
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-day photochemical evolution of organic aerosol from biomass burning emissions†
Abraham
Dearden
a,
Yicong
He
abc,
Ali
Akherati
 am,
Christopher Y.
Lim
d,
Matthew M.
Coggon
e,
Abigail R.
Koss
f,
Joost
de Gouw
gh,
Carsten
Warneke
e,
Lindsay D.
Yee
i,
John H.
Seinfeld
j,
Christopher D.
Cappa
k,
Jesse H.
Kroll
d,
Jeffrey R.
Pierce
l and
Shantanu H.
Jathar
*a
am,
Christopher Y.
Lim
d,
Matthew M.
Coggon
e,
Abigail R.
Koss
f,
Joost
de Gouw
gh,
Carsten
Warneke
e,
Lindsay D.
Yee
i,
John H.
Seinfeld
j,
Christopher D.
Cappa
k,
Jesse H.
Kroll
d,
Jeffrey R.
Pierce
l and
Shantanu H.
Jathar
*a
aDepartment of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA
bState Key Joint Laboratory of Environmental Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing, China
cState Environmental Protection Key Laboratory of Sources and Control of Air Pollution Complex, Beijing, China
dDepartment of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
eChemical Sciences Laboratory, National Oceanic and Atmospheric Administration, Boulder, CO, USA
fTofwerk, Boulder, CO, USA
gDepartment of Chemistry, University of Colorado Boulder, Boulder, CO, USA
hCooperative Institute for Research in Environmental Sciences, Boulder, CO, USA
iDepartment of Environmental Science, Policy, and Management, Berkeley, CA, USA
jDepartment of Chemical Engineering, California Institute of Technology, Pasadena, CA, USA
kDepartment of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA
lDepartment of Atmospheric Science, Colorado State University, Fort Collins, CO, USA
mRamboll, San Francisco, CA, USA
First published on 14th June 2024
Abstract
Biomass burning is an important source of primary and secondary organic aerosol (POA, SOA, and together, OA) to the atmosphere. The photochemical evolution of biomass burning OA, especially over long photochemical ages, is highly complex and there are large uncertainties in how this evolution is represented in models. Recently, Lim et al. (2019) performed and reported on photooxidation experiments of biomass burning emissions using a small environmental chamber (∼150 L) to study the OA evolution over multiple equivalent days of photochemical aging. In this work, we use a kinetic, process-level model (SOM-TOMAS; Statistical Oxidation Model-TwO Moment Aerosol Sectional) to simulate the photochemical evolution of OA in 18 chamber experiments performed on emissions from 10 different fuels. A base version of the model was able to simulate the time-dependent evolution of the OA mass concentration and its oxygen-to-carbon ratio (O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C) at short photochemical ages (0.5 to 1 equivalent days). At longer photochemical ages (>1 equivalent day), the model exhibited poor skill in predicting the OA mass concentration and significantly underestimated the OA O:C. The modeled OA after several equivalent days of photochemical aging was slightly dominated by SOA (average of 57% across all experiments) with the remainder being POA (average of 43% across all experiments). Semi-volatile organic compounds, oxygenated aromatics, and heterocyclics accounted for the majority (89%, on average) of the SOA formed. Experimental artifacts (i.e., particle and vapor wall losses) were found to be much more important in influencing the OA evolution than other processes (i.e., dilution, heterogeneous chemistry, and oligomerization reactions). Adjustments to the kinetic model seemed to improve model performance only marginally indicating that the model was missing precursors, chemical pathways, or both, especially to explain the observed enhancement in OA mass and O:C over longer photochemical ages. While far from ideal, this work contributes to a process-level understanding of biomass burning OA that is relevant for its extended evolution at regional and global scales.
:C) at short photochemical ages (0.5 to 1 equivalent days). At longer photochemical ages (>1 equivalent day), the model exhibited poor skill in predicting the OA mass concentration and significantly underestimated the OA O:C. The modeled OA after several equivalent days of photochemical aging was slightly dominated by SOA (average of 57% across all experiments) with the remainder being POA (average of 43% across all experiments). Semi-volatile organic compounds, oxygenated aromatics, and heterocyclics accounted for the majority (89%, on average) of the SOA formed. Experimental artifacts (i.e., particle and vapor wall losses) were found to be much more important in influencing the OA evolution than other processes (i.e., dilution, heterogeneous chemistry, and oligomerization reactions). Adjustments to the kinetic model seemed to improve model performance only marginally indicating that the model was missing precursors, chemical pathways, or both, especially to explain the observed enhancement in OA mass and O:C over longer photochemical ages. While far from ideal, this work contributes to a process-level understanding of biomass burning OA that is relevant for its extended evolution at regional and global scales.
Environmental significanceBiomass burning is an important source of primary and secondary organic aerosols (POA, SOA) to the atmosphere. Yet, there are large uncertainties in how the photochemical evolution of biomass burning organic aerosol (OA) is represented in models. In this work, we use a process-level model to simulate the photochemical evolution of OA in chamber experiments performed on emissions from many different wood fuels. We found that the model was able to reproduce the OA mass and its oxygen-to-carbon ratio evolution at short photochemical ages (0.5–1 equivalent days) but the model performed less optimally at longer photochemical ages (>1 equivalent day). At longer photochemical ages, the model predicted an OA that was slightly dominated by SOA formed through the oxidation of oxygenated aromatics, heterocyclic compounds, and semi-volatile organic compounds. Our work contributes to a process-level understanding of biomass burning OA that is relevant for evolution at regional and global scales. |
1 Motivation
Biomass burning, which includes wildfires, prescribed burning, agricultural burning, and residential wood combustion, is the largest combustion-related source of primary organic aerosol (POA) and volatile organic compounds (VOCs) to the atmosphere.1–4 Some of the biomass burning VOCs undergo photochemical reactions in the atmosphere to form secondary organic aerosol (SOA), which together with POA, is labeled organic aerosol (OA).5,6 Biomass burning OA contributes significantly to the global atmospheric aerosol burden7 and consequently has large impacts on climate,8,9 air quality,10 and human health.11 Although a large body of research has made significant contributions in characterizing the emissions, chemistry, microphysics, and atmospheric properties of biomass burning OA, its physicochemical evolution is extremely complex and aspects of this atmospheric evolution continue to be uncertain.In a critical review, Hodshire et al.12 primarily analyzed the ‘near-field’ evolution of wildfire OA, characterized across 13 field and 4 laboratory campaigns.13–29 They concluded that, with photochemical aging, the OA mass burden in real wildfire plumes remained relatively constant while the OA mass concentrations consistently increased in laboratory experiments. They further showed that the degree of oxygenation (i.e., oxygen-to-carbon ratio or O:C) for OA, as well as its increase with photochemical age, was larger in real wildfire plumes relative to laboratory experiments. Hodshire et al.12 laid out several hypotheses to explain differences between the field and laboratory observations. Some of these hypotheses, such as the substitution of evaporated POA with SOA production to keep the OA mass approximately constant with plume age,30,31 have been examined in some detail.32,33 Yet, other hypotheses, such as the OA evolution close to the fire or differences in the emissions composition between the field and laboratory, remain untested. The field campaign data included in Hodshire et al.12 focused on quantifying the wildfire OA evolution in diluting and chemically reacting plumes over a few hours of photochemical aging and transported over a few hundred kilometers. Understandably, this is because it is challenging to study biomass burning emissions in the real atmosphere over multiple days as the OA dilutes, mixes, and interacts with OA from other sources, and is subjected to varying photochemical and environmental conditions (e.g., temperature, relative humidity, clouds, precipitation). So, in addition to the discrepancies between field and laboratory observations outlined in Hodshire et al.,12 the multi-day evolution of biomass burning OA remains highly uncertain and continues to be a topic of significant interest. For instance, recently, Sedlacek et al.34 studied the multi-day evolution of OA coatings on black carbon particles from biomass burning and showed that the OA coating increased rapidly over the first few hours (indicating SOA formation) followed by a gradual decrease (indicating OA loss) over the course of a week.
Over the past decade, the photochemical evolution of biomass burning OA has been extensively studied in laboratory experiments using environmental chambers. Although a full literature review remains beyond the scope of this work, we highlight some of the important studies, first for residential wood combustion and then for open burning. Grieshop et al.35,36 and Heringa et al.37 studied the photochemistry of emissions from three different residential wood stoves using several different fuels and one pellet burner. Over a total of 25+ experiments, both studies found strong evidence for SOA formation where the OA mass was enhanced by factors of 1.5 to 4 after photochemical aging. Bruns et al.38 performed five experiments on emissions from a modern wood stove and measured an OA mass enhancement between ∼3 and ∼7. They reported, for the first time, that the OA mass enhancement could mostly be attributed to SOA production from oxidation of phenolic compounds and aromatic hydrocarbons. Hennigan et al.15 performed experiments on combustion emissions from fuels and fuel complexes found in the western United States (US) and saw an OA mass enhancement of 1.7 ± 0.7. They further argued that the POA mass (i.e., fresh OA) was rapidly and chemically processed such that it accounted for less than 20% of the net OA after several hours of photochemical aging. Tkacik et al.14 used a novel approach to study the oxidation chemistry of biomass burning OA. They used a dual chamber setup in which emissions were added to both chambers but only one of the chambers was perturbed (e.g., exposure to UV, exposure to O3, addition of NO to vary VOC:NOX) with the other chamber serving as a control. Although the perturbations were generally found to enhance OA mass (1.78 ± 0.91), they found that the OA mass enhancement was insensitive to the perturbation performed. Relying on the data published by Tkacik et al.14, Ahern et al.13 improved corrections for particle wall losses (average OA mass enhancement was reduced to 1.48, 17% smaller than Tkacik et al.14) and showed that the VOCs quantified using two-dimensional gas-chromatography were responsible for the most of the observed SOA. Biogenic VOCs were found to be the dominant SOA precursors for coniferous fuels and furans were found to be similarly important for grasses. Finally, Akherati et al.39 performed chamber experiments similar to Hennigan et al.15 and Tkacik et al.14 and leveraged detailed speciation of SOA precursors to show that the majority of the SOA formed in these experiments was from the oxidation of oxygenated aromatic (e.g., phenols, methoxyphenols) and heterocyclic (e.g., furans) VOCs. Although these previous chamber analyses have been extremely insightful in understanding the photochemical evolution of biomass burning OA, the experiments have only been performed over short photochemical ages (typically, <12 equivalent hours at an OH concentration of 1.5 × 106 molecule per cm3). While these data can and have been used to predict and interpret OA evolution close to the source (<100 km),33 they say little about the multi-day/week evolution, relevant for regional and global scales.
Only two studies so far have attempted to study the photochemical evolution of biomass burning OA over multiple days and up to a week in laboratory experiments. Ortega et al.29 sampled and oxidized smoke using an oxidation flow reactor (OFR, 13 L) and performed 25 different experiments on emissions from 16 different fuels. The smoke was sampled from a large room (3000 m3) that was filled by combusting a small amount of fuel (0.1–1 kg). When aged over an equivalent of five days, they found an average enhancement of 1.42 ± 0.36 in OA mass concentrations. The enhancement in OA mass was not found to correlate with the mass of speciated VOCs but did correlate with the initial POA, indicating a key role for semi-volatile organic compounds (SVOCs) as SOA precursors. Lim et al.40 sampled and oxidized smoke using a small chamber (150 L) and analyzed 20 different experiments performed on 11 different fuels; there was significant overlap with the fuels used in Ortega et al.29 The smoke was sampled from a large exhaust stack that ported biomass burning emissions from a small fire (0.25–6 kg of fuel) to the chamber. Lim et al.40 saw a much larger enhancement in OA mass concentrations (3.5 ± 1.7) compared to Ortega et al.29 as well as the chamber studies described earlier,13–15,35–39 but argued that the higher enhancement could be explained by differences in photochemical age and accounting for measurement artifacts (i.e., a declining collection efficiency in the aerosol mass spectrometer with aging). In contrast to Ortega et al.,29 Lim et al.40 found that the OA mass enhancement correlated with fresh emissions of non-methane organic compounds, specifically those that were smaller than monoterpenes (molecular weight <136 g mol−1). Both studies undertook an empirical investigation of the changes in OA mass and composition with oxidation but neither leveraged models to better understand the precursors and processes of OA with photochemical age.
Over the past few decades, chambers and OFRs have served as workhorses, providing the underlying data needed to develop, evaluate, and optimize gas/aerosol chemical mechanisms and parameterizations for use in air quality and climate models. But while chambers and OFRs aim to simulate the photochemical evolution in the real atmosphere, this oxidation chemistry and microphysics is subject to experimental artifacts. For example, chambers and flow reactors are subject to both particle and vapor wall losses, artifacts that can significantly influence measurements of aerosol mass and properties. Particle wall losses are relatively easy to correct for if size- and time-dependent loss rates can be measured.41 Vapor wall losses, on the other hand, require a kinetic treatment for the uptake and release of organic species, a process that depends on volatility and composition.42,43 Furthermore, at the unusually high oxidant concentrations encountered, especially in OFRs (factors of 20 to 100 higher than those in the ambient atmosphere), the timescales for oxidation are much shorter than those for gas/particle partitioning.44,45 This timescale difference leads to an organic species undergoing multiple generations of oxidation to form highly oxygenated, low-volatility products in the gas phase, a process that is relatively less frequent in the real atmosphere. Finally, high oxidant concentrations in OFRs also tend to elevate the importance of heterogeneous oxidation reactions (relative to gas/particle partitioning), which tend to reduce aerosol mass through fragmentation reactions followed by evaporation of the semi-volatile/volatile products.46,47 Recently, He et al.48 showed that when different aerosol processes and their corresponding timescales were appropriately simulated using a kinetic model, the model was able to reproduce observed differences in aerosol formation and composition between a chamber and OFR study for α-pinene SOA. Similar kinetic models need to be used to simulate and interpret chamber and OFR data on biomass burning OA.
In this work, we combined laboratory data40 and a process-level kinetic model (SOM-TOMAS) to simulate the photochemical evolution of OA in 18 environmental chamber experiments performed on combustion emissions from 10 different fuels. The model-simulated dilution, multigenerational gas-phase chemistry, autoxidation reactions, phase-state-influenced gas/particle partitioning, heterogeneous oxidation, oligomerization reactions, and experimental artifacts (i.e., particle and vapor wall losses). Model predictions were compared to measurements of VOCs and OA mass and composition. A suite of sensitivity simulations was performed to study the influence of individual processes on OA evolution and to assess model performance at longer photochemical ages.
In a very recently published paper, He et al.49 used a kinetic model to also simulate the experimental data from Lim et al.40 As He et al.49 and this work use the same primary dataset and because He et al.49 was published while this work was in review, we take special care to contrast the model results and interpretation from both efforts in the final section of this paper (Summary and Discussion).
2 Environmental data and modeling methods
2.1 Environmental chamber data
In this work, we simulated the photochemical evolution of biomass burning OA for the chamber experiments described in Lim et al.40 A detailed account of the physical and instrument setup, design of experiments, and chamber operation is provided in Lim et al.,40 with additional details in McClure et al.50 and Cappa et al.51 A brief description, relevant to the data used in this work, is provided below. The chamber experiments outlined in Lim et al.40 were performed at the Fire Sciences Laboratory (FSL) in Missoula, MT as part of the Fire Influence on Regional and Global Environments Experiment in 2016 (FIREX-2016).52 During these experiments, a small amount of fuel (0.25 to 6 kg) was combusted using an electric igniter and emissions from these burns were channeled into a large stack (1.6 m diameter, 17 m tall), located right above the fuel bed. A long duct (0.2 m diameter, ∼30 m long) was used to port emissions from the stack and eventually into a small PFA (perfluoroalkoxy) chamber (150 L) using an ejector-diluter system. Residence times in the duct were ∼2 s, which should have minimized losses of gases and particles during transport. Ozone (O3) was continuously added to the chamber after smoke addition and a single UV-C light with a peak at 254 nm was used to photolyze the O3 to produce high concentrations of hydroxyl radicals (OH). The photooxidation experiments were performed for a length of 30 to 60 minutes before the chamber was flushed with clean air to prepare for the next experiment.A Scanning Electrical Mobility Spectrometer (SEMS), High-Resolution Aerosol Mass Spectrometer (HR-AMS), and Single-Particle Soot Photometer (SP2) were used to characterize aerosol mass concentrations and composition. A Proton Transfer Reaction-Time of Flight-Mass Spectrometer (PTR-ToF-MS) was used to measure mixing ratios for VOCs that included key SOA precursor classes: oxygenated aromatics, heterocyclics, aromatic hydrocarbons, and biogenic VOCs.53–55 We should note that measurements from the same instrument were used in Akherati et al.39 to analyze and simulate SOA formation from biomass burning VOCs. Lim et al.40 performed a total of 56 chamber experiments but characterized the photochemical evolution in only half of those experiments. In this work, we simulated the OA evolution for 18 chamber experiments where complete VOC, aerosol, and environmental data were available. Experimental details including the fuels used are listed in Table S1.†
To orient the reader, we describe the OA data from Lim et al.40 used in this work (i.e., 18 chamber experiments). Two-thirds of the chamber experiments were performed on coniferous fuels (Douglas fir, Engelmann spruce, loblolly pine, lodgepole pine, ponderosa pine, and subalpine fir) and the remaining third were performed on a variety of different fuels (grass (bear grass), shrub (chaparral), and other (excelsior or wood shavings, dung)). The dilution and particle-wall-loss corrected OA mass concentrations and OA O:C are shown in Fig. S1† for all 18 chamber experiments. The initial OA mass concentrations and O:C varied between 16 and 330 μg m−3 and between 0.20 and 0.62, respectively. With photochemical aging, the OA mass concentrations and OA O:C increased in nearly all chamber experiments with significant variability in the enhancement across experiments (0.9–4.8× for OA mass concentrations and 1.3–4.0× for OA O:C). The final photochemical age varied between slightly less than 1 equivalent day to nearly 11 equivalent days. We did not observe a relationship between initial OA and O:C (not shown) but the initial OA mass concentrations showed a strong positive correlation with several SOA precursor classes (Fig. S2a–e†). On average, the total SOA precursors were more than a factor of two larger than the initial POA, highlighting the large potential to form SOA with oxidation (Fig. S2a†). The SOA precursor composition varied some between the chamber experiments but had an average profile of 40% heterocyclics, 29% oxygenated aromatics, 20% aromatic hydrocarbons, and 11% biogenic VOCs, roughly consistent with Akherati et al.39 SVOCs were considered to be SOA precursors but since they were estimated in this work from the initial OA measurements (see Section 2.2.2), they were expected to correlate with initial OA and hence not shown in Fig. S2.† The OA mass enhancement, calculated as an initial value subtracted from the final value, exhibited a positive correlation with the initial POA mass concentrations (R2 = 0.54) (Fig. S3a†), similar to what Ortega et al.29 discovered, and with the initial SOA precursor concentrations (R2 = 0.43) (Fig. S3c†). In contrast, the O:C enhancement, calculated as an initial value subtracted from the final value, exhibited a weaker negative correlation with the initial POA mass concentrations (R2 = 0.17) (Fig. S3e†) and with the initial SOA precursor concentrations (R2 = 0.20) (Fig. S3g†). The correlations were much weaker when the OA mass and O:C enhancement were calculated as a ratio of the final value to the initial value (Fig. S3b, S3d, S3f, and S3h†). Regardless, Fig. S3a–d† suggest that the OA enhancement might be driven by the oxidation of SVOCs, which are in equilibrium with POA, in addition to the VOCs.
2.2 Kinetic organic aerosol model
A detailed description of the SOM-TOMAS model and the governing equations can be found in previous publications.39,48 A brief overview is provided below. Within SOM, five parameters are used to track the oxidation chemistry of the VOC and its oxidation products (i.e., model species) by explicitly simulating functionalization and fragmentation reactions: (i–iv) pf,1 to pf,4, mass yields for four functionalized products that add one, two, three, and four oxygen atoms to the carbon backbone, respectively; and (v) mfrag, a parameter used to calculate the probability of fragmentation (Pfrag) based on the O:C ratio of the model species (Pfrag = (O:C)mfrag). ΔLVP, a sixth parameter, is used to quantify the decrease in volatility of the model species with the addition of an oxygen atom. Being a statistical model, the above-mentioned parameters are not always interpretable and have to be considered together to determine their ability to form SOA. The volatility (c*) and reaction rate constant with OH (kOH) are parameterized as a function of the model species' carbon (NC) and oxygen (NO) number; kOH for the VOC is based on literature values. Molecular weights for the model species are calculated from knowing the carbon, oxygen, and hydrogen (based on the remaining valence) numbers. Based on the diffusive-reactive framework described in Zaveri et al.,61 the SOM-TOMAS model simulates the kinetic gas/particle partitioning of the model species by considering the particle-phase diffusivity of the absorbing OA (Db). The SOM-TOMAS model was recently updated to simulate autoxidation reactions, heterogeneous oxidation via OH, and oligomerization reactions.48 Autoxidation is modeled by directly prescribing an empirical yield (fHOM) for highly oxygenated organic molecules (HOM), informed by previous laboratory experiments.62 Heterogeneous oxidation is modeled as a surface reaction of a model species in the particle phase with OH and assuming that the product distribution after the chemical reaction is the same as that in the gas phase; collisions between the model species and OH that lead to a chemical reaction are controlled by the uptake coefficient (γ).47 Finally, oligomerization is modeled by tracking the reversible formation and dissociation of dimers, characterized by forward and reverse reaction rates, respectively.63
The SOM-TOMAS model can simulate size-dependent losses of particles to the chamber walls. However, in this study, only a single, bulk particle wall-loss rate was available to model particle wall losses (details described in Section 2.2.2). Vapor losses to the chamber walls are modeled, following the work of Matsunaga and Ziemann.64 Here, the first-order uptake to the walls is assumed to be equal to kvap,on and the release of vapors from the walls (kvap,off) is modeled using absorptive partitioning theory with the chamber wall serving as an absorbing mass with an effective mass concentration of Cw μg m−3; Cw is parameterized as a function of the partitioning model species' c*, following Krechmer et al.42 Given the short duration of the experiments simulated here (<1 hour), we did not consider transport of the absorbed vapors through the chamber wall. Huang et al.43 have argued that this transport can influence net vapor wall loss rates during a chamber experiment but noted that this process was relevant only for experiments longer than 10 hours.
Lim et al.40 diluted the chamber with clean air doped with O3 to offset the volume sampled out by the instruments and to keep the chamber volume constant at 150 L. This dilution was significant enough (2 to 3.6 h−1) that dilution ended up being the most important process that affected concentrations inside the chamber. Therefore, we decided to explicitly model dilution in the SOM-TOMAS model since it was likely to affect gas/particle partitioning of OA. The dilution rate was calculated by first fitting a double exponential function to the mixing ratios for hydrogen cyanide (HCN) and then using the function to determine a time-dependent dilution rate, specific to each chamber experiment. We assumed HCN to be an inert tracer that did not react with OH or O3 and was not lost to the walls. Previous work has tended to use acetonitrile as an inert tracer but we did not find significant differences in the bulk dilution calculated using HCN or acetonitrile. The HCN data were preferred since they were smoother in time than those for acetonitrile. Numerically, dilution was modeled at each model time step by multiplying the chamber concentrations in the model with the incremental dilution rate. Time series data for HCN for a representative chamber experiment (ponderosa pine, Fire 38) and the corresponding time-dependent dilution rate are shown in Fig. S4.†
PTR-ToF-MS data were used to initialize mixing ratios for the SOA precursors. The PTR-ToF-MS data have been described in detail in Coggon et al.55 and the SOA precursor treatment was identical to that described in Akherati et al.39 A total of 86 unique VOCs were considered to form SOA and the full list of precursors is provided in Table S2† (reproduced from Akherati et al.39). The 86 VOCs were assigned to one of nine SOA precursor surrogates (benzene, toluene, m-xylene, naphthalene, isoprene, α-pinene, phenol/guaiacol, syringol, alkylfurans) for which SOM-TOMAS parameters have been developed from historical chamber data or were developed exclusively in this work. SOM-TOMAS parameters for these surrogates, derived from fitting SOM-TOMAS model predictions to chamber measurements of SOA mass concentrations and, when available, SOA O:C, are provided in Table S3;† note that these parameters are representative of SOA formation under low NOX conditions. Coggon et al.55 argued, based on simulating the gas-phase chemistry for a few of the Lim et al.40 fires, that peroxy radicals (RO2) from higher carbon number VOCs exclusively reacted with the hydroperoxyl radical (HO2) (∼75%) or with itself (i.e., forms gas-phase dimers) (∼25%). Accordingly, the oxidation chemistry is expected to be more representative of that encountered under low NOX conditions. Based on an analysis of wildfire plumes, two recent studies have argued that the photochemical evolution a few hours after emission shifts to being NOX sensitive or representative of low NOX conditions.65,66 Chamber walls, however, can serve as a continuous source for NOX, a pathway Coggon et al.55 did not consider, and may confound the choice between using low versus high NOX parameters to simulate SOA formation.67 Hence, we performed simulations with high NOX parameters (listed in Table S4†) to examine the sensitivity in model predictions. We should point out that since the SOM parameters only inform the trajectory of the oxidation chemistry in the carbon–oxygen grid, model predictions of the SOA mass yield for a specific precursor are likely to be different from that for the surrogate. Although the SOM-TOMAS model was run to simulate the oxidation chemistry separately for each of the 86 VOCs, VOC and SOA data were aggregated to present results across five broad precursor classes: oxygenated aromatics, heterocyclics, aromatic hydrocarbons, biogenic VOCs, and SVOCs (see next paragraph).
POA from biomass burning sources is known to be semi-volatile and reactive.68–70 Hence, we used the volatility distribution reported in May et al.70 to distribute POA and SVOCs in equilibrium with POA, within the SOM grid. This was done by first determining the POA + SVOC mass distribution in a volatility basis set (VBS) and then mapping the mass from each c* bin in the VBS to the SOM model species that had a closely matching c* and an O:C that was similar to the initial observations of the bulk POA O:C. The chemical evolution of the POA + SVOC mass was simulated assuming that the oxidation chemistry was the same as that for multi-ring aromatics (naphthalene served as the surrogate), based on the work of Akherati et al.33
Following Barmet et al.,71 dilution-corrected mixing ratios of deuterated butanol (D9) were used to calculate time-dependent OH concentrations, specific to each chamber experiment. Dilution-corrected data for D9 and the corresponding OH concentrations and exposures for a representative chamber experiment (ponderosa pine) are shown in Fig. S5.† The average OH concentration was 4 × 108 molecule per cm3 and the average OH exposure by the end of the experiment was equivalent to 6.6 days of photochemical aging at an OH concentration of 1.5 × 106 molecule per cm3. All precursors were only assumed to react with OH to form SOA. High amounts of O3 were added to the chamber before turning the lights on (with O3 continuously added during the experiment) and, hence, any biogenic VOCs from the smoke would have fully reacted with O3 before the start of the experiment. We did not model this process because, as we will show later, biogenic VOCs contributed to a small fraction of the total SOA.
Lim et al.40 determined a bulk particle wall loss (PWL) rate constant (kPWL = 0.028 min−1, τPWL = 36 min) by fitting an exponential function to dilution-corrected POA mass concentration data from a ‘dark’ chamber experiment performed on lodgepole pine; in this specific experiment, the lights were not turned on and there was no photochemistry. This particle wall-loss rate represents an upper bound estimate since the POA mass could have evaporated with dilution72 and from losses of semi-volatile vapors in equilibrium with the POA to the chamber walls.73 To account for these additional loss processes, we ran the SOM-TOMAS model for this dark experiment and simulated dilution, kinetic gas/particle partitioning, and particle and vapor wall losses; all chemical reactions were turned off. The model was run with several PWL rates (τPWL = 36, 72, 108, and ∞ min) and results are presented in Fig. S6.† We found that a PWL rate of kPWL = 0.014 min−1 (τPWL = 72 min) best reproduced the observations, indicating that the rate presented in Lim et al.40 was likely overestimated. The same bulk PWL rate was used to model losses of particles to the chamber walls for all 18 chamber experiments.
The vapor wall loss rate has not been quantified for the chamber used by Lim et al.40 Assuming that the vapor wall loss rate scales as a linear function of the surface area to volume ratio for the chamber,74 we used historical estimates from five separate environmental chambers to calculate a kvap,on of ∼2 × 10−2 s−1 (τGWE = 0.4 min) for the Lim et al.40 chamber; Paul Scherrer Institute (5.5 m3, kvap,on of 3.3 × 10−3 s−1, τGWE = 2.5 min),75 University of Colorado Boulder (8 m3, kvap,on of 5.2 × 10−4 s−1, τGWE = 16 min),42 Colorado State University (10 m3, kvap,on of 1.3 × 10−3 s−1, τGWE = 6.5 min),58 Georgia Institute of Technology (13 m3, kvap,on of 10−4 s−1, τGWE = 83 min),76 and California Institute of Technology (30 m3, kvap,on of 4 × 10−4 s−1, τGWE = 21 min).43 The τGWE values were calculated based on the time required to achieve gas–wall equilibrium for a species with a c* of 103 μg m−3.
| Simulation | Dilution | Particle wall losses | Vapor wall losses | Semi-volatile POA | Heterogeneous chemistry | Oligomerization | Phase state | NOX |
|---|---|---|---|---|---|---|---|---|
| a Does not include autoxidation reactions. | ||||||||
| Base | Yes | Yes | Yes | Yes | Yes | Yes | Liquid | Low |
| Dilution off | No | Yes | Yes | Yes | Yes | Yes | Liquid | Low |
| PWL off | Yes | No | Yes | Yes | Yes | Yes | Liquid | Low |
| PWL × 2 | Yes | Yes; 2× | Yes | Yes | Yes | Yes | Liquid | Low |
| VWL off | Yes | Yes | No | Yes | Yes | Yes | Liquid | Low |
| Non-volatile POA | Yes | Yes | Yes | No | Yes | Yes | Liquid | Low |
| Het. Chem. off | Yes | Yes | Yes | Yes | No | Yes | Liquid | Low |
| Oligomerization off | Yes | Yes | Yes | Yes | Yes | No | Liquid | Low |
| High NOXa | Yes | Yes | Yes | Yes | Yes | Yes | Liquid | High |
| D b = 10−19 m2 s−1 | Yes | Yes | Yes | Yes | Yes | Yes | Viscous | Low |
We also performed the following simulations with modifications to the model inputs or chemical schemes. First, the heterogeneous oxidation scheme was simplified such that the model species' reaction with OH was assumed to form a single oxidation product with four oxygen atoms added to the reactant. This scheme, which simulated an aggressive uptake of oxygen into the particle phase, was used to run simulations with two different uptake coefficients (γ = 1, 6). While an uptake coefficient of 1 reflects an oxidation reaction at the OH collision limit, a value greater than 1 assumes that a single OH collision with a particle will result in multiple oxidation reactions; a γ of 6 was used to represent an extreme upper bound case (Nah et al., 2013).77 Second, Coggon et al.55 were able to speciate more than 150 VOCs using the PTR-ToF-MS, of which only 86, as identified by Akherati et al.,39 were considered to form SOA. To investigate whether some of the smaller carbon number VOCs were responsible for SOA formation (Table S5†), we modeled the oxidation chemistry from 20 additional VOCs using n-dodecane as a surrogate (since most were acyclic organic compounds). Third, we explored how an average SOA precursor profile based on an average of 57 fires sampled during FIREX-2016 affected model predictions.53 Initial SOA precursor concentrations for each chamber experiment were determined by multiplying the averaged emissions ratio of the SOA precursor with acetonitrile with the initial experiment-specific acetonitrile concentrations. Finally, we performed 100 Monte-Carlo simulations where we randomized the distribution of POA + SVOC mass in the SOM grid while ensuring that the mass distribution reproduced the volatility behavior observed by May et al.70 and matched the initial OA O:C. All of the simulations described above were performed on all 18 chamber experiments.
3 Results
3.1 Example results from a ponderosa pine chamber experiment
Predictions from simulations performed with the SOM-TOMAS model for a representative fuel from the western US and experiment (ponderosa pine, Fire 38 in Table S1†) are compared against measurements in Fig. 1. This specific experiment was chosen since it was broadly illustrative of the model performance across all 18 chamber experiments (described in Section 3.2 and showcased in Fig. 3, 4, and 5). Model predictions in Fig. 1a and b are from the Base simulation (solid line) and from sensitivity simulations (dash-dotted lines) performed by systematically turning off or adjusting a specific process. Predictions and measurements of OA mass concentrations presented in panels (a), (c), and (d) have been corrected for dilution and PWL; being a ratio, the O:C data in panel (b) remain uncorrected and represent those for the suspended OA.
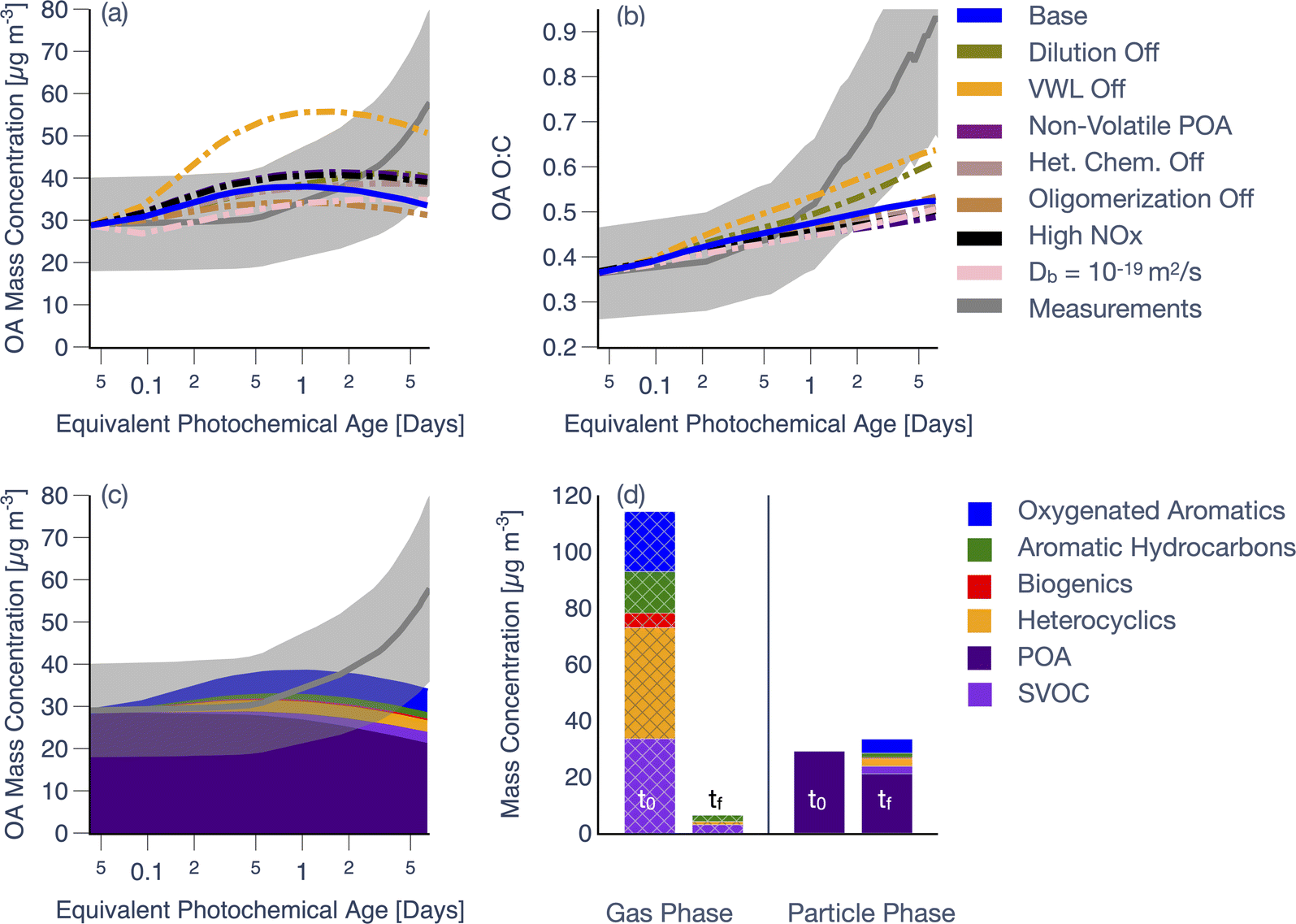 | ||
| Fig. 1 SOM-TOMAS model predictions for (a) OA mass concentration and (b) OA O:C compared against measurements as a function of photochemical age (assuming an OH concentration of 1.5 × 106 molecule per cm3) for the ponderosa pine chamber experiment (Fire 38). (c) ‘Base’ model predictions resolved by precursor class compared against bulk measurements of OA mass concentration. (d) Initial (t0) and final (tf) gas- and particle-phase predictions resolved by precursor class from the ‘Base’ simulation. In panels (a–c), mean measurements are shown with a gray solid line with the shaded region representing the 2-sigma uncertainty. Sensitivity simulation results in (a) and (b) are shown with dash-dotted lines. The hatching in (d) shows precursor concentrations in the gas-phase. The modeled and measured mass concentrations in panels (a, c, and d) have been corrected for dilution and additionally for PWL for OA. | ||
As shown in Fig. 1a, the model-measurement comparison for OA mass concentrations varied with photochemical age. For instance, the model produced a small increase in OA mass concentrations through 0.5–1 equivalent day of photochemical aging when the measurements were relatively flat. At photochemical ages longer than 0.5–1 equivalent day, the model produced a small decrease in the OA mass concentrations (net increase of 13% compared to the initial OA), which deviated from the measurements that continued to rise (net increase of 100% compared to the initial OA). Model predictions of OA O:C, as shown in Fig. 1b, closely followed the measurements up to one equivalent day of photochemical aging. While model predictions of OA O:C continued to increase thereafter, the modeled increase with respect to the initial OA O:C was dwarfed (net increase of 42%) relative to the increase experienced by the measurements (net increase of 155%). The change in the predicted OA composition (Fig. 1c) and O:C (Fig. 1b) stemmed from the production of SOA, primarily driven by the oxidation of SVOCs, oxygenated aromatics, and heterocyclics, with minor contributions from aromatic hydrocarbons and biogenic VOCs. After slightly more than six equivalent days of photochemical aging, the OA was dominated by POA (63%) in the model with a smaller contribution from SOA (37%).
The temporal trends in modeled OA mass concentrations, shown in Fig. 1a, can be explained as follows. The VOC reactivities (i.e., reaction rate constant with OH × OH concentration) for the key SOA precursor classes (i.e., SVOCs, oxygenated aromatics, heterocyclics) were high enough that the precursor mixing ratios should have been depleted within 1 equivalent day of photochemical aging. Model predictions and measurements of precursor decay, grouped by precursor class as a function of photochemical age, are shown in Fig. S7† for this experiment; SVOCs were not included in Fig. S7† since there were no direct measurements to compare against. Model predictions of precursor decay seemed to agree the most with observations for aromatic hydrocarbons but were largely inconsistent with the measurements for the three other precursor classes. This was most likely because there were significant interferences at the mass-to-charge ratios used to track the precursors, from fragmentation of oxidation products during ionization in the PTR-ToF-MS.55 The high VOC reactivities and rapid precursor decay resulted in the small burst in SOA production initially (Fig. 1a). However, with few precursors left after one equivalent day, the model was unable to produce much more SOA. This also indicated that there were minor contributions to SOA from slower reacting precursors (i.e., aromatic hydrocarbons) and from the multigenerational chemistry of the oxidation products left behind after the initial SOA burst. Furthermore, any SOA production past one equivalent day of photochemical aging was partially balanced by evaporation of POA and SOA from dilution of the chamber volume (2.4 h−1). The end result was that the predictions of OA mass concentrations increased earlier in the experiment but continued to slightly decrease with aging, inconsistent with the shape of the measurements.
Results from the sensitivity simulations provided additional insight on the relative importance of the various processes. Simulations performed with a non-reactive POA, using high NOX parameters, semi-solid OA, or without dilution, heterogeneous oxidation, or oligomerization, individually, resulted in no significant change in OA mass concentrations (±20%) relative to the Base simulation. We note that the use of a semi-solid OA flattened the OA evolution, relative to the Base simulation, by slowing down the loss of POA to evaporation and the addition of SOA via condensation. The use of a slower dimer formation (kf = 10−25 cm3 per molecule per s) and dissociation (kr = 1.1 × 10−3 s−1) rate, informed by the work of He et al.48, did not produce large swings in the model predictions, relative to the Base simulation (not shown). Simulations performed with vapor wall losses turned off (VWL Off), however, seemed to produce a much stronger change in OA mass concentrations (±50%), relative to the Base simulation. The VWL Off simulation predicted a large spike in OA mass concentrations at short photochemical ages highlighting that vapor wall losses are an important artifact to consider for this small chamber. We should note that VWL likely plays a dynamic role in these experiments since vapors, lost to the walls initially when the concentrations are higher, may be released from the walls later in the experiment when the concentrations are depleted from continuous dilution of the chamber volume; our modeling accounts for this process.
As varying the PWL rate affected the PWL-correction, these results, including those from the Base simulation, are presented separately in Fig. 2. Simulations performed with particle wall losses turned off (PWL off) and particle wall loss rates doubled (PWL × 2) seemed to produce a very small increase or decrease (<1%), respectively, in OA mass concentrations, relative to the Base simulation. This meant that over the range of PWL rates examined here, the loss of particles to the wall did not alter the photochemistry and microphysics inside the chamber in ways that resulted in a different PWL-corrected prediction for OA mass concentrations. In contrast, assumptions about the particle wall loss rate had a significant influence on PWL-corrected estimates of OA mass concentration, especially at photochemical ages longer than 1 equivalent day – clearly visualized in the PWL × 2 case. These results highlight the important role PWL rates likely play in small environmental chambers. While the PWL-off simulation produced the best agreement amongst the three simulations with the OA mass concentration measurements near the end of the experiment, it is unlikely that there were no losses of particles to the chamber walls. Rather, this suggests that a bulk, time-independent particle wall-loss rate informed by a single dark experiment and used to correct for losses in all experiments, may add to the uncertainty in simulating the experiment-specific trends in OA evolution.
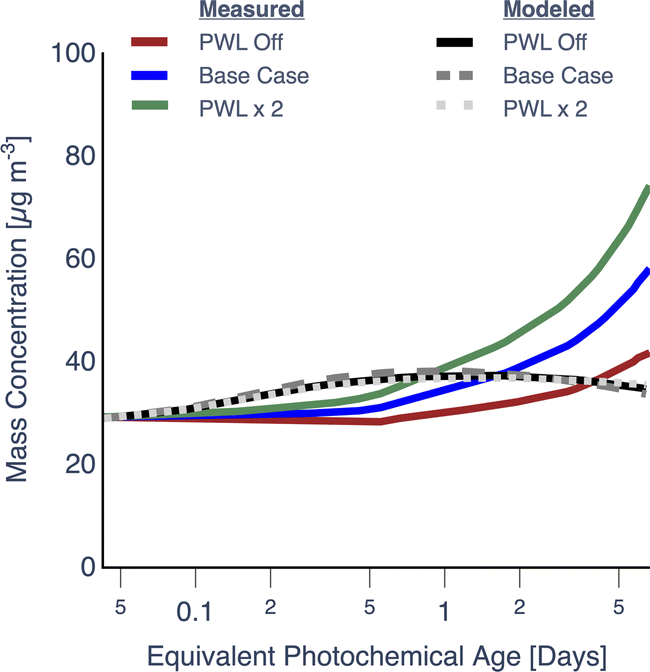 | ||
| Fig. 2 SOM-TOMAS model predictions and measurements for OA mass concentrations assuming no particle wall losses (PWL off), τPWL = 72 min (Base), and τPWL = 36 min (PWL × 2) for the ponderosa pine chamber experiment (Fire 38). The modeled and measured OA mass concentrations have been corrected for dilution and PWL. | ||
Most of the sensitivity simulations produced a smaller spread in the OA O:C evolution and even when they did (i.e., dilution off, VWL off), the predictions significantly underestimated the change in measured OA O:C after one equivalent day of photochemical aging (Fig. 1b). We were surprised by the no heterogeneous oxidation result since, in recent work from our group on studying photooxidation of α-pinene in OFR experiments, we found heterogeneous oxidation to be much more important in controlling the SOA O:C over long photochemical ages.48
To highlight the highly oxidizing environment inside the chamber, we plot the initial and final precursor (and OA) concentrations in Fig. 1d. Over the course of six equivalent days of photochemical aging (∼35 min of clock time), nearly all of the primary precursors (SVOCs, oxygenated aromatics, heterocyclics, aromatic hydrocarbons, and biogenic VOCs) were consumed. Based on the net loss of precursors and net production of SOA, we calculated an end-of-experiment SOA mass yield of 11% for this experiment.
3.2 Photochemical evolution of OA from eighteen different fires
Model predictions of OA mass concentrations and O:C are compared against measurements for 18 chamber experiments in Fig. 3 and 4, respectively. Results are shown for the Base simulations and for simulations that used the high NOX parameters. In addition, predictions of the normalized OA composition from the Base simulations for the 18 chamber experiments are shown in Fig. S8† at two different time points: 0.5 equivalent days and at the end of the experiment. In Fig. 3 and 4, the model-measurement comparison was found to be highly variable (more so for OA mass concentrations) and a few of the key findings are outlined below. First, very broadly, model predictions of OA mass concentration seemed to agree with the measurements at shorter photochemical ages but the model-measurement comparison varied quite significantly at longer photochemical ages. In addition, the model was not able to reproduce the shape of the OA mass concentration with time for many of the experiments. As the heterogeneity in photochemical ages across the chamber experiments makes this hard to notice in Fig. 3, we reexamined this comparison, resolved by photochemical age, in Fig. 5 (described later in this section). In 8 out of 18 chamber experiments, the model-predicted OA mass concentration was clearly outside the measured uncertainty range by the end of the experiment; overestimated in 3 and underestimated in 5 chamber experiments. The modeled OA mass concentration profile with photochemical age was contained within the measurement uncertainty envelope in 7 out of the 18 chamber experiments and in only 3 out of 18 chamber experiments did the model predictions appear to follow the mean in the measurements.
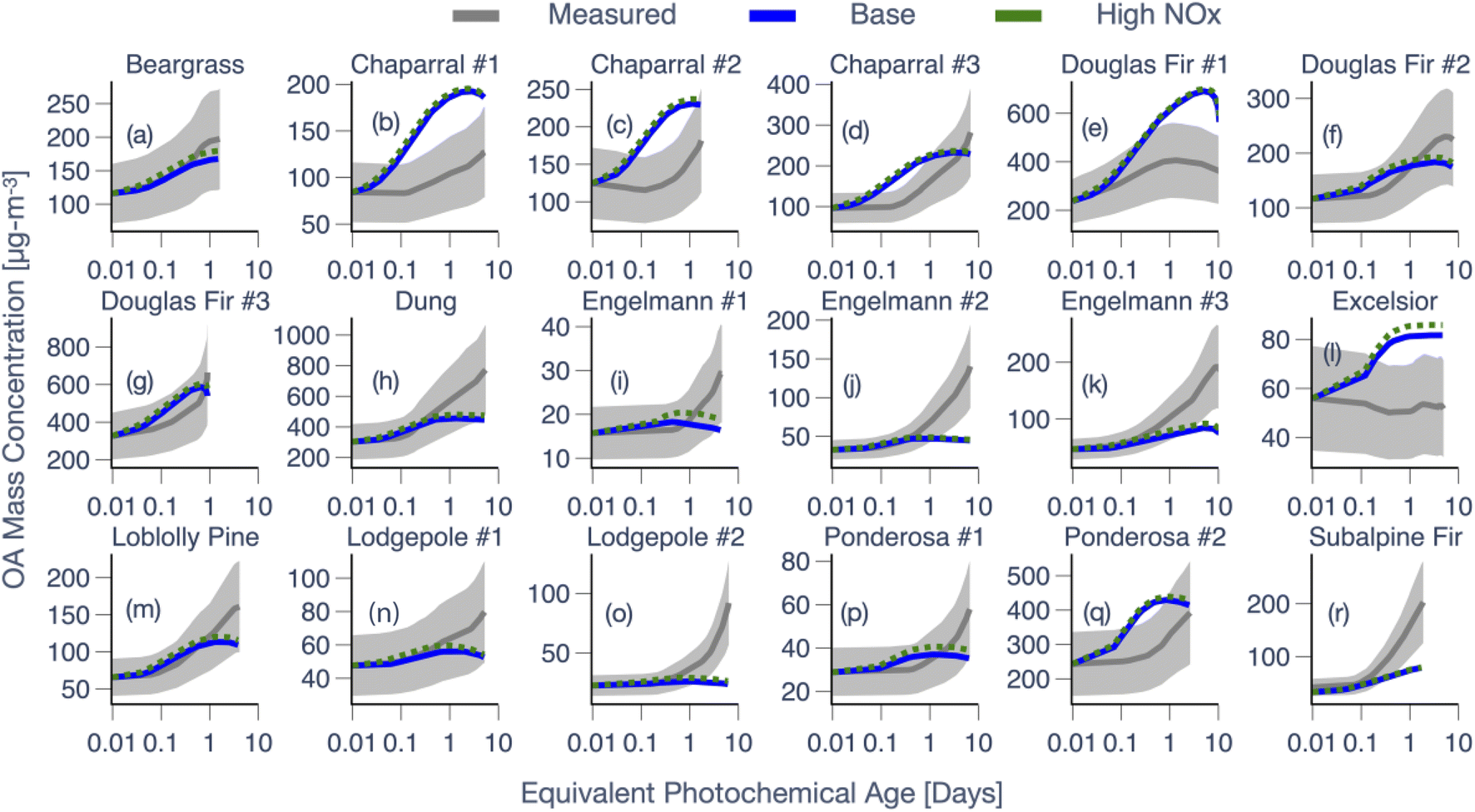 | ||
| Fig. 3 SOM-TOMAS model predictions from the Base and high NOX simulations compared against measurements of OA mass concentration for all 18 experiments (a–r). Each panel has a unique y-axis scale but the same log-transformed x-axis scale. Experiments based on the same fuel have been numbered serially and the panels have been organized alphabetically by fuel. Mean measurements are shown with a gray solid line with the shaded region representing the 2-sigma uncertainty. All OA mass concentrations have been corrected for dilution and PWL. | ||
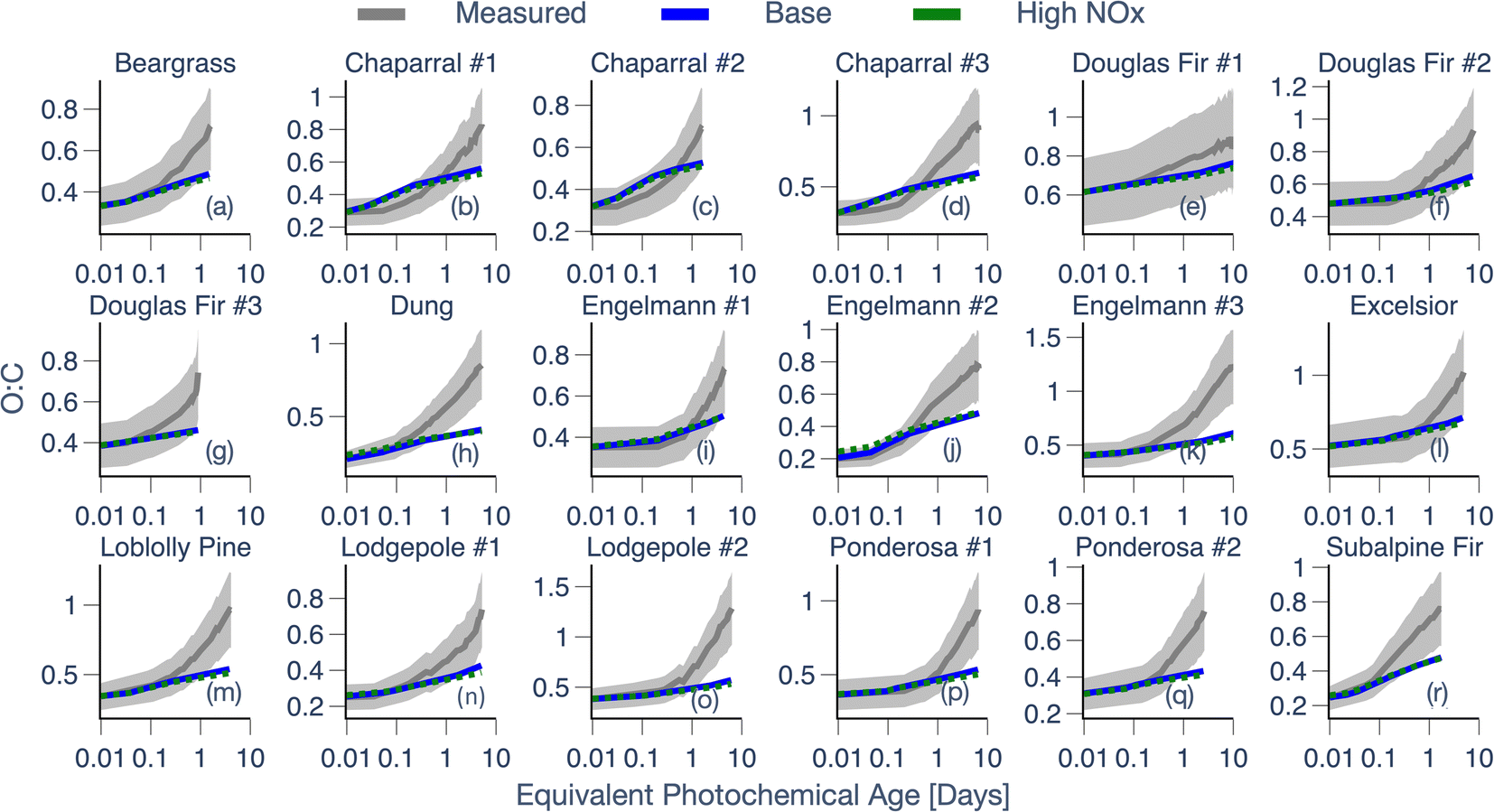 | ||
| Fig. 4 Same as Fig. 3 but for OA O:C for all 18 experiments (a–r). The OA O:C is calculated for the suspended OA and it did not require corrections for dilution or PWL. | ||
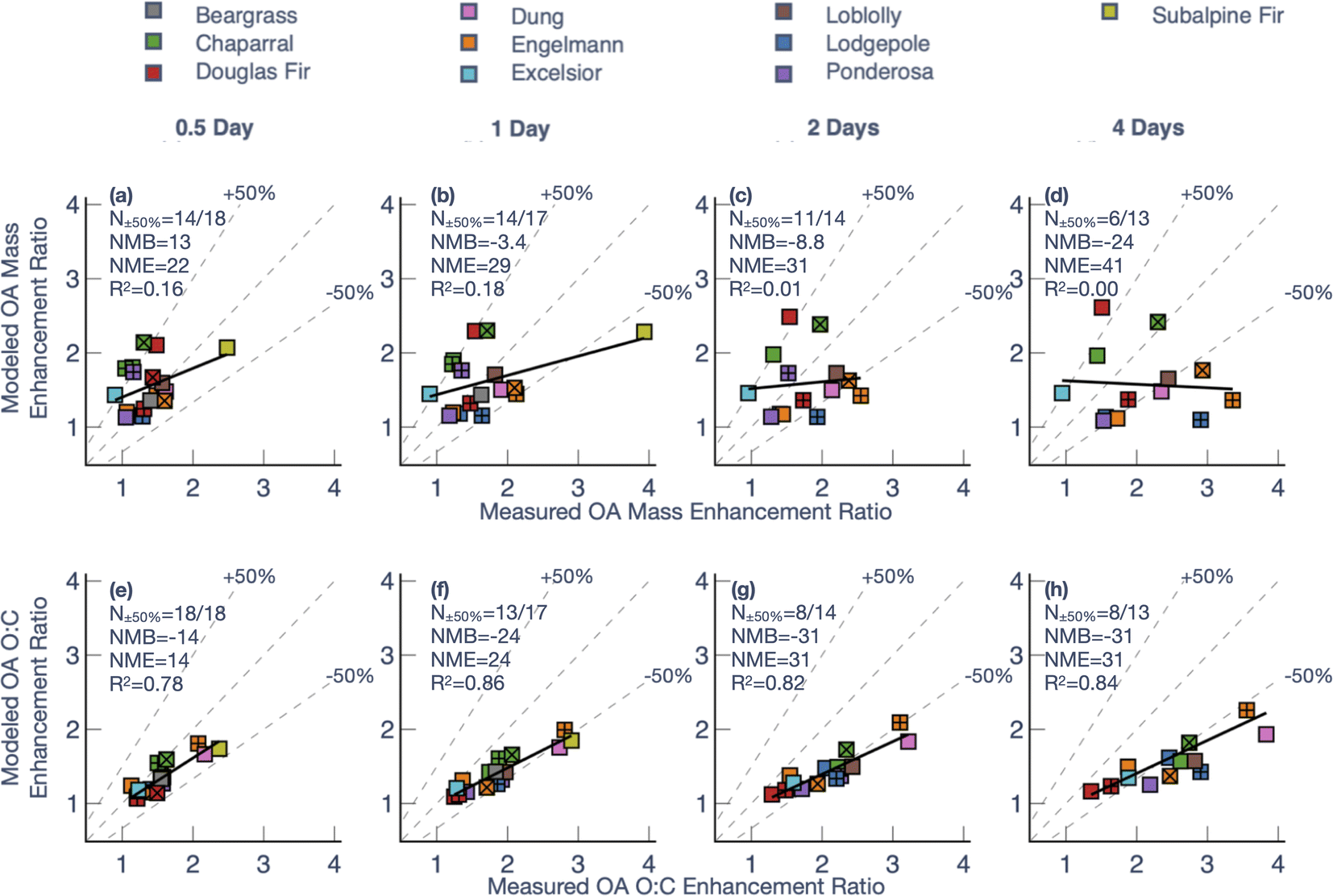 | ||
| Fig. 5 Model predictions of (a–d) OA mass enhancement ratio and (e–h) OA O:C enhancement ratio compared to measurements at four different photochemical ages (a and e) 0.5 days, (b and f) 1 day, (c and g) 2 days, and (d and h) 4 days. Different colors are used to denote different fuels with the patterns (plus, cross) used to separate multiple experiments performed on the same fuel. N±50% indicates the number of experiments where model predictions were within ±50% of the measurements and NMB and NME are the Normalized Mean Bias and Normalized Mean Error, respectively. | ||
Second, VOC oxidation led to SOA formation in all 18 experiments and contributed to changes in the OA composition and the increase in model predictions of OA mass concentrations and O:C. While there was some variability in SOA formation across chamber experiments, SOA contributed to 25% to 60% of the total OA at 0.5 equivalent days of photochemical age and 37% to 70% of the total OA by the end of the chamber experiment. On average, SOA accounted for 47% and 57% of the total OA at those two time points, respectively. The end-of-experiment SOA mass yield across all 18 chamber experiments ranged from 10% to 45%. Similar to the conclusions made by Akherati and coworkers who simulated the OA evolution in laboratory fires39 and wildfire plumes,33 the SOA formation was dominated by oxidation of SVOCs (average contribution of 21%), oxygenated aromatics (primarily, phenols and methoxyphenols; average contribution of 19%), and heterocyclics (primarily, furanic VOCs; average contribution of 10%). Biogenic VOCs also contributed to SOA formation, but it was limited to a handful of chamber experiments performed on coniferous fuels; 3 out of 18 where biogenic SOA was ∼8% of total SOA. This biogenic SOA contribution is likely to be overestimated since the O3 added to the chamber is expected to entirely deplete all biogenic VOCs before the lights were turned on. Differences in the OA mass composition between 0.5 equivalent days (Fig. S8a†) and the end of the experiment (Fig. S8b†) were marginal with the exception that the SOA contribution from aromatic hydrocarbons at 0.5 equivalent days was much smaller (average contribution of 1.5%), relative to that at the end of the experiment (average contribution of 3.7%). Similar to the model predictions shown in Fig. 1, modeled SOA formation slowed after one equivalent day of photochemical aging in nearly all chamber experiments, and this was linked to the near depletion of modeled SOA precursors over that timescale. Overall, SOA could be up to half of biomass burning OA with prolonged aging, which aligns well with recent findings from empirical analyses of aircraft measurements,5 numerical modeling,12,78 and studies that have combined both33 for wildfire OA.
Third, while there were a couple of exceptions (e.g., dung, subalpine fir), model predictions of OA O:C from the Base simulations matched the observed increase in OA O:C over 0.5 to 1 equivalent days of photochemical aging but systematically underestimated the observed O:C increase with additional aging. Except for Douglas fir #1, the model heavily underestimated the OA O:C for 17 fires by the end of the experiment. Across all 18 chamber experiments, the model-predicted increase in OA O:C was partly linked to SOA production. This was because the fresh POA had an initial O:C of 0.35 ± 0.1 and the SOA added from oxidation of SVOCs, oxygenated aromatics, and heterocyclics had a higher O:C (0.4–0.6, 0.8–1, and 0.85, respectively).58,79 Aside from SOA condensation, model predictions of OA O:C continued to increase with photochemical age, especially after the dominant SOA precursors were depleted, from multigenerational aging and heterogeneous oxidation of OA.
Fourth, the model predictions for OA mass concentrations and O:C did not vary much for simulations performed with the high NOX parameters. On close observation, the use of high NOX parameters resulted in slightly higher OA mass concentrations and slightly lower OA O:C, relative to the Base simulations. We found this to be a little surprising for OA mass concentrations since, broadly, lower NOX conditions have been linked to higher SOA mass yields for certain precursor classes (e.g., aromatic hydrocarbons, monoterpenes). In this case, the model predictions were found to be insensitive to NOX presumably because some of the precursor classes important for biomass burning (e.g., oxygenated aromatics, SVOCs modeled as naphthalene) do not exhibit a strong dependence on NOX. The Base (i.e., low NOX) simulations predicted a slightly higher OA O:C compared to the high NOX simulations as autoxidation reactions, which result in production of highly oxygenated products (O:C∼1), were only considered in the Base simulations.
Finally, we make two additional comments about the model-measurement comparisons for multiple experiments performed on the same fuel. First, the model appeared to do well on OA mass concentrations for two of the three chamber experiments performed on Douglas fir (#2 and #3) and one of the three chamber experiments performed on chaparral (#3). Second, the model systematically underestimated the OA mass concentrations for all chamber experiments performed on Engelmann spruce and lodgepole pine. Regardless, the model did not perform better or worse for a particular fuel type (conifer, shrub, grass, and dung) although we should note that our ability to distinguish model performance as a function of fuel type was affected by the small sample size of the dataset.
To investigate the model performance at the same photochemical age, we compared model predictions of the OA mass and O:C enhancement ratios (final to initial) against measurements at four different photochemical ages – 0.5, 1, 2, and 4 days – in Fig. 5. Since there was significant variability in the maximum photochemical age achieved across the 18 chamber experiments, the model-measurement comparisons had to be performed on a sample size that decreased with photochemical age (from N = 18 at 0.5 days to N = 13 at 4 days). For OA mass enhancement ratios at 0.5 equivalent days of photochemical aging, the model exhibited a moderately small bias (Normalized Mean Bias (NMB) = 13%) and error (Normalized Mean Error (NME) = 22%), alongside poor model skill (correlation coefficient (R2) = 0.16). A small bias and error meant that the average and range of the OA mass enhancement ratios predicted by the model (average = 1.5, range = 1.1–2.1) were very similar to that for the measurements (average = 1.4, range = 0.9–2.5). The error (i.e., NME), and skill (i.e., R2) in model predictions got progressively worse with photochemical aging. At 4 equivalent days of photochemical aging, the model exhibited a negative bias (NMB = −24%), a larger error (NME = 41%), and little to no model skill (R2 < 0.01). While most of the model predictions for the OA mass enhancement ratio were within 50% of the measurements at 0.5 and 1 equivalent days (14/18 and 14/17, respectively), only 6 of the 13 chamber experiments were within the 50% envelope at 4 equivalent days. The model performance for OA mass enhancement ratios at the shorter photochemical ages (0.5 and 1 equivalent days) was consistent with two previous modeling studies based on chamber experiments specifically performed over short photochemical ages (<12 equivalent hours).13,39
In contrast, the model performance for OA O:C enhancement ratios was much easier to interpret. The model exhibited good skill in predicting the enhancement in OA O:C, with an R2 that creeped higher at longer photochemical ages (R2 of 0.67 at 0.5 days to R2 of 0.78 at 4 days). As shown in Fig. 4, the model was able to generally reproduce the observed O:C enhancement up to one equivalent day of photochemical aging (NMB = −14%, NME = 14%) but the model steadily underestimated the observed OA O:C enhancement with additional aging. By 4 equivalent days of photochemical aging, the model underestimated the OA O:C enhancement ratio by ∼50% (NMB = −31%, NME = 31%). All or most of the model predictions for the OA O:C enhancement ratio were within 50% of the measurements at 0.5 and 1 equivalent days (18/18 and 13/17, respectively) while only 8 of the 13 chamber experiments were within the 50% envelope at 4 equivalent days. The comparisons in Fig. 5 suggest that the model is missing precursors, processes, or both that may be relevant for OA mass and O:C enhancement linked to POA processing and SOA production at longer photochemical ages (>1 day).
We probed the model's underestimation and poor skill by performing a variance analysis on the OA mass concentrations at 4 equivalent days of photochemical aging. By fixing the photochemical age to 4 equivalent days, we were analyzing the model performance at an aging time where it performed poorly, noting that our analysis was limited to a subset of experiments (N = 13). Using values from the end of the experiment would have increased the sample size to 18 but would have made it difficult to account for differences in photochemical age. Results from the variance analysis, which included variables present within the model (initial POA, SOA precursors, and OA O:C) and those not considered in the model (modified combustion efficiency, fuel moisture content, fuel type), are shown in Fig. 6. The model error in OA mass, expressed as a ratio of modeled to measured OA, showed a weak positive correlation with initial concentrations of SOA precursors (R2 = 0.25) (Fig. 6b). Specifically, the model underestimated the OA mass when the initial SOA precursor concentrations were lower suggesting that a different set of SOA precursors than those measured by the PTR-ToF-MS, as noted by Hatch et al.,80 might be responsible for SOA formation in these experiments. Defining the model error as [modeled OA–measured OA] or a ratio of the modeled ΔOA to measured ΔOA produced a variance analysis that was identical to that shown in Fig. 6 (not shown). The model error exhibited a poor relationship, or the absence of one, with the modified combustion efficiency (R2 = 0.03), fuel moisture content (R2 = 0.07), and fuel type. The model error in OA O:C did not correlate with any of the variables, except for initial O:C (Fig. S9†). We admit that we do not know how to interpret the weak positive correlation of the model error in OA mass and OA O:C with the initial O:C.
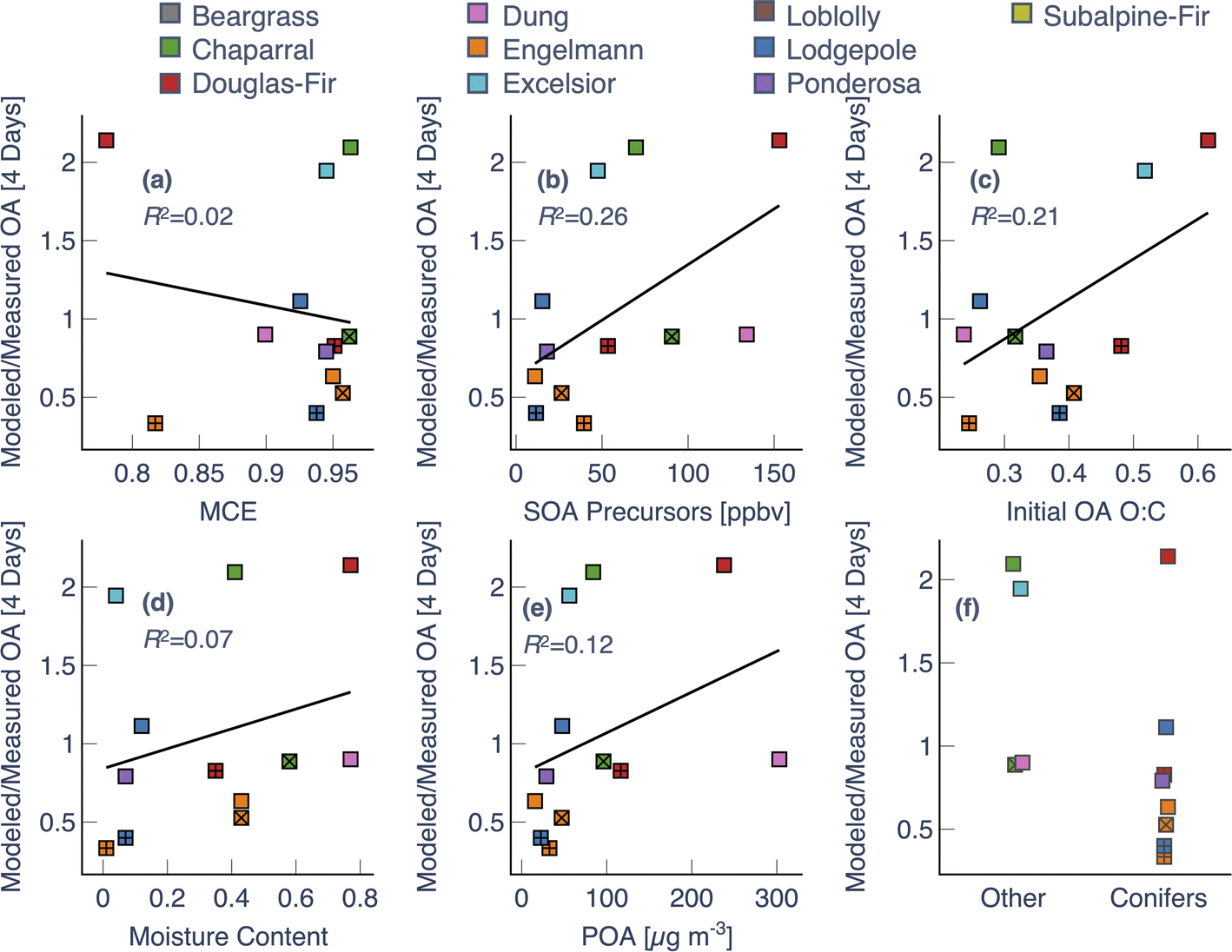 | ||
| Fig. 6 Model variance in OA mass expressed as a ratio of the modeled to measured OA compared against (a) MCE, (b) initial SOA precursors, (c) initial OA O:C, (d) fuel moisture content, (e) initial POA, and (f) fuel type (conifers versus others). Variance analysis is performed at a photochemical age of 4 equivalent days. Multiple experiments performed with the same fuel are denoted with either a + or × inside the symbol. | ||
3.3 Model sensitivities
We studied sensitivity in model predictions to individual processes and results from this analysis for all 18 chamber experiments are shown in Fig. S10.† Here, we compared model predictions of OA mass concentration and O:C from the sensitivity simulations to those from the Base simulations. The presentation of the sensitivity analysis (N = 13) was done at four equivalent days of photochemical aging as one of the reasons for performing the sensitivity analysis was to study the reasons for the model's poor performance at longer photochemical ages. Analogous to the findings in Fig. 1, adjusting PWL (PWL off or PWL × 2) and turning vapor wall losses off resulted in the largest change in OA mass concentrations (median change of 45%, 30%, and 30%, respectively), relative to the Base results. In contrast, turning dilution, oligomerization, or heterogeneous oxidation off, using high NOX parameters, treating POA as non-volatile and non-reactive, or assuming a semi-solid OA individually, resulted in much smaller changes in the OA mass concentrations (median change of 7%, 10%, 6%, 3%, 9%, and 2%, respectively). We should note that the model response (i.e., sensitivity) to a particular process varied modestly across the 18 chamber experiments, despite comparisons of the model output at the same photochemical age. Given that particle and vapor wall losses influenced model predictions significantly, one potential reason for the poor model performance at longer photochemical ages might be the use of a single, time-independent value to determine those wall loss rates. Interestingly, model predictions of OA O:C were only mildly sensitive to turning dilution and vapor wall losses off (median increase of 14% and 9%, respectively) and much less sensitive to all other processes. Hence, an experiment-specific particle or vapor wall loss rate is unlikely to influence the model performance for OA O:C.
Results from simulations performed with model adjustments for the ponderosa pine chamber experiment are shown in Fig. 7. Similar to the motivation for performing the sensitivity analysis described earlier, these simulations were used to study reasons for the model's poor performance at longer photochemical ages. The heterogeneous oxidation schemes that rapidly produced lower-volatility and more oxygenated products and the inclusion of smaller VOCs, both increased model predictions of OA mass concentrations by up to 37%, relative to the Base results (Fig. 7a). The heterogeneous oxidation scheme with a higher uptake coefficient was the only configuration that produced a proportionately strong increase (24%) in O:C (Fig. 7c). The use of an average SOA precursor profile increased OA mass concentrations, relative to the Base simulation, but produced no change in the OA O:C (Fig. 7a and c). OA mass concentrations were enhanced because the average SOA precursor profile, based on measurements in the stack,53 resulted in higher initial precursor concentrations compared to the direct chamber measurements described in Coggon et al.55 and Lim et al.40 We did not find this difference to be surprising as Akherati et al.39 (c.f. Fig. S6†) had shown that there were meaningful losses of SOA precursors in the duct used to port smoke from the stack to their larger chamber. Randomly varying the distribution of POA + SVOC mass in the SOM grid (i.e., Monte-Carlo simulations) seemed to produce some variability in model predictions of OA mass concentrations (+20%) (Fig. 7b) and O:C (±7%) (Fig. 7d) but these variations were not significant enough to explain the underestimation seen for OA O:C at longer photochemical ages.
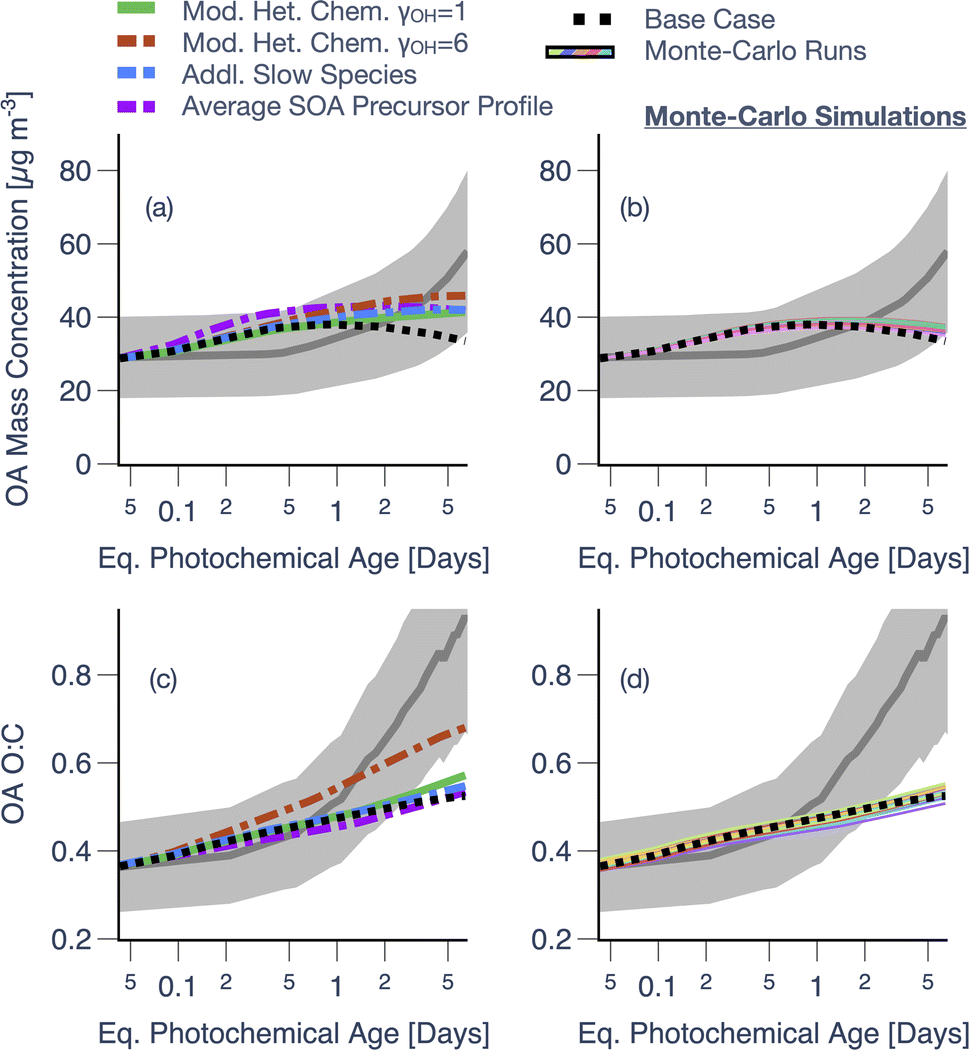 | ||
| Fig. 7 Model predictions of (a and b) OA mass concentrations and (c and d) OA O:C compared against measurements for the ponderosa pine chamber experiment (Fire 38). Predictions are from simulations performed with the Base configuration, modified treatment for heterogeneous oxidation (a and c), SOA formation from smaller and slower-reacting VOCs (a and c), use of an average SOA precursor profile (a and c), and Monte-Carlo simulations (b and d). | ||
Overall, of the model adjustments undertaken here, the use of an average VOC profile and aggressive heterogeneous oxidation scheme, both marginally improved the model performance (i.e., NMB) at longer photochemical ages without significantly affecting the performance at shorter photochemical ages (Fig. S11†). Broadly, the sensitivity simulations and model adjustments discussed in this section strengthen the argument that the model is missing precursors, pathways, or both that can help explain the strong increase in OA mass concentrations and O:C at longer photochemical ages.
4 Summary and discussion
In this work, we simulated the photochemical evolution of biomass burning OA in 18 separate chamber experiments performed in a small environmental chamber. The simulations were performed with a process-level kinetic model (i.e., SOM-TOMAS) that simulated dilution, multigenerational gas-phase chemistry, autoxidation reactions, phase-state-influenced gas/particle partitioning, heterogeneous oxidation, oligomerization reactions, and chamber artifacts (i.e., particle and vapor wall losses). Broadly, we found that the model was able to generally reproduce the OA mass and O:C evolution at short photochemical ages (0.5–1 equivalent days), consistent with earlier work, but the model performed less optimally at longer photochemical ages (>1 equivalent day). For instance, at four equivalent days of photochemical aging, the model underestimated both the enhancement in OA mass and O:C but showed good skill in predicting the increase in OA O:C. Neither the variance analyses nor the sensitivity simulations were able to provide conclusive evidence for why the model performance deteriorated with photochemical age. We tentatively argue that the model performance would have been better if we had access to experiment-specific and time-dependent particle and vapor wall loss rates although neither of these would have affected the model's ability to accurately predict the increase in OA O:C. Simulations performed with adjustments made to the heterogeneous oxidation scheme offered a slight improvement in model performance at longer photochemical ages.
Although our work and that of He et al.49 both use kinetic models and rely on the same primary laboratory data, they differ in some of their analysis and conclusions. Hence, it becomes important to directly compare conclusions from our work against those from He et al.49 Broadly, both studies seemed to agree that the two different models can modestly reproduce the OA mass concentration evolution and that SVOCs, oxygenated aromatics, and heterocyclics are important SOA precursors; we should note that the model performance for OA mass concentrations does seem to be slightly better for He et al.49 relative to our work. The big difference, however, is in the ability of the two studies to reproduce the change in OA O:C. He et al.49 closely reproduce the change in OA O:C for all photochemical ages and experiments while this work significantly underestimates that change. This is very likely because He et al.,49 within the two-dimensional volatility basis set (2D-VBS) framework, use an explicit chemical mechanism (i.e., GECKO-A) to inform the distribution of first-generation oxidation products and use an aggressive fragmentation kernel, features that produce higher O:C species relative to the functionalization and fragmentation schemes used in the SOM-TOMAS model. Another aspect that distinguishes our work from that of He et al.49 is that we investigated the model-measurement comparison for individual experiments, examined the sensitivity to physical and chemical processes, and assessed model performance across photochemical age. We will admit that differences between the two studies likely point to the superiority of the He et al.49 approach in simulating the multigenerational aging of SOA.
The SOM-TOMAS model can be used to determine aerosol mass yields for SOA precursors that would be relevant for photochemical evolution over multiple days. The dilution- and particle-wall-loss-corrected SOA mass yields from the end of the experiment varied between 10% and 45% over the 18 chamber experiments. If these were further corrected for vapor wall losses (simulated by turning vapor wall losses off), the SOA mass yields increased to between 22% and 49%. In these chamber experiments where the SOA precursors were 2.4× the initial POA emissions (Fig. S2a†), these yields were found to enhance the OA mass burden even when some of the POA was lost to dilution-driven evaporation. However, Akherati et al.33 discovered that SOA precursor emissions were similar to POA emissions (i.e., ∼1×) in real wildfire plumes sampled using a research aircraft in the western US. Hence, the SOA mass yields estimated in this work are likely to boost OA mass burdens by at least ∼30% in real wildfire plumes over regional scales without considering the fate of POA subject to evaporation and oxidation. The SOA produced is bound to alter the chemical, microphysical, and optical properties of biomass burning aerosol with photochemical age. Biomass burning is an important source of SOA precursor emissions, just like it is for POA emissions, and these precursors have significantly large yields that they need to be considered in air quality climate models if we are to simulate SOA production and the physicochemical evolution of biomass burning OA.
The SOM-TOMAS model currently does not account for aqueous processing or photolysis of organic compounds in the particle phase. Both of these processes have been shown to alter SOA production and loss rates in model systems.81,82 We do not anticipate the lack of aqueous chemistry to be a severe limitation here since the Lim et al.40 chamber experiments were performed under low relative humidity conditions (25–40%). However, we cannot fully discount the effects from small amounts of water taken up by a rapidly evolving OA. Coggon et al.55 argued that except for a few photolabile species (e.g., furfural), the 254 nm UV-C light used by Lim et al.40 had a negligible impact on the photooxidation of biomass burning VOCs. Yet, the impact from absorption of UV-C light by OA remains uncertain. Aqueous processing and photolysis will need to be considered as important processes in future biomass burning OA modeling work.
Our work was motivated to better understand the chemistry and microphysics of biomass burning OA at long photochemical ages, which could shed light on its evolution in the real atmosphere at regional and global scales. Results from this work provided key insights on the transformation of OA mass and composition with aging while indicating that we may be missing key precursors, processes, or both that become more important at longer photochemical ages. The missing precursors could take the form of primary species and/or oxidation products that are not easily measured by the PTR-ToF-MS,83 which highlights the need for multi-instrument approaches to characterize SOA precursors in complex mixtures such as those emitted from biomass burning. We recommend that future work should continue to combine kinetic modeling with laboratory and field datasets to improve our understanding of biomass burning aerosol. Such modeling will be important in developing mechanisms and parameterizations to represent multi-day aging of biomass burning aerosol in three-dimensional models, a prerequisite to studying the impact of biomass burning on climate, air quality, and human health.
Data availability
The environmental chamber data used in this work are archived by the Chemical Sciences Laboratory at the National Oceanic and Atmospheric Administration (https://csl.noaa.gov/groups/csl7/measurements/2016firex/FireLab/DataDownload/). The latest version of the SOM-TOMAS model is archived on Github (https://github.com/yicongh/SOM-TOMAS-v2).Author contributions
AD and SHJ designed the modeling study. YH and AA developed the model with help from SHJ and JRP. AD developed model inputs, performed the simulations, and analyzed the data. CYL, MMC, ARK, JG, CW, LDY, JHS, CDC, and JHK provided the experimental data. AD and SHJ wrote the paper with contributions from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was developed under Assistance Agreement No. R840008 awarded by the U.S. Environmental Protection Agency (EPA) to Colorado State University (SHJ) and support from the National Oceanic and Atmospheric Administration (NA17OAR4310003, NA21OAR4310128, NA16OAR4310112, NA16OAR4310111) and the U.S. Department of Energy (DOE), Office of Science (DE-SC0017975). It has not been formally reviewed by EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the Agency. EPA does not endorse any products or commercial services mentioned in this publication. The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the EPA. We thank several anonymous reviewers at NOAA for reviewing this manuscript.References
- J. S. Reid, R. Koppmann, T. F. Eck and D. P. Eleuterio, Atmos. Chem. Phys., 2005, 5, 799–825 CrossRef CAS.
- J.-F. Lamarque, T. C. Bond, V. Eyring, C. Granier, A. Heil, Z. Klimont, D. Lee, C. Liousse, A. Mieville, B. Owen, M. G. Schultz, D. Shindell, S. J. Smith, E. Stehfest, J. Van Aardenne, O. R. Cooper, M. Kainuma, N. Mahowald, J. R. McConnell, V. Naik, K. Riahi and D. P. van Vuuren, Atmos. Chem. Phys., 2010, 10, 7017–7039 CrossRef CAS.
- C. Granier, B. Bessagnet, T. Bond, A. D'Angiola, H. Denier van der Gon, G. J. Frost, A. Heil, J. W. Kaiser, S. Kinne, Z. Klimont, S. Kloster, J.-F. Lamarque, C. Liousse, T. Masui, F. Meleux, A. Mieville, T. Ohara, J.-C. Raut, K. Riahi, M. G. Schultz, S. J. Smith, A. Thompson, J. van Aardenne, G. R. van der Werf and D. P. van Vuuren, Clim. Change, 2011, 109, 163–190 CrossRef CAS.
- M. O. Andreae, Atmos. Chem. Phys., 2019, 19, 8523–8546 CrossRef CAS.
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29469–29477 CrossRef CAS PubMed.
- W. Permar, Q. Wang, V. Selimovic, C. Wielgasz, R. J. Yokelson, R. S. Hornbrook, A. J. Hills, E. C. Apel, I.-T. Ku, Y. Zhou, B. C. Sive, A. P. Sullivan, J. L. Collett Jr, T. L. Campos, B. B. Palm, Q. Peng, J. A. Thornton, L. A. Garofalo, D. K. Farmer, S. M. Kreidenweis, E. J. T. Levin, P. J. DeMott, F. Flocke, E. V. Fischer and L. Hu, J. Geophys. Res., 2021, 126(11), e2020JD033838 CrossRef CAS.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Rev. Geophys., 2017, 55, 509–559 CrossRef.
- Y. Jiang, Z. Lu, X. Liu, Y. Qian, K. Zhang, Y. Wang and X.-Q. Yang, Atmos. Chem. Phys., 2016, 16, 14805–14824 CrossRef CAS.
- N. Bellouin, J. Quaas, E. Gryspeerdt, S. Kinne, P. Stier, D. Watson-Parris, O. Boucher, K. S. Carslaw, M. Christensen, A.-L. Daniau, J.-L. Dufresne, G. Feingold, S. Fiedler, P. Forster, A. Gettelman, J. M. Haywood, U. Lohmann, F. Malavelle, T. Mauritsen, D. T. McCoy, G. Myhre, J. Mülmenstädt, D. Neubauer, A. Possner, M. Rugenstein, Y. Sato, M. Schulz, S. E. Schwartz, O. Sourdeval, T. Storelvmo, V. Toll, D. Winker and B. Stevens, Rev. Geophys., 2020, 58, e2019RG000660 CrossRef CAS PubMed.
- C. D. McClure and D. A. Jaffe, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7901–7906 CrossRef CAS PubMed.
- B. Ford, M. Val Martin, S. E. Zelasky, E. V. Fischer, S. C. Anenberg, C. L. Heald and J. R. Pierce, Geohealth, 2018, 2, 229–247 CrossRef CAS PubMed.
- A. L. Hodshire, A. Akherati, M. J. Alvarado, B. Brown-Steiner, S. H. Jathar, J. L. Jimenez, S. M. Kreidenweis, C. R. Lonsdale, T. B. Onasch, A. M. Ortega and J. R. Pierce, Environ. Sci. Technol., 2019, 53, 10007–10022 CrossRef CAS PubMed.
- A. T. Ahern, E. S. Robinson, D. S. Tkacik, R. Saleh, L. E. Hatch, K. C. Barsanti, C. E. Stockwell, R. J. Yokelson, A. A. Presto, A. L. Robinson, R. C. Sullivan and N. M. Donahue, J. Geophys. Res., 2019, 124, 3583–3606 CrossRef CAS.
- D. S. Tkacik, E. S. Robinson, A. Ahern, R. Saleh, C. Stockwell, P. Veres, I. J. Simpson, S. Meinardi, D. R. Blake, R. J. Yokelson, A. A. Presto, R. C. Sullivan, N. M. Donahue and A. L. Robinson, J. Geophys. Res., 2017, 122, 6043–6058 CrossRef CAS.
- C. J. Hennigan, M. A. Miracolo, G. J. Engelhart, A. A. May, A. A. Presto, T. Lee, A. P. Sullivan, G. R. McMeeking, H. Coe, C. E. Wold, W. M. Hao, J. B. Gilman, W. C. Kuster, J. Gouw, B. A. Schichtel, J. L. Collett Jr, S. M. Kreidenweis and A. L. Robinson, Atmos. Chem. Phys., 2011, 11, 7669–7686 CrossRef CAS.
- P. V. Hobbs, P. Sinha, R. J. Yokelson, T. J. Christian, D. R. Blake, S. Gao, T. W. Kirchstetter, T. Novakov and P. Pilewskie, J. Geophys. Res., 2003, 108(D13) DOI:10.1029/2002jd002352.
- X. Liu, Y. Zhang, L. G. Huey, R. J. Yokelson, Y. Wang, J. L. Jimenez, P. Campuzano-Jost, A. J. Beyersdorf, D. R. Blake, Y. Choi, J. M. St. Clair, J. D. Crounse, D. A. Day, G. S. Diskin, A. Fried, S. R. Hall, T. F. Hanisco, L. E. King, S. Meinardi, T. Mikoviny, B. B. Palm, J. Peischl, A. E. Perring, I. B. Pollack, T. B. Ryerson, G. Sachse, J. P. Schwarz, I. J. Simpson, D. J. Tanner, K. L. Thornhill, K. Ullmann, R. J. Weber, P. O. Wennberg, A. Wisthaler, G. M. Wolfe and L. D. Ziemba, J. Geophys. Res., 2016, 121, 7383–7414 CrossRef CAS.
- P. F. DeCarlo, E. J. Dunlea, J. R. Kimmel, A. C. Aiken, D. Sueper, J. Crounse, P. O. Wennberg, L. Emmons, Y. Shinozuka, A. Clarke, J. Zhou, J. Tomlinson, D. R. Collins, D. Knapp, A. J. Weinheimer, D. D. Montzka, T. Campos and J. L. Jimenez, Atmos. Chem. Phys., 2008, 8, 4027–4048 CrossRef CAS.
- V. Vakkari, V.-M. Kerminen, J. P. Beukes, P. Tiitta, P. G. Zyl, M. Josipovic, A. D. Venter, K. Jaars, D. R. Worsnop, M. Kulmala and L. Laakso, Geophys. Res. Lett., 2014, 41, 2644–2651 CrossRef CAS.
- A. A. May, T. Lee, G. R. McMeeking, S. Akagi, A. P. Sullivan, S. Urbanski, R. J. Yokelson and S. M. Kreidenweis, Atmos. Chem. Phys., 2015, 15, 6323–6335 CrossRef CAS.
- S. K. Akagi, J. S. Craven, J. W. Taylor, G. R. McMeeking, R. J. Yokelson, I. R. Burling, S. P. Urbanski, C. E. Wold, J. H. Seinfeld, H. Coe, M. J. Alvarado and D. R. Weise, Atmos. Chem. Phys., 2012, 12, 1397–1421 CrossRef CAS.
- M. J. Cubison, A. M. Ortega, P. L. Hayes, D. K. Farmer, D. Day, M. J. Lechner, W. H. Brune, E. Apel, G. S. Diskin, J. A. Fisher, H. E. Fuelberg, A. Hecobian, D. J. Knapp, T. Mikoviny, D. Riemer, G. W. Sachse, W. Sessions, R. J. Weber, A. J. Weinheimer, A. Wisthaler and J. L. Jimenez, Atmos. Chem. Phys., 2011, 11, 12049–12064 CrossRef.
- J. Brito, L. V. Rizzo, W. T. Morgan, H. Coe, B. Johnson, J. Haywood, K. Longo, S. Freitas, M. O. Andreae and P. Artaxo, Atmos. Chem. Phys., 2014, 14, 12069–12083 CrossRef.
- H. Forrister, J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. Thornhill, B. Anderson, G. Diskin, A. Perring, J. Schwarz, P. Campuzano-Jost, D. Day, B. Palm, J. Jimenez, A. Nenes and R. Weber, Geophys. Res. Lett., 2015, 42(11), 4623–4630 CrossRef CAS.
- S. Collier, S. Zhou, T. B. Onasch, D. A. Jaffe, L. Kleinman, A. J. Sedlacek 3rd, N. L. Briggs, J. Hee, E. Fortner, J. E. Shilling, D. Worsnop, R. J. Yokelson, C. Parworth, X. Ge, J. Xu, Z. Butterfield, D. Chand, M. K. Dubey, M. S. Pekour, S. Springston and Q. Zhang, Environ. Sci. Technol., 2016, 50, 8613–8622 CrossRef CAS PubMed.
- M. D. Jolleys, H. Coe, G. McFiggans, G. Capes, J. D. Allan, J. Crosier, P. I. Williams, G. Allen, K. N. Bower, J. L. Jimenez, L. M. Russell, M. Grutter and D. Baumgardner, Environ. Sci. Technol., 2012, 46, 13093–13102 CrossRef CAS PubMed.
- M. D. Jolleys, H. Coe, G. McFiggans, J. W. Taylor, S. J. O’shea, M. Le Breton, S.-B. Bauguitte, S. Moller, P. Di Carlo, E. Aruffo, P. I. Palmer, J. D. Lee, C. J. Percival and M. W. Gallgher, Atmos. Chem. Phys., 2015, 15, 3077–3095 CrossRef CAS.
- M. D. Jolleys, H. Coe, G. McFiggans, G. Capes, J. D. Allan, J. Crosier, P. I. Williams, G. Allen, K. N. Bower, J. L. Jimenez, L. M. Russell, M. Grutter and D. Baumgardner, Environ. Sci. Technol., 2012, 46, 13093–13102 CrossRef CAS PubMed.
- A. M. Ortega, D. A. Day, M. J. Cubison, W. H. Brune, D. Bon, J. A. De Gouw and J. L. Jimenez, Atmos. Chem. Phys., 2013, 13, 11551–11571 CrossRef.
- L. A. Garofalo, M. A. Pothier, E. J. T. Levin, T. Campos, S. M. Kreidenweis and D. K. Farmer, ACS Earth Space Chem., 2019, 3, 1237–1247 CrossRef CAS.
- Y. Liang, C. N. Jen, R. J. Weber, P. K. Misztal and A. H. Goldstein, Atmos. Chem. Phys., 2021, 21, 5719–5737 CrossRef CAS.
- L. A. Garofalo, Y. He, S. H. Jathar, J. R. Pierce, C. D. Fredrickson, B. B. Palm, J. A. Thornton, F. Mahrt, G. V. Crescenzo, A. K. Bertram, D. C. Draper, J. L. Fry, J. Orlando, X. Zhang and D. K. Farmer, Environ. Sci. Technol., 2021, 55, 15637–15645 CrossRef CAS PubMed.
- A. Akherati, Y. He, L. A. Garofalo, A. L. Hodshire, D. K. Farmer, S. M. Kreidenweis, W. Permar, L. Hu, E. V. Fischer, C. N. Jen, A. H. Goldstein, E. J. T. Levin, P. J. DeMott, T. L. Campos, F. Flocke, J. M. Reeves, D. W. Toohey, J. R. Pierce and S. H. Jathar, Environ. Sci.: Atmos., 2022, 2, 1000–1022 CAS.
- A. J. Sedlacek 3rd, E. R. Lewis, T. B. Onasch, P. Zuidema, J. Redemann, D. Jaffe and L. I. Kleinman, Environ. Sci. Technol., 2022, 56, 14315–14325 CrossRef PubMed.
- A. P. Grieshop, N. M. Donahue and A. L. Robinson, Atmos. Chem. Phys., 2009, 9, 2227–2240 CrossRef CAS.
- A. P. Grieshop, J. M. Logue, N. M. Donahue and A. L. Robinson, Atmos. Chem. Phys., 2009, 9, 1263–1277 CrossRef CAS.
- M. F. Heringa, P. F. DeCarlo, R. Chirico, T. Tritscher, J. Dommen, E. Weingartner, R. Richter, G. Wehrle, A. S. H. Prévôt and U. Baltensperger, Atmos. Chem. Phys., 2011, 11, 5945–5957 CrossRef CAS.
- E. A. Bruns, I. El Haddad, J. G. Slowik, D. Kilic, F. Klein, U. Baltensperger and A. S. H. Prévôt, Sci. Rep., 2016, 6, 27881 CrossRef CAS PubMed.
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. de Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Environ. Sci. Technol., 2020, 54, 8568–8579 CrossRef CAS PubMed.
- C. Y. Lim, D. H. Hagan, M. M. Coggon, A. R. Koss, K. Sekimoto, J. de Gouw, C. Warneke, C. D. Cappa and J. H. Kroll, Atmos. Chem. Phys., 2019, 19, 12797–12809 CrossRef CAS.
- A. T. Lambe, A. T. Ahern, L. R. Williams, J. G. Slowik, J. P. S. Wong, J. P. D. Abbatt, W. H. Brune, N. L. Ng, J. P. Wright, D. R. Croasdale, D. R. Worsnop, P. Davidovits and T. B. Onasch, Atmos. Meas. Tech., 2011, 4(3), 445–461 CrossRef CAS.
- J. E. Krechmer, D. Pagonis, P. J. Ziemann and J. L. Jimenez, Environ. Sci. Technol., 2016, 50, 5757–5765 CrossRef CAS PubMed.
- Y. Huang, R. Zhao, S. M. Charan, C. M. Kenseth, X. Zhang and J. H. Seinfeld, Environ. Sci. Technol., 2018, 52(4), 2134–2142 CrossRef CAS PubMed.
- B. B. Palm, S. S. de Sá, D. A. Day, P. Campuzano-Jost, W. Hu, R. Seco, S. J. Sjostedt, J.-H. Park, A. B. Guenther, S. Kim, J. Brito, F. Wurm, P. Artaxo, R. Thalman, J. Wang, L. D. Yee, R. Wernis, G. Isaacman-VanWertz, A. Goldstein, Y. Liu, S. R. Springston, R. Souza, M. Newburn, M. L. Alexander, S. Martin and J. Jimenez, Atmos. Chem. Phys., 2018, 18, 467–493 CrossRef CAS.
- S. Eluri, C. D. Cappa, B. Friedman, D. K. Farmer and S. H. Jathar, Atmos. Chem. Phys., 2018, 18, 13813–13838 CrossRef CAS.
- J. D. Smith, J. H. Kroll, C. D. Cappa, D. L. Che, C. L. Liu, M. Ahmed, S. R. Leone, D. R. Worsnop and K. R. Wilson, Atmos. Chem. Phys., 2009, 9, 3209–3222 CrossRef CAS.
- J. H. Kroll, J. D. Smith, D. L. Che, S. H. Kessler, D. R. Worsnop and K. R. Wilson, Phys. Chem. Chem. Phys., 2009, 11, 8005–8014 RSC.
- Y. He, A. T. Lambe, J. H. Seinfeld, C. D. Cappa, J. R. Pierce and S. H. Jathar, Environ. Sci. Technol., 2022, 56, 6262–6273 CrossRef CAS PubMed.
- Y. He, B. Zhao, S. Wang, R. Valorso, X. Chang, D. Yin, B. Feng, M. Camredon, B. Aumont, A. Dearden, S. H. Jathar, M. Shrivastava, Z. Jiang, C. D. Cappa, L. D. Yee, J. H. Seinfeld, J. Hao and N. M. Donahue, Nat. Geosci., 2024, 17(2), 124–129 CrossRef CAS.
- C. D. McClure, C. Y. Lim, D. H. Hagan, J. H. Kroll and C. D. Cappa, Atmos. Chem. Phys., 2020, 20(3), 1531–1547 CrossRef CAS.
- C. D. Cappa, C. Y. Lim, D. H. Hagan, M. Coggon, A. Koss, K. Sekimoto, J. de Gouw, T. B. Onasch, C. Warneke and J. H. Kroll, Atmos. Chem. Phys., 2020, 20, 8511–8532 CrossRef CAS.
- V. Selimovic, R. J. Yokelson, C. Warneke, J. M. Roberts, J. De Gouw, J. Reardon and D. W. T. Griffith, Atmos. Chem. Phys., 2018, 18, 2929–2948 CrossRef CAS.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. Roberts, C. Warneke, R. Yokelson and J. Gouw, Atmos. Chem. Phys., 2018, 18, 3299–3319 CrossRef CAS.
- K. Sekimoto, A. R. Koss, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, C. Warneke, R. Yokelson, J. Roberts and J. Gouw, Atmos. Chem. Phys., 2018, 18, 9263–9281 CrossRef CAS.
- M. M. Coggon, C. Y. Lim, A. R. Koss, K. Sekimoto, B. Yuan, J. B. Gilman, D. H. Hagan, V. Selimovic, K. J. Zarzana, S. S. Brown, J. Roberts, M. Müller, R. Yokelson, A. Wisthaler, J. Krechmer, J. Jimenez, C. Cappa, J. Kroll, J. Gouw and C. Warneke, Atmos. Chem. Phys., 2019, 19, 14875–14899 CrossRef CAS.
- C. D. Cappa and K. R. Wilson, Atmos. Chem. Phys., 2012, 12, 9505–9528 CrossRef CAS.
- P. J. Adams and J. H. Seinfeld, J. Geophys. Res., D: Atmos., 2002, 107, AAC4–1–AAC4–23 CrossRef.
- Y. He, B. King, M. Pothier, L. Lewane, A. Akherati, J. Mattila, D. K. Farmer, R. L. McCormick, M. Thornton, J. R. Pierce, J. Volckens and S. H. Jathar, Environ. Sci.: Processes Impacts, 2020, 22, 1461–1474 RSC.
- Y. He, A. Akherati, T. Nah, N. L. Ng, L. A. Garofalo, D. K. Farmer, M. Shiraiwa, R. A. Zaveri, C. D. Cappa, J. R. Pierce and S. H. Jathar, Environ. Sci. Technol., 2021, 55, 1466–1476 CrossRef CAS PubMed.
- S. O’Donnell, A. Akherati, Y. He, A. L. Hodshire, J. E. Shilling, C. Kuang, J. D. Fast, F. Mei, S. Schobesberger, J. A. Thornton, J. N. Smith, S. H. Jathar and J. R. Pierce, J. Geophys. Res.: Atmos., 2023, 128(3), e2022JD037525 CrossRef.
- R. A. Zaveri, R. C. Easter, J. E. Shilling and J. H. Seinfeld, Atmos. Chem. Phys., 2014, 14, 5153–5181 CrossRef.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- E. R. Trump and N. M. Donahue, Atmos. Chem. Phys., 2014, 14, 3691–3701 CrossRef.
- A. Matsunaga and P. J. Ziemann, Aerosol Sci. Technol., 2010, 44, 881–892 CrossRef CAS.
- L. Xu, J. D. Crounse, K. T. Vasquez, H. Allen, P. O. Wennberg, I. Bourgeois, S. S. Brown, P. Campuzano-Jost, M. M. Coggon, J. H. Crawford, J. P. DiGangi, G. S. Diskin, A. Fried, E. M. Gargulinski, J. B. Gilman, G. I. Gkatzelis, H. Guo, J. W. Hair, S. R. Hall, H. A. Halliday, T. F. Hanisco, R. A. Hannun, C. D. Holmes, L. G. Huey, J. L. Jimenez, A. Lamplugh, Y. R. Lee, J. Liao, J. Lindaas, J. A. Neuman, J. B. Nowak, J. Peischl, D. A. Peterson, F. Piel, D. Richter, P. S. Rickly, M. A. Robinson, A. W. Rollins, T. B. Ryerson, K. Sekimoto, V. Selimovic, T. Shingler, A. J. Soja, J. M. S. Clair, D. J. Tanner, K. Ullmann, P. R. Veres, J. Walega, C. Warneke, R. A. Washenfelder, P. Weibring, A. Wisthaler, G. M. Wolfe, C. C. Womack and R. J. Yokelson, Sci. Adv., 2021, 7, eabl3648 CrossRef CAS PubMed.
- M. A. Robinson, Z. C. J. Decker, K. C. Barsanti, M. M. Coggon, F. M. Flocke, A. Franchin, C. D. Fredrickson, J. B. Gilman, G. I. Gkatzelis, C. D. Holmes, A. Lamplugh, A. Lavi, A. M. Middlebrook, D. M. Montzka, B. B. Palm, J. Peischl, B. Pierce, R. H. Schwantes, K. Sekimoto, V. Selimovic, G. S. Tyndall, J. A. Thornton, P. Van Rooy, C. Warneke, A. J. Weinheimer and S. S. Brown, Environ. Sci. Technol., 2021, 55, 10280–10290 CrossRef CAS PubMed.
- F. Rohrer, B. Bohn, T. Brauers, D. Brüning, F.-J. Johnen, A. Wahner and J. Kleffmann, Atmos. Chem. Phys., 2005, 5, 2189–2201 CrossRef CAS.
- J. A. Huffman, K. S. Docherty, A. C. Aiken, M. J. Cubison, I. M. Ulbrich, P. F. DeCarlo, D. Sueper, J. T. Jayne, D. R. Worsnop, P. J. Ziemann and J. L. Jimenez, Atmos. Chem. Phys., 2009, 9, 7161–7182 CrossRef CAS.
- L. E. Hatch, A. Rivas-Ubach, C. N. Jen, M. Lipton, A. H. Goldstein and K. C. Barsanti, Atmos. Chem. Phys., 2018, 18, 17801–17817 CrossRef CAS.
- A. A. May, E. J. T. Levin, C. J. Hennigan, I. Riipinen, T. Lee, J. L. Collett Jr, J. L. Jimenez, S. M. Kreidenweis and A. L. Robinson, J. Geophys. Res., 2013, 118, 11327–11338 CAS.
- P. Barmet, J. Dommen, P. F. DeCarlo, T. Tritscher, A. P. Praplan, S. M. Platt, A. S. H. Prévôt, N. M. Donahue and U. Baltensperger, Atmos. Meas. Tech., 2012, 5, 647–656 CrossRef CAS.
- A. P. Grieshop, N. M. Donahue and A. L. Robinson, Geophys. Res. Lett., 2007, 34(14), L14810 CrossRef.
- Q. Bian, A. A. May, S. M. Kreidenweis and J. R. Pierce, Atmos. Chem. Phys., 2015, 15, 11027–11045 CrossRef CAS.
- W. H. Brune, Environ. Sci. Technol., 2019, 53, 3645–3652 CrossRef CAS PubMed.
- A. Bertrand, G. Stefenelli, S. M. Pieber, E. A. Bruns, B. Temime-Roussel, J. G. Slowik, H. Wortham, A. S. H. Prévôt, I. El Haddad and N. Marchand, Atmos. Chem. Phys., 2018, 18, 10915–10930 CrossRef CAS.
- T. Nah, R. C. McVay, X. Zhang, C. M. Boyd, J. H. Seinfeld and N. L. Ng, Atmos. Chem. Phys., 2016, 16, 9361–9379 CrossRef CAS.
- T. Nah, S. H. Kessler, K. E. Daumit, J. H. Kroll, S. R. Leone and K. R. Wilson, J. Phys. Chem. A, 2014, 118(23), 4106–4119 CrossRef CAS PubMed.
- Q. Bian, S. H. Jathar, J. K. Kodros, K. C. Barsanti, L. E. Hatch, A. A. May, S. M. Kreidenweis and J. R. Pierce, Atmos. Chem. Phys., 2017, 17, 5459–5475 CrossRef CAS.
- P. S. Chhabra, N. L. Ng, M. R. Canagaratna, A. L. Corrigan, L. M. Russell, D. R. Worsnop, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2011, 11, 8827–8845 CrossRef CAS.
- L. E. Hatch, R. J. Yokelson, C. E. Stockwell, P. R. Veres, I. J. Simpson, D. R. Blake, J. J. Orlando and K. C. Barsanti, Atmos. Chem. Phys., 2017, 17, 1471–1489 CrossRef CAS.
- M. A. Zawadowicz, B. H. Lee, M. Shrivastava, A. Zelenyuk, R. A. Zaveri, C. Flynn, J. A. Thornton and J. E. Shilling, Environ. Sci. Technol., 2020, 54, 3861–3870 CrossRef CAS PubMed.
- L. Yu, J. Smith, A. Laskin, C. Anastasio, J. Laskin and Q. Zhang, Atmos. Chem. Phys., 2014, 14, 13801–13816 CrossRef.
- K. J. Nihill, M. M. Coggon, C. Y. Lim, A. R. Koss, B. Yuan, J. E. Krechmer, K. Sekimoto, J.-L. Jimenez, J. de Gouw, C. D. Cappa, C. L. Heald, C. Warneke and J. H. Kroll, Atmos. Chem. Phys., 2023, 23(14), 7887–7889 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of environmental chamber data, additional SOM-TOMAS results and comparisons with measurements, and sensitivity simulation results. See DOI: https://doi.org/10.1039/d3ea00111c |
| This journal is © The Royal Society of Chemistry 2024 |