
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of Mg(AlMe4)2 towards neutral tris(pyrazolyl)alkanes†
Felix
Kracht
,
Christoph
Stuhl
,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
 *
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen (EKUT), Auf der Morgenstelle 18, 72076, Germany. E-mail: reiner.anwander@uni-tuebingen.de
First published on 21st October 2024
Abstract
Various new tris(pyrazolyl)alkanes of the class R′CTp3-R (R′ = Me, Et, nPr, iBu; R = Et, cyPr, Cy, p-tBuPh, Ph; cyPr = cyclopropyl, Cy = cyclohexyl, p-tBuPh = para-tert-butylphenyl) were synthesised and their reactivity towards Mg(AlMe4)2 was examined. Along with new examples of recurring structural motifs, such as separated ion pairs and “metal in a box” complexes, e.g., [(MeCTp3-Et)2Mg][AlMe4]2, several magnesium complexes with new structural features/compositions were obtained. Treatment of the “metal in a box” species [(MeCTp3-R)Mg][AlMe4]2 with THF donor gave the terminal methyl complex [(MeCTp3-cyPr)MgMe(thf)2][AlMe4]. Variation of the backbone alkyl substituent R′ in the tris(pyrazolyl)alkane R′CTp3-Ph gave ionic liquids (R′ = Et, nPr) and the methyl-bridged dimagnesium complex [({iBuCTp3-Ph}Mg{AlMe4})2(μ-Me)][AlMe4]. The bulky Cy and p-tBuPh moieties at the pyrazolyl 3 position gave the new structural motif [MeCTp3-RMg(ηn-AlMe4)][AlMe4] (η3, R = Cy; η2, R = p-tBuPh), stabilising a “[Mg(AlMe4)]+” entity. The AlMe3 group can be reversibly displaced under reduced pressure affording the new separated ion pair [(MeCTp3-p-tBuPh)MgMe][AlMe4] with a terminal “[MgMe]+” moiety. Moderate thermal treatment of both [(MeCTp3-p-tBuPh)MgMe][AlMe4] and [(MeCTp3-p-tBuPh)Mg(η2-AlMe4)][AlMe4] resulted in selective C–H-bond activation in the 5 position of one of the pyrazolyl moieties and the formation of an AlMe3-modified anionic tris(pyrazolyl)alkane and hence the neutral complex [MeC(pz3-p-tBuPh)2(pz3-p-tBuPh,5-AlMe3)]MgMe.
Introduction
While Grignard compounds RMgX (R = hydrocarbyl; X = Br, Cl) are among the most eminent reagents in organic synthesis,1–8 the elucidation of the Schlenk equilibrium has clearly opened new avenues in organometallic chemistry. Crucially, manipulation of the Schlenk equilibrium by donor solvents features a viable path to dialkyl and diaryl magnesium species.9,10 As early as 1964 Weiß structurally characterised [MgMe2]n, which was obtained in this way.11 Since then, several donor-stabilised monomeric and dimeric complexes of [MgMe2]n have been reported (e.g., donor = quinuclidine, THF, TMEDA).12–17The seminal discoveries of tris(pyrazolyl)methane (HCTp) by Hückel und Bretschneider in 1935 (ref. 18) and subsequently of the trispyrazolylborato (Tp) scorpionate ligands in 1966 by Trofimenko paved the way for a new class of highly versatile ancillary ligands applicable in various areas of coordination chemistry.19–27 Such polypyrazolyl scorpionate ligands can be easily modified both at the pyrazole carbon atoms (Tp3-R,5-R) and in the case of HCTp additionally at the apical carbon atom (R′CTp3-R,4-R,5-R). Neutral R′CTp (R = H, alkyls) have been repeatedly used to stabilise highly reactive magnesium alkyl species.28 Also, several complexes of magnesium bearing a mono-anionic CTp ligand have been structurally characterised (see Scheme 1).29–34 “Metal in a box” complexes such as (CTp3-R,4-R,5-R)2Mg (I) were first described by Mountford and Breher in 2008.29 Here, the application of magnesium dialkyl species led to the deprotonation of the backbone hydrogen. Similarly, such carbanion formation can be triggered in the presence of highly basic [AlMe4] moieties, which can subsequently form trialkylaluminium adducts (or heteroaluminato-type species) to afford complexes (R3AlCTp3-R)2Mg (II).30,35 By using an alkylated backbone or by protonating the apical carbanion with HOTf, separated ion pair “metal in a box” complexes [(R′CTp3-R,5-R)2Mg][A]2 (III, A = OTf, AlMe4) can be obtained.29,35–37 When HCTp is reacted with a bulky magnesium base MgR2 (R = Ph or [N(SiMe3)2]) or has a bulky substituent at the 3 position (adamantyl), deprotonation occurs as well but only one monoanionic CTp ligand coordinates at the magnesium centre to yield (CTp3-Me,5-R)MgR(do) (IV).29,30,34,35 Re-protonation of complexes of the type IV afforded (HCTpMe,Me)MgX2(do) (V) with the neutral HCTp donor.29,30,32
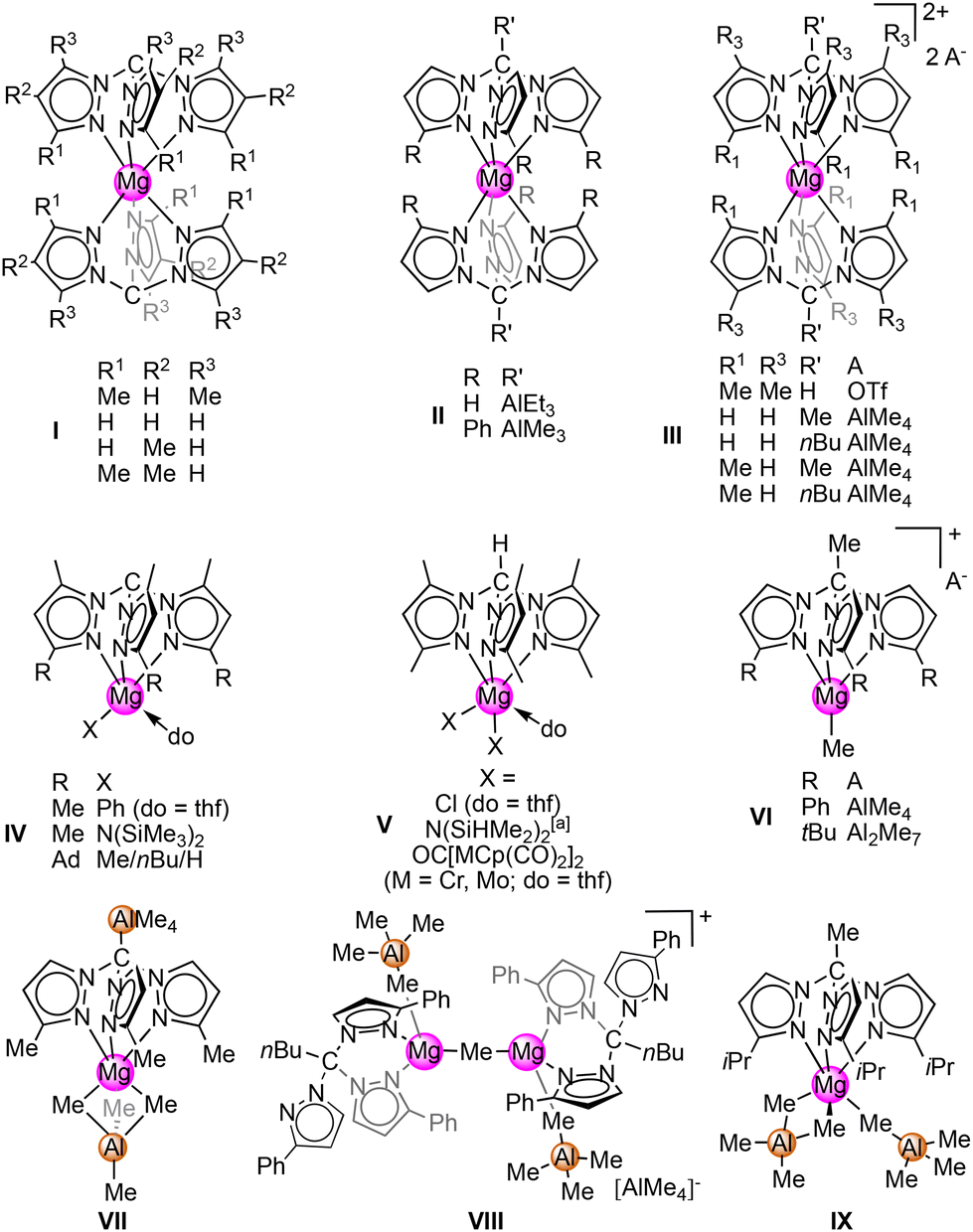 | ||
| Scheme 1 Structural motifs of reported magnesium tris(pyrazolyl)alkane and methanide complexesa κ2(N,N′). | ||
We have recently reported on several new magnesium tris(pyrazolyl)alkane complexes that resulted from the treatment of homoleptic tetramethylaluminate Mg(AlMe4)2 with differently substituted R′CTp3-R.35 The isolated complexes revealed species of types II and III as recurring structural motifs. Sterically demanding MeCTp3-R (R = tBu, Ph) ligands were able to stabilise a [MgMe]+ fragment in compounds [(MeCTp3-R)MgMe][A] (VI, A = AlMe4, Al2Me7). Use of silyl-substituted Me3SiCTp resulted in SiMe4 elimination and formation of a monoanionic heteroaluminate species “Me3AlCTp”. Accordingly, a “metal in a box” complex could be prevented and the neutral Me3AlCTp3-MeMg(AlMe4) (VII) was isolated with one [AlMe4] unit remaining at the magnesium centre. Longer alkyl chains in the backbone led to the dimagnesium complex [({nBuCTp3-Ph}Mg{AlMe4})2(μ-Me)][AlMe4] (VIII) with the rare κ2(N,N′) coordination mode of the RCTp ligand. Finally, the MeCTp3-iPr ligand provided the appropriate steric bulk to accommodate two [AlMe4] units at the magnesium centre, namely MeCTp3-iPrMg(AlMe4)2 (IX).35
In the present study, we further expanded the tris(pyrazolyl)alkane ligand library [R′CTp3-R] to gain an even better understanding of the steric influence of both the position 3 substituents on the pyrazolyl groups and the apical carbon atom substitution on the stabilisation of magnesium alkyl fragments.
Results and discussion
R′CTp3-R ligand synthesis
Several new R′CTp3-R derivatives were synthesized to investigate the feasibility of additional bis(aluminate) complexes of the favoured type IX (Scheme 1). The HCTp3-R derivatives 2 were obtained from the corresponding 3-R-pyrazoles 1 applying the phase-transfer catalysis reaction of Elguero et al., which was optimised by Reger et al.38,39 To separate the desired product from the three other formed isomers, the mixture was refluxed with p-toluenesulfonic acid (Scheme 2). The apical carbon atom was alkylated in a one-pot synthesis by initial lithiation of the carbon atom with Li[N(SiMe3)2], followed by the addition of alkyl halides, which gives the neutral tris(pyrazolyl)alkane R′CTp3-R (3, Scheme 2 and Fig. 1). | ||
| Scheme 2 Preparation of ligands R′CTp3-R3a–g. (i) (nBu)4NBr, CHCl3, Na2CO3, H2O, reflux, 3–7 d. (ii) pTsOH, toluene, 120 °C, 1 d. (iii) Li[N(SiMe3)2], THF, −78 °C, 30 min. (iv) RI, THF, −78 °C to rt, 16 h. | ||
 | ||
| Fig. 1 Representative solid-state structure of 3a. For the crystal structures of compounds 3b, 3c (connectivity) and 3e–g, see the ESI.† | ||
Considering the successful formation of the donor-stabilised bis(tetramethyl)aluminate magnesium complex IX employing MeCTp3-iPr, tris(pyrazolyl)alkanes 3a–c were synthesised to examine the steric demand of pyrazolyl substituents (R = Et, cyPr, Cy) comparable to the iPr group and to complete the series of previously employed substituents Me and iPr.35 The bulky 3d (R = 3-p-tBuPh) was selected not only to prevent the formation of a “metal in a box” complex but also to assess the feasibility of methyl activation and hence the formation of a methylidene species. For comparison, the potassium salt of isoelectronic monoanionic Tp3-p-tBuPh formed the neutral complex Tp3-p-tBuPhMgMe when reacted with MgMe2.40 We previously noticed a pronounced variability of the reactivity of monomeric Mg(AlMe4)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41,42 towards R′CTp3-Ph depending on the chain length of R′, affording either separated ion pairs VI or the unusual methyl-bridged dimagnesium complex VIII. To further examine the effect of distinct backbone alkylation, the tris(pyrazolyl)alkanes 3e–g (R′ = Et, nPr, iBu) were targeted. All ligands were purified by recrystallization or column chromatography. Solid-state structures for 3a–c and 3e–g could be determined by single-crystal X-ray diffraction (SCXRD) analysis (Fig. 1 or ESI†). Tris(pyrazolyl)alkane backbone moieties other than alkyl chains (e.g. H, SiMe3 or Bn = benzyl) were ruled out since we found previously that their reaction with Mg(AlMe4)2 either led to decomposition or carbanion formation via the displacement of the hydrogen atom at the apical carbon atom with an AlMe3 moiety.
41,42 towards R′CTp3-Ph depending on the chain length of R′, affording either separated ion pairs VI or the unusual methyl-bridged dimagnesium complex VIII. To further examine the effect of distinct backbone alkylation, the tris(pyrazolyl)alkanes 3e–g (R′ = Et, nPr, iBu) were targeted. All ligands were purified by recrystallization or column chromatography. Solid-state structures for 3a–c and 3e–g could be determined by single-crystal X-ray diffraction (SCXRD) analysis (Fig. 1 or ESI†). Tris(pyrazolyl)alkane backbone moieties other than alkyl chains (e.g. H, SiMe3 or Bn = benzyl) were ruled out since we found previously that their reaction with Mg(AlMe4)2 either led to decomposition or carbanion formation via the displacement of the hydrogen atom at the apical carbon atom with an AlMe3 moiety.
Magnesium R′CTp3-R complexes
Homoleptic Mg(AlMe4)2 was previously identified as a suitable precursor preventing the decomposition of R′CTp3-R ligands as observed in the case of MgMe2.35 For the synthesis of the new discrete magnesium complexes, a solution of Mg(AlMe4)2 in n-hexane was added dropwise to a solution of 2d or 3a–g in toluene (for an overview see Scheme 3). | ||
| Scheme 3 Reaction of Mg(AlMe4)2 with 2d, 3a–d and 3g in a toluene/n-hexane mixture. | ||
To assess whether the sterically demanding HCTp3-p-tBuPh (2d) can prevent methanide formation and any concomitant functionalization of the apical carbon atom, Mg(AlMe4)2 was treated with the crude product of 2d. However, SCXRD analysis of the crystallized product revealed the recurring structural motif of the neutral “metal in a box” bis(methanide) complex (Me3AlCTp3-p-tBuPh)2Mg (4) (see the ESI†). Even the bulky p-tBuPh groups protrude into the coordination sphere of the other scorpionate ligand and thus cannot prevent the accommodation of two monoanionic heteroaluminato ligands. The magnesium centre in 4 is coordinated by the six nitrogen atoms of the two methanide ligands in a distorted octahedral fashion with N–Mg–N angles in the range of 85.45(9) to 102.49(14)°. The apical carbon–magnesium–apical carbon axis is close to linear (178.58°).
Treatment of Mg(AlMe4)2 with 3a/b instantly gave a white precipitate indicative of the formation of a separated ion pair with a “metal in a box” structure. This very structural motif could be confirmed by the solid-state structure of complex [(MeCTp3-Et)2Mg][AlMe4]2 (5a) (Fig. 2). Two MeCTp3-Et ligands coordinate the magnesium centre in the κ3(N,N′,N′′) mode forming a dicationic entity with two [AlMe4]− anions as counter ions. The six-coordinated magnesium centre adopts a slightly distorted octahedral geometry with longer distances to the equatorial nitrogen atoms (Mg1–N2, 2.1666(10) Å; Mg1–N4, 2.1676(10) Å; Mg1–N6, 2.1458(10) Å). All observed Mg–N distances are in the expected range of magnesium complexes bearing two tris(pyrazolyl)methane ligands (2.113(1)–2.196(1) Å).29–31,33,35 The CH2 groups of the ethyl moiety are almost co-planar with the magnesium centre and the CH3 groups are displaced only marginally from this plane. The apical carbon atom (C01) adopts an almost ideal tetrahedral geometry (109.21(9)–109.93(9)°). The NMR spectra of 5a match those found in the reference literature, with ligand signals only slightly shifted.
 | ||
| Fig. 2 Crystal structure of 5a. Hydrogen atoms, one lattice THF molecule and the second [AlMe4]− are omitted for clarity and atomic displacements are set at a 50% probability level. | ||
In the 13C NMR spectrum, the two [AlMe4]− ions appear as a non-binominal sextet at δ = −4.2 ppm due to the interaction with the 27Al nucleus (I = 5/2, 1J(Al,C) = 70.4 Hz) (Fig. S29†).43 In the 1H NMR spectrum, the proton signals of the [AlMe4]− moieties appear at δ = −1.28 ppm but the expected sextet is not fully resolved (Fig. S28†).
Given the similar reaction behaviour of the Mg(AlMe4)2/3b mixture involving product precipitation, the formation of putative complex [(MeCTp3-cyPr)2Mg][AlMe4]2 (5b) seemed obvious. However, a crystalline material could only be obtained by heating a solid sample in a J. Young valve NMR tube to 150 °C in toluene. The structural motif of a separated ion pair [(MeCTp3-cyPr)2Mg][AlMe4]2 could be confirmed by a connectivity structure of crystals obtained under these harsh conditions (Fig. S6†). The formation of the “metal in a box” complex came as a surprise since the steric difference of cyPr and iPr is marginal. It is likely that the conformational/rotational flexibility of the iPr methyl moieties counteracts the coordination of a second MeCTp ligand. Because of the high insolubility of the precipitated product in aliphatic and aromatic solvents, THF was used as a solvent for NMR measurements. The 1H NMR spectrum indicated that the separated ion pair converted to a complex of the type [(MeCTp3-R)MgMe(thf)2][AlMe4] as proposed previously by NMR spectroscopy.35 Crystallisation could be achieved by cooling the THF solution to −40 °C revealing the solid-state structure of methyl complex [MeCTp3-cyPrMgMe(thf)2][AlMe4] (6) (Fig. 3). The [MgMe]+ fragment is coordinated by one MeCTp3-cyPr ligand and two THF donor molecules, which are fully separated from the charge-balancing [AlMe4]− counter ion. The magnesium centre adopts a distorted octahedral geometry with angles ranging from 78.18(10) to 105.39(17)°.
 | ||
| Fig. 3 Crystal structure of 6. Hydrogen atoms are omitted for clarity and atomic displacements are set at a 50% probability level. | ||
Unsurprisingly, the Mg1–C001 distance of 2.182(4) Å is slightly longer than those of 4-coordinate terminal methyl magnesium complexes [MeCTp3-RMgMe][AlMe4] (R = tBu: 2.115(2) Å and R = Ph: 2.091(2) Å) and TpR,MeMgMe (R = Me: 2.097(4) Å and R = tBu: 2.119(3) Å) but distinctly shorter than the Mg–C distance involving the bridging position of MgMe2 with 2.234(2) and 2.24(3) Å.11,35,44,45 However, the Mg1–C001 distance in 6 is in line with other six-coordinate terminal methyl complexes [MeMg(15-crown-5)][A] (A = Me5Mg2: 2.140(7) Å and A = Cp: 2.124(2) Å) and [MeMg(thf)(dme)2]I (2.162(6) Å).46–48 It appears that the cyPr substituents of 6 provide enough steric flexibility so that the magnesium centre can accommodate two THF molecules, which is in contrast to complexes [MeCTp3-RMgMe][AlMe4] (VI; R = tBu, Ph) mentioned above, in which the THF solvent is not coordinated to magnesium.
For further probing the impact of the apical carbon substitution, Mg(AlMe4)2 was reacted with R′CTp3-Ph (R′ = Et (3e), nPr (3f), and iBu (3g)). Note that complexes [MeCTp3-PhMgMe][AlMe4] (R′ = Me, type VI) and [({nBuCTp3-Ph}Mg{AlMe4})2(μ-Me)][AlMe4] (R′ = nBu, VIII) were identified previously.35 Accordingly, instant oil formation was observed in the case of tris(pyrazolyl)alkanes 3e and 3f. The crude products showed characteristic behaviour of an ionic liquid, impeding their crystallographic characterization. Moreover, the 1H NMR spectra showed broadened signals that were not indicative of any structural motif detected so far. However, the Mg(AlMe4)2/3g reaction produced colourless crystals suitable for SCXRD analysis. The connectivity structure revealed that the methyl-bridged dimagnesium complex [({iBuCTp3-Ph}Mg{AlMe4})2(μ-Me)][AlMe4] (7, Fig. S8†) is isostructural to the previously identified n-butyl derivative VIII.
Further increasing the steric demand of the substituent in the 3 position of the pyrazole rings (compared to R′ = iPr, compound IX), Mg(AlMe4)2 was reacted with 3c bearing cyclohexyl (Cy) groups. Crystals suitable for SCXRD analysis were obtained by recrystallisation in 1,2-difluorobenzene. The solid-state structure revealed the new structural motif of the separated ion pair [(MeCTp3-Cy)Mg(AlMe4)][AlMe4] (8) (Fig. 4).
 | ||
| Fig. 4 Crystal structure of 8. Hydrogen atoms are omitted for clarity and atomic displacements are set at a 50% probability level. | ||
The MeCTp3-Cy ligand stabilises a [Mg(AlMe4)]+ fragment via κ3(N,N′,N′′) coordination while the [AlMe4] ligand interacts with the magnesium centre in an η3 fashion. The monoanionic [AlMe4] counter ion is fully separated. The magnesium centre adopts a slightly distorted octahedral geometry with both ligands coordinating in a facial manner. The C01–Mg1–Al1 axis running through the ligand backbone is almost linear (176.45°). As expected the cyclohexyl groups adopt the chair conformation pointing away from the magnesium centre just like the cyPr groups in 6 and the iPr groups in IX.35
Crystallisation of the reaction product formed by the treatment of Mg(AlMe4)2 with the bulky 3d (with R′ = p-tBuPh) was achieved by redissolving the residue in benzene and dropwise addition of n-pentane to the solution at 40 °C. As detected for 8, the solid-state structure revealed the formation of a mono-cationic complex entity in [(MeCTp3-p-tBuPh)Mg(AlMe4)][AlMe4] (9) (Fig. 5) with the magnesium centre also coordinated with one tris(pyrazolyl)ethane ligand in the κ3(N,N′,N′′) mode. However, due to the enhanced steric demand of 3d compared to 3c, this time the [AlMe4] unit is η2-coordinated. The now 5-coordinate magnesium centre adopts a strongly distorted square pyramidal geometry with two nitrogen atoms (N2 and N4) of the scorpionate ligand and two carbon atoms of the [AlMe4] unit (C40 and C41) located in the equatorial and one ligand nitrogen (N6, the “sting”) in the apical position. Hence, the coordination of the MeCTp3-p-tBuPh ligand itself appears highly asymmetrical, very much scorpionate-like with two pincers and one sting. The putative sixth coordination position for octahedral geometry is approached by ortho-carbon atoms of two different phenyl rings (C18: 3.592, C9: 3.700 Å). The overall asymmetric coordination of the scorpionate ligand is also seen in the distance of the phenyl rings, measured by the distance of the ortho-carbon atoms closest to each other (C9–C18 4.118 Å, C5–C31 6.187 Å, and C22–C35 8.567 Å) and furthermore in the N–N distances between the three coordinating nitrogen atoms (N2–N4 2.717 Å, N2–N6 2.877 Å, and N4–N6 3.110 Å), as well as by the N–Mg–N angles (N2–Mg1–N4 76.64(5)°, N2–Mg1–N6 83.24(6)°, and N4–Mg1–N6 91.91(6)°). Finally, the C01–Mg1–Al1 axis deviates considerably from linearity (165.43°).
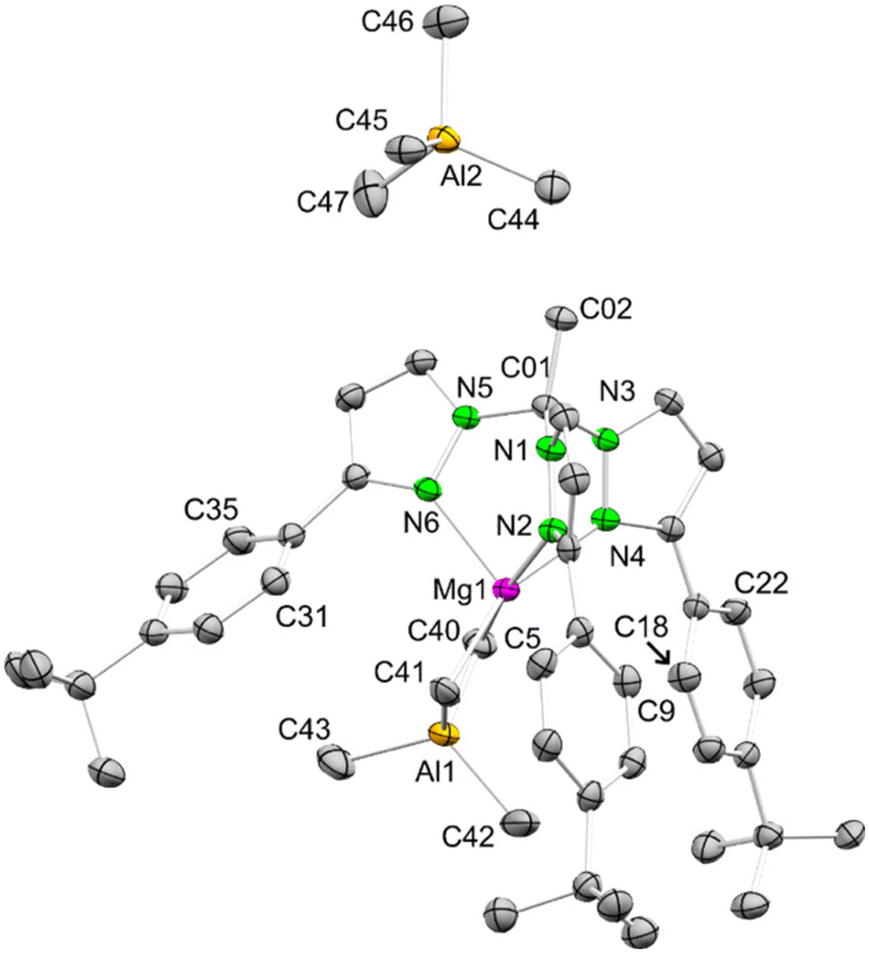 | ||
| Fig. 5 Crystal structure of 9. Hydrogen atoms, the lattice [D6]benzene molecules and the disorder in one tBu group are omitted for clarity and atomic displacements are set at a 50% probability level. | ||
The pronounced asymmetric tris(pyrazolyl)ethane coordination in complex 9 is not detected in the ambient temperature 1H NMR spectrum, which is similar to that of complex 8. The two pyrazole hydrogen atoms in the 4- and 5 positions appear as multiplets in the aromatic region at δ = 8.03/7.84 ppm and δ = 6.28/5.80 ppm, respectively, with an integral of each being three. Also, there is only one sharp signal found for the bridging and terminal methyl groups of the coordinated [AlMe4] unit and for the methyl groups of the displaced [AlMe4] at δ = −0.19 (8)/−0.34 ppm (9), which indicates fast exchange of the coordinated and free tetramethylaluminato groups. Because of this dynamic behaviour VT 1H NMR spectroscopy experiments were performed on both complexes 8 and 9 (Fig. S34/38†). Accordingly, the signals at δ = −0.19 ppm and δ = −0.34 ppm broaden as the temperature decreases. For 8, the signal splits into two separated singlets at −50 °C while for 9 this behaviour is detected at −60 °C. At −80 °C, these two signals are located at δ = 0.18 and −0.06 ppm (8) and at δ = 0.24 and −0.79 ppm (9), both in a ratio of 2:1. All other signals ascribed to 8 split into two signals at −80 °C even that of the backbone methyl group, which means that the [AlMe4] group exchange is more complicated. The same signal splitting is observed for 9 (at −80 °C) except that the signal of the backbone methyl group appears as a very broad signal. This could indicate that for 9 only one R′CTp3-R ligand species is present during the exchange of the [AlMe4] groups, but acts in a hemilabile fashion with one arm off. The splitting of the signals is completely reversible for both complexes upon warming the solutions to ambient temperatures. Complex 9 also remains intact when heated to 80 °C and re-cooled to ambient temperature.
Behaviour of [(MeCTp3-p-tBuPh)Mg(AlMe4)][AlMe4] (9) at elevated temperatures and under vacuum
As revealed by 1H NMR spectroscopy, further heating of complex 9 to 90 °C led to a selective activation of the scorpionate ancillary ligand. Full conversion was indicated by a new set of signals showing the pyrazolyl moieties in a ratio of 1:2 along with the backbone methyl group, which appeared slightly shifted to lower field. Slow evaporation of the solvent generated crystals suitable for SCXRD analysis. The solid-state structure revealed C–H-bond activation of one pyrazole ring at the 5 position forming the neutral complex [MeC(pz3-p-tBuPh)2(pz3-p-tBuPh,5-AlMe3)]MgMe (10) (Scheme 4 and Fig. 6). Notably, heating a solution of 10 to 100 °C resulted in complete decomposition. Complex 10 adopts a distorted tetrahedral geometry with the magnesium centre accommodating the new scorpionate ligand in the routine η3 fashion and a terminal methyl group. The Mg1–C43 distance of 2.090(2) Å matches other terminal Mg–C distances of tetrahedral magnesium methyl complexes.35,44 Due to the metalation of one pyrazolyl ring the scorpionate ligand is monoanionic. Because of this and the less bulkier methyl group the overall geometry of the ligand scaffold is more symmetrical in comparison with that of complex 9. As a consequence, the N–N distances of the three coordinating nitrogen atoms lie between 2.842 and 2.873 Å (cf., 9Δmax = 0.393 Å) and the C01–Mg1–C43 axis is close to linear (177.65°). Since the scorpionate ligand in 10 is monoanionic, complex 10 is now comparable to the Tp3-p-tBuPhMgMe complex proposed by Parkin.40 A striking feature of the 1H NMR spectrum of 10 is the considerable low-field shift at δ = 7.32 ppm of the singlet of the hydrogen atom in the 4 position of the activated pyrazolyl ring. In contrast, the doublet signal of the hydrogen atom in the 4 position of the non-activated pyrazolyl rings appears at δ = 5.90 ppm. The methyl ligand and the AlMe3 group were detected at δ = −0.99 ppm and 0.16 ppm, respectively.
 | ||
| Scheme 4 Thermal activation and reversible coordination of AlMe3 in complex 9. R = p-tBuPh. | ||
 | ||
| Fig. 6 Crystal structure of 10. Hydrogen atoms and two solvent toluene molecules are omitted for clarity and atomic displacements are set at a 50% probability level. | ||
By applying reduced pressure, AlMe3 can be displaced from complex 9 forming the new separated ion pair [(MeCTp3-p-tBuPh)MgMe][AlMe4] (11) (Scheme 4). The 1H NMR spectrum of 11 shows slightly shifted signals of the ancillary ligand while the counter ion [AlMe4] and the methyl group coordinated to the magnesium centre appear as two distinct signals at δ = −0.09 ppm and −1.04 ppm, respectively (Fig. S42†). Adding a stoichiometric amount of AlMe3 to 11 re-formed complex 9 quantitatively, thus proving full reversibility. This behaviour is reminiscent of the TpMe,MeMg(AlMe4)/TpMe,MeMgMe system engaging in donor-induced tetraalkylaluminate cleavage.42 Heating complex 11 to 90 °C also led to the formation of complex 10, suggesting that the C–H-bond activation in the 5 position of the pyrazolyl moiety is caused by the attack of the separated [AlMe4] anion via the release of methane.
Cone angle calculations
The mathematically exactly calculated cone angles49 of the scorpionate ligands of the newly synthesised complexes are shown in Table 1. The cone angles featured by [(MeCTp3-Et)2Mg][AlMe4]2 (5a, Θ° = 247.95°) and [MeCTp3-cyPrMgMe(thf)2][AlMe4] (6, Θ° = 250.80°) are very close to that observed for the bis(tetramethylaluminato) complex [(MeCTp3-iPr)Mg(AlMe4)2] (Θ = 248.76°).35 Despite this similarity, completely different coordination environments exist even though all complexes exhibit a coordination number of six. It seems that the slightly higher steric demand of the iPr group in comparison to Et and cyPr, caused by the flexibility of the two Me groups, is sufficient to prevent the formation of the “metal in a box” complex. As expected the cone angle calculated for [(MeCTp3-Cy)Mg(AlMe4)][AlMe4] (8) (Θ° = 260.64°) is distinctly higher than that enforced by the iPr, Et or cyPr substituents, which allows the stabilization of the Mg(η3-AlMe4)+ fragment with three methyl groups coordinated to the metal centre. The tentative cone angles of the two disordered species of 9 are determined as Θ° = 289.60° and Θ° = 291.34°. The only magnesium complexes with a cone angle comparable to 9 are [(MeCTp3-Ph)MgMe][AlMe4] (Θ° = 282.84°) and the extremely bulky [(CTpAd,Me)MgMe] (Θ° = 301.47°).35,50 Upon thermal C–H-bond activation and formation of [MeC(pz3-p-tBuPh)2(pz3-p-tBuPh,5-AlMe3)]MgMe (10), the cone angle increased even further to Θ° = 299.10°. Now that the scorpionate ligand has a negative charge it is better comparable to the adamantyl methanide complex [(CTpAd,Me)MgMe] and both angles are indeed very close. Comparing the cone angles of (Me3AlCTp3-p-tBuPh)2Mg (4) and 10, which both feature anionic scorpionate ligands with p-tBuPh moieties in the 3 position, there is a difference of 11.08°. Therefore, the ligand coordinated to the metal centre opposite to the examined/calculated R′CTp3-R ligand has a vast influence on the cone angle too.| 4 | 5a | 6 | 8 | 9 | 10 |
|---|---|---|---|---|---|
| 288.02 | 247.95 | 250.80 | 260.64 | 289.60 | 299.10 |
| 291.34 |
Conclusions
The scope of the tris(pyrazolyl)alkane R′CTp3-R library, here with varying substituents in the pyrazolyl 3 position and at the apical carbon atom, could be successfully expanded by derivatives MeCTp3-Et, MeCTp3-cyPr, MeCTp3-Cy, MeCTp3-p-tBuPh, EtCTp3-Ph, nPrCTp3-Ph and iBuCTp3-Ph (cyPr = cyclopropyl, Cy = cyclohexyl, 3-p-tBuPh = para-tert-butylphenyl). As anticipated, the reaction of bis(tetramethylaluminato) magnesium Mg(AlMe4)2 with neutral tris(pyrazolyl)alkanes R′CTp3-R is strongly dependent on the steric effect of such scorpionate ligands, resulting in the partial or full displacement of the aluminato ligands. The smaller Et and cyPr substituents in the 3 position favour the formation of the recurring “undesired” structural motifs of a “metal in a box” complex [(MeCTp3-R)2Mg][AlMe4]2 (R = Et, cyPr). The bulky Cy and p-tBuPh moieties led to the separated ion pairs [(MeCTp3-R)Mg(AlMe4)][AlMe4] (R = Cy, p-tBuPh) with distinct η2- and η3-coordination modes of the AlMe4 moiety. Variation of the alkyl substituent R′ at the apical carbon atom of R′CTp3-Ph (R′ = Et, nPr, iBu) led to the isolation of the dimagnesium species [({iBuCTp3-Ph}Mg{AlMe4})2(μ-Me)][AlMe4] with a κ2(N,N′) coordination mode of the tris(pyrazolyl)alkane ligand. Separated ion pairs of the type [(MeCTp3-R)Mg(AlMe4)][AlMe4] are prone to AlMe3 separation, when exposed to a vacuum, and a selective pyrazolyl deprotonation, upon thermal treatment. The latter C–H-bond activation is triggered by the non-coordinating [AlMe4] anion and generates a new monoanionic scorpionate ligand in [MeC(pz3-p-tBuPh)2(pz3-p-tBuPh,5-AlMe3)]MgMe featuring a terminal Mg–CH3 moiety.Experimental
General considerations
All manipulations were performed under rigorous exclusion of air and moisture under an argon atmosphere (<1 ppm O2, <1 ppm of H2O) in an MB200B glovebox (MBraun) or according to standard Schlenk techniques and in oven-dried glassware. Solvents (THF, n-pentane, n-hexane, Et2O and toluene) were purified by using Grubbs-type columns (MBraun SPS-800, solvent purification system) and stored inside a glovebox. THF was dried further over molecular sieves. [D6]Benzene, [D8]toluene and [D8]THF were obtained from Sigma Aldrich and dried over a Na/K alloy and filtered prior to use. 1,4-Dioxane was dried over sodium metal, distilled and degassed and stored inside a glovebox. 1,2-Difluorobenzene was purchased from Sigma Aldrich, dried over CaH2, distilled and degassed prior to use. Acetophenone (>98%), benzophenone (>98%), n-BuLi (2.5 M in n-hexane), cyclopropyl methyl ketone (99%), 1,4-dihydro-2H-pyrane (>97%), ethyl bromide (>99%), ethyl iodide (99%, copper stabilized), hexamethyldisilazane (>99%), hydrazine dihydrochloride (>98%), methylmagnesium bromide (3 M in Et2O), n-propyl iodide (99%), 1-iodo-2-methylpropane (97%, copper stabilized), pyrazole (>98%) and tetra-n-butylammonium bromide (98%) were purchased from Sigma Aldrich and used as received. Iodomethane (>99%) and methyl tert-butyl ether (MTBE) were purchased from Acros Organics and used as received. N,N-Dimethylformamide dimethylacetate (>97%), 4′-tert-butylacetophenone (98%) and trimethylaluminium (98%) were purchased from abcr and used as received. Cyclohexyl methyl ketone (95%) was purchased from Alfa Aesar and used as received. Li[N(SiMe3)2],51 MgMe29,10 and Mg(AlMe4)241 were synthesized according to the literature procedures. The 3-R-pyrazoles 1 were synthesized according to standard procedures (R = Et,52cyPr,53 Cy,53 Ph,53p-tBuPh53). Argon was supplied by Westfalen AG. 1H and 13C NMR spectra were recorded on a Bruker AVII+400 spectrometer (1H: 400.11 MHz; 13C: 100.61 MHz) at 299 K. The chemical shifts compiled in the Experimental section are referenced to solvent residual resonances in parts per million relative to tetramethylsilane. The variable temperature 1H NMR spectra of 8 and 9 were recorded in a J. Young valve NMR tube on a Bruker AVII+500 spectrometer (1H: 500.13 MHz; 13C: 125.76 MHz) at 299 K. IR spectra were recorded on a NICOLET 6700 FTIR spectrometer (Thermo Fisher Scientific). The samples were mixed with KBr powder and measured in a DRIFT cell with KBr windows. DRIFT data were converted by using the Kubelka–Munk refinement. Elemental analyses were performed on an Elementar Vario MICRO cube. Single crystals were grown from saturated solutions of [D6]benzene, 1,2-difluorobenzene, Et2O, MTBE, n-pentane, or THF by standard techniques. Suitable single crystals for X-ray structure studies were selected in a glovebox and coated with Parabar 10312 (Hampton Research). Crystallographic data were measured on a Bruker APEX II DUO instrument equipped with an IμS micro focus sealed tube and QUAZAR optics for MoKα radiation (λ = 0.71073 Å). The structures were solved by direct methods using SHELXT software packages and refined on F2 (with all independent reflections) using the SHELXL software package. The non-hydrogen atoms were refined anisotropically. All hydrogen atoms of C–H bonds were located using a riding model at the expected position for hydrogen with a fixed interatomic distance.
:undesired isomer = 1:0.4.
:undesired isomer = 1:0.6.
:undesired isomer = 1:0.2.
:undesired isomer = 1:0.1.
:undesired isomer = 1:0.1.
Preparation and characterization of compounds 3, 4, 5, 6, 8, 9, 10 and 11
:1 petroleum ether and Et2O. The fractions containing the desired product were combined and the solvent was removed under reduced pressure, which gave 3a as a colourless powder (670 mg, 2.14 mmol, 28%). Crystals suitable for SCXRD analysis could be obtained by recrystallization from MTBE. 1H NMR (400.1 MHz, CDCl3, 26 °C): δ = 6.52 (d, 3H, 3JHH = 2.5 Hz; 5-H(pz)), 6.09 (d, 3H, 3JHH = 2.5 Hz; 4-H(pz)), 2.91 (s, 3H; CH3CTp), 2.67 (q, 6H, 3JHH = 7.6 Hz; CH2CH3), 1.24 ppm (t, 3H, 3JHH = 7.6 Hz; CH2CH3). 13C{1H} NMR (100.6 MHz, CDCl3, 26 °C): δ = 156.7 (3-C(pz)), 129.6 (5-C(pz)), 104.8 (4-C(pz)), 90.1 (CH3CTp), 25.9 (CH3CTp), 21.6 (CH2CH3), 13.8 ppm (CH2CH3). DRIFTS: ṽmax = 3134w, 3115w, 2969s, 2935m, 2873m, 2641vw, 1703vw, 1526s, 1467m, 1454m, 1371s, 1312m, 1260s, 1209s, 1112m, 1049s, 1008w, 976m, 952w, 796s, 759s, 659w, 632w, 578vw, 439vw cm−1. Elemental analysis calcd (%) for C17H24N6 (312.42 g mol−1): C 65.36, H 7.74, N 26.90; found: C 65.49, H 7.47, N 26.94.
:1 mixture of petroleum ether and Et2O with subsequent recrystallization from Et2O to afford 3b as colourless crystals (983 mg, 2.82 mmol, 31%) suitable for SCXRD analysis. 1H NMR (400.1 MHz, CDCl3, 26 °C): δ = 6.46 (d, 3H, 3JHH = 2.6 Hz, 5-H(pz)), 5.85 (d, 3H, 3JHH = 2.6 Hz, 4-H(pz)), 2.84 (s, 3H, CH3CTp), 1.92 (m, 3H, (CH2)2CH), 0.88 (m, 6H, (CH2)2CH), 0.67 ppm (m, 6H, (CH2)2CH). 13C{1H} NMR (100.6 MHz, CDCl3, 26 °C): δ = 157.2 (3-C(pz)), 129.7 (5-C(pz)), 102.7 (4-C(pz)), 90.1 (CTp), 25.8 (CH3CTp), 9.4 ((CH2)2CH), 8.2 ppm ((CH2)2CH). DRIFTS: ṽmax = 3142w, 3124w, 3087vw, 3003m, 2952w, 2852vw, 2469vw, 2075vw, 1708w, 1613vw, 1531vs, 1480m, 1444m, 1397s, 1371s, 1305s, 1259vs, 1220s, 1208vs, 1177s, 1113m, 1045s, 988s, 883m, 763s, 709vw, 561vw, 631vw, 488vw cm−1. Elemental analysis calcd (%) for C20H24N6 (348.21 g mol−1): C 68.94, H 6.94, N 24.12; found C 68.86, H 6.85, N 23.99.
:1 mixture of petroleum ether and Et2O to afford 3d as a colourless powder (671 mg, 10.7 mmol, 33%). 1H NMR (400.1 MHz, CDCl3, 26 °C): δ = 7.79 (m, 6H; o-H(Ph)), 7.45 (m, 6H; m-H(Ph)), 6.82 (d, 3H, 3JHH = 2.5 Hz; 5-H(pz)), 6.58 (d, 3H, 3JHH = 2.5 Hz; 4-H(pz)), 3.16 (s, 3H; CH3CTp), 1.36 ppm (s, 27H; C(CH3)3). 13C{1H} NMR (100.6 MHz, CDCl3, 26 °C): δ = 153.2 (3-C(pz)), 151.3 (p-C(Ph)), 130.5 (5-C(pz)), 130.1 (i-C(Ph)), 125.7 (o-C(Ph)), 125.5 (m-C(Ph)), 103.8 (4-C(pz)), 91.1 (MeCTp), 34.6 (C(CH3)3), 31.3 (C(CH3)3), 26.0 ppm (CH3CTp). DRIFTS: ṽmax = 3127vw, 2963m, 2902w, 2865w, 1719vw, 1668vw, 1605vw, 1501m, 1450w, 1387w, 1365w, 1268m, 1232s, 1124w, 1089vw, 1047w, 1017vw, 978vw, 948vw, 839m, 793m, 763m, 742w, 700vw, 642vw, 560w, 501vw cm−1. Elemental analysis calcd (%) for C41H48N6 (624.88 g mol−1): C 78.81, H 7.74, N 13.45; found: C 79.01, H 7.85, N 13.35.
:1 mixture of n-hexane and MTBE to afford 3e as colourless crystals (1.55 g, 3.28 mmol, 73%) suitable for SCXRD analysis. 1H NMR (400.1 MHz, CDCl3, 26 °C): δ = 7.86 (m, 6H, o-H(Ph)), 7.38 (m, 9H, m-H(Ph), p-H(Ph)), 7.07 (d, 3H, 3JHH = 2.8 Hz, 5-H(pz)), 6.62 (d, 3H, 3JHH = 2.7 Hz, 4-H(pz)), 3.54 (q, 2H, 3JHH = 7.2 Hz, CH2CTp), 1.38 ppm (t, 3H, 3JHH = 7.2 Hz, CH3CH2C(pz)3). 13C{1H} NMR (100.6 MHz, CDCl3, 26 °C): δ = 153.1 (3-C(pz)), 133.1 (i-C(Ph)), 131.6 (5-C(pz)), 128.8 (m-C(Ph)), 128.3 (p-C(Ph)), 126.1 (o-C(Ph)), 103.7 (4-C(pz)), 93.1 (CTp), 34.1 (CH2CTp), 9.3 ppm (CH3CH2). DRIFTS: ṽmax = 3145vw, 3064vw, 3004vw, 2982vw, 2951vw, 2953w, 1604vw, 1582vw, 1528m, 1497m, 1457s, 1390s, 1358m, 1298m, 1275w, 1247w, 1217vs, 1180vw, 1160vw, 1100w, 1075m, 1051m, 1028m, 859s, 758s, 750s, 520vw cm−1. Elemental analysis calcd (%) for C30H26N6 (470.22 g mol−1): C 76.57, H 5.57, N 17.86; found C 76.30, H 5.97, N 17.62.
:1 mixture of n-hexane and MTBE to afford 3f as colourless crystals (2.13 g, 4.40 mmol, 97%) suitable for SCXRD analysis. 1H NMR (400.1 MHz, CDCl3, 26 °C): δ = 7.87 (m, 6H, o-H(Ph)), 7.39 (m, 9H, m-H(Ph), p-H(Ph)), 7.06 (d, 3H, 3JHH = 2.7 Hz, 5-H(pz)), 6.62 (d, 3H, 3JHH = 2.7 Hz, 4-H(pz)), 3.46 (m, 2H, CH2CTp), 1.89 (m, 2H, CH2CH2CTp), 1.08 (t, 3H, 3JHH = 7.4 Hz, CH3CH2) ppm. 13C{1H} NMR (100.6 MHz, CDCl3, 26 °C): δ = 153.1 (3-C(pz)), 133.1 (i-C(Ph)), 131.5 (5-C(pz)), 128.8 (m-C(Ph)), 128.3 (p-C(Ph)), 126.1 (o-C(Ph)), 103.7 (4-C(pz)), 92.8 (C(pz)3), 42.5 (CH2CTp), 17.8 (CH2CH2CTp), 14.4 (CH3CH2) ppm. DRIFTS: ṽmax = 3059vw, 2961w, 2869vw, 1604vw, 1528m, 1499vs, 1455vs, 1435s, 1385s, 1357s, 1301s, 1279w, 1210vs, 1100w, 1074w, 1044w, 1028w, 946vw, 903m, 855m, 800w, 752s, 695s, 686m, 659vw, 627w, 511vw cm−1. Elemental analysis calcd (%) for C31H28N6 (484.2 g mol−1): C 76.83, H 5.82, N 17.34; found C 76.35, H 5.69, N 17.43.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for the compounds have been deposited in the Cambridge Crystallographic Data Centre (2386219–2386230).†Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors thank Dr Klaus Eichele and Kristina Heß for recording VT NMR spectra.References
- V. Grignard, C. R. Hebd. Seances Acad. Sci., 1900, 130, 1322–1325 CAS.
- E. C. Ashby, Q. Rev., Chem. Soc., 1967, 21, 259 RSC.
- F. Bickelhaupt, Angew. Chem., Int. Ed. Engl., 1974, 13, 419–420 CrossRef CAS.
- E. C. Ashby, J. Laemmle and H. M. Neumann, Acc. Chem. Res., 1974, 7, 272–280 CrossRef CAS.
- F. Bickelhaupt, Angew. Chem., 1987, 99, 1020–1035 CrossRef CAS.
- M. Orchin, J. Chem. Educ., 1989, 66, 586 CrossRef CAS.
- P. R. Markies, R. M. Altink, A. Villena, O. S. Akkerman, F. Bickelhaupt, W. J. J. Smeets and A. L. Spek, Adv. Organomet. Chem., 1991, 402, 289–312 CrossRef CAS.
- G. S. Silverman and P. E. Rakita, Handbook of Grignard Reagents, CRC Press, 1996 Search PubMed.
- W. Schlenk and W. Schlenk Jr., Chem. Ber., 1929, 62, 920–924 CrossRef.
- A. C. Cope, J. Am. Chem. Soc., 1935, 57, 2238–2240 CrossRef CAS.
- E. Weiss, J. Organomet. Chem., 1964, 2, 314–321 CrossRef CAS.
- J. Toney and G. D. Stucky, J. Organomet. Chem., 1970, 22, 241–249 CrossRef CAS.
- T. Greiser, J. Kopf, D. Thoennes and E. Weiss, J. Organomet. Chem., 1980, 191, 1–6 CrossRef CAS.
- H. Viebrock and E. Weiss, J. Organomet. Chem., 1994, 191, 121–126 CrossRef.
- R. I. Yousef, B. Walfort, T. Rüffer, C. Wagner, H. Schmidt, R. Herzog and D. Steinborn, J. Organomet. Chem., 2005, 690, 1178–1191 CrossRef CAS.
- O. Michel, C. Meermann, K. W. Törnroos and R. Anwander, Organometallics, 2009, 28, 4783–4790 CrossRef CAS.
- A. K. Bartholomew, L. M. Guard, N. Hazari and E. D. Luzik, Aust. J. Chem., 2013, 66, 1455–1458 CrossRef CAS.
- W. Hückel and H. Bretschneider, Ber. Dtsch. Chem. Ges., 1937, 70, 2024–2026 CrossRef.
- S. Trofimenko, J. Am. Chem. Soc., 1966, 88, 1842–1844 CrossRef CAS.
- S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 3170–3177 CrossRef CAS.
- S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 6288–6294 CrossRef CAS.
- S. Trofimenko, J. C. Calabrese, J. K. Kochi, S. Wolowiec, F. B. Hulsbergen and J. Reedijk, Inorg. Chem., 1992, 31, 3943–3950 CrossRef CAS.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- A. L. Rheingold, R. L. Ostrander, B. S. Haggerty and S. Trofimenko, Inorg. Chem., 1994, 33, 3666–3676 CrossRef CAS.
- C. López, D. Sanz, R. M. Claramunt, S. Trofimenko and J. Elguero, J. Organomet. Chem., 1995, 503, 265–276 CrossRef.
- S. Trofimenko, A. L. Rheingold and L. M. Liable Sands, Inorg. Chem., 2002, 41, 1889–1896 CrossRef CAS PubMed.
- S. Trofimenko, in Progress in Inorganic Chemistry, John Wiley & Sons, Ltd, 2007, pp. 115–210 Search PubMed.
- S. Krieck, A. Koch, K. Hinze, C. Müller, J. Lange, H. Görls and M. Westerhausen, Eur. J. Inorg. Chem., 2016, 2016, 2332–2348 CrossRef CAS.
- H. R. Bigmore, J. Meyer, I. Krummenacher, H. Rüegger, E. Clot, P. Mountford and F. Breher, Chem. – Eur. J., 2008, 14, 5918–5934 CrossRef CAS.
- M. G. Cushion, J. Meyer, A. Heath, A. D. Schwarz, I. Fernández, F. Breher and P. Mountford, Organometallics, 2010, 29, 1174–1190 CrossRef CAS.
- J. Meyer, I. Kuzu, S. González-Gallardo and F. Breher, Z. Anorg. Allg. Chem., 2013, 639, 301–307 CrossRef CAS.
- J. Meyer, S. González-Gallardo, S. Hohnstein, D. Garnier, M. K. Armbruster, K. Fink, W. Klopper and F. Breher, Chem. – Eur. J., 2015, 21, 2905–2914 CrossRef CAS PubMed.
- C. Müller, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Inorg. Chem., 2015, 54, 635–645 CrossRef PubMed.
- R. Lalrempuia, A. Stasch and C. Jones, Chem. – Asian J., 2015, 10, 447–454 CrossRef CAS PubMed.
- C. Stuhl, C. Maichle-Mössmer and R. Anwander, Chem. – Eur. J., 2018, 24, 14254–14268 CrossRef CAS PubMed.
- M. Veith, Eur. J. Inorg. Chem., 2000, 2000, 1883–1899 CrossRef.
- S. Harder, F. Feil and T. Repo, Chem. – Eur. J., 2002, 8, 1991–1999 CrossRef CAS.
- S. Juliá, J. M. del Mazo, L. Avila and J. Elguero, Org. Prep. Proced. Int., 1984, 16, 299–307 CrossRef.
- D. L. Reger, T. C. Grattan, K. J. Brown, C. A. Little, J. J. S. Lamba, A. L. Rheingold and R. D. Sommer, J. Organomet. Chem., 2000, 607, 120–128 CrossRef CAS.
- P. Ghosh and G. Parkin, Inorg. Chem., 1996, 35, 1429–1430 CrossRef CAS PubMed.
- K. Ziegler and E. Holzkamp, Liebigs Ann. Chem., 1957, 605, 93–97 CrossRef CAS.
- J. L. Atwood and G. D. Stucky, J. Am. Chem. Soc., 1969, 91, 2538–2543 CrossRef CAS.
- B. Wrackmeyer and E. V. Klimkina, Z. Naturforsch., B: J. Chem. Sci., 2014, 63, 923–928 CrossRef.
- O. Michel, H. M. Dietrich, R. Litlabø, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Organometallics, 2012, 31, 3119–3127 CrossRef CAS.
- C. Stuhl and R. Anwander, Dalton Trans., 2018, 47, 12546–12552 RSC.
- A. D. Pajerski, M. Parvez and H. G. Richey, J. Am. Chem. Soc., 1988, 110, 2660–2662 CrossRef CAS.
- M. Vestergren, J. Eriksson and M. Håkansson, Chem. – Eur. J., 2003, 9, 4678–4686 CrossRef CAS PubMed.
- A. Jaenschke, J. Paap and U. Behrens, Z. Anorg. Allg. Chem., 2008, 634, 461–469 CrossRef CAS.
- J. A. Bilbrey, A. H. Kazez, J. Locklin and W. D. Allen, J. Comput. Chem., 2013, 34, 1189–1197 CrossRef CAS PubMed.
- R. Lalrempuia, A. Stasch and C. Jones, Chem. – Asian J., 2015, 10, 447–454 CrossRef CAS PubMed.
- U. Wannagat and H. Niederprüm, Angew. Chem., 1959, 71, 574–574 CrossRef CAS.
- B. M. Ahmed and G. Mezei, RSC Adv., 2015, 5, 24081–24093 RSC.
- A.-K. Pleier, H. Glas, M. Grosche, P. Sirsch and W. R. Thiel, Synthesis, 2001, 55–62 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2386219–2386230. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02722a |
| This journal is © The Royal Society of Chemistry 2024 |