
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of the stabilizing/leaving group in palladium catalysed cross-coupling reactions†
Lorenzo
Palio‡
 ab,
Francis
Bru‡
a,
Tommaso
Ruggiero
a,
Laurens
Bourda
a,
Kristof
Van Hecke
a,
Catherine
Cazin
*a and
Steven P.
Nolan
*a
ab,
Francis
Bru‡
a,
Tommaso
Ruggiero
a,
Laurens
Bourda
a,
Kristof
Van Hecke
a,
Catherine
Cazin
*a and
Steven P.
Nolan
*a
aDepartment of Chemistry, Center for Sustainable Chemistry, Ghent University, Krijgslaan 281 (S-3), 9000, Ghent, Belgium
bUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 – UCCS Unité de Catalyse et Chimie Solide, F-59000, Lille, France
First published on 19th October 2024
Abstract
Despite the widespread use of well-defined PdII complexes as pre-catalysts for cross-coupling processes, the role of the throw-away ligand is still underexplored. In this work we focused on the complexes of the type [Pd(NHC)(η3-R-allyl)Cl] (NHC = N-heterocyclic carbene) and we investigated the influence of the R substitution on the allyl moiety. Starting from the already described [Pd(IPr)(η3-cinnamyl)Cl] and [Pd(IPr*)(η3-cinnamyl)Cl] (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, IPr* = N,N′-1,3-bis[2,6-bis(diphenylmethyl)-4-methylphenyl]imidazol-2-ylidene) we prepared eight new complexes bearing new substitutions on the cinnamyl motif and we tested them in the C–N bond formation to evaluate the effect of the throw-away ligand modification in the catalytic activity. In addition, we studied the undesired formation of the less active off-cycle [PdI2(NHC)2(η3-R-allyl)(μ-Cl)] dimers from the corresponding PdII complexes to evaluate the role of the new throw-away ligands on the inhibition of this process.
Introduction
Palladium N-heterocyclic carbenes (NHCs) have been widely studied in the last decades.1–5 They have been ubiquitously investigated for applications in homogeneous catalysis, especially in cross-coupling reactions.6–13 This class of organometallic compounds have the advantage to be air and moisture stable and permit a strict control on the metal–ligand stoichiometry, for which the ideal value has been proven to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.14,15 This well-defined approach to catalysis allows a deeper knowledge of the catalytic system and has been embraced by other research groups in recent years.16–19 The steric bulk around the Pd center given by the NHCs plays an important role in the reductive elimination and the transmetallation process, overall increasing the catalytic activity of the complex.15,20,21
:1.14,15 This well-defined approach to catalysis allows a deeper knowledge of the catalytic system and has been embraced by other research groups in recent years.16–19 The steric bulk around the Pd center given by the NHCs plays an important role in the reductive elimination and the transmetallation process, overall increasing the catalytic activity of the complex.15,20,21
One aspect of the well-defined Pd–NHCs systems that deserves to be studied more thoroughly is the role of the “throw-away” ligands. These species are responsible for the stability of the well-defined systems by occupying the coordination sites ensuring a stable oxidation state. These ligands are reported to disconnect from the complex during the activation process from the PdII to the Pd0 active species.22,23 Therefore, the nature of the throw-away ligand plays a key role in the activity of the complex. One of the most widespread Pd–NHC pre-catalyst structures is the [Pd(NHC)(η3-R-allyl)Cl] type, developed for the first time by us in 2002, in which the allyl-based throw-away ligand ensures a fast and easy activation of the catalyst23–25 (Fig. 1).
 | ||
| Fig. 1 Examples of well-defined Pd–NHC allyl-based complexes. | ||
Palladium(II) complexes of the type of [Pd(NHC)(η3-R-allyl)Cl] system (R = allyl, crotyl, cinnamyl, indenyl) have shown great activity for a wide range of C–C and C–heteroatom bond formation reactions.24–33 Particularly, the design of the allyl-modified moiety [Pd(NHC)(η3-cin)Cl] (cin = cinnamyl) complex class by 2006 was examined in order to achieve a faster activation to the Pd(0) active species at lower temperatures.24,34,35 This latter generation of allyl systems was designed with the help of teaching from Trost and Tsuji on allylic alkylation and the relative stability/reactivity of substituted allyl fragments bound to Pd. Later, this was further elaborated by Colacot and Grasa at Johnson Matthey in their construction of Pd–allyl systems where the Buchwald ligands are the ancillary supporting ligands.16
Substitution at the allyl terminus leads to destabilisation of the allyl fragment bound to palladium and therefore to easier activation leading to the putative “Pd–NHC” species. This ease of activation in such systems, a pre-catalytic step whose importance is too often ignored, leads to Suzuki–Miyaura and the Buchwald–Hartwig cross-couplings involving unactivated aryl halides that can be performed at room temperature. The effect of bulky substituents on the other palladium ligand also profoundly affects catalytic activity. Indeed, systems bearing a bulky NHC such as in [Pd(IPr*)(η3-R-allyl)Cl] (IPr* = N,N′-1,3-bis[2,6-bis(diphenylmethyl)-4-methylphenyl]imidazol-2-ylidene) and [Pd(anti-(2,7))-(SICyoctNap)(η3-cin)Cl] permit difficult cross-coupling reactions under very mild conditions. The typical example is their use in the formation of tetra-ortho-substituted biaryls.36,37 An interesting observation here is that crowded environments about the Pd pre-catalysts architecture allow for highly congested junctions in C–C and C–N bond making reactions to be formed.
Among the plethora of cross-coupling reactions, the Buchwald–Hartwig amination of aryl halides is one of the most widespread examples representing an efficient tool for the synthesis of several pharmaceutical agents.38–43 Several salient examples of the use of palladium N-heterocyclic carbene complexes in this C–N coupling exist in the literature.24,28,29,44–47
Hazari and co-workers proposed the activation mechanism of the [Pd(NHC)(η3-R-allyl)Cl] species in presence of a weak base and suggested the presence of an equilibrium between the Pd0–NHC active species and the less active [PdI2(NHC)2(μ-η3-R-allyl)(μ-Cl)] species48,49 (Fig. 2). In the report, it was shown that the presence of a R substituent on the allyl moiety raises the kinetic barrier of the comproportionating reaction between the PdII and the Pd0 species, thereby increasing the concentration of the active monoligated species.49
 | ||
| Fig. 2 Scheme of the activation and the dimerization via comproportionation presented by the Hazari group.49 | ||
Results and discussion
Taking from these teachings, we have now prepared new substituted [Pd(NHC)(η3-R-allyl)Cl] complexes to explore their activity in the C–N bond formation. Efforts were also aimed at minimizing or entirely blocking the decomposition route that leads to inactive dimer formation. To achieve this, four new dimeric synthons were designed bearing bulkier allyl substituents and synthesised (Fig. 3). These were then converted to well-defined [Pd(NHC)(η3-R-allyl)Cl] complexes to further explore the role of the allyl throw-away ligand and its role in the decomposition route and define productive reactivity in a model palladium-mediated cross-coupling reaction. | ||
| Fig. 3 Novel dimers (1–4) synthesized for this study. | ||
Synthesis of [Pd(NHC)(η3-R-cin)Cl] complexes
The synthesis of the new complexes involves the cleavage of the novel [Pd(η3-R-cin)(μ-Cl)]2 dimer (1–4) with the corresponding imidazolium salt in the presence of an excess of potassium carbonate in acetone at 60 °C for 5 h. The [Pd(η3-R-allyl)(μ-Cl)]2 dimers were prepared starting from the corresponding allyl chloride following the reported procedure (Fig. 4).50 | ||
| Fig. 4 Structures of the newly synthesised [(NHC)Pd(η3-R-allyl)Cl] pre-catalysts. | ||
The corresponding [Pd(NHC)(η3-R-allyl)Cl] complexes are obtained with good yields (>80%) using the weak base route.51 As an alternative synthetic route, the [Pd(NHC)(η3-R-allyl)Cl] complexes can be obtained through oxidative addition of the R-allyl chloride with our previously reported [Pd(NHC)(PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)] synthon.52 This route allows rapid functionalization and derivatization from a common synthon, the [Pd(NHC)(η3-allyl)Cl]. (See the Experimental section and ESI† for further details) (Fig. 5A and B).
CPh)] synthon.52 This route allows rapid functionalization and derivatization from a common synthon, the [Pd(NHC)(η3-allyl)Cl]. (See the Experimental section and ESI† for further details) (Fig. 5A and B).
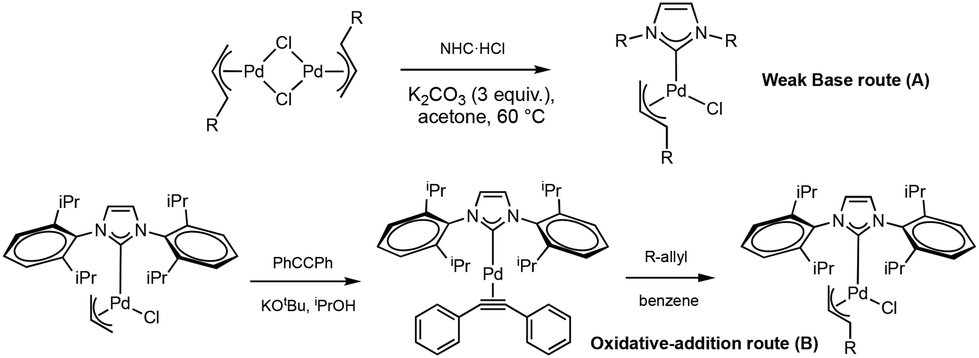 | ||
| Fig. 5 The two possible synthetic routes leading to the novel pre-catalysts. | ||
Both palladium dimers and the [Pd(NHC)(η3-R-allyl)Cl] series are air- and moisture-stable and can be stored on a shelf in air for an indefinite amount of time. Single crystals for X-ray analysis were obtained for all the [Pd(η3-R-allyl)(μ-Cl)]2 dimers by slow diffusion of hexane vapour into a saturated solution of the complex in dichloromethane.
Room temperature Buchwald–Hartwig amination of aryl chlorides
To compare the activity of the newly designed [Pd(NHC)(η3-R-allyl)Cl] complexes, these were tested under identical conditions as those already mentioned in the Buchwald–Hartwig amination, permitting an evaluation of the effect of the substitution on the phenyl ring of the cinnamyl moiety with both IPr and IPr* ligands. The reaction between morpholine and 2,6-dimethylchlorobenzene was chosen as a model reaction (Table 1). Initially, the four new [Pd(IPr)(η3-R-allyl)Cl] species were tested alongside the [Pd(IPr)(η3-cin)Cl] standard. The cross-coupling was performed at room temperature with 1 mol% of Pd and KOtBu as base in anhydrous DME. The reaction with [Pd(IPr)(η3-cin)Cl] is reported to reach completion after 2 hours.24 Complete conversion was reached by all catalysts after 2.5 h, with the only exception of IPr-2 for which the conversion stopped at 84%, showing the lowest activity compared to the others. The conversion was also monitored after 30 minutes to evaluate the difference in activity. After this time, the aryl chloride conversion ratio was 21% when [Pd(IPr)(η3-cin)Cl] was used. A similar value of 20% was obtained with IPr-1, while IPr-2 gave only 11% confirming the previous observation. IPr-3 and IPr-4 showed to be more active, giving respectively 42% and 38% conversions after 30 minutes. After 2 hours, IPr-3 is the only species that gave 100% conversion of the starting material, while IPr-4 reached 85%. IPr-1 and IPr-2 showed 81% and 65% conversion of the aryl chloride, following the trend previously observed. From these initial results, the substitution at the C4 of the phenyl ring of the cinnamyl moiety does not appear to improve catalyst activity, but rather leads to lower conversions. However, the modification on the ortho positions of the cinnamyl moiety, oriented towards the Pd center, leads to improved catalytic activity.
|
|
||
|---|---|---|
| Catalyst | Time [h] | Conversiona |
| Conditions: aryl chloride (0.5 mmol, 1 eq.), morpholine (0.55 mmol, 1.1 eq.), 0.5 mL of DME, reaction under inert atmosphere.a Average result of two separate experiments. | ||
| [Pd(IPr)(cin)Cl] | 0.5/2.5 | 21/100 |
| IPr-1 | 0.5/2/2.5 | 20/81/100 |
| IPr-2 | 0.5/2/2.5 | 11/65/84 |
| IPr-3 | 0.5/2/2.5 | 38/100/100 |
| IPr-4 | 0.5/2/2.5 | 42/85/100 |
| [Pd(IPr*)(cin)Cl] | 0.5/2 | 55/92 |
| IPr*-1 | 0.5/2 | 21/78 |
| IPr*-2 | 0.5/2 | 37/75 |
| IPr*-3 | 0.5/2 | 87/100 |
| IPr*-4 | 0.5/2 | 65/97 |

We next move to the more sterically congested IPr* analogues in the same cross-coupling reaction under identical conditions. The already described [Pd(IPr*)(η3-cin)Cl] structure bearing the unmodified cinnamyl group was used as standard. As in the previous case, all species reached full conversion after 2.5 hours.
After 30 minutes, IPr*-3 showed the highest conversion value with 87%, followed by IPr*-4 (65%), and by [Pd(IPr*)(cin)Cl] (55%). On the other hand, IPr*-1 and IPr*-2 gave respectively 21% and 37% conversions. These conversion values achieved with the bulkier IPr* series are generally higher than the ones obtained with the IPr series, and concomitantly follow the same activity trend as a function of the cinnamyl substitution pattern. The substitution of the phenyl group with the mesityl showed the greatest increase in catalytic performance. The 2-naphthyl modification also leads to a higher catalytic activity in comparison with the unsubstituted cinnamyl analogue. On the other hand, the IPr*-1 and IPr*-2 pre-catalysts that only bear substitution at the C4 of the cinnamyl moiety, showed a greater decrease in the conversion than the ones observed for the IPr series. After 2 hours, IPr*-3 converted 100% of the starting material, confirming it is the best pre-catalyst overall, while IPr*-4 provided almost full conversion at 97%, proving a greater activity than [Pd(IPr*)(η3-cin)Cl] that reached a 92% conversion. IPr*-1 and IPr*-2 converted respectively 80% and 75% of the starting material in 2 hours.
So, the steric congestion about the palladium center can be appreciated, single crystals of IPr*-3 were grown for diffraction studies. A graphical representation of the structure of IPr*-3 is presented in Fig. 6. It is clear from the metrical parameters that there exists a significant elongation of the Pd–C allyl bond when this carbon is substituted with a mesityl group (Callyl3), going from 2.104(2) Å at the unsubstituted allylic terminus to the central allyl carbon (2.159(4) Å) to the mesityl bearing allylic terminus 2.305(4) Å. This compares to values of 2.110(7) Å, 2.270(10) Å and 2.155(11) Å for these three positions found for the related [Pd(IPr*)(cin)Cl] complex (Table 2).37 The substitution on the aryl fragment of the allyl evidently causes a major distortion of the allyl fragment effectively creating significantly more access to the palladium center in IPr*-3.
 | ||
| Fig. 6 X-ray structure of the IPr*-3 pre-catalysts showing the thermal displacement ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Pd–Cl 2.3605(6), Pd–CNHC 2.032(2), Pd–Callyl1 2.104(2), Pd–Callyl2 2.159(4), Pd–Callyl3 2.305(4); CNHC–Pd–Cl 92.46(6), CNHC–Pd–Callyl1 100.74(9), Callyl1–Pd–Callyl3 67.14(11), Callyl3–Pd–Cl 99.98(9). | ||
| Å | IPr*-3 | [Pd(IPr*)(cin)Cl] | |
|---|---|---|---|
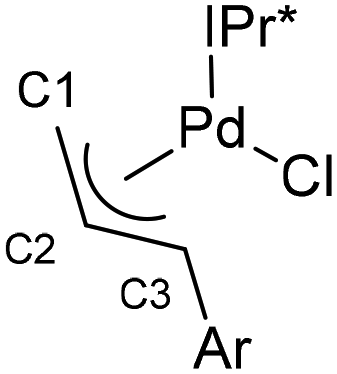
|
Pd–C1 | 2.104(2) | 2.110(7) |
| Pd–C2 | 2.159(4) | 2.270(10) | |
| Pd–C3 | 2.305(4) | 2.155(11) |
 | ||
| Fig. 7 Scope of the Buchwald–Hartwig reaction mediated by IPr*-3. Isolated yields reported as an average of two separate experiments. | ||
A factor that can be responsible for the increase in the catalytic activity of our new systems is the diminution of the formation of the off-cycle and catalytically inactive [PdI2(NHC)2(η3-R-allyl)(μ-Cl)] species. The preparation of these dimers can be achieved by the reaction of the [Pd(NHC)(η3-R-allyl)Cl] complex with an excess of potassium carbonate in degassed EtOH under inert atmosphere.56 Different PdI dimers bearing various NHCs are reported within a maximum time of 5 hours. In this work, we aimed to investigate the ease of formation of the PdI dimers starting from the new [Pd(NHC)(η3-R-allyl)Cl] pre-catalysts.
Initially, the four IPr-bearing palladium complexes were investigated. The corresponding [Pd(NHC)(η3-R-allyl)Cl] complex was reacted with 3 eq. of K2CO3 in EtOH and the ratio between the formed dimer and the remaining PdII complex was evaluated every hour for 5 hours.
The central proton peak of the allyl system is reported to be abnormally shifted upfield, around 1.70 ppm (ref. 49) due to the high electronic density present on the allyl fragment caused by the back-bonding from the d orbitals of the Pd–Pd bonding to the π* orbital of the allyl moiety.49,57,58 Therefore, the signal of this central proton was selected to monitor dimer formation. For IPr-1, already after 1 h of reaction, a sharp decrease of the signals of the allyl system of the monomeric species was observed, along with the appearance of a peak around 1.85 ppm. The signal of acetaldehyde, formed from the oxidation of ethanol, is detected starting from 4 h. From the reaction involving IPr-2, the complete decomposition of the monomeric complex occurs already after 1 h. Many species that are difficult to detect are formed during the experiment, the PdI dimer appears after 1 h, but decomposes during the time. The aldehydic proton of the acetaldehyde and the olefin signal of the species that is formed in the reductive elimination step that leads to the monoligated Pd0 species disconnected from the Pd are also noticeable (see the ESI† for further details). For IPr-3, after 1 h, no PdII precursor remains. On the other hand, significant amounts of acetaldehyde were detected during the 5-hour reaction time starting already after 1 h. This result suggests that this catalyst is easily activated, forming the monoligated L-Pd0 fragment, but has less of a tendency to form the PdI dimer due to the steric hindrance of the mesityl group on the allyl moiety. The experiment with IPr-4 is the only one that shows that after 1 h the precursor is still not completely decomposed, suggesting a slower PdI dimer formation for this species. A summary of these results is reported in Table 3. On the other hand, the IPr* complexes did not show any PdI dimer formation. This we suspect is due to the significant steric hindrance of the IPr* ligand.56
|
|
Complete decomposition time (h) | PdI dimer formation starting time | Acetaldehyde formation starting time (h) |
|---|---|---|---|
| IPr-1 | 1 | 2 | 4 |
| IPr-2 | 1 | 1 | 2 |
| IPr-3 | 1 | 1 | 1 |
| IPr-4 | 2 | 1 | 1 |

Experimental
Synthetic procedures
CPh)]52 (1 equiv.) and benzene. This mixture is stirred at room temperature for 15 minutes after which time the vial is removed from the glovebox for the work-up. The solvent is evaporated in vacuo and the remaining solid is dissolved in a minimal amount of DCM. The desired product is then recrystallised by adding pentane and sonication, and isolated by filtration and washed with cold pentane.
Conclusions
We have successfully prepared a new series of [Pd(NHC)(η3-R-allyl)Cl] complexes bearing a substituted-cinnamyl moiety, with the metal center decorated with IPr or IPr* ligands. The influence of the bulkier throw-away ligands was studied by testing the pre-catalysts in the Buchwald–Hartwig cross-coupling reaction. The results showed that the naphthyl and the mesityl modifications on the allyl motif led to an increase in catalytic activity when compared to the unsubstituted cinnamyl-based complexes. On the other hand, the substitution with p-tol and especially with the p-tBu-phenyl groups resulted in a decrease in catalytic activity. This reactivity difference is significantly more pronounced when the NHC is IPr*. The best catalyst among the series examined is [Pd(IPr*)(η3-mes-allyl)Cl] (IPr*-3). The results highlight how this composition is particularly active for the preparation of bulky secondary and tertiary amines with 2,6-di-ortho-substituted starting materials. The higher reactivity is due to the complex stability, and this has been probed by attempting to transform the [Pd(NHC)(η3-R-allyl)Cl] complexes into catalytically inactive Pd(I) dimers. For the IPr-containing series this transformation clearly occurs under the specific reaction conditions examined. As for the IPr* analogue, a complete lack of reactivity in this manner is observed. In this sterically more demanding series, the Pd(I) dimer formation is completely inhibited.The series of experiments performed here on both modifying the allyl and NHC moieties teach us that to target a very active, yet stable pre-catalyst, one needs steric protection on the NHC hemisphere of the complex while also having a high degree of substitution on the allyl which serves two purposes: (1) it destabilises the Pd–allyl bond as it is highly substituted and (2) it hinders the role of the liberated allyl to act as a bridging ligand in comproportionation reactions to stabilise the Pd(I) dimer structure. More work along these lines is presently ongoing in our laboratories.
Author contributions
LP and FB performed experimental work, contributed to writing the manuscript draft. TR, LB and KVH performed diffraction work. CC and SPN directed the work, contributed to the writing of the draft and final manuscript as well as secured funding.Data availability
All data included and leading to conclusions presented in this manuscript are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Research Foundation – Flanders (FWO) (G0A6823N to SPN and G0C5423N to CSJC and G0A8723N to KVH) for financial support. Umicore AG are gratefully thanked for gifts of materials.References
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523–1533 CrossRef PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC.
- C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- R. D. J. Froese, C. Lombardi, M. Pompeo, R. P. Rucker and M. G. Organ, Acc. Chem. Res., 2017, 50, 2244–2253 CrossRef CAS PubMed.
- M. S. Viciu, R. A. Kelly, E. D. Stevens, F. Naud, M. Studer and S. P. Nolan, Org. Lett., 2003, 5, 1479–1482 CrossRef CAS PubMed.
- P. Lei, G. Meng, Y. Ling, J. An and M. Szostak, J. Org. Chem., 2017, 82, 6638–6646 CrossRef CAS PubMed.
- S. Shi and M. Szostak, Chem. Commun., 2017, 53, 10584–10587 RSC.
- C. M. Zinser, F. Nahra, M. Brill, R. E. Meadows, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2017, 53, 7990–7993 RSC.
- S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589–2599 CrossRef CAS PubMed.
- S. Ostrowska, T. Scattolin and S. P. Nolan, Chem. Commun., 2021, 57, 4354–4375 RSC.
- K. Wang, R. Fan, X. Wei and W. Fang, Green Synth. Catal., 2022, 3, 327–338 CrossRef CAS.
- S. Ostrowska, L. Palio, A. Czapik, S. Bhandary, M. Kwit, K. Van Hecke and S. P. Nolan, Catalysts, 2023, 13, 559 CrossRef CAS.
- G. A. Grasa, M. S. Viciu, J. Huang and S. P. Nolan, J. Org. Chem., 2001, 66, 7729–7737 CrossRef CAS PubMed.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- L. L. Hill, J. L. Crowell, S. L. Tutwiler, N. L. Massie, C. C. Hines, S. T. Griffin, R. D. Rogers, K. H. Shaughnessy, G. A. Grasa, C. C. C. Johansson Seechurn, H. Li, T. J. Colacot, J. Chou and C. J. Woltermann, J. Org. Chem., 2010, 75, 6477–6488 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, S. L. Parisel and T. J. Colacot, J. Org. Chem., 2011, 76, 7918–7932 CrossRef CAS PubMed.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC.
- N. C. Bruno, N. Niljianskul and S. L. Buchwald, J. Org. Chem., 2014, 79, 4161–4166 CrossRef CAS PubMed.
- G. Bastug and S. P. Nolan, Organometallics, 2014, 33, 1253–1258 CrossRef CAS.
- X. Tian, J. Lin, S. Zou, J. Lv, Q. Huang, J. Zhu, S. Huang and Q. Wang, J. Organomet. Chem., 2018, 861, 125–130 CrossRef CAS.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366–374 CrossRef CAS PubMed.
- N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 0025 CrossRef CAS PubMed.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed.
- M. S. Viciu, R. F. Germaneau and S. P. Nolan, Org. Lett., 2002, 4, 4053–4056 CrossRef CAS PubMed.
- M. S. Viciu, R. F. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Organometallics, 2002, 21, 5470–5472 CrossRef CAS.
- M. S. Viciu, O. Navarro, R. F. Germaneau, R. A. Kelly, W. Sommer, N. Marion, E. D. Stevens, L. Cavallo and S. P. Nolan, Organometallics, 2004, 23, 1629–1635 CrossRef CAS.
- M. J. Cawley, F. G. N. Cloke, R. J. Fitzmaurice, S. E. Pearson, J. S. Scott and S. Caddick, Org. Biomol. Chem., 2008, 6, 2820–2825 RSC.
- A. Chartoire, A. Boreux, A. R. Martin and S. P. Nolan, RSC Adv., 2013, 3, 3840–3843 RSC.
- P. R. Melvin, A. Nova, D. Balcells, W. Dai, N. Hazari, D. P. Hruszkewycz, H. P. Shah and M. T. Tudge, ACS Catal., 2015, 5, 3680–3688 CrossRef CAS.
- P. R. Melvin, N. Hazari, H. M. C. Lant, I. L. Peczak and H. P. Shah, Beilstein J. Org. Chem., 2015, 11, 2476–2486 CrossRef CAS PubMed.
- C. M. Zinser, K. G. Warren, R. E. Meadows, F. Nahra, A. M. Al-Majid, A. Barakat, M. S. Islam, S. P. Nolan and C. S. J. Cazin, Green Chem., 2018, 20, 3246–3252 RSC.
- Y. Liu, T. Scattolin, A. Gobbo, M. Beliš, K. Van Hecke, S. P. Nolan and C. S. J. Cazin, Eur. J. Inorg. Chem., 2022, 2022, e202100840 CrossRef CAS.
- P. Lei, G. Meng, Y. Ling, J. An, S. P. Nolan and M. Szostak, Org. Lett., 2017, 19, 6510–6513 CrossRef CAS PubMed.
- G. Li, P. Lei, M. Szostak, E. Casals-Cruañas, A. Poater, L. Cavallo and S. P. Nolan, ChemCatChem, 2018, 10, 3096–3106 CrossRef CAS.
- L. Wu, E. Drinkel, F. Gaggia, S. Capolicchio, A. Linden, L. Falivene, L. Cavallo and R. Dorta, Chem. – Eur. J., 2011, 17, 12886–12890 CrossRef CAS PubMed.
- A. Chartoire, M. Lesieur, L. Falivene, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem. – Eur. J., 2012, 18, 4517–4521 CrossRef CAS PubMed.
- B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599–1626 CrossRef CAS.
- J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS.
- P. A. Forero-Cortés and A. M. Haydl, Org. Process Res. Dev., 2019, 23, 1478–1483 CrossRef.
- R. Emadi, A. Bahrami Nekoo, F. Molaverdi, Z. Khorsandi, R. Sheibani and H. Sadeghi-Aliabadi, RSC Adv., 2023, 13, 18715–18733 RSC.
- G. Le Duc, S. Meiries and S. P. Nolan, Organometallics, 2013, 32, 7547–7551 CrossRef CAS.
- S. Meiries, G. Le Duc, A. Chartoire, A. Collado, K. Speck, K. S. A. Arachchige, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2013, 19, 17358–17368 CrossRef CAS PubMed.
- Y.-C. Hsu and M.-T. Chen, Eur. J. Inorg. Chem., 2022, 2022, e202100828 CrossRef CAS.
- D.-Z. Zheng, H.-G. Xiong, A.-X. Song, H.-G. Yao and C. Xu, Org. Biomol. Chem., 2022, 20, 2096–2101 RSC.
- P. R. Melvin, D. Balcells, N. Hazari and A. Nova, ACS Catal., 2015, 5, 5596–5606 CrossRef CAS.
- D. P. Hruszkewycz, D. Balcells, L. M. Guard, N. Hazari and M. Tilset, J. Am. Chem. Soc., 2014, 136, 7300–7316 CrossRef CAS PubMed.
- Y. Tatsuno, T. Yoshida, S. Otsuka, N. Al-Salem and B. L. Shaw, Inorg. Synth., 1990, 342–345 CrossRef CAS.
- E. A. Martynova, N. V. Tzouras, G. Pisanò, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 RSC.
- F. Bru, M. Lesieur, A. Poater, A. M. Z. Slawin, L. Cavallo and C. S. J. Cazin, Chem. – Eur. J., 2022, 28, e202201917 CrossRef CAS PubMed.
- M. S. Viciu, R. M. Kissling, E. D. Stevens and S. P. Nolan, Org. Lett., 2002, 4, 2229–2231 CrossRef CAS PubMed.
- A. Chartoire, X. Frogneux and S. P. Nolan, Adv. Synth. Catal., 2012, 354, 1897–1901 CrossRef CAS.
- Y. N. Timsina, G. Xu and T. J. Colacot, ACS Catal., 2023, 13, 8106–8118 CrossRef CAS.
- Y. Liu, V. A. Voloshkin, T. Scattolin, L. Cavallo, B. Dereli, C. S. J. Cazin and S. P. Nolan, Dalton Trans., 2021, 50, 5420–5427 RSC.
- S. Sakaki, K. Takeuchi, M. Sugimoto and H. Kurosawa, Organometallics, 1997, 16, 2995–3003 CrossRef CAS.
- H. Kurosawa, Pure Appl. Chem., 1998, 70, 1105–1110 CrossRef CAS.
- W.-F. Wang, J.-B. Peng, X. Qi, J. Ying and X.-F. Wu, Chem. – Eur. J., 2019, 25, 3521–3524 CrossRef CAS PubMed.
- W. Lölsberg, S. Ye and H.-G. Schmalz, Adv. Synth. Catal., 2010, 352, 2023–2031 CrossRef.
- N. W. J. Ang, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 12842–12847 CrossRef CAS PubMed.
- C. I. Jette, Z. J. Tong, R. G. Hadt and B. M. Stoltz, Angew. Chem., Int. Ed., 2020, 59, 2033–2038 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and XRD data. CCDC 2367688–2367692. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02533d |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |