
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemporal changes in thirdhand cigarette smoke film composition and oxidation of co-existing surface film chemicals†
April M.
Hurlock
and
Douglas B.
Collins
 *
*
Department of Chemistry, Bucknell University, Lewisburg, PA 17837, USA. E-mail: dbc007@bucknell.edu
First published on 5th February 2024
Abstract
The composition of air-exposed surfaces can have a strong impact on air quality and chemical exposure in the indoor environment. Third hand smoke (THS), which includes surface-deposited cigarette smoke residue along with the collection of gases evolved from such residues, is becoming increasingly recognized as an important source of long-term tobacco smoke exposure. While studies have described gas/surface partitioning behaviour and some multiphase reaction systems involving THS, the possibility of time-dependent changes in chemical composition due to chemical reactivity that is endogenous to the deposited film has yet to be investigated. In this study, sidestream cigarette smoke was allowed to deposit on glass surfaces that were either clean or pre-coated with chemicals that may be oxidized by reactive oxygen species found in the smoke. Surface films included a low volatility antioxidant, tris(2-carboxyethyl)phosphine (TCEP), and two compounds relevant to surface films found within buildings, oleic acid (OA) and squalene (SQ). Upon deposition, oxidation products of nicotine, TCEP, OA, and SQ were formed over time periods of hours to weeks. The inherent oxidative potential of cigarette smoke deposited as a THS film can therefore initiate and sustain oxidation chemistry, transforming the chemical composition of surface films over long periods of time after initial smoke deposition. An interpretation of the THS oxidation results is provided in the context of other types of deposited particulate air pollutants with known oxidative potential that may be introduced to indoor environments. Continued study of THS and deposited surface films found indoors should consider the concept that chemical reservoirs found on surfaces may be reactive, that the chemical composition of indoor surface films may be time-dependent, and that the deposition of aerosol particles can act as a mechanism to initiate oxidation in surface films.
Environmental significanceThirdhand smoke (THS) – the residue and associated gases left behind by prior smoking activities – represents an important route of passive tobacco smoke exposure. Some reactions that can alter the composition and toxicity of THS have been explored, but the inherent oxidative potential of smoke suggests that oxidation reactions may occur in THS without other inputs. This study shows that THS residues become oxidized over time without added reactive gases, and that deposited cigarette smoke can oxidize co-existing films of chemicals commonly found indoors. These results suggest that surface contamination by types of particulate matter that exhibit oxidative potential could lead to transformations of surface film composition, and perhaps toxicity, that are not currently being considered. |
Introduction
Air-exposed environmental films can be formed through the deposition of organic, inorganic, and biological material to surfaces. Processes that participate in film formation generally include gas-surface partitioning,1–5 particulate matter deposition,3,5,6 residue transfer by direct contact, and the application of consumer products such as cleaning agents, decorative coatings, pesticides, or protective finishes.7 The chemical composition of indoor surface films,4,8 urban grime,9 and other environmental films10 are topics of current research, as they are being recognized as important participants in atmospheric and multiphase chemistry11,12 and constitute important reservoirs for chemical exposure indoors.6,13,14 For instance, indoor surfaces have been known to be an important sink for reactive gases15 and an important source for volatile and semi-volatile organic compounds (S/VOCs).16,17 However, more recent research has characterized the dynamic chemical nature of indoor surfaces as reservoirs for bidirectional partitioning of compounds across a range of volatility, acidity, and octanol-air partition coefficient values.17–20While the composition of a surface film may be driven, to first order, by the array of aerosol particle and SVOC sources, it may also be important to understand the role of reactivity within the film as an additional controlling factor, especially as films reside on surfaces over extended periods of time. The impact of multiphase chemistry on surfaces continues to be thoroughly studied, and many studies focus on reactive gas removal and associated chemical kinetics.7,11,12 A somewhat smaller but growing group of studies has investigated the chemical changes of the condensed phase (e.g., surface films and aerosol particles) resulting from multiphase reactions.21–24 Reactive trace gases including ozone (O3)15,22,25,26 and the hydroxyl radical (·OH)21,24,27 have been most thoroughly investigated due to their critical roles in the multiphase oxidation of aerosol particles and interfacial films. A growing understanding of the role of autoxidation24,28,29 and reactive intermediate production22,30,31 in the condensed phase of multiphase chemical systems has been taking shape. The presence and persistence of reactive intermediates may lead to long-term changes in aerosol particle composition and also may lead to adverse health effects.12
Aerosol particles have been shown to exhibit an oxidative potential (OP) that has been associated with negative health effects.12,32–34 In general, OP is detected and measured using assays in which samples are exposed to antioxidants, the decay of which is then quantified.35 Similar to the impact of oxidative potential on biological systems,36 the deposition of aerosol particles that exhibit OP onto surfaces may represent a means to introduce oxidants to an air-exposed surface film. Recent and emerging research has been indicating that reactive oxygen intermediates likely form within surface films31 and may exhibit a persistent free radical character.37
Deposited cigarette smoke residue and tobacco-related gases that are in pseudo-equilibrium with indoor surfaces have collectively been termed “thirdhand smoke” (THS). A long-lived reservoir of cigarette smoke contaminants, THS can act as a source of SVOCs, polycyclic aromatic hydrocarbons, tobacco-specific nitrosamines, and a variety of other compounds to indoor environments.6,16,38,39 In this study, the term “THS films” will be used with reference to the surface-deposited fraction of THS material. THS is a vector for substantial environmental exposures to tobacco smoke via inhalation, dermal uptake, and ingestion.6,14 The magnitude of THS exposure is similar to or greater than second-hand smoke, with disproportionate effects on children and people with lower income levels.40,41 Research on THS is expanding,6 including studies of its chemistry, physicochemical properties, exposure routes, health effects, and approaches to mitigation. Specific examples of known chemical transformations of THS residue include multiphase reactions with nitrous acid (HONO) to form nicotine-specific nitrosamines,39 and the reaction of THS residue components with gaseous O3.25,26,42 Schick and co-workers38 have shown that both partitioning and reactivity can occur on the 1 hour timescale, including the removal of several cigarette smoke components from THS films to the gas phase. Those authors also demonstrated the in situ formation of tobacco-specific nitrosamines in THS films,38 likely owing to a reaction with HONO derived from surface uptake of ambient NO2.
More recent studies have probed the dynamic nature of THS in indoor environments, mainly focusing on phase partitioning processes. Components of THS, through partitioning between surfaces and the gas phase, can migrate from outdoor smoking areas to the indoors by partitioning to ambient aerosol particles that are transported by air handling systems20 or by the smokers themselves.43 Viscosity and pH can drive chemically-selective partitioning processes to inorganic and organic aerosol particle surfaces.19 If the condensed phase is aqueous, it has been observed that Henry's Law can describe both uptake and off-gassing, for instance from simulated lung lining fluid.44 Therefore, the chemical composition of THS films controls the supply of tobacco-related SVOCs to the gas phase over long periods of time in THS contaminated environments.16,19,20,45
Chemical characterization of fresh cigarette smoke has been widely conducted and thoroughly documented.46 While studies have sought to perform broad chemical characterizations of THS films44 and gases,16,44 along with the consequences of multiphase chemistry on THS films,25,26,39,42 reactive processes that are endogenous to condensed-phase THS have not garnered focus. It has been known for decades that freshly collected cigarette smoke exhibits a persistent free radical character, which likely involves semiquinones associated with (mono- and poly-) aromatic compounds in cigarette smoke.47,48 Radicals, reactive intermediates, and redox-active metals are associated with the induction of oxidative stress both internally47,49 and externally to humans.50–52
While the OP of particulate matter is normally focused on a relationship with human health effects (via inhalation or deposition to skin), the present study explores the potential for oxidants present in the condensed phase to transform abiotic air-exposed surface films upon dry deposition, wherein the deposited particles act as an oxidant delivery vehicle. The composition, viscosity, and porosity of the substrate material has been implicated in the partitioning behaviour of compounds found in THS.44 In this study, we intend to show that exposure of surfaces to sidestream cigarette smoke can induce oxidation in surface films, altering their composition to produce various oxidation products, including hydroperoxides. A non-volatile antioxidant (tris(2-carboxyethyl)phosphine; TCEP) was used to demonstrate oxidative action of THS films. Since many indoor surfaces are commonly coated with reservoirs of organic compounds,18,53 the effect of THS film oxidation on the composition of surfaces that were pre-coated with low volatility olefinic organic compounds was also investigated. Detailed qualitative analysis of oxidized surface film proxies was conducted with an eye toward a better understanding of oxidant identity, mechanism, and time-evolving chemical exposures on THS-contaminated surfaces of increasing chemical complexity.
Experimental
Study design
Sidestream cigarette smoke was generated, entrained in a sweep gas flow of ultrapure air, and then injected into a glass chamber. The chamber contained sample substrates oriented horizontally near the bottom of the chamber, resting on a glass microscope slide. After injection, the smoke was allowed to deposit on surfaces for 30 minutes before the first sample substrate was removed for analysis. To the naked eye, the optical thickness of particles in the chamber was significantly reduced during the 30 minutes waiting time. Two types of experiments were conducted in the present study: (1) pooled extractions of multiple sample substrates for qualitative analysis, and (2) time-resolved experiments in which one sample substrate was extracted at a time across the period of approximately one week. In each case, triplicate measurements of each substrate extract were conducted; results are reported as the mean of replicate measurements and uncertainties reported in the study are equal to the standard deviation (σ).Chemicals and materials
Glass substrates were manually cut from soda-lime glass microscope slides (plain pre-cleaned; VWR Micro Slides) into 1.0 × 2.5 cm rectangles. Research cigarettes were used in the smoking apparatus (1R6F, University of Kentucky). Squalene (SQ), tris(2-carboxyethyl)phosphine (TCEP), oleic acid (OA), and bis(2-ethylhexyl)sebacate (BES) were obtained from Sigma Aldrich. Acetaminophen (Sigma Aldrich) was used as a global internal standard. Water was obtained directly from a deionization system (Aries ARS-102, ResinTech, Inc.; Camden, NJ) with a resistance of 18.2 MΩ. Formic acid (Agilent Technologies), methanol (MeOH; Fisher Optima), isopropanol (IPA; BDH Analytical), and acetonitrile (ACN; Fisher Optima) were LC-MS grade. Ammonium formate (Alfa Aesar) buffer was prepared in deionized water to a concentration of 10 mM and was then adjusted to pH = 8.5 using 6 M ammonium hydroxide solution. The background gas used in all experiments was ultra-pure air generated in the laboratory (Aadco 737-30).Sample preparation
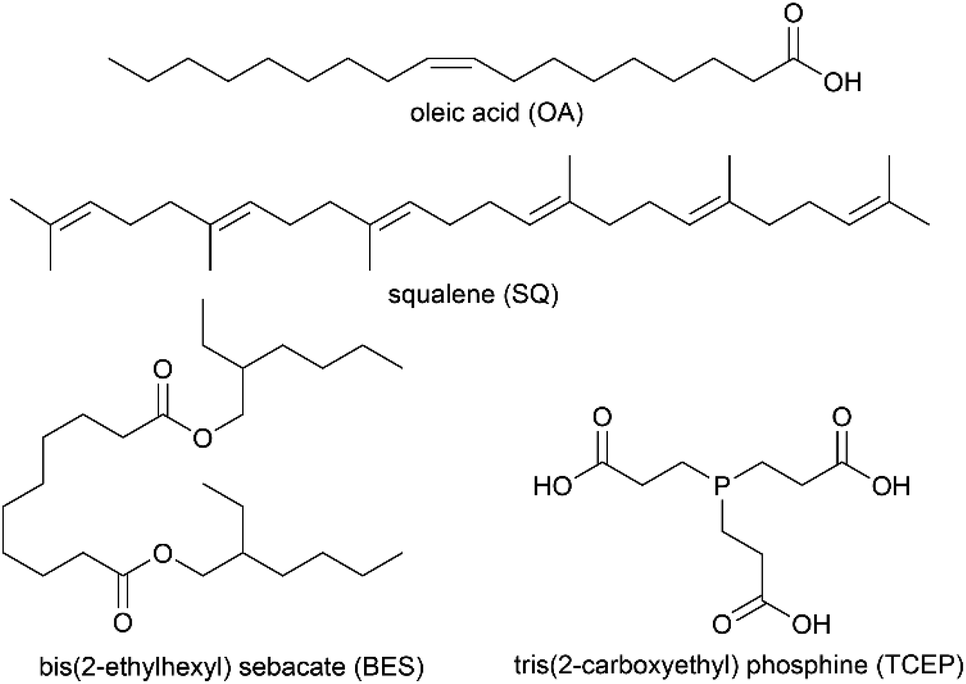 | ||
| Chart 1 Compounds used to make organic surface films. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 acetonitrile/water mixture. The vial was placed on its side and extracted on an analog rocker table (VWR) for 10 min, then the substrate was turned over and extracted for another 10 min. Extract solution was transferred to an amber glass 2.0 mL autosampler vial (Agilent Technologies) for LC-MS analysis. Extraction efficiencies were not assessed. It is assumed that the extraction efficiency for each chemical system was consistent throughout each experiment. Absolute quantitation was not used in this study, and since all samples were normalized to the first sample in each experiment, we feel the assumption about extraction recovery is reasonable.
:50 acetonitrile/water mixture. The vial was placed on its side and extracted on an analog rocker table (VWR) for 10 min, then the substrate was turned over and extracted for another 10 min. Extract solution was transferred to an amber glass 2.0 mL autosampler vial (Agilent Technologies) for LC-MS analysis. Extraction efficiencies were not assessed. It is assumed that the extraction efficiency for each chemical system was consistent throughout each experiment. Absolute quantitation was not used in this study, and since all samples were normalized to the first sample in each experiment, we feel the assumption about extraction recovery is reasonable.
For qualitative analysis of reaction products in OA + BES and SQ experiments, five substrates were extracted sequentially in the same aliquot of extraction solvent to maximize film extraction into a minimal volume without the need to concentrate the samples further. Five substrates could be extracted into 2.0 mL of the appropriate extraction volume (vide supra) by placing two substrates in an extraction vial at once for 10 min on each side. Those two substrates were then removed and replaced with the remaining three substrates in the vial for 10 min on each side. All of the extraction solvent was then transferred to an autosampler vial for LC-MS analysis.
Chemical analysis
Relative quantitation and qualitative analysis were performed using LC coupled to high resolution mass spectrometry (LC-HRMS) and/or tandem mass spectrometry (LC-MS/MS) techniques. All analytical separations were performed with reverse-phase high performance liquid chromatography using a core–shell C18 column (Kinetex C18 EVO; 100 × 3.00 mm, 5 μm diameter particles; Phenomenex, Inc.) Different method parameters were used based on the desired analytes (see Table S1†). A gradient method using two mobile phases was used. For the determination of tobacco alkaloids and TCEP, mobile phase A contained 10% methanol in 10 mM ammonium formate at pH = 8.5, and mobile phase B contained 100% methanol. For the determination of OA and its oxidation products, an isocratic separation using 5% of aqueous 0.1% formic acid and 95% acetonitrile was used. SQ and its oxidation products were separated with a gradient method using aqueous 0.1% formic acid as mobile phase A and acetonitrile as mobile phase B.HRMS measurements were conducted with either an Exactive Orbitrap (Thermo Scientific) or a quadrupole/time-of-flight (Q-ToF; Agilent 6560) mass spectrometer. The Orbitrap MS was capable of high-resolution/accurate mass acquisition of mass-to-charge (m/z) ratios (m/Δm = 60000 at m/z 400). The Q-ToF MS was capable of a lower mass resolving power (m/Δm = 25000 at m/z 400), but enabled tandem mass spectrometry (MS/MS) with product ion analysis that still allows for molecular formula prediction, which was used for qualitative analysis in this study. Electrospray ionization (ESI) was used in all analyses except for SQ, in which atmospheric pressure chemical ionization (APCI) was implemented to enable ionization of analytes with lower polarity. OA and its oxidation products were analyzed in negative ion mode, but all other analyses were performed in positive ion mode. The Orbitrap HRMS instrument was fitted with a heated electrospray ionization (Ion Max HESI) ion source and liquid handling was performed with an Accela 1260 quaternary pump. Samples were manually injected into a 10 μL sample loop and separations were performed at room temperature (20–22 °C). The Q-ToF MS instrument used was capable of drift tube ion mobility measurements, but for this study it was used in “Q-ToF only” mode. The instrument was fitted with an Agilent Dual Jet Stream ion source for ESI and a standard Agilent APCI source for SQ experiments. Analytical separations on the LC-Q-ToF system were performed with an Agilent 1290 Infinity II liquid chromatograph with a quaternary pump, automated multisampler with a variable volume loop set to 5 μL, and temperature-controlled column compartment held at 20 °C. See Table S2† for ion source settings for all analyses. LC-MS/MS was performed with quadrupole isolation width set at 1.3 mass-to-charge units followed by collision induced dissociation in ultrapure N2 (Grade 5.0UH; Praxair). Further details on MS/MS settings can be found in Table S3.†
Standards for nicotelline, N-formylnornicotine, (S)-cotinine, and (1S′, 2S′)-nicotine 1′-N-oxide were obtained from Toronto Research Chemicals/LGC Standards. A standard for (S)-nicotine was obtained from Cerilliant. Standards for cis-9,10-epoxyoctadecanoic acid (the epoxide of oleic acid) and 2,3-epoxidosqualene were obtained from Cayman Chemical.
Data analysis
Qualitative analysis of Q-ToF MS and MS/MS data was performed with MassHunter Qualitative Analysis 10.0 (Agilent Technologies). Compound discovery was performed with molecular feature extraction (MFE) in MassHunter, followed by molecular formula annotation based on comparison of measured exact mass with predicted exact mass, along with fitting the intensities of two or more isotopologues. Preparation of graphical representations of data was performed with Igor Pro 9. Relative quantitation based on Q-ToF and Orbitrap measurements was performed by directly importing raw data files into Skyline version 22,54,55 wherein extracted chromatographic peaks were integrated and normalized to the internal standard. Time series experiments were quantified on a relative basis using triplicate LC-MS injections of each sample that was extracted at different incubation times in the glass chamber. For each sample extracted at time point (t), the peak area (A) for each compound (j) was first normalized to the peak area of the internal standard (IS) for each repeated measurement (i). The normalized areas were then averaged over the total number of repeat injections (n) to obtain an averaged, IS-normalized signal (Sj,t).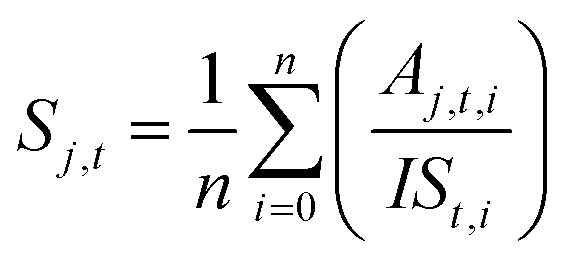 | (1) |
Results and discussion
Chemical transformations of THS films on clean glass
THS films were generated via dry deposition of sidestream cigarette smoke. An initial suspect screening analysis was conducted using HRMS measurements of THS film extracts from clean glass surfaces. Exact mass matches to molecular formulae (with support from two or more stable isotopologues) with mass accuracy better than 5 ppm were obtained for a list of 12 tobacco-related compounds listed in Table S4.† Most of the matched exact masses had extracted ion chromatograms exhibiting a single chromatographic peak. Such was the case for m/z 161.1079, although several isomeric compounds with the molecular formula C10H12N2 (computed m/z = 161.1073 [M + H]+) are known to be found in tobacco smoke. Two sets of isobaric, but chromatographically separable compounds were observed: one pair of chromatographic peaks with m/z 177.1026 and another pair with m/z 179.1181. Tandem MS analysis enabled assignment of cotinine and N-formylnornicotine to each of the chromatographic peaks that appear in the extracted ion chromatogram for m/z 177.1026 (Fig. S3†). The MS/MS spectra for the two chromatographic peaks with m/z 179.1181 exhibited high similarity (Fig. S4†) and corresponded with the detailed ESI-MS/MS studies of nicotine oxides by Smyth et al.,56 suggesting that a pair of nicotine 1′-N-oxide diastereomers were formed and detected.Several of the compounds in Table S4† were removed from the THS film over time relative to the t = 0 sample. Four of the matched suspect compounds had consistent relative signal levels throughout one week of aging under whole sidestream cigarette smoke. As noted above, MS/MS experiments support putative annotations of the four compounds in Fig. 1B as cotinine (mz177a), N-formylnornicotine (mz177b), nicotelline (mz234), and nornicotine (mz149). Removal of nornicotine was observed in some replicates and to some degree between 149.5 and 167.5 hours in the experiment depicted in Fig. 1, and perhaps could be grouped with compounds shown in panel C.
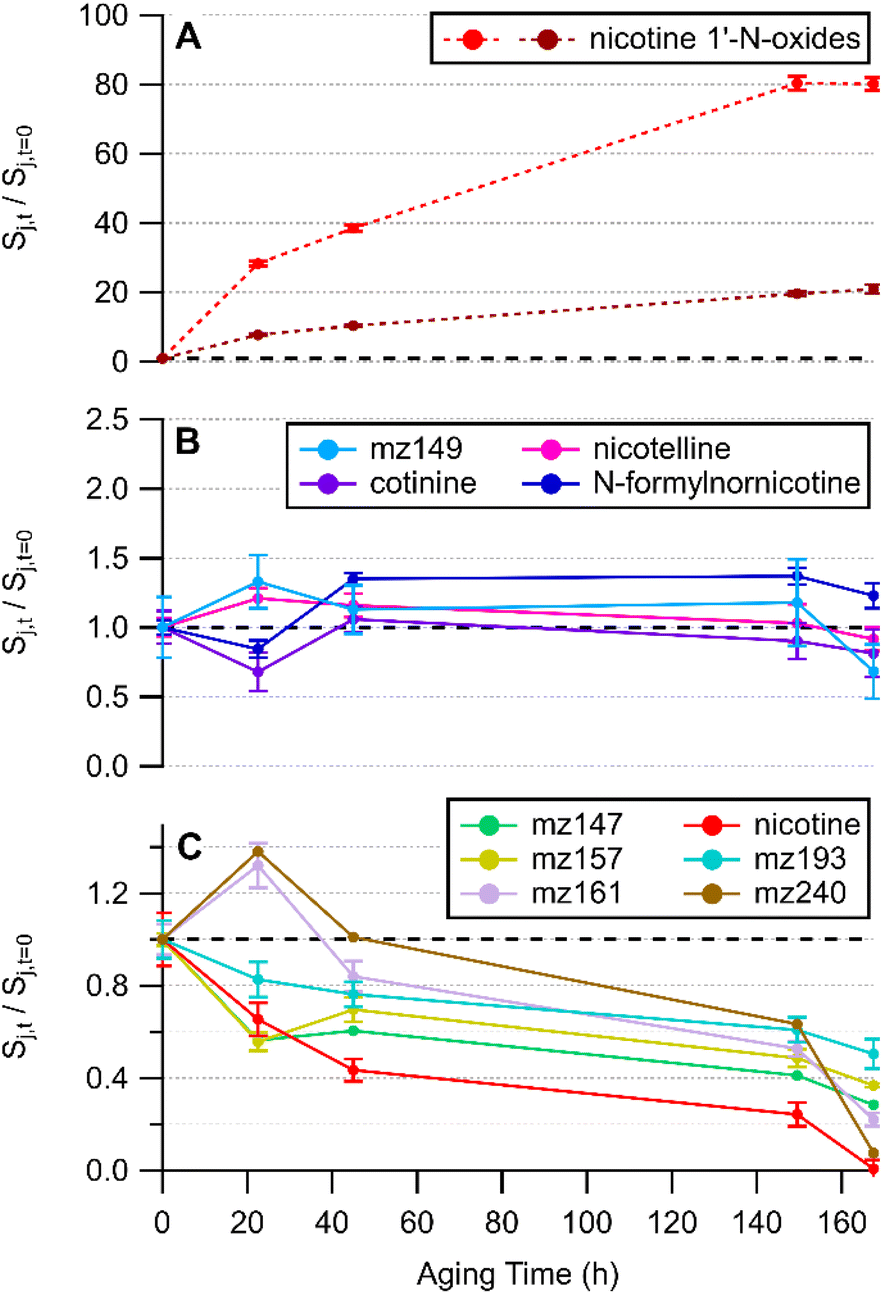 | ||
| Fig. 1 Time series of compounds listed in Table S4† grouped by observed behaviour in THS film extracts. Species are labelled with a chemical name if they have been verified beyond exact mass and stable isotope matching. Compounds that increased in abundance are shown in (A), those that were consistent during the experiment are shown in (B), and those that decreased in abundance are shown in (C). The substrate was clean glass. A dashed back line is shown at where Sj,t/Sj,t=0 = 1 on each vertical axis. All panels share a horizontal time axis. Error bars represent the standard deviation of triplicate LC-HRMS injections. Replicate experiments are shown in Fig. S5.† | ||
Some compounds were removed during the study period while others were steady in abundance relative to a sample collected approximately 30 minutes after smoke generation (Fig. 1). Nicotine was the most readily observed compound in the sample and decreased by at least 80% after 150 hours across all experimental replicates. It is possible that evaporative losses could contribute to the decreasing trend in nicotine and other SVOCs. However, the appearance of nicotine 1′-N-oxides provided evidence that nicotine removal was at least partially due to oxidation. The earlier-eluting diastereomer had a smaller peak area than the later peak and the relative rate of change in the earlier peak was greater than for the later-eluting nicotine oxide peak (Fig. 1A). Removal of nicotine from the film via partitioning to the gas phase was not out of the question; however, the samples were incubated under whole smoke so the extent of removal via film-to-gas partitioning would not be as important as for a film bathed in ventilated (nicotine-poor) air. The total mass of nicotine in the chamber (surface-sorbed + gaseous) should be largely consistent across the entirety of the experiment.
In a more detailed temporal trend analysis, focus will be placed on the behaviour of nicotine, N-formylnornicotine, cotinine, nicotelline, and the sum of both nicotine 1′-N-oxide diastereomers (Chart 2). MS/MS data for each compound in Chart 2 has been verified against an authentic standard (Fig. S3 and S4†). Nicotelline has been identified as a reliable tracer for tobacco smoke and THS,57,58 and in this study it provided a measure of consistency between samples in each experiment. In the present study, cotinine and N-formylnornicotine exhibited different behaviour depending on the conditions of the surface onto which smoke was deposited. On clean glass, cotinine and N-formylnornicotine were largely unchanged in relative abundance through 170 hours of incubation (Fig. 1B), despite the fact that both have been demonstrated as products of nicotine oxidation.42,59
 | ||
| Chart 2 Main tobacco alkaloid target compounds. | ||
THS deposition on a low-volatility organic antioxidant film
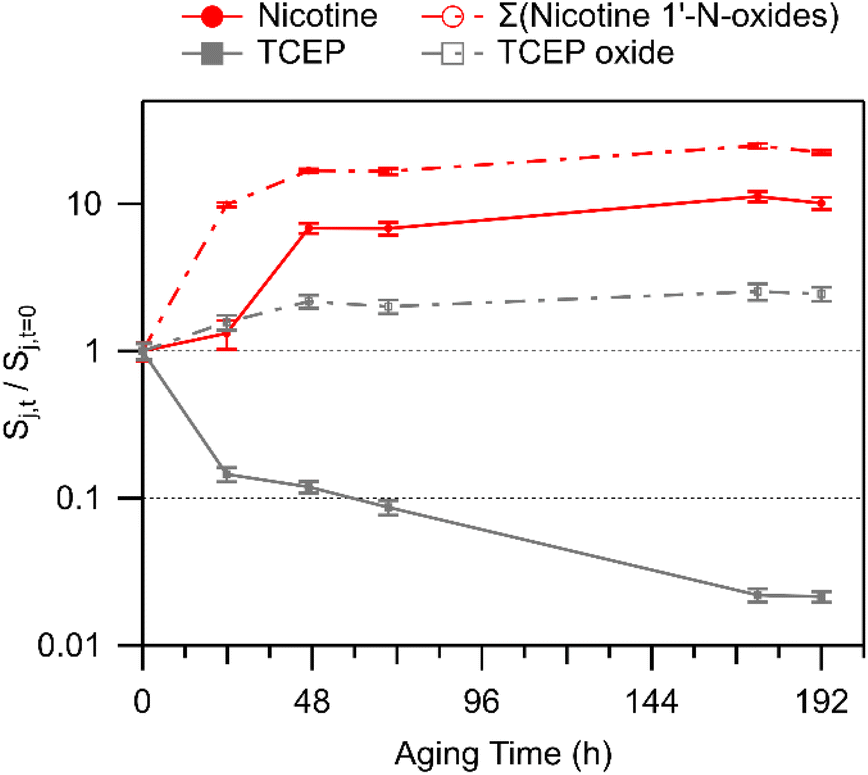 | ||
| Fig. 2 Temporal changes in nicotine, nicotine 1′-N-oxides (sum of diastereomers), TCEP, and TCEP oxide. Values above unity indicate an increase in abundance on the glass substrates, while values less than unity indicate net removal. Error bars represent one standard deviation based on triplicate LC-HRMS injections. Experimental replicates are shown in Fig. S9.† | ||
Nicotine oxide formation was observed coincident with TCEP oxide formation, indicating that TCEP did not fully quench the oxidative capacity of the deposited smoke. It is also possible that deposition was locally inhomogeneous on the TCEP surface film, precluding TCEP from mixing with and quenching oxidants in every deposited particle, or perhaps even across the entire mass of an individual deposited particle. For instance, when considering the deposition of an individual smoke particle onto a TCEP film, it may be that oxidants near the surface of the particle – at the TCEP/particle interface – proceeded to oxidize the antioxidant probe, but material buried more deeply within individual particles still reacted with abundant reduced organic compounds that are characteristic of the smoke sample itself, of which nicotine is an especially abundant example.
Koa = 13.5) that is approximately 260000 times larger than that of nicotine (logKoa = 8.08; Koa values were predicted using KOAWIN, EpiSuite, US EPA). Its equilibrium gas–phase mixing ratio would be much smaller than that of nicotine, cotinine, and N-formylnornicotine (if each compound was in equal abundance), so the potential for re-partitioning from deposited smoke found elsewhere in the chamber to the TCEP film was expected to be minimal. The significantly lower volatility of nicotelline allows for the attribution of a vast majority of its signal to direct aerosol particle dry deposition, while abundances of nicotine, cotinine, and N-formylnornicotine (and other alkaloids with similar volatilities) may be more substantially influenced by SVOC partitioning. To further investigate the strength of SVOC partitioning to the TCEP film, the behaviour of cotinine (logKoa = 9.93) and N-formylnornicotine (logKoa = 9.98) was compared with two other SVOCs that have similar Koa. Scopoletin (logKoa = 9.62) and 2,3′-bipyridine (logKoa = 8.99) both decreased slightly in abundance in film extracts of TCEP-coated glass substrates, while cotinine and N-formylnornicotine showed relative increases in abundance in the same extracts (especially cotinine; Fig. 3). The relationships between the measured compounds therefore suggest that nicotine oxidation was an important contributor to the increasing trends in cotinine and N-formylnornicotine on TCEP films. It was interesting that nicotine oxidation continued to produce nicotine 1′-N-oxide, cotinine, and N-formylnornicotine in the presence of TCEP due to the reducing environment of the film. Localization of oxidation chemistry within deposited particulate matter that is not in direct contact with the solid TCEP coating is a tempting explanation. However, such an explanation would seem to necessitate a similar type of observation in extracts from clean glass, wherein the relative abundances of cotinine and N-formylnornicotine were not observed to increase.
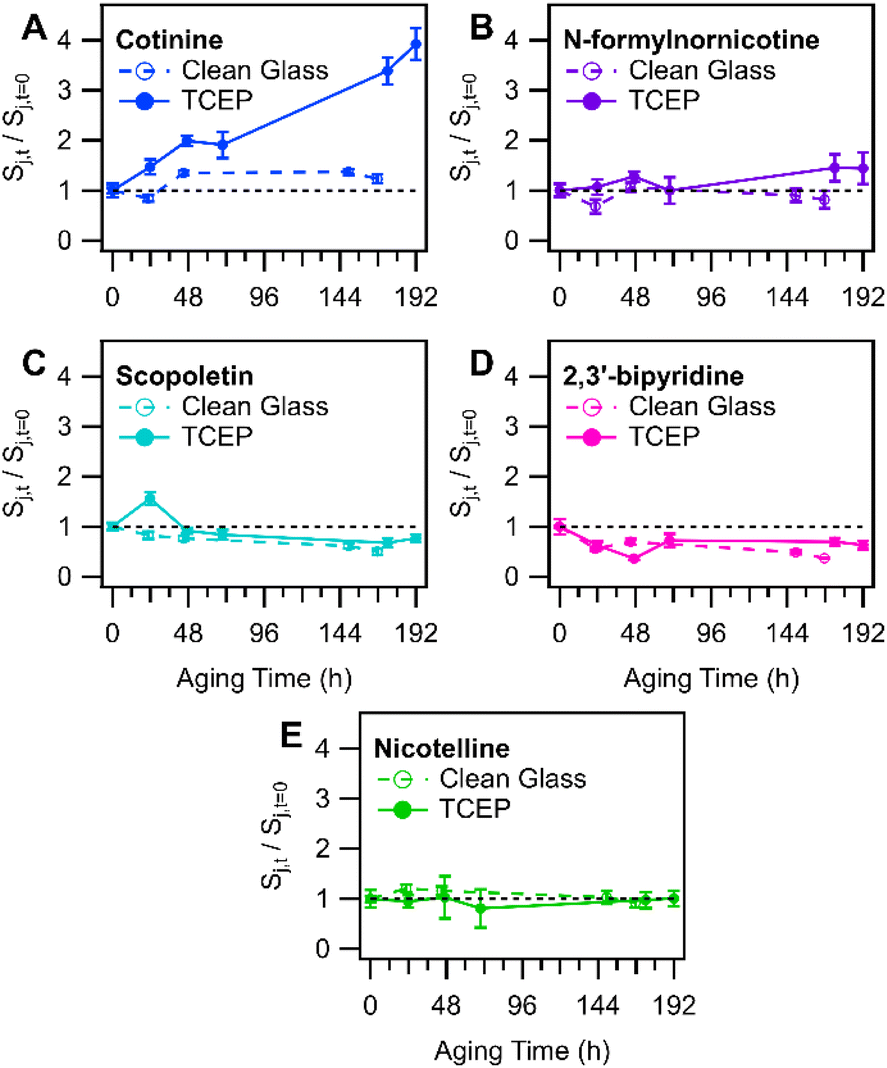 | ||
| Fig. 3 Comparison of temporal profiles of tobacco-related compounds extracted from clean glass substrates versus glass substrates (in a separate experiment) that were coated with TCEP before exposure to tobacco smoke. Panels depict data for tobacco-related compounds (A) cotinine, (B) N-formylnornicotine, (C) scopoletin, (D) 2,3′-bipyridine, (E) nicotelline. Error bars represent one standard deviation based on triplicate LC-HRMS injections of each sample. | ||
Effects of cigarette smoke on organic surface film composition
In order to investigate the chemical outcomes of THS mixing with other organic film-forming materials on indoor surfaces, a series of trial films were deposited on glass and exposed to sidestream cigarette smoke, just as in the clean glass and TCEP cases. Three compounds, in addition to TCEP (Chart 2), were tested as films: a weakly-reactive oil (bis(2-ethylhexyl)sebacate; BES), a monounsaturated fatty acid (oleic acid; OA), and a polyisoprenoid (squalene; SQ). Since OA did not deposit evenly on the surface by itself, it was co-deposited with BES. The two compounds were deposited successively: BES followed by OA.Upon exposure to smoke and subsequent incubation, nicotine oxide formation was observed in BES film extracts, just as in experiments with other surface films (and on clean glass), but observable changes in nicotine, cotinine, and N-formylnornicotine abundance were most similar to clean glass (Fig. S10†). The lack of observable uptake of nicotine to BES on the multi-day timescale may be related to rapid equilibration with the low-viscosity film material. If gas-film SVOC equilibrium was largely complete by the time the first sample was collected and extracted, approximately 30 minutes after smoke was first introduced to the glass chamber, the trend in nicotine would be normalized after equilibration occurred. The results of BES experiments, considered in concert with TCEP experiment results, suggest the importance of kinetic limitations to SVOC partitioning (slow diffusion and gradient formation)8,19,62 in viscous organic films, along with the participation of the film in the formation of reactive species or intermediates.
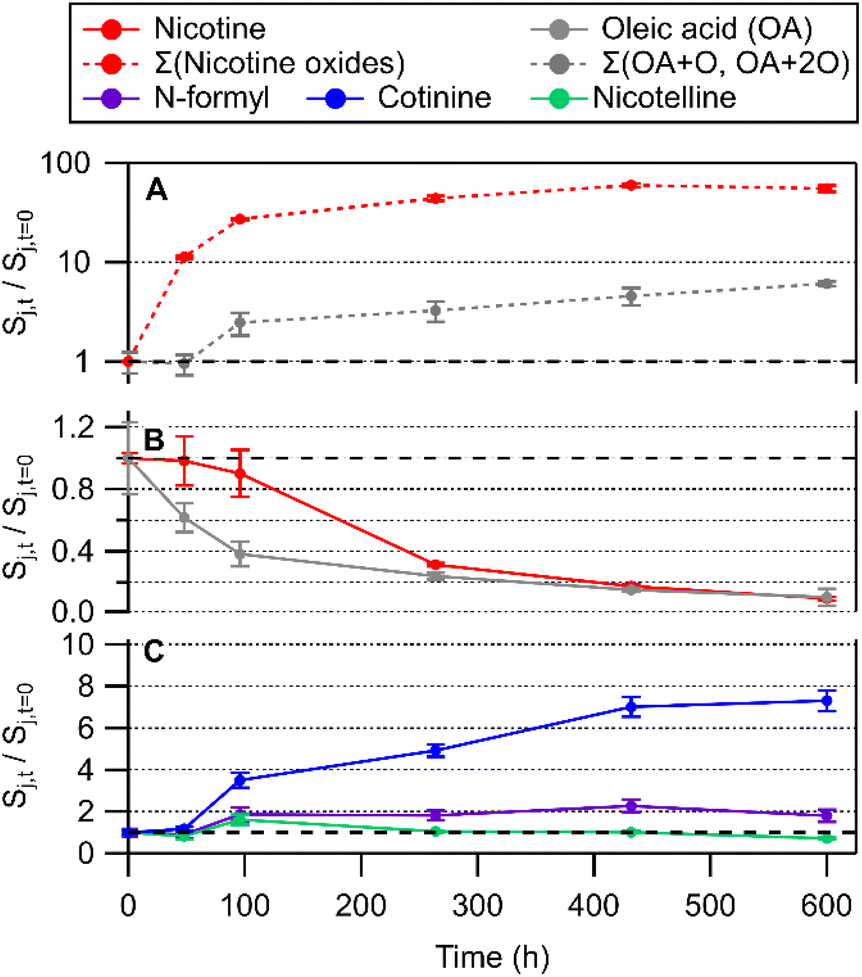 | ||
| Fig. 4 Time series of chemical transformations in an OA/BES film upon cigarette smoke deposition, including (A) nicotine oxides and TCEP oxide, (B) nicotine and oleic acid, and (C) other tobacco related alkaloids. Panel (A) is shown with a logarithmic axis to display a wide degree of change in relative abundance in oxidation products. Oxidation product traces represent the sum of nicotine 1′-N-oxide diastereomers (Σ(nicotine oxides)) and the sum of all peak areas from OA + O and OA + 2O (Σ(OA + O, OA + 2O)). OA + O and OA + 2O are shown in detail in Fig. 5. Error bars represent the standard deviation of three repeat injections. | ||
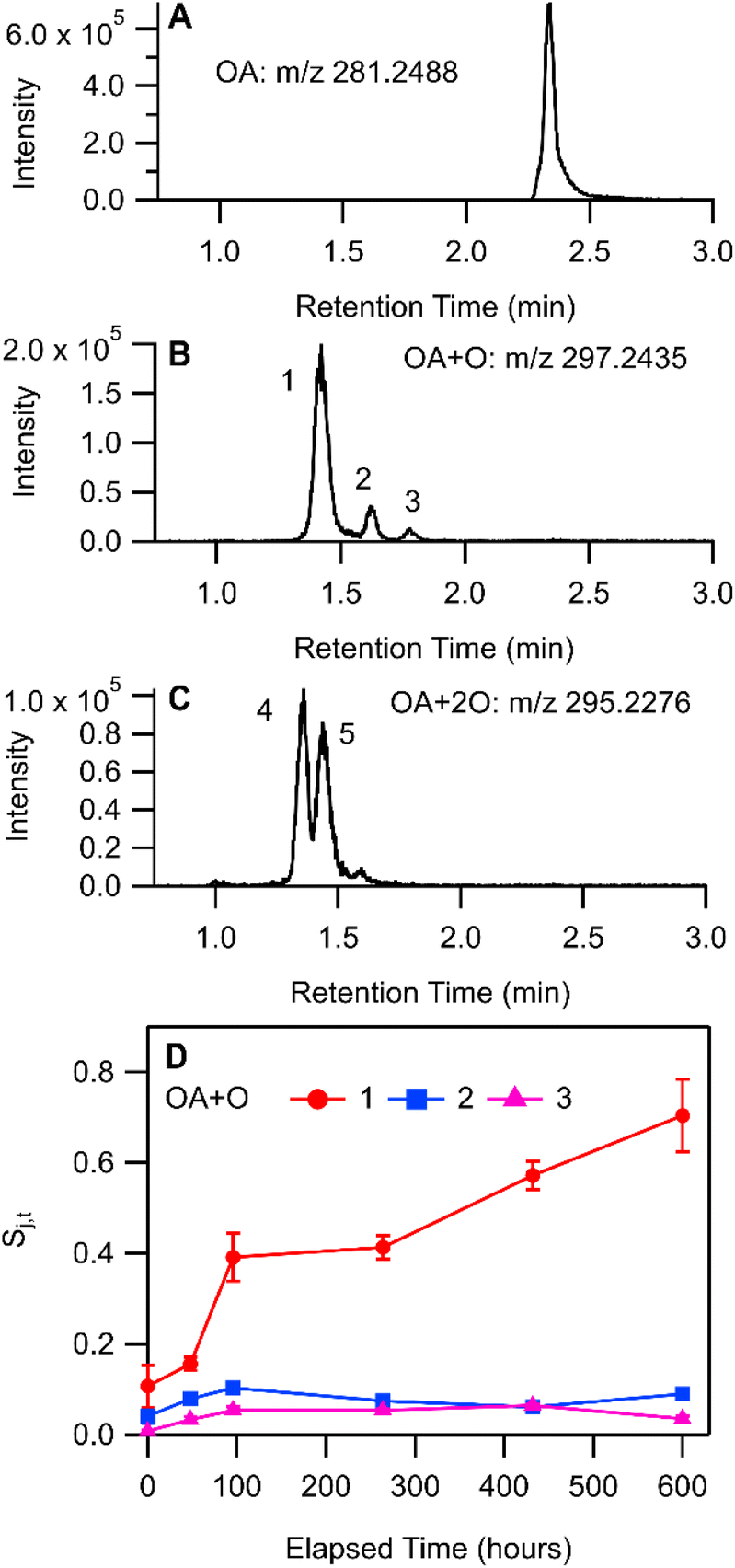 | ||
| Fig. 5 LC-HRMS product analysis of OA/BES reaction with deposited THS. Chromatograms from the 600 h sample for (A) oleic acid, and its oxidation products (B) OA + O and (C) OA + 2O, represented by the extracted ion chromatograms for mass-to-charge ratios shown. Panel (D) shows the mean of internal standard-normalized areas of chromatographic peaks labelled in panels (B) and (C). Error bars in (D) represent the standard deviation of three repeat LC-MS injections. | ||
Qualitative analysis of LC-HRMS data for THS + OA extracts was processed with non-targeted molecular feature extraction using MassHunter. Several chromatographically separable oxidation products of OA were observed in LC-HRMS measurements with negative-mode ESI. Three chromatographic peaks (labelled 1, 2, and 3 in Fig. 5B) were observed in the extracted ion chromatogram (EIC) for m/z 297.2438 [C18H34O3–H]− and two peaks (labelled 4 and 5 in Fig. 5C) were observed in the EIC for m/z 295.2280 [C18H34O4–H2O–H]−. Measured stable isotope abundance ratios were compared with computed isotope abundances using the idotp metric (a vector-based similarity metric having a scale similar to a dot product) in Skyline.54,55 Molecular formulae reported had idotp values greater than 0.9, which support the molecular formula assignments for both [C18H34O4–H2O–H]− and [C18H34O3–H]− across all chromatographic peaks shown in panels B and C in Fig. 5. While some chromatographic peaks associated with OA + O and OA + 2O overlap to some degree, retention times identified at peak centroids differ at least slightly (Table S5†). None of the chromatographic peaks of interest for m/z 295.2280 or m/z 297.2438 co-eluted with the OA starting material (m/z 281.2488 [C18H34O2–H]−; Fig. 5A) nor did they perfectly co-elute with one another. Therefore, in-source oxidation, fragmentation, and other possible ion chemistry artefacts involving the starting material were likely not a factor associated with the observation of OA + O or OA + 2O features by LC-HRMS.
The formation of C18H34O3 compounds based on the addition of a single oxygen atom to oleic acid (OA + O) may include oleic acid epoxide, ketostearic acid, or hydroxyoleic acid. Indeed, three chromatographic peaks appear in the EIC for m/z 297.2438 (Fig. 5B). Peak 1 was the largest OA + O signal throughout the experiment and also increased the most overall, while Peaks 2 and 3 increased for approximately 100 h and then were stable or showed a decrease in area (Fig. 5D). Measurement of an OA epoxide standard produced a single chromatographic peak at 1.64 min, overlapping well with Peak 2 (Fig. S11†). Collision-induced dissociation of the chromatographically-resolved oxidized OA precursor ions was not successful, so further structural characterization was not possible for OA oxidation products. Still we speculate that OA + O Peaks 1 and 3 may correspond with molecules such as ketostearic acid and/or hydroxyoleic acid.
When OA/BES films were incubated under ultra-pure air (see ESI†), the formation of OA + 2O signals were similar in magnitude and temporal trend to signals observed in the THS treatment so Peaks 4 and 5 will not be discussed in greater detail. It is worthwhile to note that OA + 2O species would be expected to form on air-exposed surfaces in indoor environments.
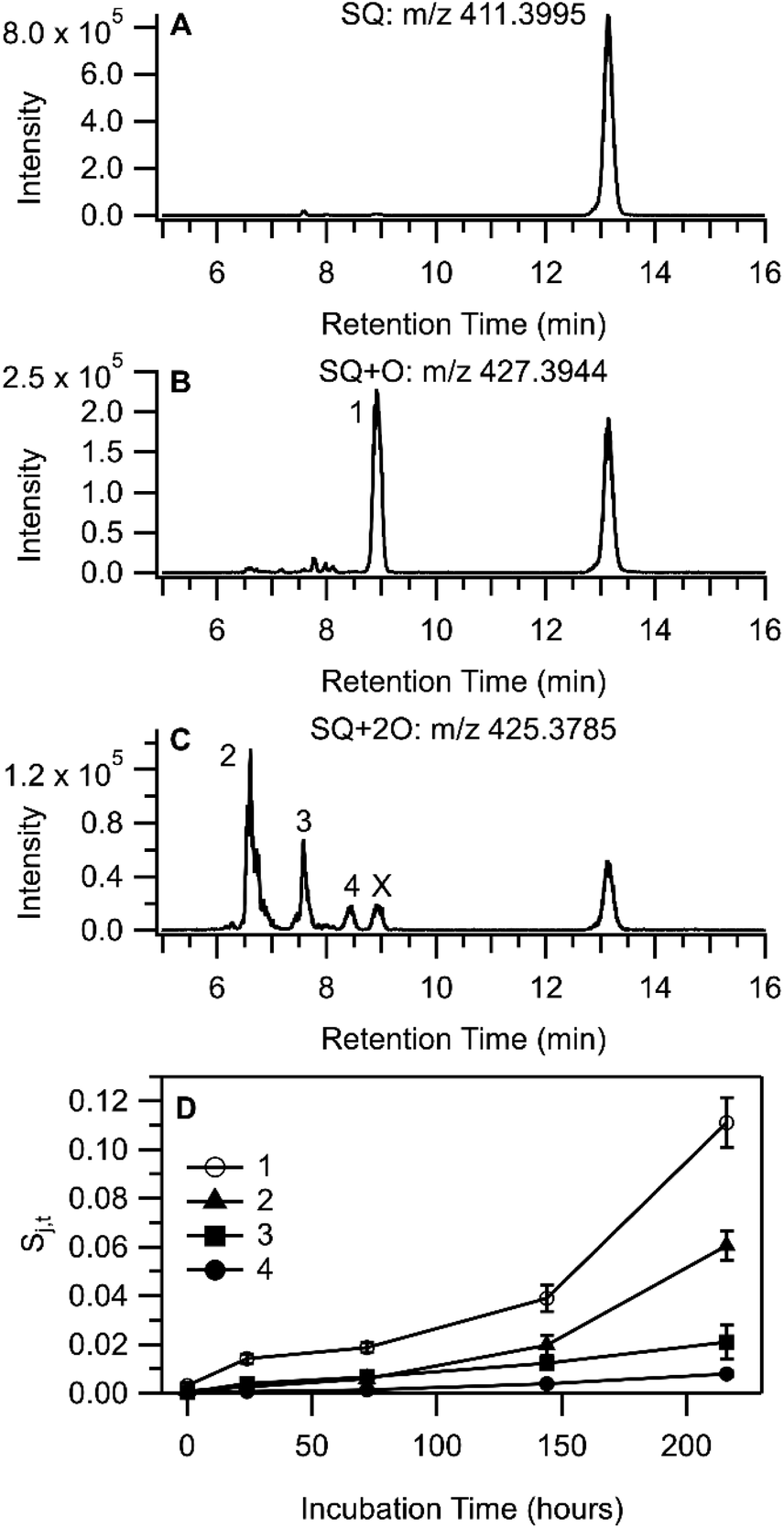 | ||
| Fig. 6 LC-HRMS product analysis of SQ reaction with THS residue. Extracted ion chromatograms from the sample incubated for 216 h corresponding to (A) SQ, (B) SQ + O, and (C) SQ + 2O are shown. The peak labelled “X” in (C) overlaps exactly with peak 1 and was not considered further. Intensities are the sum of the three most abundant isotopologues. Panel (D) shows the temporal profile of SQ + O and SQ + 2O peak area (after internal standard normalization). Chromatographic peaks labelled with numerals in (B) and (C) match with legend entries in (D). | ||
Molecular formula and adduct assignments for LC-HRMS features in Fig. 6 were supported by comparing calculated and measured peak area ratios of the three most abundant isotopologues using the idotp function in Skyline;54,55 signals obtained each chromatographic peak found in an EIC had idotp >0.9. Oxidation of SQ leading to the addition of a single O atom suggests the formation of SQ monoepoxide, similar to OA. The MS and MS/MS spectra were consistent with prior reports of SQ epoxide detection,63,64 and the SQ + O signal from a THS + SQ sample collected at 216 h of incubation was a close match to the SQ epoxide standard (Fig. S17†). The presence of a single chromatographic peak in the EIC for m/z 427.3935 (labelled 1 in Fig. 6B) was somewhat surprising given the variety of possible SQ + O isomers, although based on prior studies,63,65 it is likely that reverse phase liquid chromatography may not be capable of resolving isomers of SQ + O.
The detection of SQ + 2O compounds occurred consistently as a proton adduct with a neutral loss of H2O [M + H–H2O]+ (m/z 425.3779), the EIC of which shows at least three chromatographic peaks (labelled 2, 3, and 4 in Fig. 6C). Other related ions were detected in MS1 spectra collected for peaks 2 and 3 (Fig. S16 and Table S7†), including a small signal at m/z 443.3890 [C30H50O2 + H]+ which corresponded with intact, protonated SQ + 2O. Neutral loss of H2O from SQ + 2O65,66 along with the likelihood of free radical-initiated autoxidation may suggest the existence of a SQ hydroperoxide (SQ-OOH) in the sample extracts, although other oxygenated functional groups are possible. MS/MS spectra for m/z 425 were used to gain further insight into the structural similarities and differences between molecules eluting in peaks 2, 3, and 4 (Fig. S18†). Further loss of H2O to form a fragment at m/z 407.3812 was observed in all three MS/MS spectra. Peaks 2 and 4 show an MS/MS fragment at m/z 339.3034, a close exact mass match (3.6 ppm) to the [C25H38 + H]+ ion, which corresponds to the collision-induced loss of one isoprene unit and an oxygen atom. The MS1 spectra for Peaks 2 and 4 were also similar, with m/z 425.3779 and m/z 407.3678 as the most prominent features, which bear striking similarity to MS1 spectra for SQ–OOH substituted at odd-numbered positions as reported by Shimizu et al.67 Peak 3 has a somewhat different MS/MS spectrum and a markedly different MS1 spectrum compared to Peaks 2 and 4. In-source fragmentation was a prominent feature of the Peak 3 MS1 spectrum (Fig. S16†), suggesting a relatively labile SQ + 2O species. Among the prominent signals in the MS1 spectrum of Peak 3 were in-source fragments m/z 425.3779 from the neutral loss of H2O from SQ + 2O, and also m/z 411.3629 from the neutral loss of CH3OH from SQ + 2O (a more detailed dissection of the data is provided in Section S7 of the ESI†). The MS/MS spectrum for Peak 3 contained a signal at m/z 269.2263, which corresponded to only four contiguous isoprene monomer units of SQ (loss of two 5-carbon subunits). Taken together, the two different neutral losses from SQ + 2O along with the MS/MS observation of the loss of two 5-carbon subunits from SQ + 2O suggest the oxidation of SQ at two different olefinic sites, perhaps at either terminus of the SQ chain (the 2/3 and 22/23 positions). Assignment of the compound that eluted at Peak 3 to SQ–OOH is therefore not so compelling and may instead arise from an SQ + 2O product in which two separate hydroxyl or carbonyl groups are present.68
Insights on the nature of endogenous oxidation in THS films
Tobacco smoke aerosol is known to contain iron, so Fenton chemistry is often implicated in reactions leading to smoke-related oxidative stress.34 While the active reactive oxygen species (ROS) were likely OH and HO2 radicals, longer-lived aromatic and polycyclic aromatic semiquinones in cigarette smoke ‘tar’ are thought to sustain the oxidative potential of tobacco smoke34,47,49 and are notable as environmentally persistent free radicals (EPFRs). Based on the literature, along with oxidation product analysis of SQ and OA film oxidation by THS, the confluence of semiquinones and metals likely led to HOx-driven oxidative action within THS films.Evidence for autoxidation was present in the THS + SQ samples. The present study corroborates prior SQ oxidation studies performed with complementary types of experiments.52,69 Analysis of HRMS and MS/MS data suggested that the SQ + 2O Peak 3 was indicative of a compound with multiple sites of oxidation on the same molecule. Installation of oxidized functional groups at multiple sites on SQ, as suggested by Peak 3, could be supported by hydroperoxide decomposition in the SQ film environment, and/or that peroxides or peracids may be active condensed phase oxidants within THS films in addition to reactive free radical species.
The impact of persistent autoxidation in THS-contaminated surface films does not appear to be restricted to transformations of the surface film material. The observation of pronounced cotinine formation in the TCEP and OA/BES cases (no observed in clean glass or BES cases) suggests that reactive intermediates formed in association with the surface film oxidation process could be oxidizing nicotine to cotinine, whereas in clean glass and BES cases, nicotine oxidation appeared to mainly lead to nicotine 1′-N-oxide formation. A larger relative increase in nicotine 1′-N-oxides was observed in the OA/BES film compared to clean glass. The increased cotinine (and to a lesser extent, N-formylnornicotine) production on TCEP and OA/BES coated glass compared to clean glass may result from a difference in the identity of the oxidant(s) involved, but may also arise from additional nicotine available based on uptake from the gas phase and re-partitioning from other surfaces in the chamber.
Environmental significance
Indoor surfaces harbor chemical films that develop based on the uptake of gaseous and particulate material;1,4,5 they act as a reservoir for semivolatile organic compounds, along with inorganic acids and bases.17,18 Recent studies have also highlighted that surface films and household dust contain EPFRs and ROS,31,37 but the time-evolution of surface film composition due to the presence of condensed phase oxidants has not been thoroughly studied. The findings of the present study highlight the dynamic nature of surface film chemical composition upon the deposition of cigarette smoke, which is known to possess an inherent oxidative potential.47,70 EPFRs can activate the formation of highly reactive oxidants (e.g., hydroxyl radicals), and some EPFRs have lifetimes of months or longer in ambient atmospheric aerosol particle samples.71 Aerosol particles that contain EPFRs and become deposited on surfaces could influence the composition of environmental surface films gradually over long timescales, as indicated by Filippi et al.37 and also shown in the present study. The co-presence of (metallic) redox couples (e.g., Fe2+/Fe3+) with EPFRs could lead to the formation of highly reactive (short-lived) oxidants in the condensed phase with subsequent regeneration of EPFR species.72 Cyclic mechanisms for ROS production would support the prolonged presence of EPFRs, all the while consistently producing more highly reactive species that can transform surface film composition.The ability of deposits formed by other types of aerosol particles that typically exhibit OP,73 such as biomass burning (BB),33,36 traffic related air pollutants (TRAP),32 mineral dust,74 or secondary organic aerosol (SOA),75 to introduce the degree of oxidation chemistry that was conferred by THS in the present study is not yet known, but is likely. It is important to bear in mind that OP assays may respond differently from one another based on the exact nature of the (bio)chemical processes involved in the assay readout,73 so one must retain a critical eye when attempting to predict surface oxidation chemistry using OP assay results. Tobacco smoke could be considered a subset of BB smoke and some assays indicate similar OP.73 Wong et al.33 showed that atmospheric aging of BB aerosol particles can increase their oxidative potential, so aged or ambient BB aerosol may have more pronounced effects than lab-generated fresh BB. Similarities between THS and TRAP composition include polycyclic aromatic compounds and metals,76 suggesting that TRAP reactivity upon surface deposition could be similar to what has been demonstrated in the present study of THS deposits. In fact, SQ oxidation has been demonstrated in solutions containing metals and PAHs that were meant to serve as a proxy for exposure to urban pollutants.69 The surface substrate composition may also play a role in driving condensed phase oxidation of surface films, especially if the substrate can provide ingredients for catalytic oxidation, such as Fe2+/Fe3+ or Cu+/Cu2+. Recent indoor measurements of EPFRs on surfaces strongly suggest that reactivity in films have strong potential,37 and the delivery of key reactive components might be critical in driving the overall rate of surface film oxidation.
Conclusions
The effects of cigarette smoking on the indoor environment are continuing to be clarified. Environmental tobacco smoke and second-hand smoke effects are relatively well established, while the importance of THS is still being investigated. The present study provides information on the chemically dynamic nature of THS films over week-long timescales resulting from oxidative transformations of chemicals found in THS and those that may co-exist on indoor surfaces with cigarette smoke residue. The formation of oxidized tobacco alkaloids along with oxidation products of alkenes found on surfaces demonstrates the ability of THS to transform the composition of contaminated indoor surfaces over time. The exact chemical outcomes of THS-driven reactive transformations within real indoor surface films will result from time-dependent behaviour in concert with (and in competition with) bidirectional gas/surface partitioning processes already known to be important for THS and indoor environments more generally.Author contributions
The authors, AMH and DBC, shared a variety of roles (based on the CRediT taxonomy) in the present study, including formal analysis, methodology, data curation, conceptualization, validation, visualization, writing, and editing. AMH performed most of the investigations. DBC was also responsible for funding acquisition, project administration, resources, and supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project was supported by a grant from the Alfred P. Sloan Foundation (G-2019-12365) through the SURF-CIE Consortium (via the University of California, San Diego) and the Office of the Provost at Bucknell University. Support was provided by the National Science Foundation for instrumentation (CHE/MRI-2018547), the Physical Sciences Scholars program at Bucknell (DUE/S-STEM-1742124), and the STEM Scholars program at Bucknell (DUE/STEP-1317446). The authors acknowledge the operational support for instrumentation from Dr Morgan Olsen and Dr Peter Findeis. Maggie E. Young and Naomi R. Douek assisted with executing some investigations.References
- C. M. A. Eichler, J. Cao, G. Isaacman-VanWertz and J. C. Little, Indoor Air, 2019, 29, 17–29 CrossRef CAS.
- C. J. Weschler and W. W. Nazaroff, Indoor Air, 2017, 27, 1101–1112 CrossRef CAS PubMed.
- Q.-T. Liu, R. Chen, B. E. McCarry, M. L. Diamond and B. Bahavar, Environ. Sci. Technol., 2003, 37, 2340–2349 CrossRef CAS PubMed.
- C. Y. Lim and J. P. Abbatt, Environ. Sci. Technol., 2020, 54, 14372–14379 CrossRef CAS PubMed.
- V. W. Or, M. Wade, S. Patel, M. R. Alves, D. Kim, S. Schwab, H. Przelomski, R. O'Brien, D. Rim, R. L. Corsi, M. E. Vance, D. K. Farmer and V. H. Grassian, Environ. Sci.: Processes Impacts, 2020, 22, 1698–1709 RSC.
- P. Jacob, N. L. Benowitz, H. Destaillats, L. Gundel, B. Hang, M. Martins-Green, G. E. Matt, P. J. E. Quintana, J. M. Samet, S. F. Schick, P. Talbot, N. J. Aquilina, M. F. Hovell, J.-H. Mao and T. P. Whitehead, Chem. Res. Toxicol., 2017, 30, 270–294 Search PubMed.
- A. P. Ault, V. H. Grassian, N. Carslaw, D. B. Collins, H. Destaillats, D. J. Donaldson, D. K. Farmer, J. L. Jimenez, V. F. McNeill, G. C. Morrison, R. E. O'Brien, M. Shiraiwa, M. E. Vance, J. R. Wells and W. Xiong, Chem, 2020, 6, 3203–3218 CAS.
- R. E. O'Brien, Y. Li, K. J. Kiland, E. F. Katz, V. W. Or, E. Legaard, E. Q. Walhout, C. Thrasher, V. H. Grassian, P. F. DeCarlo, A. K. Bertram and M. Shiraiwa, Environ. Sci.: Processes Impacts, 2021, 23, 559–568 RSC.
- M. L. Diamond, S. E. Gingrich, K. Fertuck, B. E. McCarry, G. A. Stern, B. Billeck, B. Grift, D. Brooker and T. D. Yager, Environ. Sci. Technol., 2000, 34, 2900–2908 CrossRef CAS.
- J. S. Grant, Z. Zhu, C. R. Anderton and S. K. Shaw, ACS Earth Space Chem., 2019, 3, 305–313 CrossRef CAS.
- D. B. Collins and V. H. Grassian, in Physical Chemistry of Gas-Liquid Interfaces, Elsevier, 2018, pp. 271–313 Search PubMed.
- U. Pöschl and M. Shiraiwa, Chem. Rev., 2015, 115, 4440–4475 CrossRef.
- R. Habre, D. C. Dorman, J. Abbatt, W. P. Bahnfleth, E. Carter, D. Farmer, G. Gawne-Mittelstaedt, A. H. Goldstein, V. H. Grassian, G. Morrison, J. Peccia, D. Poppendieck, K. A. Prather, M. Shiraiwa, H. M. Stapleton, M. Williams and M. E. Harries, Environ. Sci. Technol., 2022, 56, 10560–10563 CrossRef CAS.
- V. Bahl, P. J. Iii, C. Havel, S. F. Schick and P. Talbot, PLoS One, 2014, 9, e108258 CrossRef.
- G. Morrison, Environ. Sci. Technol., 2008, 42, 3495–3499 CrossRef PubMed.
- M. Sleiman, J. M. Logue, W. Luo, J. F. Pankow, L. A. Gundel and H. Destaillats, Environ. Sci. Technol., 2014, 48, 13093–13101 CrossRef CAS PubMed.
- D. M. Lunderberg, K. Kristensen, Y. Tian, C. Arata, P. K. Misztal, Y. Liu, N. Kreisberg, E. F. Katz, P. F. DeCarlo, S. Patel, M. E. Vance, W. W. Nazaroff and A. H. Goldstein, Environ. Sci. Technol., 2020, 54, 6751–6760 CrossRef CAS PubMed.
- C. Wang, D. B. Collins, C. Arata, A. H. Goldstein, J. M. Mattila, D. K. Farmer, L. Ampollini, P. F. DeCarlo, A. Novoselac, M. E. Vance, W. W. Nazaroff and J. P. D. Abbatt, Sci. Adv., 2020, 6, eaay8973 CrossRef CAS PubMed.
- D. B. Collins, C. Wang and J. P. D. Abbatt, Environ. Sci. Technol., 2018, 52, 13195–13201 CrossRef CAS PubMed.
- P. F. DeCarlo, A. M. Avery and M. S. Waring, Sci. Adv., 2018, 4, eaap8368 CrossRef PubMed.
- I. J. George and J. P. D. Abbatt, Nat. Chem., 2010, 2, 713–722 CrossRef CAS PubMed.
- M. Shiraiwa, Y. Sosedova, A. Rouvière, H. Yang, Y. Zhang, J. P. D. Abbatt, M. Ammann and U. Pöschl, Nat. Chem., 2011, 3, 291–295 CrossRef CAS PubMed.
- T. Berkemeier, A. Mishra, C. Mattei, A. J. Huisman, U. K. Krieger and U. Pöschl, ACS Earth Space Chem., 2021, 5, 3313–3323 CrossRef CAS.
- T. Nah, S. H. Kessler, K. E. Daumit, J. H. Kroll, S. R. Leone and K. R. Wilson, Phys. Chem. Chem. Phys., 2013, 15, 18649 RSC.
- L. M. Petrick, M. Sleiman, Y. Dubowski, L. A. Gundel and H. Destaillats, Atmos. Environ., 2011, 45, 4959–4965 CrossRef CAS.
- X. Tang, N. R. González, M. L. Russell, R. L. Maddalena, L. A. Gundel and H. Destaillats, Environ. Res., 2020, 110462 Search PubMed.
- R. Alwarda, S. Zhou and J. P. D. Abbatt, Indoor Air, 2018, 28, 655–664 CrossRef CAS PubMed.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- T. Berkemeier, S. S. Steimer, U. K. Krieger, T. Peter, U. Pöschl, M. Ammann and M. Shiraiwa, Phys. Chem. Chem. Phys., 2016, 18, 12662–12674 RSC.
- G. Morrison, R. Moravec and Z. Yao, Environ. Sci. Technol. Lett., 2023, 10(6), 528–532 CrossRef CAS.
- S. Biswas, V. Verma, J. J. Schauer, F. R. Cassee, A. K. Cho and C. Sioutas, Environ. Sci. Technol., 2009, 43, 3905–3912 CrossRef CAS PubMed.
- J. P. S. Wong, M. Tsagkaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes and R. J. Weber, Environ. Sci. Technol., 2019, 53, 6747–6756 CrossRef CAS PubMed.
- A. Valavanidis, T. Vlachogianni and K. Fiotakis, Int. J. Environ. Res. Public Health, 2009, 6, 445–462 CrossRef CAS PubMed.
- J. G. Charrier and C. Anastasio, Atmos. Chem. Phys., 2012, 12, 9321–9333 CrossRef CAS.
- W. Y. Tuet, F. Liu, N. de Oliveira Alves, S. Fok, P. Artaxo, P. Vasconcellos, J. A. Champion and N. L. Ng, Environ. Sci. Technol. Lett., 2019, 6, 126–132 CrossRef CAS.
- A. Filippi, R. Sheu, T. Berkemeier, U. Pöschl, H. Tong and D. R. Gentner, Environ. Sci.: Atmos., 2022, 2, 128–136 CAS.
- S. F. Schick, K. F. Farraro, C. Perrino, M. Sleiman, G. van de Vossenberg, M. P. Trinh, S. K. Hammond, B. M. Jenkins and J. Balmes, Tob. Control, 2014, 23, 152–159 CrossRef PubMed.
- M. Sleiman, L. A. Gundel, J. F. Pankow, P. Jacob, B. C. Singer and H. Destaillats, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 6576–6581 CrossRef CAS PubMed.
- G. E. Matt, P. J. E. Quintana, M. F. Hovell, J. T. Bernert, S. Song, N. Novianti, T. Juarez, J. Floro, C. Gehrman, M. Garcia and S. Larson, Tob. Control, 2004, 13, 29 CrossRef CAS PubMed.
- G. E. Matt, A. L. Merianos, P. J. E. Quintana, E. Hoh, N. G. Dodder and E. M. Mahabee-Gittens, JAMA Netw. Open, 2022, 5, e2147184 CrossRef PubMed.
- A. Wylie, Master's thesis, University of Toronto, 2020.
- R. Sheu, C. Stönner, J. C. Ditto, T. Klüpfel, J. Williams and D. R. Gentner, Sci. Adv., 2020, 6, eaay4109 CrossRef CAS PubMed.
- R. Sheu, T. Hass-Mitchell, A. Ringsdorf, T. Berkemeier, J. Machesky, A. Edtbauer, T. Klüpfel, A. Filippi, B. A. M. Bandowe, M. Wietzoreck, P. Kukučka, H. Tong, G. Lammel, U. Pöschl, J. Williams and D. R. Gentner, Environ. Sci.: Atmos., 2022, 2(5), 943–963 Search PubMed.
- K. Yeh, L. Li, F. Wania and J. P. D. Abbatt, Environ. Int., 2022, 160, 107063 CrossRef CAS PubMed.
- A. Rodgman and T. A. Perfetti, The Chemical Components of Tobacco and Tobacco Smoke, CRC Press, Boca Raton, FL, 2nd edn, 2013 Search PubMed.
- D. F. Church and W. A. Pryor, Environ. Health Perspect., 1985, 64, 111–126 CrossRef CAS PubMed.
- M. Shein and G. Jeschke, Chem. Res. Toxicol., 2019, 32, 1289–1298 Search PubMed.
- J. B. Wooten, S. Chouchane and T. E. McGrath, in Cigarette Smoke and Oxidative Stress, ed. B. Halliwell and H. E. Poulsen, Springer, Berlin, 2006, pp. 5–46 Search PubMed.
- M. Egawa, Y. Kohno and Y. Kumano, Int. J. Cosmet. Sci., 1999, 21, 83–98 CrossRef CAS PubMed.
- Q. Zhang, S. Tang, G. Huang and H. Liu, J. Cosmet. Dermatol., 2022, 21, 3085–3094 CrossRef PubMed.
- G. Percoco, A. Patatian, F. Eudier, M. Grisel, T. Bader, E. Lati, G. Savary, C. Picard and P. Benech, Exp. Dermatol., 2021, 30, 1610–1618 CrossRef CAS PubMed.
- B. L. Deming and P. J. Ziemann, Indoor Air, 2020, 30, 914–924 CrossRef CAS PubMed.
- B. X. MacLean, B. S. Pratt, J. D. Egertson, M. J. MacCoss, R. D. Smith and E. S. Baker, J. Am. Soc. Mass Spectrom., 2018, 29, 2182–2188 CrossRef CAS PubMed.
- K. J. Adams, B. Pratt, N. Bose, L. G. Dubois, L. St John-Williams, K. M. Perrott, K. Ky, P. Kapahi, V. Sharma, M. J. MacCoss, M. A. Moseley, C. A. Colton, B. X. MacLean, B. Schilling and J. W. Thompson, Alzheimer's Disease Metabolomics Consortium, J. Proteome Res., 2020, 19, 1447–1458 CrossRef CAS PubMed.
- T. J. Smyth, V. N. Ramachandran, A. McGuigan, J. Hopps and W. F. Smyth, Rapid Commun. Mass Spectrom., 2007, 21, 557–566 CrossRef CAS PubMed.
- P. Jacob, M. L. Goniewicz, C. M. Havel, S. F. Schick and N. L. Benowitz, Chem. Res. Toxicol., 2013, 26, 1615–1631 Search PubMed.
- N. J. Aquilina, C. M. Havel, P. Cheung, R. M. Harrison, K.-F. Ho, N. L. Benowitz and P. Jacob III, Environ. Int., 2021, 150, 106417 CrossRef CAS PubMed.
- P. A. Crooks, in Nicotine and Related Alkaloids, ed. J. W. Gorrod and J. Wahren, 1993, pp. 81–109 Search PubMed.
- S. Zhou, B. C. H. Hwang, P. S. J. Lakey, A. Zuend, J. P. D. Abbatt and M. Shiraiwa, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 11658–11663 CrossRef CAS PubMed.
- Z. Zhou, S. Zhou and J. P. D. Abbatt, Environ. Sci. Technol., 2019, 53, 12467–12475 CrossRef CAS PubMed.
- M. Shiraiwa, M. Ammann, T. Koop and U. Pöschl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11003–11008 CrossRef CAS PubMed.
- E. Camera, M. Ludovici, M. Galante, J.-L. Sinagra and M. Picardo, J. Lipid Res., 2010, 51, 3377–3388 CrossRef CAS PubMed.
- K. A. Mountfort, H. Bronstein, N. Archer and S. M. Jickells, Anal. Chem., 2007, 79, 2650–2657 CrossRef CAS PubMed.
- K. Nakagawa, D. Ibusuki, Y. Suzuki, S. Yamashita, O. Higuchi, S. Oikawa and T. Miyazawa, J. Lipid Res., 2007, 48, 2779–2787 CrossRef CAS PubMed.
- B. N. Dorakumbura, F. Busetti and S. W. Lewis, Forensic Chem., 2019, 100166 CrossRef CAS.
- N. Shimizu, H. Bersabe, J. Ito, S. Kato, R. Towada, T. Eitsuka, S. Kuwahara, T. Miyazawa and K. Nakagawa, J. Oleo Sci., 2017, 66, 227–234 CrossRef CAS PubMed.
- N. Shimizu, J. Ito, S. Kato, Y. Otoki, M. Goto, T. Eitsuka, T. Miyazawa and K. Nakagawa, Sci. Rep., 2018, 8, 9116 CrossRef PubMed.
- F. Eudier, N. Hucher, C. Picard, G. Savary and M. Grisel, Chem. Res. Toxicol., 2019, 32, 285–293 Search PubMed.
- W. A. Pryor, B. J. Hales, P. I. Premovic and D. F. Church, Science, 1983, 220, 425–427 CrossRef CAS PubMed.
- W. Gehling and B. Dellinger, Environ. Sci. Technol., 2013, 47, 8172–8178 CrossRef CAS PubMed.
- R. Chen and J. J. Pignatello, Environ. Sci. Technol., 1997, 31, 2399–2406 CrossRef CAS.
- H. J. Forman and C. E. Finch, Free Radicals Biol. Med., 2018, 117, 202–217 CrossRef CAS PubMed.
- C. Nishita-Hara, H. Kobayashi, K. Hara and M. Hayashi, GeoHealth, 2023, 7, e2022GH000736 CrossRef PubMed.
- W. Y. Tuet, Y. Chen, L. Xu, S. Fok, D. Gao, R. J. Weber and N. L. Ng, Atmos. Chem. Phys., 2017, 17, 839–853 CrossRef CAS.
- G. C. Lough, J. J. Schauer, J.-S. Park, M. M. Shafer, J. T. DeMinter and J. P. Weinstein, Environ. Sci. Technol., 2005, 39, 826–836 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed methodological information, information on control studies, and qualitative LC-MS/MS analysis. See DOI: https://doi.org/10.1039/d3ea00142c |
| This journal is © The Royal Society of Chemistry 2024 |