
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization and chemical bonding analysis of the quaternary cyanamides Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6†
Hicham
Bourakhouadar
,
Juan
Medina-Jurado
,
Peter C.
Müller
,
Alex J.
Corkett
 * and
Richard
Dronskowski
*
* and
Richard
Dronskowski
*
Chair of Solid-State and Quantum Chemistry, Institute of Inorganic Chemistry, RWTH Aachen University, 52056 Aachen, Germany. E-mail: drons@HAL9000.ac.rwth-aachen.de; alexander.corkett@ac.rwth-aachen.de
First published on 30th October 2024
Abstract
Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6 were prepared via solid-state metathesis reactions either via a more exothermic direct reaction between Li2NCN, MnCl2 and HfCl4 or a milder two-step reaction in which ternary Li2Zr(NCN)3 was first prepared and subsequently reacted with MnF2. Their crystal structures were determined from powder X-ray diffraction data and found to crystallize isotypically in low-symmetry variants of the [NiAs]-type MNCN structure with P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1m symmetry and comprise corundum-like [T2(NCN)3]2+ layers (T = Hf4+, Zr4+) alternating with [Li2Mn(NCN)3]2− layers. In-depth chemical bonding analysis was undertaken using LOBSTER to calculate the Löwdin charges which reveal significant differences in covalency between the two metal layers that is also reflected in the crystal orbital bond indices (COBI) of the metal–nitrogen bonds as well as the carbon–nitrogen bonds that show distinct single and triple bond character, which is also evident from infrared spectroscopy measurements. A geometric analysis of all known quaternary cyanamides with the general formula A2MT2(NCN)6 (A: an alkali metal, M: a divalent metal and T: a tetravalent metal) demonstrates the adaptation of the NCN unit to cation size differences, expressed as total distortion δtotal, by tilting away from the stacking axis. This tilting impacts the octahedral environment of the three different metal sites causing a distortion, quantified by means of the quadratic elongation λoct, revealing that the divalent and alkali metal sites are strongly dependent on δtotal whilst the tetravalent site is less influenced by the total distortion. Electronic structure calculations reveal Li2MnHf2(NCN)6 to have an indirect band gap with a wide band gap of approximately 2.4 eV, in good agreement with the measured value of 2.1 eV. Furthermore, SQUID magnetometry measurements reveal predominantly antiferromagnetic interactions, but no transition to a long-range ordered state, presumably as a result of the magnetic dilution of the octahedral site, in which only 1/6 of the interstices are occupied by paramagnetic cations.
1m symmetry and comprise corundum-like [T2(NCN)3]2+ layers (T = Hf4+, Zr4+) alternating with [Li2Mn(NCN)3]2− layers. In-depth chemical bonding analysis was undertaken using LOBSTER to calculate the Löwdin charges which reveal significant differences in covalency between the two metal layers that is also reflected in the crystal orbital bond indices (COBI) of the metal–nitrogen bonds as well as the carbon–nitrogen bonds that show distinct single and triple bond character, which is also evident from infrared spectroscopy measurements. A geometric analysis of all known quaternary cyanamides with the general formula A2MT2(NCN)6 (A: an alkali metal, M: a divalent metal and T: a tetravalent metal) demonstrates the adaptation of the NCN unit to cation size differences, expressed as total distortion δtotal, by tilting away from the stacking axis. This tilting impacts the octahedral environment of the three different metal sites causing a distortion, quantified by means of the quadratic elongation λoct, revealing that the divalent and alkali metal sites are strongly dependent on δtotal whilst the tetravalent site is less influenced by the total distortion. Electronic structure calculations reveal Li2MnHf2(NCN)6 to have an indirect band gap with a wide band gap of approximately 2.4 eV, in good agreement with the measured value of 2.1 eV. Furthermore, SQUID magnetometry measurements reveal predominantly antiferromagnetic interactions, but no transition to a long-range ordered state, presumably as a result of the magnetic dilution of the octahedral site, in which only 1/6 of the interstices are occupied by paramagnetic cations.
Introduction
Research into NCN-containing compounds has received particular and increasing attention in recent years.1 From a HSAB “hard and soft (Lewis) acids and bases” perspective, the NCN2− anion lies between oxide O2− and sulfide S2−, thus prompting it to be considered as a pseudochalcogenide or “divalent nitride”. Being also metastable and more covalent than their oxide analogues, carbodiimides often exhibit divergent properties and therefore hold great potential for a whole range of practical applications. Specifically, metal carbodiimides have been exploited as negative electrode materials in Na- or Li-ion batteries,2,3 and also in photovoltaic devices,4 fluorescent light sources5,6 and light-emitting diodes.7,8 From a structural point of view, many of these compounds can be derived from their oxide-analogues by replacing O2− with the extended NCN2− anion, either in its symmetric carbodiimide (−N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN−) or asymmetric cyanamide form (N
CN−) or asymmetric cyanamide form (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–N2−), with structures generally consisting of layers of the NCN unit in either a hexagonal closed arrangement, derived from NiAs, or in a cubic closed arrangement, derived from NaCl. The metals in turn occupy the octahedral holes, so that an alternating anion-metal layered arrangement is obtained.
C–N2−), with structures generally consisting of layers of the NCN unit in either a hexagonal closed arrangement, derived from NiAs, or in a cubic closed arrangement, derived from NaCl. The metals in turn occupy the octahedral holes, so that an alternating anion-metal layered arrangement is obtained.
Unlike their respective oxide analogues, metal carbodiimides and cyanamides are generally not prepared by the conventional ceramic method due to their often endothermic nature. Synthesis of these compounds requires milder conditions, such as those offered by solid-state metathesis (SSM) reactions,9 which have proven particularly effective. Such reactions are highly exothermic and promoted by the release of the lattice energy of the co-produced metathesis salt. Indeed, the application of the SSM reaction has given rise to a wide variety of binary, ternary, quaternary metal carbodiimides/cyanamides. The simplest examples of this group of compounds are binary representatives (MNCN) which adopt either a rhombohedral structure that derives from NaCl (this is the case for MnNCN, MgNCN, CaNCN, CdNCN, SrNCN) or a hexagonal structure,10–13 which relates to NiAs (for FeNCN, CoNCN, NiNCN).14,15 The ternary family of compounds AMIII(NCN)2 (A = Li or Na, M = Al, In, Yb, Y, Sc) adopt a low-symmetry orthorhombic modification of NiAs, with a zigzag arrangement of the two metal channels,16–18 and there are also perovskite-like phases of the MIIHfIV(NCN)3 (M = Mn, Fe) type.19 Common to all these compounds is the fact that they can all be described by a close relation to their oxide or chalcogenide analogues. However, the NCN unit distinguishes itself with its ability to tilt away from the stacking axis in order to accommodate metals of different sizes into the cationic layers, in a distortion that is not accessible to the spherical oxygen anion. In this way, the sizes of ions incorporated in the structures of metal carbodiimides/cyanamides play an important role in the stability of the structures, and can even allow for vacancies in the layers, as is seen in Hf(NCN)2 and Zr(NCN)2 which adopt vacancy-ordered NiAs derived structures with an open ‘wine-rack’ like framework.20
These observations highlighting a similarity to binary metal oxides and chalcogenides are less apparent when it comes to the quaternary family A2MIISnIV2(NCN)6 (A = Li or Na, M = Mn, Fe, Co, Ni and Mg),21–23 where vacancy and cation ordering schemes are combined, giving rise to structures unique to carbodiimide chemistry. The crystal structures of these compounds comprise layers of metal cations alternating with NCN2− ions, with an intra- and inter-layer cation ordering schemes. Motivated by this work, and with the intention of expanding this family, the new quaternary cyanamides Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6 have been prepared. In addition, characterization by powder X-ray diffraction, infrared spectroscopy and chemical bonding analysis by means of DFT calculations are presented together with a detailed analysis of how the replacement of Sn4+ ions by Hf4+ or Zr4+ ions affect the coordination environments of the metals.
Experimental
Synthesis of Li2MnHf2(NCN)6
In an argon filled glovebox a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 stoichiometric ratio of Li2NCN (prepared as described in ref. 24), MnCl2 (ThermoFisher, 99.9%) and HfCl4 (ThermoFisher, 98%, sublimed under dynamic vacuum at 200 °C for further purification) was homogenized, on the 0.4 g scale, in an agate pestle and mortar and loaded into a silica tube and sealed under vacuum. The sample was heated in a tubular furnace up to 700 °C and held at this temperature for 8 hours with heating and cooling rates of 2 °C min−1. The accrued grayish colored sample (main yield 95%) proved stable to air. The product was washed with water and ethanol, before being dried at 100 °C.
:1:2 stoichiometric ratio of Li2NCN (prepared as described in ref. 24), MnCl2 (ThermoFisher, 99.9%) and HfCl4 (ThermoFisher, 98%, sublimed under dynamic vacuum at 200 °C for further purification) was homogenized, on the 0.4 g scale, in an agate pestle and mortar and loaded into a silica tube and sealed under vacuum. The sample was heated in a tubular furnace up to 700 °C and held at this temperature for 8 hours with heating and cooling rates of 2 °C min−1. The accrued grayish colored sample (main yield 95%) proved stable to air. The product was washed with water and ethanol, before being dried at 100 °C.
Synthesis of Li2MnZr2(NCN)6
A reaction mixture of the ternary compound Li2Zr(NCN)3 (prepared as described in ref. 25) and MnF2 (ThermoFisher, 99%) was homogenized in an agate pestle and mortar, then loaded into a silica tube and sealed. The mixture was heated to 550 °C and the sample remained at this temperature for 8 hours with heating and cooling rates of 2 °C min−1. The brown quaternary compound (main yield 92%) proved stable to air. The product was washed with water and ethanol, before being dried at 100 °C.PXRD analysis
Powder X-ray diffraction (PXRD) data were recorded on a washed Li2MnHf2(NCN)6 sample at room temperature using a calibrated STOE STADI-MP powder diffractometer with a flat sample holder (Mo Kα1, linear PSD, 2θ range 3–65° with individual steps of 0.005°) and on washed Li2MnZr2(NCN)6 using a calibrated STOE STADI-P powder diffractometer with a flat sample holder (Cu Kα1, linear PSD, 2θ range 3–65° with individual steps of 0.005°). Rietveld refinements were performed using GSAS with the EXPGUI interface.26 Full details concerning the structure determination including all intensity data are available in CIF format and have been deposited under the CCDC entry numbers 2382982 for Li2MnHf2(NCN)6 and 2382983 for Li2MnZr2(NCN)6.†Infrared measurements
The infrared spectra of both quaternary cyanamides were measured on a Shimadzu IRSpirit FT-IR spectrometer.CHN measurements
The chemical compositions of Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6 were determined by CHN analysis using a Heraeus CHN-O Rapid analyser.UV-Vis measurements
UV–Vis spectra of Li2MnHf2(NCN)6 were recorded on a Shimadzu UV-2006 spectrophotometer. Tauc plots were calculated via the Kubelka–Munk function F(R) = (1 − R)2/2R to determine the bandgap size.Computational details
The experimental structure of Li2MnHf2(NCN)6 was optimized using Blöchl's projector augmented wave method27 as implemented in the Vienna ab initio simulation package (VASP).28–31 The exchange–correlation energy was modeled with the GGA-type functional PBEsol,32 and an additional D3 term with Becke–Johnson damping33 was employed to correct for dispersive interactions. The k-point meshes with a 11 × 11 × 7 point density was generated using the Monkhorst–Pack34 scheme and integrated using Blöchl's tetrahedron method.35 The kinetic-energy cutoff was chosen as 500 eV and convergence criteria were set to 5 × 10−3 eV Å−1 (10−6 eV) for the ionic (electronic) iterations. Onsite correlation were corrected with a Hubbard U-parameter36 of 3.90 eV, close to the self-consistent value used earlier (4.33 eV) for modelling the binary MnNCN.37 For an accurate description of the electronic ground state of manganese, spin-polarization was enabled that resulted in a d5 high-spin configuration of Mn.After the structural optimization, a single point calculation was performed with the same setup as described above. The wave function generated thereby was unitarily transformed (projected) onto a local-orbital basis set using the LOBSTER program.38–40 This atomic-orbital basis set enables the calculation of Löwdin charges41 and bond orders by means of the crystal orbital bond index (COBI)42 that is a generalization of the molecular Wiberg/Mayer bond index for periodic materials.43–45 For deeper insight, an additional band-structure calculation was performed using Gaussian smearing for k-space integration due to technical reasons. All other parameters were kept constant.
SQUID-magnetometry
The magnetic properties were analysed with a Quantum Design (MPMS-5S, Quantum Design, San Diego) SQUID magnetometer. 10 mg of Li2MnHf2(NCN)6 were placed in PTFE containers. Experimental data were collected in the temperature range between 2 K and 400 K at H = 1000 Oe. All data were corrected for diamagnetic contributions of the sample holder and the intrinsic contribution of the compound (χm,dia = –2.85 × 10−4 cm3 mol−1).Results and discussion
The first quaternary transition metal cyanamide, namely Li2MnSn2(NCN)6 was discovered in a quite unexpected way, since initial synthetic efforts were focused on the synthesis of ternary MnSn(NCN)3.21 However, the serendipitous incorporation of lithium from the precursor Li2NCN resulted in the successful formation of Li2MnSn2(NCN)6. With the aim of varying the tetravalent cation from Sn4+ to Hf4+ or Zr4+, the quaternary Li2MnHf2(NCN)6 was targeted via a solid-state metathesis reaction between Li2NCN as a carbodiimide source, MnCl2 and HfCl4 at 700 °C as given in eqn. (1). | (1) |
Attempts to synthesize the zirconium-containing analog via an equivalent direct ‘three-component’ reaction of Li2NCN with the corresponding chlorides proved possible at 650 °C, leading to the desired quaternary, Li2MnZr2(NCN)6, along with an as yet unknown by-product and a particularly large quantity of zirconium oxide. Therefore, an alternative, milder two-step reaction, which proved effective for the preparation of sodium-containing quaternaries Na2MSn2(NCN)6 phases, was used.22 Here the ternary metal carbodiimide was first prepared (eqn. (2))25 and then reacted with the transition metal halide (eqn. (3)).
| 3 Li2NCN + ZrCl4 → Li2Zr(NCN)3 + 4 LiCl | (2) |
| 2 Li2Zr(NCN)3 + MnF2 → Li2MnZr2(NCN)6 + 2 LiF | (3) |
The observed powder X-ray reflections of Li2MnHf2(NCN)6 were indexed to a hexagonal unit cell with lattice parameters a = 5.90691 Å, c = 9.6076 Å. A starting model of Li2MnHf2(NCN)6 was generated from that of Li2MnSn2(NCN)6 (P1m) by replacing Sn4+ with Hf4+ at the 2c position.21 The tetravalent cation Zr4+ (0.72 Å, sixfold coordination)46 has an almost equivalent size to that of Hf4+ (0.71 Å, sixfold coordination)46 making hafnium easily replaced by zirconium at the 2c positions to obtain Li2MnZr2(NCN)6 which crystallizes isotypically with lattice parameters a = 5.9479 Å, c = 9.6403 Å. The chemical formula of both quaternaries was verified by CHN analysis, which shows good agreement between experimental and calculated C and N contents (Table S1†). Rietveld refinements were then performed for both Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6 phases starting from fully ordered P1m models which resulted in good agreements to the observed data. However, further refinements permitting partial disorder between Li and Hf/Zr positions, whilst constraining both sites to be fully occupied, resulted in improved fits to the observed data and cation site disorder of 3.0(1) % and 5.6(1)% for the Hf and Zr analogues, respectively. In the final refinements, the results of which are summarized in Tables 1, 2 and S2,† the thermal displacement parameters (Uiso) of Li and Hf/Zr were constrained to be equal as were the Uisos for all non-metal atoms (C and N) (Fig. 1).
 | ||
| Fig. 1 Rietveld fit of Li2MnHf2(NCN)6 to PXRD data, showing observed (red), calculated (black) and difference (blue) intensities. Bragg positions of Li2MnHf2(NCN)6 (purple) and 7.80(7) wt.% HfO2 baddeleyite P21/c (green) are denoted by vertical markers.47 | ||
| Atom | Wyckoff site | x | y | z | occ. | Uiso (102 Å2) |
|---|---|---|---|---|---|---|
| Trigonal, P1m (No. 162), Z = 1, a = 5.90691(6) Å, c = 9.6076(2) Å; Rwp = 3.69%, Rp = 2.83%, χ2 = 3.649, Rwpb = 4.25%. |
||||||
| Li1 | 2d | ⅓ | ⅔ | ½ | 0.970(1) | 0.574(7) |
| Hf2 | 2d | ⅓ | ⅔ | ½ | 1 – occ.(Li1) | " |
| Mn | 1b | 0 | 0 | ½ | 1 | 0.460(7) |
| Hf1 | 2c | ⅓ | ⅔ | 0 | occ.(Li1) | U iso(Li1) |
| Li2 | 2c | ⅓ | ⅔ | 0 | 1 – occ.(Li1) | U iso(Li1) |
| N1 | 6k | 0.689(1) | 0 | 0.3834(6) | 1 | 1.5(1) |
| N2 | 6k | 0.590(1) | 0 | 0.1298(6) | 1 | " |
| C | 6k | 0.642(2) | 0 | 0.2672(7) | 1 | " |
| Li2MnT2(NCN)6 | T = Hf | T = Zr |
|---|---|---|
| a (Å) | 5.90691(6) | 5.94790(12) |
| c (Å) | 9.6076(2) | 9.6403(3) |
| V (Å3) | 290.313(7) | 295.358(13) |
| Bond lengths (Å) | ||
| Li–N1 [6×] | 2.326(4) | 2.303(5) |
| Mn–N1 [6×] | 2.151(6) | 2.212(8) |
| T–N2 [6×] | 2.179(4) | 2.206(5) |
| C–N1 | 1.151(9) | 1.202(13) |
| C–N2 | 1.355(9) | 1.295(13) |
| Bond angles (°) | ||
| N1–Li–N1 | 90.49(18), 77.0(2), 159.3(3), 105.8(3) | 98.7(2), 83.6(3), 177.0(4), 98.7(2) |
| N2–T–N2 | 98.75(17), 77.02(3), 174.3(3), 85.7(2) | 90.4(2), 77.8(3), 160.6(4), 104.9(5) |
| N1–C–N2 | 179.1(9) | 174.9(15) |
The crystal structure of both new quaternary cyanamides derive from NiAs and relate to the aristotypic FeNCN by a group–subgroup relation.14,48 Starting from FeNCN with P63/mmc symmetry, through a translationengleiche transition (t2) the Fe site splits into two metal positions. A subsequent klassengleiche (k3) operation leads to a cell expansion and symmetry lowering from Pm1 to P1m resulting in four new metal positions (1a, 1b, 2c, 2d). The metal atoms arrange in two different layers, with both inter- and intra-layer cation ordering, one of fully occupied [Li2Mn(NCN)3]2− sheets and the other of only partially filled [T2(NCN)3]2+ sheets with T as either Zr or Hf. These metal layers are linked by a hexagonal closed packing of cyanamide anions (Fig. 2).
 | ||
| Fig. 2 Crystal structure of Li2MnHf2(NCN)6 (a), view along the c direction of the vacancy-ordered [Hf2(NCN)3]2+ layer (b), the [Li2Mn(NCN)3]2− layer (c) and metal-N6 octahedra (d). | ||
The TN6 octahedra with (T = Hf, Zr) have T–N bond lengths of 2.179(4) Å and 2.206(5) Å for hafnium and zirconium respectively, similar to the average distances in Li2T(NCN)3 (2.16 Å) (T = Hf, Zr) and therefore consistent with the presence of either a tetravalent hafnium or zirconium.25 The LiN6 (2.326(4) and 2.303(5) Å) and MnN6 (2.151(6) and 2.212(8) Å) octahedra in both compounds are also similar to those seen in Li2MnSn2(NCN)6 and indicative of Mn2+ and Li+.21
A single crystallographically distinct NCN moiety is present in Li2MnHf2(NCN)6 (Fig. 3, right), which is slightly bent with a bond angle of N1–C–N2 = 179.1(9)° and a tilting angle of θ = 13.5(3)°. However, two different C–N bond lengths are observed (C–N1 = 1.151(9) Å and C–N2 = 1.355(9) Å), which clearly evidence strong triple and single bond character as seen in Li2MnSn2(NCN)6.21 This is a result of the highly asymmetric coordination environment of the NCN moiety, which coordinates to two Hf4+ or Zr4+ cations at one end and to one Mn2+ cation and two Li+ cations at the other, reducing the symmetry of the NCN anion shape to give a clear cyanamide character. This result is corroborated by IR measurements which show deformation vibrations (δ) between 600 and 700 cm−1, asymmetric vibrations (νas) around 2100 cm−1 and a symmetric vibration (νs) around 1250 cm−1 that is IR forbidden for symmetric carbodiimide forms.
 | ||
| Fig. 3 Infrared spectra of Li2MnZr2(NCN)6 (top left) and Li2MnHf2(NCN)6 (bottom left). Coordination environment of the NCN unit in both quaternary structures (right). | ||
To better understand the chemical bonding in quaternary cyanamides, crystal orbital bond indices (COBI) as well as wave function-based Löwdin charges were calculated using LOBSTER for Li2MnHf2(NCN)6.40,42 The integrated COBI (ICOBI) characterizes the covalent bond in the solid state by referring directly to the classical bond order. Löwdin charges as well as ICOBIs of all respective atoms are shown in Fig. 4.41
 | ||
| Fig. 4 Ionic and covalent bonding properties in Li2MnHf2(NCN)6 by means of Löwdin atomic charges and integrated crystal orbital bond indices. | ||
The oxidation states of Li+, Mn2+ and Hf4+ correlate with Löwdin charges of +0.69e for Li, +1.31e for Mn and +1.51e for Hf. The hafnium charge of +1.51e is significantly smaller than +4e, rather surprising in the first place but explained by the energy-dependent COBI value showing significantly larger covalency for the Hf–N interaction than for the covalently almost negligible Mn–N and Li–N interactions, indicating a higher ionicity for the latter two. Indeed, the ICOBI (Hf–N) is equal to 0.54 for each of the six Hf–N distances, indicating a strongly covalent contribution. By contrast, ICOBI is only 0.18 and 0.07 for Mn–N and Li–N respectively. Such a behavior of the tetravalent metal atom was similarly observed for Ti4+ in Ti–O bonds in BaTiO3.42 Despite the difficulties normally encountered by DFT functionals in dealing with the cyanamide form of NCN, the orientation of single and triple bonds is mirrored by DFT calculations with bond orders of C–N1 = 2.26 and C–N2 = 1.55, in line with both the coordinative expectations and the experimental results.
The one-dimensionally extended NCN unit contrasts with the spherical oxide anion in its ability to tilt and bend, giving it a valuable advantage in terms of flexibility, allowing it to accommodate significant cation size differences. Our group recently developed an efficient method to describe these size differences within layers of NCN-containing compounds which can be expressed as a total distortion (δtotal).49 In the present case of quaternary A2MT2(NCN)6 compounds (A: an alkali metal, M: a divalent metal and T: a tetravalent metal) the total distortion is δtotal = (rA − rT) + rM. Given that a tilting of the NCN unit is a consequence of size differences, it is possible to establish a positive correlation between the tilting angle and the total distortion, δtotal. This can be clearly seen in Fig. 5 for quaternary compounds with composition A2MT2(NCN)6 in which as the total distortion increases, the tilting angle of the NCN unit also increases in an approximately linear manner. In the two novel quaternary compounds, the NCN units are tilted away from the stacking axis with angles of 13.5(3)° and 11.5(3)° for Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6 respectively, consistent with the rest of the reported quaternary cyanamide compounds.
 | ||
| Fig. 5 Total distortion versus tilting angle encountered in all the reported quaternary compounds, Hf(NCN)2, (a) Li2MnZr2(NCN)6 and (b) Li2MnHf2(NCN)6. The total distortion of the quaternaries was calculated with δtotal = (rA − rT) + rM, that of the binary Hf(NCN)2 was calculated with δtotal = 2rA.17,49 | ||
These tilt distortions in turn have an influence on the nitrogen positions, which affects the regularity of the metal octahedra. This can be expressed quantitatively by the quadratic elongation parameter (dimensionless λoct, equal to 1 for the ideal octahedron).50 By setting this parameter against the total distortion δtotal individually on the three different metal sites of the quaternary compounds (Fig. 6), it appears that in the divalent metal site, Fig. 6(a), upon increasing total distortion, the octahedron tends rapidly towards a more regular one. A similar, albeit less pronounced, decreasing linear trend is also observed in the alkali metal site, Fig. 6(b). This tendency to rapidly approach the divalent site can be explained geometrically by the fact that the nitrogen atoms move along the distortion axis toward the divalent site. At the tetravalent metal site, however, the distortion appears to be independent from the total distortion as no trend is noticed. One explanation could also be of a steric nature, since hafnium shares space in its layer with a vacant site, the latter behaving like a huge cation augmenting the position of the nitrogen atoms, thus preventing the adoption of a regular octahedron and explaining why, as yet, only d0 (Zr4+ and Hf4+) or d10 (Sn4+) cations have been realized at this position. Nonetheless, it is clear at the outset that a model based on geometrical criteria (such as ionic radii) is hardly justified for the tetravalent site (Hf) since the associated chemical bonding is largely covalent in nature, as shown before (Fig. 4).
 | ||
Fig. 6 Total distortion vs. quadratic elongation (calculated with 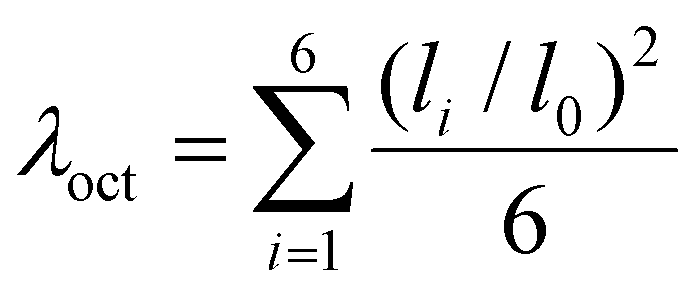 ) in quaternary cyanamides with a structure of A2MT2(NCN)6 (a) divalent metal site (b) alkali metal site (c) tetravalent metal site. ) in quaternary cyanamides with a structure of A2MT2(NCN)6 (a) divalent metal site (b) alkali metal site (c) tetravalent metal site. | ||
As such, this analysis reveals a delicate balance between geometrical (size of the ions) and electronic (nature of the bond) requirements. By modifying these factors, one could target the synthesis, perhaps by SSM, of a whole host of other quaternary A2MT2(NCN)6 compounds incorporating a complementary balance of metals over the three different sites.
The band structure of Li2MnHf2(NCN)6 was calculated for the geometry-optimized crystal structure. The static calculation yields an indirect zero-temperature band gap of approximately 2.4 eV similar to that calculated for Na2MnSn2(NCN)6. In order to further investigate the electronic structure, density of states (DOS) and (fat) band structure calculations were analyzed. The total DOS is dominated by Mn 3d levels, Hf 5d levels and N 2p levels as was observed previously in other quaternaries.22 Li, N 2s, and the remaining Mn levels show no significant contributions in the chosen energy range of −10 eV to +5 eV (Fig. 7).
 | ||
| Fig. 7 Spin-polarized total DOS and contributing local DOS for Li2MnHf2(NCN)6 (left). Calculated band structures with highlighted minority/majority spin channels of Mn 3d and Hf 5d (right). | ||
The optical band gap was experimentally determined by UV–vis spectroscopy, yielding a value of 2.1 eV for the grey colored sample which is in very good accordance with the calculated data. Future work will look at varying the nature of the transition metal species at the ‘M’ site in order to engender electronic band gaps and band edge positions suitable for solar energy harvesting (Fig. 8).
 | ||
| Fig. 8 Tauc plot of Li2MnHf2(NCN)6 for indirect allowed transition (r = 1/2). | ||
The magnetic susceptibility of Li2MnHf2(NCN)6 was measured between 2–400 K in an applied field of 1000 Oe. Accounting for a 7.8 wt% of HfO2 impurity suggested from Rietveld refinement, the data were corrected leading to the results shown in Fig. 9. These data exhibit paramagnetic behavior down to the lowest temperature measured with a progressive increase in χm with decreasing T and no signature for the onset of long-range magnetic order. At temperatures above 300 K Curie–Weiss behavior is observed, and a linear fit to the inverse susceptibility data (χm−1) yields C = 3.89 cm3 K mol−1, which is a little smaller than that expected for a single non-interacting high-spin Mn2+ center (3.99–4.65 cm3 K mol−1),51 and a negative Weiss temperature θ = −58.5 K indicative of predominantly antiferromagnetic interactions. This absence of a transition to a long-range magnetically order state is likely a result of the magnetic dilution of the octahedral metal positions, since in Li2MnHf2(NCN)6 only 1/6 of the sites are occupied by paramagnetic cations. In contrast [NaCl]-derived MnNCN and [NiAs]-derived FeNCN, with fully occupied octahedral interstices, display Néel temperatures of 28 K and 350 K,13,52 respectively, whilst Cr2(NCN)3 with 1/3 vacancies exhibits a transition to an ordered ferromagnetic state below 114 K.53,54
 | ||
| Fig. 9 Magnetic molar susceptibility (χm) versus temperature (T) plot for Li2MnHf2(NCN)6 (left) measured at an applied field of H = 1000 Oe and inverse molar susceptibility (χm−1) versus temperature (T) for Li2MnHf2(NCN)6 (right). | ||
Conclusions
Herein we report the solid-state metathetic synthesis of two new quaternary cyanamides, Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6. Both phases crystallize isostructurally to Li2MnSn2(NCN)6 in a trigonal unit cell with P1m symmetry, and both are stabilized by the extended nature of the NCN2− anion which offers an additional flexibility, compared with O2−, through the tilting of the cyanamide unit that is impossible in oxide chemistry. This helps to explain the absence of any comparable oxide analogues. The different coordination requirements of the metal cations result in an ordered design over the octahedral sites with corundum-like [T2(NCN)3]2+ layers (T = Hf, Zr) alternating with [Li2Mn(NCN)3]2− layers. Meanwhile, a computational study using LOBSTER affords in-depth analysis of the bonding situation, revealing a higher degree of covalency in the Hf–N bonds, compared with more ionic Li–N and Mn–N bonds, which leads to the asymmetry of the NCN2− moiety. In addition, a survey of all existing A2MT2(NCN)6 quaternaries shows the capacity of the NCN unit to adapt to cation size differences through cooperative tilt distortions. Furthermore, analysis of the quadratic elongation of individual metal octahedra provides deeper insight into the subtle balance of geometric and electronic requirements of the constituent cations. The optical band gap was measured by UV-vis, giving a value of 2.1 eV, in very good agreement with the computed value. Magnetic susceptibility measurements suggest predominantly antiferromagnetic interactions (θ = −58.5 K), however, no signature for a transition to a long-range ordered state was observed, presumably resulting from the magnetic dilution of the octahedral metal sites, only 1/6 of which are occupied by paramagnetic Mn2+ cations. Future work will seek to vary the nature of the divalent transition metal species to engender band gaps suitable for photochemical applications and explore the electrochemical properties of this family.
Data availability
Crystallographic data for Li2MnHf2(NCN)6 and Li2MnZr2(NCN)6 have been deposited at the CCDC under 2382982 and 2382983,† respectively and can be obtained from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr T. Storp for assistance with PXRD measurements and Dr Ing. Dr rer. nat. Jan van Leusen for the magnetic correction. AJC would also like to acknowledge the financial support of the Deutsche Forschungsgemeinschaft (Project number 441856704).References
- A. J. Corkett, O. Reckeweg, R. Pöttgen and R. Dronskowski, Chem. Mater., 2024, 36, 9107–9125 CrossRef CAS.
- M. T. Sougrati, A. Darwiche, X. Liu, A. Mahmoud, R. P. Hermann, S. Jouen, L. Monconduit, R. Dronskowski and L. Stievano, Angew. Chem., Int. Ed., 2016, 55, 5090–5095 CrossRef CAS.
- A. Eguia-Barrio, E. Castillo-Martinez, X. Liu, R. Dronskowski, M. Armand and T. Rojo, J. Mater. Chem. A, 2016, 4, 1608–1611 RSC.
- Q. Liu, Y. Liu, G. Dai, L. Tian, J. Xu, G. Zhao, N. Zhang and Y. Fang, Appl. Surf. Sci., 2015, 357, 745–749 CrossRef CAS.
- M. Kubus, R. Heinicke, M. Ströbele, D. Enseling, T. Jüstel and H.-J. Meyer, Mater. Res. Bull., 2015, 62, 37–41 CrossRef CAS.
- W. P. Clark and R. Niewa, Z. Anorg. Allg. Chem., 2020, 646, 114–119 CrossRef CAS.
- M. Krings, G. Montana, R. Dronskowski and C. Wickleder, Chem. Mater., 2011, 23, 1694–1699 CrossRef CAS.
- S. Yuan, Y. Yang, F. Chevire, F. Tessier, X. Zhang and G. Chenz, J. Am. Ceram. Soc., 2010, 93, 3052–3055 CrossRef CAS.
- H.-J. Meyer, Dalton Trans., 2010, 39, 5973–5982 RSC.
- X. Liu, M. Krott, P. Müller, C. Hu, H. Lueken and R. Dronskowski, Inorg. Chem., 2005, 44, 3001–3003 CrossRef CAS.
- U. Berger and W. Schnick, J. Alloys Compd., 1994, 206, 0925–8388 CrossRef.
- A. Frank and N. Caro, Deutsches Reichspatent, 88363, 1895 Search PubMed.
- G. Baldinozzi, B. Malinowska, M. Rakib and G. Durand, J. Mater. Chem., 2002, 12, 268–272 RSC.
- X. Liu, L. Stork, M. Speldrich, H. Lueken and R. Dronskowski, Chem. – Eur. J., 2009, 15, 1558–1561 CrossRef CAS PubMed.
- M. Krott, X. Liu, B. P. T. Fokwa, M. Speldrich, H. Lueken and R. Dronskowski, Inorg. Chem., 2007, 46(6), 2204–2207 CrossRef CAS.
- M. Kubus, R. Heinicke, M. Ströbele, D. Enseling, T. Jüstel and H.-J. Meyer, Mater. Res. Bull., 2015, 62, 37–41 CrossRef CAS.
- L. Unverfehrt, M. Ströbele and H.-J. Meyer, Z. Anorg. Allg. Chem., 2013, 639, 22–24 CrossRef CAS.
- A. J. Corkett, K. Chen and R. Dronskowski, Eur. J. Inorg. Chem., 2020, 27, 2596–2602 CrossRef.
- H. Bourakhouadar, J. Hempelmann, J. van Leusen, A. Drichel, L. Bayarjargal, A. Koldemir, M. K. Reimann, R. Pöttgen, A. Slabon, A. J. Corkett and R. Dronskowski, J. Am. Chem. Soc., 2024, 146, 26071–26080 CrossRef CAS PubMed.
- K. Dolabdjian, A. Kobald, C.-P. Romao and H.-J. Meyer, Dalton Trans., 2018, 47, 10249–10255 RSC.
- A. J. Corkett and R. Dronskowski, Dalton Trans., 2019, 48, 15029–15035 RSC.
- A. J. Corkett, Z. Chen, C. Ertural, A. Slabon and R. Dronskowski, Inorg. Chem., 2022, 45, 18221–18228 CrossRef.
- X. Qiao, A. J. Corkett, R. P. Stoffel and R. Dronskowski, Z. Anorg. Allg. Chem., 2021, 647, 2162 CrossRef CAS.
- J. Glaser, H. Bettentrup, T. Jüstel and H.-J. Meyer, Inorg. Chem., 2010, 49(6), 2954–2959 CrossRef CAS.
- K. Dolabdjian, C. Castro and H.-J. Meyer, Eur. J. Inorg. Chem., 2018, 44, 1624–1630 CrossRef.
- B. H. J. Toby, Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS , 0927-0256.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(23), 16223–16233 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- R. Nelson, P. M. Konze and R. Dronskowski, J. Phys. Chem. A, 2017, 121, 7778–7786 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2013, 34(29), 2557–2567 CrossRef CAS.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37(11), 1030–1035 CrossRef CAS PubMed.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, J. Comput. Chem., 2020, 41(21), 1931–1940 CrossRef CAS.
- C. Ertural, S. Steinberg and R. Dronskowski, RSC Adv., 2019, 9(51), 29821–29830 RSC.
- P. C. Müller, C. Ertural, J. Hempelmann and R. Dronskowski, J. Phys. Chem. C, 2021, 125(14), 7959–7970 CrossRef.
- K. B. Wiberg, Tetrahedron Lett., 1968, 24(3), 1083–1096 CrossRef CAS.
- I. Mayer, Chem. Phys. Lett., 1983, 97(3), 270–274 CrossRef CAS.
- I. Mayer, J. Comput. Chem., 2007, 28(1), 204–221 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, 25, 751–767 CrossRef.
- H. J. Goldschmidt and M. J. Walker, J. Appl. Crystallogr., 1969, 2, 281 CrossRef.
- R. Pöttgen, A. Corkett and R. Dronskowski, Z. Kristallogr., 2023, 238, 95–103 Search PubMed.
- J. Medina-Jurado, A. J. Corkett and R. Dronskowski, Z. Kristallogr. – Cryst. Mater., 2024, 239, 1–2 CrossRef.
- K. Robinson, Science, 1971, 172, 567–570 CrossRef CAS PubMed.
- H. Lueken, Magnetochemie, Teubner Verlag, Stuttgart, 1999 Search PubMed.
- M. Krott, A. Houben, P. Müller, W. Schweika and R. Dronskowski, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 024117 CrossRef.
- X. Tang, H. Xiang, X. Liu, M. Speldrich and R. Dronskowski, Angew. Chem., Int. Ed., 2010, 49, 4738–4742 CrossRef CAS PubMed.
- K. B. Sterri, C. Besson, A. Houben, P. Jacobs, M. Hoelzel and R. Dronskowski, New J. Chem., 2016, 40, 10512–10519 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2382982 and 2382983. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02615b |
| This journal is © The Royal Society of Chemistry 2024 |