and d).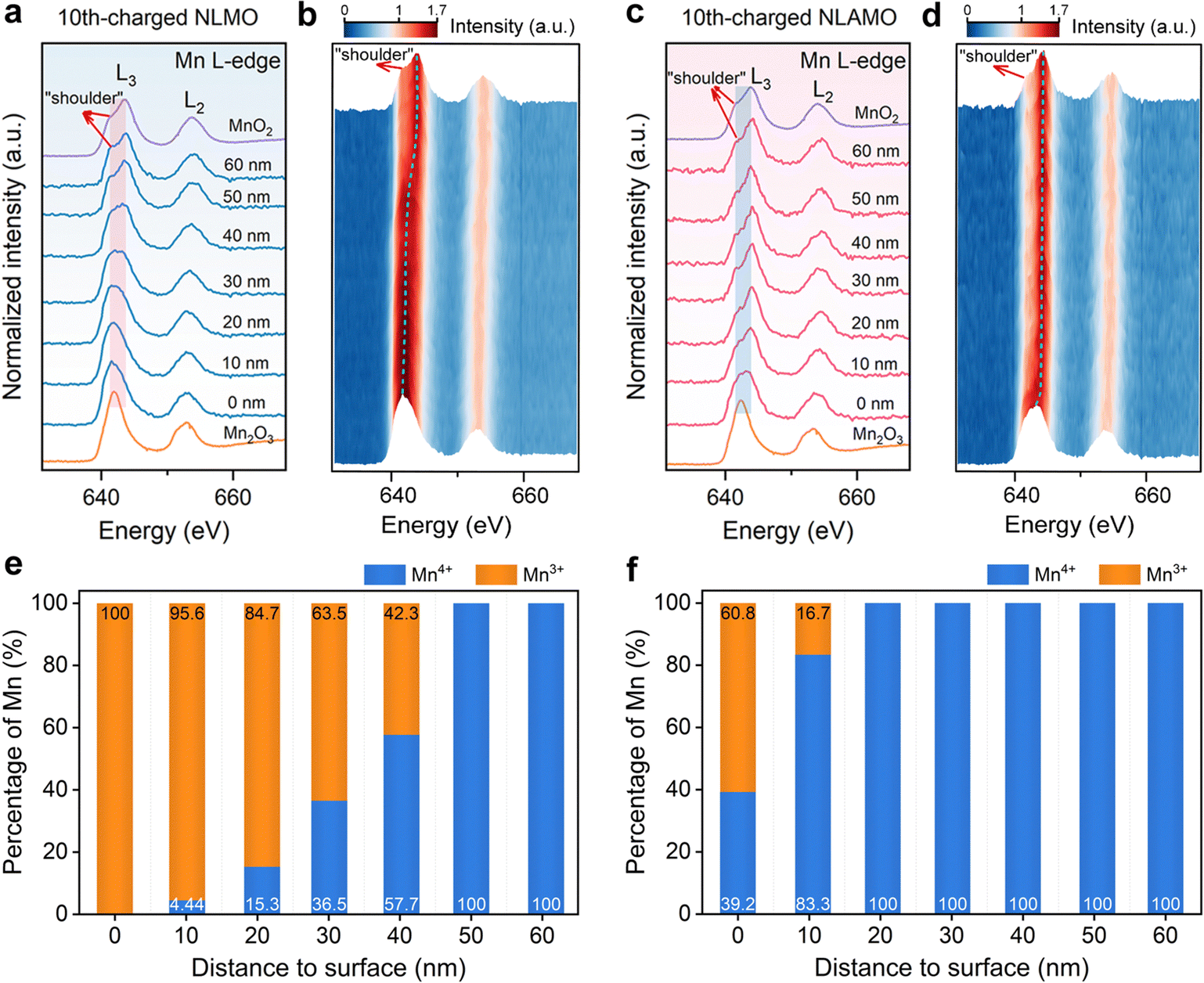 | ||
| Fig. 3 Mn oxidation state distribution within a single particle. (a)–(d) Mn L-edge EELS spectra of the 10th-charged NLMO (a) and (b) and NLAMO (c) and (d). All the spectra in (a)–(d) are normalized with the Mn L2-edge. (e) and (f) Percentage of Mn3+ and Mn4+ for the 10th-charged NLMO (e) and NLAMO (f). | ||
Furthermore, a quantitative analysis of Mn valence is performed by the multiple linear least-square (MLLS) fitting method (Fig. S26 and S27, ESI†). For the 10th-charged NLMO, the Mn oxidation state in the interior differs from that at the surface, and it is astonishing that Mn4+ are completely reduced to Mn3+ at the particle surface (Fig. 3e). This unusual phenomenon is attributed to the charge compensation for the oxygen loss, which results in heterogeneous redox of Mn upon charging.38 However, aside from a handful of Mn3+ within 10 nm to the surface, a uniform Mn4+ distribution is found throughout the 10th-charged NLAMO particle, implying its homogeneous Mn redox (Fig. 3f). The above outcomes have been also testified by the data of the L3/L2 intensity ratio in EELS spectra (Fig. S28–S30, ESI†).34,39 These intriguing results show that transition metal cations can be reduced even at the highly desodiated state for oxygen-redox cathodes owing to the oxygen loss.
The averaged electronic structure change is investigated by X-ray absorption near-edge structure (XANES) characterization (Fig. 4a–c). The Mn oxidation states are determined by the integral method (Fig. S31, ESI†), and the quantitative results are shown in Fig. 4d.40 In the first cycle, NLMO and NLAMO show nearly overlapped Mn K-edges at the charged and discharged states, indicating the analogous Mn redox. Upon cycling, the Mn K-edge of charged NLMO persistently shifts to lower energy, and the Mn valence varies from +4 (1st) to +3.85 (10th) and then to +3.74 (30th). This confirms that the Mn oxidation state is heterogeneous within the cycled NLMO single particle at the charged state. Comparatively, even after 30 cycles, the Mn valence of charged NLAMO is well maintained at around +4, verifying the uniform distribution of Mn4+. It is clear that the accumulation of Mn3+ at the charged state reduces the overall Mn valence and lowers the reduction potential of NLMO, thus resulting in voltage decay. Through the prevention of Mn reduction in the charging process, this electrochemical issue can be well overcome.
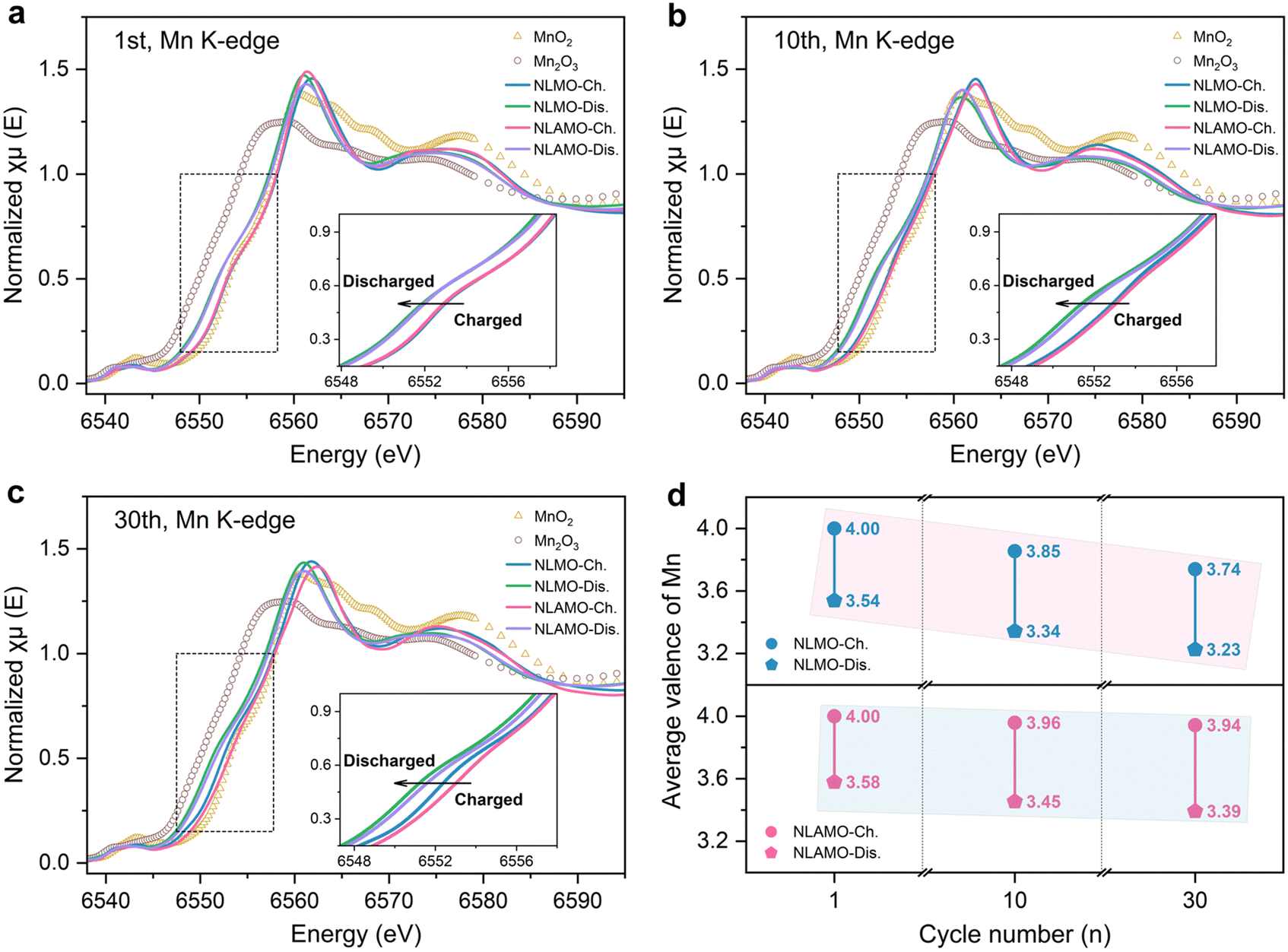 | ||
| Fig. 4 Ensemble-averaged analysis of the Mn oxidation state. (a)–(c) Normalized Mn K-edge XANES spectra of NLMO and NLAMO collected after the 1st (a), 10th (b) and 30th (c) cycles. (d) The quantification results of the Mn average oxidation state at different electrochemical states. | ||
The role of Al in electronic structure regulation
Charge density difference analysis is conducted to study the electronic structure (Fig. 5a and b). For both NLMO and NLAMO, charges accumulate around the lattice oxygen. However, Al plays a critical role in the charge distribution for NLAMO. Through providing more charges to the adjacent oxygen atoms, the electron localization is facilitated, which improves the reversibility of the oxygen redox. Bader charge analysis is further carried out to evaluate the charge transfer (Fig. 5c). Note that the label value represents the transferred electron number instead of the absolute charge value.41 Benefitting from extra electrons provided by Al atoms, the average charge around the O atoms in NLAMO is more negative than that in NLMO, confirming the electron localization. The larger negative charges on oxygen effectively inhibit the excessive oxidation of lattice oxygen and the formation of oxygen vacancies. More importantly, the electron localization effect still sustains in the charged NLAMO (Fig. S32 and S33, ESI†). In addition, the density of states (DOS) for both cathodes are calculated (Fig. S34, ESI†).42–44 In contrast to NLMO, the O 2p orbital of NLAMO exhibits an elevated energy level toward the Fermi level. Though the elevated O 2p level facilitates the oxygen redox, the electron localization in the NLAMO cathode can suppress the O2 release and the surface oxygen vacancy formation, which effectively enhances the structural stability and the oxygen redox reversibility. Based on the above calculation results, it is demonstrated that the strong Al–O bond weakens the neighboring covalent Mn–O bond through the inductive effect, thus alleviating the variation of the oxygen 2p band to ameliorate the oxygen stability during cycling.45–47 Therefore, the introduction of Al can tune both the cationic Mn and anionic O redox reactions by electronic structure regulation, thus significantly improving the electrochemical performance.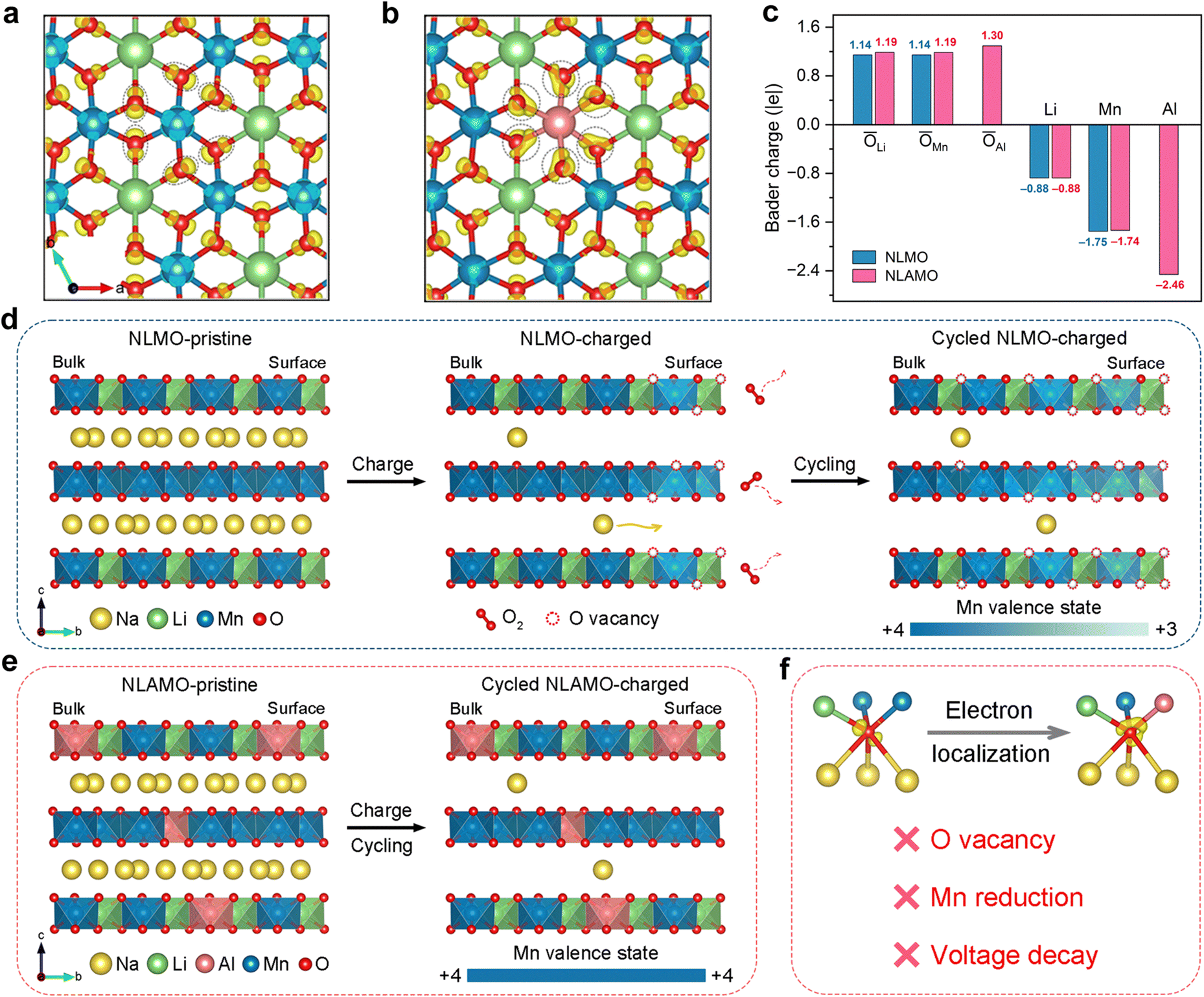 | ||
| Fig. 5 The origin of the voltage decay and the role of Al in electronic structure regulation to suppress voltage decay. (a) and (b) Charge density difference of NLMO (a) and NLAMO (b). (c) Charge transfer number of different atoms for NLMO and NLAMO. (d) and (e) Schematic illustration of the oxygen redox behaviors and the Mn oxidation state evolutions for NLMO (d) and NLAMO (e). (f) The role of Al in oxygen electron localization to suppress the voltage decay. | ||
The nature and suppression of voltage decay
Excluding the influences of the long-range P2–O2 phase transition and Li loss, the irreversible oxygen redox behavior and the resultant local structure degradation are imperative to be considered for voltage decay (Fig. S35–S38 and Table S7, ESI†). Though the oxygen redox features during the initial cycle, such as O–O dimerization and oxygen loss, have been reported, the mechanism over the extended cycles and the corresponding local structural changes are still overlooked, which hinders the fundamental understanding of voltage attenuation.16,48,49 Here, combined with spatially local STEM-EELS and ensemble-averaged XAS, the origin of voltage decay in NLMO has been unambiguously elucidated (Fig. 5d). Owing to the irreversible oxygen loss during the charge process, oxygen vacancies are initially formed on the particle surface and then progressively injected into the bulk during electrochemical cycling. The original Mn4+ ions around the oxygen vacancies are apt to be reduced at the charged state, thus generating Mn3+ ions on the particle surface. Concomitantly, with the oxygen vacancies growing inwards in the bulk, a gradient distribution of Mn3+ ions is delivered within the whole particle, which finally leads to the heterogeneity of redox reactions, giving rise to the noticeable voltage decay.On the basis of the above analysis, the P2-Na0.8Li0.24Al0.03Mn0.73O2 cathode with negligible voltage decay is well designed by electronic structure regulation. With the help of the enhanced electron localization around oxygen by Al, NLAMO effectively suppresses the irreversible oxygen loss and the pervasive oxygen vacancy formation, ultimately avoiding the partial Mn reduction upon full charge (Fig. 5e and f). As a result, NLAMO achieves a superior voltage retention of 99.5% after 50 cycles, and the average discharge voltage is still maintained at 2.64 V even after 200 cycles.
Conclusions
To summarize, we demonstrate that the voltage decay in ORA cathode materials is correlated with the steric nonuniformity of the electrochemical reactions. Specifically, the overoxidation of oxygen anions at high voltage induces lattice oxygen loss and oxygen vacancy formation from the surface to the bulk, and these oxygen vacancies further reduce the local Mn ions, leading to the spatially heterogeneous Mn valence. During cycling, this infaust process continuously accumulates, which causes the gradual decrease of the output voltage (i.e., voltage decay). Introducing strong Al–O bonds is proved to be effective in hampering the generation of oxygen defects and the reduction of Mn cations via the oxygen electron localization effect. The well-architected NLAMO possesses highly stable operation voltage over 200 cycles, showing negligible voltage decay. Our work enables the voltage degradation mechanism to be profoundly understood and provides a perspective for the electronic structure design of high-capacity cathode materials.Author contributions
K. Zhang conceived the concept and supervised the work. R. Sun and Z. Wu designed the experiments and performed the material synthesis and electrochemical tests. Y. Ni performed the theoretical calculations and contributed to the overall interpretation of the results. M. Hou, H. Sun, W. Zhang and H. Li collected and interpreted the microscopy images and EELS spectra. Z. Wu performed and interpreted the XANES measurements. L. Zhang performed and interpreted the soft XAS measurements. P. Jiao performed the DEMS analyses. R. Sun, Z. Wu, K. Zhang, F. Cheng and J. Chen wrote the manuscript. All the authors contributed to the discussion and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22121005, 22005155 and 52072186), the National Key Research and Development Program of China (2022YFB2402200 and 2019YFA0705600) and the Fundamental Research Funds for the Central Universities of China (63233017, 63231002 and 63231198). We acknowledge the valuable time from beamline BL11B and 02B02 of the Shanghai Synchrotron Radiation Facility.References
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- J.-M. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- R. Usiskin, Y. Lu, J. Popovic, M. Law, P. Balaya, Y.-S. Hu and J. Maier, Nat. Rev. Mater., 2021, 6, 1020–1035 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sánchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y.-S. Hu, Science, 2020, 370, 708–711 CrossRef CAS.
- J. Xiao, Y. Xiao, J. Li, C. Gong, X. Nie, H. Gao, B. Sun, H. Liu and G. Wang, SmartMat, 2023, e1211 CrossRef.
- X. Rong, E. Hu, Y. Lu, F. Meng, C. Zhao, X. Wang, Q. Zhang, X. Yu, L. Gu, Y.-S. Hu, H. Li, X. Huang, X.-Q. Yang, C. Delmas and L. Chen, Joule, 2019, 3, 503–517 CrossRef CAS.
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M. A. Pérez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Lu, D. E. McNally, A. V. Chadwick, F. Giustino, T. Schmitt, L. C. Duda, M. R. Roberts and P. G. Bruce, Nat. Chem., 2018, 10, 288–295 CrossRef CAS.
- Y. Wang, L. Wang, H. Zhu, J. Chu, Y. Fang, L. Wu, L. Huang, Y. Ren, C.-J. Sun, Q. Liu, X. Ai, H. Yang and Y. Cao, Adv. Funct. Mater., 2020, 30, 1910327 CrossRef CAS.
- A. Tsuchimoto, X.-M. Shi, K. Kawai, B. Mortemard de Boisse, J. Kikkawa, D. Asakura, M. Okubo and A. Yamada, Nat. Commun., 2021, 12, 631 CrossRef CAS PubMed.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- Z. Wu, Y. Ni, S. Tan, E. Hu, L. He, J. Liu, M. Hou, P. Jiao, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2023, 145, 9596–9660 CrossRef CAS.
- L. Yang, R. Chen, Z. Liu, Y. Gao, X. Wang, Z. Wang and L. Chen, Battery Energy, 2022, 1, 20210015 CrossRef CAS.
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS PubMed.
- K. McColl, R. A. House, G. J. Rees, A. G. Squires, S. W. Coles, P. G. Bruce, B. J. Morgan and M. S. Islam, Nat. Commun., 2022, 13, 5275 CrossRef CAS.
- K. Zhang, D. Kim, Z. Hu, M. Park, G. Noh, Y. Yang, J. Zhang, V. W. Lau, S.-L. Chou, M. Cho, S.-Y. Choi and Y.-M. Kang, Nat. Commun., 2019, 10, 5203 CrossRef CAS PubMed.
- M. Sathiya, A. M. Abakumov, D. Foix, G. Rousse, K. Ramesha, M. Saubanère, M. L. Doublet, H. Vezin, C. P. Laisa, A. S. Prakash, D. Gonbeau, G. VanTendeloo and J.-M. Tarascon, Nat. Mater., 2015, 14, 230–238 CrossRef CAS PubMed.
- D. Eum, B. Kim, S. J. Kim, H. Park, J. Wu, S.-P. Cho, G. Yoon, M. H. Lee, S.-K. Jung, W. Yang, W. M. Seong, K. Ku, O. Tamwattana, S. K. Park, I. Hwang and K. Kang, Nat. Mater., 2020, 19, 419–427 CrossRef CAS PubMed.
- E. Hu, X. Yu, R. Lin, X. Bi, J. Lu, S. Bak, K.-W. Nam, H. L. Xin, C. Jaye, D. A. Fischer, K. Amine and X.-Q. Yang, Nat. Energy, 2018, 3, 690–698 CrossRef CAS.
- J. Zhang, J.-B. Kim, J. Zhang, G.-H. Lee, M. Chen, V.-W. Lau, K. Zhang, S. Lee, C.-L. Chen, T.-Y. Jeon, Y.-W. Kwon and Y.-M. Kang, J. Am. Chem. Soc., 2022, 144, 7929–7938 CrossRef CAS.
- L. Yang, X. Li, J. Liu, S. Xiong, X. Ma, P. Liu, J. Bai, W. Xu, Y. Tang, Y.-Y. Hu, M. Liu and H. Chen, J. Am. Chem. Soc., 2019, 141, 6680–6689 CrossRef CAS.
- X. Cao, X. Li, Y. Qiao, M. Jia, F. Qiu, Y. He, P. He and H. Zhou, ACS Energy Lett., 2019, 4, 2409–2417 CrossRef CAS.
- N. Voronina, M.-Y. Shin, H.-J. Kim, N. Yaqoob, O. Guillon, S. H. Song, H. Kim, H.-D. Lim, H.-G. Jung, Y. Kim, H.-K. Lee, K.-S. Lee, K. Yazawa, K. Gotoh, P. Kaghazchi and S.-T. Myung, Adv. Energy Mater., 2022, 12, 2103939 CrossRef CAS.
- S. Kumakura, Y. Tahara, K. Kubota, K. Chihara and S. Komaba, Angew. Chem., Int. Ed., 2016, 55, 12760–12763 CrossRef CAS.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS PubMed.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A1225–A1229 CrossRef CAS.
- K. Wang, Z. Zhang, S. Cheng, X. Han, J. Fu, M. Sui and P. Yan, eScience, 2022, 2, 529–536 CrossRef.
- S. Hwang, S. M. Kim, S.-M. Bak, S. Y. Kim, B.-W. Cho, K. Y. Chung, J. Y. Lee, E. A. Stach and W. Chang, Chem. Mater., 2015, 27, 3927–3935 CrossRef CAS.
- T. Zhou, H. Wang, Y. Wang, P. Jiao, Z. Hao, K. Zhang, J. Xu, J.-B. Liu, Y.-S. He, Y.-X. Zhang, L. Chen, L. Li, W. Zhang, Z.-F. Ma and J. Chen, Chem, 2022, 8, 2817–2830 CAS.
- P. Yan, J. Zheng, Z.-K. Tang, A. Devaraj, G. Chen, K. Amine, J.-G. Zhang, L.-M. Liu and C. Wang, Nat. Nanotechnol., 2019, 14, 602–608 CrossRef CAS PubMed.
- P. M. Csernica, S. S. Kalirai, W. E. Gent, K. Lim, Y.-S. Yu, Y. Liu, S.-J. Ahn, E. Kaeli, X. Xu, K. H. Stone, A. F. Marshall, R. Sinclair, D. A. Shapiro, M. F. Toney and W. C. Chueh, Nat. Energy, 2021, 6, 642–652 CrossRef CAS.
- Q. Zhang, Q. F. Gu, Y. Li, H.-N. Fan, W.-B. Luo, H.-K. Liu and S.-X. Dou, iScience, 2019, 19, 244–254 CrossRef CAS.
- F. Yan, X. Feng, Y.-S. Liu, L. C. Kao, P.-A. Glans, W. Yang and J. Guo, Energy Environ. Mater., 2021, 4, 139–157 CrossRef.
- A. K. Shukla, Q. M. Ramasse, C. Ophus, H. Duncan, F. Hage and G. Chen, Nat. Commun., 2015, 6, 8711 CrossRef CAS.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- J. Zhang, Q. Wang, S. Li, Z. Jiang, S. Tan, X. Wang, K. Zhang, Q. Yuan, S.-J. Lee, C. J. Titus, K. D. Irwin, D. Nordlund, J.-S. Lee, P. Pianetta, X. Yu, X. Xiao, X.-Q. Yang, E. Hu and Y. Liu, Nat. Commun., 2020, 11, 6342 CrossRef CAS PubMed.
- C. Ma, J. Alvarado, J. Xu, R. J. Clément, M. Kodur, W. Tong, C. P. Grey and Y. S. Meng, J. Am. Chem. Soc., 2017, 139, 4835–4845 CrossRef CAS.
- H. Dau, P. Liebisch and M. Haumann, Anal. Bioanal. Chem., 2003, 376, 562–583 CrossRef CAS PubMed.
- C. Zhang, B. Wei, M. Wang, D. Zhang, T. Uchiyama, C. Liang, L. Chen, Y. Uchimoto, R. Zhang, P. Wang and W. Wei, Energy Storage Mater., 2022, 46, 512–522 CrossRef.
- H. Yang, T. Xiong, Z. Zhu, R. Xiao, X. Yao, Y. Huang and M.-S. Balogun, Carbon Energy, 2022, 4, 820–832 CrossRef CAS.
- Y. Huang, H. Yang, T. Xiong, D. Adekoya, W. Qiu, Z. Wang, S. Zhang and M.-S. Balogun, Energy Storage Mater., 2020, 25, 41–51 CrossRef.
- S. Zhou, P. Huang, T. Xiong, F. Yang, H. Yang, Y. Huang, D. Li, J. Deng and M.-S. Balogun, Small, 2021, 17, 2100778 CrossRef CAS.
- K. Kawai, X.-M. Shi, N. Takenaka, J. Jang, B. Mortemard de Boisse, A. Tsuchimoto, D. Asakura, J. Kikkawa, M. Nakayama, M. Okubo and A. Yamada, Energy Environ. Sci., 2020, 15, 2591–2600 RSC.
- X. Wang, X. Fan, X. Yu, S. Bak, Z. Shadike, I. Waluyo, A. Hunt, S. D. Senanayake, H. Li, L. Chen, C. Wang, R. Xiao, E. Hu and X.-Q. Yang, Adv. Funct. Mater., 2020, 31, 2001633 CrossRef.
- C. Cai, X. Li, P. Hu, T. Zhu, J. Li, H. Fan, R. Yu, T. Zhang, S. Lee, L. Zhou and L. Mai, Adv. Funct. Mater., 2023, 33, 2215155 CrossRef CAS.
- X. Rong, D. Xiao, Q. Li, Y. Niu, F. Ding, X. Hou, Q. Wang, J. Xu, C. Zhao, D. Zhou, R. Xiao, X. Yu, W. Yin, L. Gu, H. Li, X. Huang, L. Chen and Y.-S. Hu, eScience, 2023, 3, 100159 CrossRef.
- C. Li, F. Geng, B. Hu and B. Hu, Mater. Today Energy, 2020, 17, 100474 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S38 and Tables S1–S7. See DOI: https://doi.org/10.1039/d3ee02817h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |