
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual phosphorescent emissions from conformers of iridium complex rotors†
Yu-Ting
Hsu
,
Chandni
Bhagani
,
Juan A.
Aguilar
 ,
Mark A.
Fox
,
Dmitry
Yufit
,
Ross J.
Davidson
* and
Andrew
Beeby
*
,
Mark A.
Fox
,
Dmitry
Yufit
,
Ross J.
Davidson
* and
Andrew
Beeby
*
Department of Chemistry, Durham University, South Rd, Durham, DH1 3LE, UK. E-mail: ross.davidson@durham.ac.uk; andrew.beeby@durham.ac.uk
First published on 11th October 2024
Abstract
The chiral iridium rotors Ir(ppy)2(pyX)Cl (X = C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–SiR3, R = alkyl) remarkably contain two distinct rotational conformers in the ground (S0) and excited (T1) states that can be detected by NMR and emission measurements respectively at variable temperatures. The observed phosphorescent emissions, vibronic (involving L = ppy) and broad (L = pyX), arise from different triplet ligand to metal charge transfers from the two rotational conformers at distinct 3MLCT excited states. Both conformers exist in these Ir(ppy)2(pyX)Cl rotors due to the electron-withdrawing, conjugated substituent X.
C–SiR3, R = alkyl) remarkably contain two distinct rotational conformers in the ground (S0) and excited (T1) states that can be detected by NMR and emission measurements respectively at variable temperatures. The observed phosphorescent emissions, vibronic (involving L = ppy) and broad (L = pyX), arise from different triplet ligand to metal charge transfers from the two rotational conformers at distinct 3MLCT excited states. Both conformers exist in these Ir(ppy)2(pyX)Cl rotors due to the electron-withdrawing, conjugated substituent X.
Introduction
Luminescent transition-metal complexes have been investigated intensively in the last two decades1 particularly in the area of optoelectronics2 where iridium complexes remain as the industrial standards for green and red pixels in commercial OLED displays.3 Iridium complexes have recently received attention as molecular sensors due to their excellent photophysical properties and simple synthetic procedures.4 Luminescent molecular rotors are also of current interest as distinct rotational conformers result in different emissions.5 Iridium complexes as such rotors are described here for the first time.The Ir(N^C)2(py-X)Cl family of complexes consists of an iridium coordinated to a cyclometallated ligand (N^C), such as 2-phenylpyridine (ppy), and 4-X-py is a pyridine-based ligand that coordinates trans to one N^C nitrogen. The first example of such a system was reported in 1998 using 2-phenylbenzothiazole as the N^C ligand and unsubstituted pyridine with vibronic emission maxima at 556 and 596 nm and quantum yield at near-unity.6 Related iridium complexes have since been reported using facile synthetic procedures and also show vibronic emissions.7 These reasons make such complexes ideal for incorporation into more complex systems, for example, polymer-based oxygen sensors and multimetallic assembles.8–12 These compounds are chiral where one enantiomer (Λ or Δ) contains two non-equivalent N^C ligands as shown in Fig. 1 for the complex with the general formula Ir(ppy)2(py-X)Cl. They are considered as molecular rotors where the rotation of the py-X ligand could be free. This rotation could also be restricted to result in non-equivalent hydrogen and carbon atoms at E2/E2′ and E3/E3′ positions.
 | ||
| Fig. 1 Iridium-complex rotors discussed in this study. | ||
In 2003, photophysical studies8,13 on the iridium complexes A and B with X = –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and –CH2CH2SiMe2Ph in Fig. 1 revealed that the phosphorescent emission for A is significantly broader and more red-shifted than the vibronic emissions observed for B. It was speculated then that the broad emission for A is due to the conjugation of the vinyl group.
CH2 and –CH2CH2SiMe2Ph in Fig. 1 revealed that the phosphorescent emission for A is significantly broader and more red-shifted than the vibronic emissions observed for B. It was speculated then that the broad emission for A is due to the conjugation of the vinyl group.
In 2013, the NMR spectra of the iridium complex C with X = 2-pyridyl were investigated in detail. The broad proton and carbon-13 NMR peaks for C observed at ambient temperature were assigned to the pyridyl group E in Fig. 1.14 At 213 K, the proton peaks were resolved as E2, E2′, E3 and E3′ due to the presumed restricted rotation about the Ir–N(py) bond and the chirality of the molecule. There are other reports of broad proton peaks in Ir(ppy)2(py-X)Cl like the parent10 R = H (D, Fig. 1) and related7,8 derivatives at ambient temperatures indicating that restricted rotation about the Ir–N(py) bond is typical for these chiral complexes even though these observations were not explained in the reports.
Here, iridium complexes 1–3 are synthesised where X is a substituted ethynyl group (Fig. 1) to investigate their rotations and emissions at variable temperatures. The choice of an ethynyl group was to emulate the vinyl group (an electron-withdrawing conjugated group) where a broad emission was found in A and to explore the impact of rotational conformers on the vibronic or broad emission behaviours in complexes 1–3.
Results and discussion
The pyridine ligands, 4-(triisopropylsilylethynyl)pyridine, 4-(triethylsilylethynyl)pyridine and 4-(trimethylsilylethynyl)pyridine, were reacted with the complex μ-[Ir(ppy)2Cl]2 to obtain the corresponding Ir(ppy)2Cl(py-X) complexes 1, 2 and 3 with yields of 83%, 32% and 23% respectively. The X-ray crystal structure of complex 1 (Fig. S10†) reveals the Λ enantiomer with a Cl–Ir–N(py)–C(py) torsion angle of −37.4°.Proton NMR spectra for the complexes at 298 K in deuterated dichloromethane (DCM-d2) contained broad peaks for protons at E2/E2′ positions in each complex with estimated full width at half-height (FWHH) values of 400, 500 and 300 Hz for 1, 2 and 3 respectively (Fig. S8†). These broad peak observations due to restricted rotations about the Ir-pyridine bond are in accord with the reported data for complex C where a much shorter peak width for the E2/E2′ protons was found at 15 Hz.14 Broad proton peaks were also present for E3/E3′ protons which had peak widths of 16, 22 and 16 Hz for 1, 2 and 3 respectively. 13C{1H} NMR spectra for 1–3 also showed broad 13C peaks corresponding to the carbons at E2/E2′ positions with widths of 12, 17 and 16 Hz for 1, 2 and 3 (Fig. S9†).
Variable temperature (VT) proton NMR spectra were recorded for 1–3 in DCM-d2 from 298 K to 213 K. Each broad peak for E2/E2′ protons becomes two distinct peaks for the non-equivalent protons in the pyridine ligand at 253 K and as well-resolved doublets at 213 K. Likewise, each broad peak for E3/E3′ is changed to two broad peaks for the non-equivalent protons at 253 K and as doublets at 213 K. Deuterated tetrahydrofuran (THF-d8) was also used to obtain proton spectra for 1 at lower temperatures to 193 K to search for a second rotational barrier (Fig. 2).
 | ||
| Fig. 2 (a) Variable temperature 1H NMR spectra of 1 in THF-d8. (b) Arrhenius plot of the E3 and E3′ proton signals recorded in THF-d8. | ||
The line broadening technique was employed by measuring the change of full width at half-height (FWHH) for the peaks widening from exchange effects to determine the thermodynamic parameters of this motion.15 The signals from the hydrogens at E3 and E3′ were used as they remained unobstructed by signal overlap for the temperature range in THF-d8 and coincide with the region where slow exchange is observed, i.e., k ≪ Δν, so the rate constant (k) is approximated by k = π(he − ho) where he is the FWHH for peaks widened from exchange effects and ho is the FWHH for the peaks showing no exchange effects (Ha of the ppy ligand used here).
The Arrhenius plot revealed two distinct domains, with the activation energy (Ea) at 49 (E3) and 53 (E3′) kJ mol−1 above 230 K similar to the ‘stiff’ rotors reported elsewhere16 and with Ea at 4.9 (E3) and 5.2 (E3′) kJ mol−1 indicative of a near ‘free’ rotator below 230 K. The Eyring plots (Fig. S31–S33†) provides insight as to the origin of the two observed domains, ΔH‡ = 47.5 (E3), 51.5 (E3′) kJ mol−1 and ΔS‡ = −22.9 (E3), −6.5 (E3′) J mol−1 K−1 for the high Ea domain, while ΔH‡ = 3.3 (E3), 3.5 (E3′) kJ mol−1 and ΔS‡ = −217.3 (E3), −216.9 (E3′) J mol−1 K−1 for the low Ea domain. This shows that the low Ea domain motion is entropically driven meaning it corresponds to a large number of conformers.
Conformers for 1 were explored at PBE0/LANL2DZ:3-21G* with the IEF-PCM(THF) solvent model to understand the origin of the two rotational domains. The Cl–Ir–N(py)–C(py) dihedral angle was fixed for every degree to establish the energy barrier for the rotation about the Ir–N(py) bond and optimised. The energy barrier for a complete rotation was estimated at 41.9 kJ mol−1 based on the fixed angle plot (Fig. 3). The transition state geometry TS1 was located with a barrier energy of 59.9 kJ mol−1. These values are in accord with 47.5–51.5 kJ mol−1 observed for the high Ea domain. This energy barrier is the result of steric hindrance between the py ring (hydrogen at E2) and the ppy pyridyl ring (hydrogen at D6). In addition to this large energy barrier, a much smaller energy barrier of 1.0 kJ mol−1 was observed in the −30 to 30° region corresponding to the interaction between the py ring and the chlorine atom. The unfavourable interaction between the py ring E and the phenyl ring at A also contributes to the barrier here. The transition state geometry TS2 was located with an identical energy barrier. This small barrier accounts for the second domain observed in the VT NMR measurements.
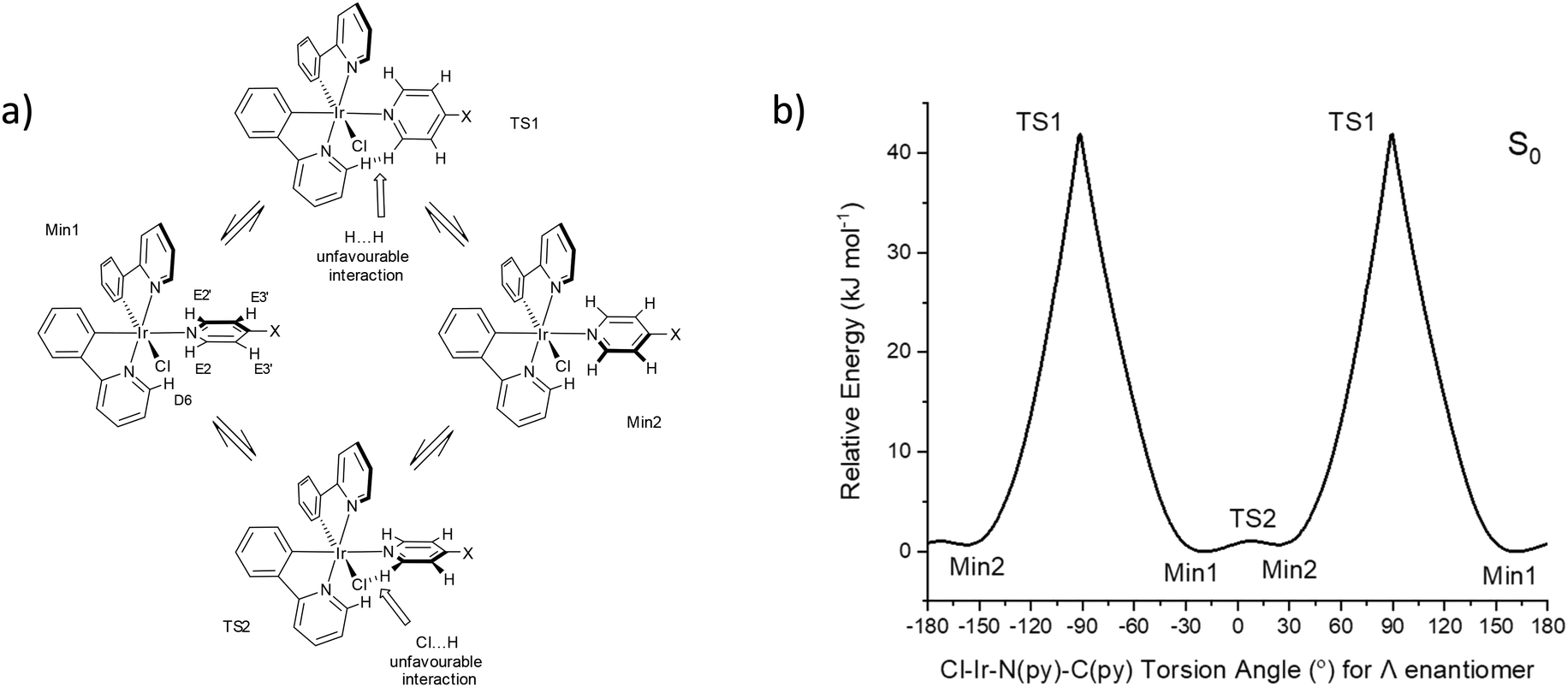 | ||
| Fig. 3 (a) Rotational conformers for 1 located by computations, X = CC–SiiPr3. (b) Relative ground state (S0) energies of 1 with respect to fixed Cl–Ir–N–C dihedral angles. | ||
The conformer at the most stable minimum, Min1, in the ground state with a Cl–Ir–N(py)–C(py) torsion angle of −20.2° for the Λ enantiomer is also present in the crystal structure for 1. The same conformer exists in other reported crystal structures of related Ir(N^C)2(py-X)Cl complexes (Table S4†).8–11,14,17,18
A similar conformer to the second minimum, Min2, with a Cl–Ir–N(py)–C(py) torsion angle of 20.6° for the Λ enantiomer is present in the crystal structure of a related Ir(N^C)2Cl(py-X) complex where N^C is 1-phenylisoquinoline and X = H with a Cl–Ir–N(py)–C(py) torsion angle at 7.0° along with the expected conformer (Min1) containing a Cl–Ir–N(py)–C(py) torsion angle at −27.8° (Fig. S11†).10 This crystal structure confirms that two conformers with different pyridine ligand orientations can exist in Ir(N^C)2(py-X)Cl complexes.
The rotation barriers found from the variable temperature NMR data for 1–3 here suggest that similar rotation barriers occur in other Ir(ppy)2(py-X)Cl complexes where broad proton peaks at E2/E3 positions at ambient temperatures have been reported.7,8,10,14 No broad peaks are observed for Ir(F2ppy)2(py-X)Cl [F2ppy = 2-(2′,4′-difluorophenyl)pyridine] complexes where all four distinct proton peaks at the E2, E2′, E3 and E3′ positions are present instead.9,11,17 Restricted rotations of the pyridyl ligand with the F2ppy ligands exist at room temperatures for Ir(F2ppy)2(py-X)Cl complexes with the first rotation barrier energies expected to be higher than Ea = 47 kJ mol−1 based on 1 here.
The UV–visible absorption spectrum of each complex (1–3) in dichloromethane (DCM) showed the expected 3MLCT bands of 400–500 nm with 1MLCT at 350–400 nm, with absorptions <350 nm attributed to π → π* transitions (Fig. S35†). The emission spectra of the complexes (1–3) in degassed DCM at room temperatures revealed apparent vibronic emissions with λmax = 504 and 532 nm, emission lifetimes (τ) of 0.16 (1), 0.21 (2) and 0.23 (3) μs and photoluminescence quantum yields (PLQY, Φ) of 5.7 (1), 4.7 (2) and 2.5 (3) %. The pure radiative lifetimes (τ0) of the complexes were determined to be 2.82 (1), 4.58 (2) and 3.09 (3) μs indicating phosphorescence from the 3MLCT excited state (Fig. S36 and Table S5†). The lifetimes and PLQYs are similar to that for Ir(ppy)2(py-X)Cl complexes A (τ = 0.06, Φ = 6; in toluene), B (0.43, 5; in toluene) and D (0.20, 13; in DCM).8,10
Intriguingly, the emission spectra for complex 1 recorded in tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MeTHF) revealed subtly broader and more red-shifted emissions to that found for 1 in DCM (Fig. S37†). These observations suggest subtle changes in the conformer make-up in different solvents even though these solvents have similar polarities (dielectric constant, ε; DCM 8.93; THF 7.43; MeTHF 6.97). The viscosities (cP at 20 °C) of these solvents are more varied with DCM at 0.44, THF 0.55 and MeTHF 0.85 thus may be responsible for different conformer populations of 1 in these solvents. Viscosity has been discussed as the reason for different properties observed in other molecular rotors.19
Variable temperature steady-state emission spectra were recorded to examine the impact of the pyridine rotations on the emission properties of complexes 1–3 (Fig. 4 and Fig. S39–S48†). In DCM, the solutions revealed higher peak resolutions and emission intensities as temperatures are lowered in steps from 300 K to 180 K.
 | ||
| Fig. 4 Variable temperature (VT) emission spectra for 1 recorded in (a) DCM and (b–d) MeTHF solutions. | ||
In MeTHF, the emissions from solutions of complexes 1–3 showed remarkable changes as temperatures are lowered. The vibronic and broad emissions at 300 K increase in intensities when lowered to 250 K as found for the vibronic emissions in DCM solutions. From 240 K to 130 K however, the vibronic emission intensities go down while the broad emission intensities go up in MeTHF. When the solutions were further cooled from 120 K to 80 K, the spectra were resolved into vibronic emissions similar to other 80 K spectra for Ir(ppy)2(py-X)Cl complexes10 such as D while the broad emissions were absent at temperatures below 115 K. Deconvolution analyses were carried out on emission spectra of 1 in MeTHF at 300 K, 240 K and 130 K to show the two distinct emissions, vibronic and broad (Fig. 5). The maximum of the broad emission is estimated at 580 nm with the highest energy onset at 500 nm.
 | ||
| Fig. 5 Deconvolution curves from emission spectra of 1 in MeTHF at (a) 300 K, (b) 240 K and (c) 130 K. | ||
The conformers for 1 at the triplet excited states (T1) responsible for the dual emissions were examined by fixing the Cl–Ir–N(py)–C(py) dihedral angle and optimising the geometry at PBE0/LANL2DZ:3-21G* in an IEF-PCM(THF) solvent field (Fig. 6). Again, two minima were located (Min1 and Min2) but Min2 is the more stable conformer at the T1 state by 6.6 kJ mol−1. Vibronic calculations20 on these T1 conformers at PBE0/LANL2DZ:3-21G* revealed maxima for the vibronic emission at 504 and 539 nm and the maximum for the broad emission at 576 nm in excellent agreement with the vibronic (504 and 532 nm) and broad (580 nm) emission spectra observed experimentally.
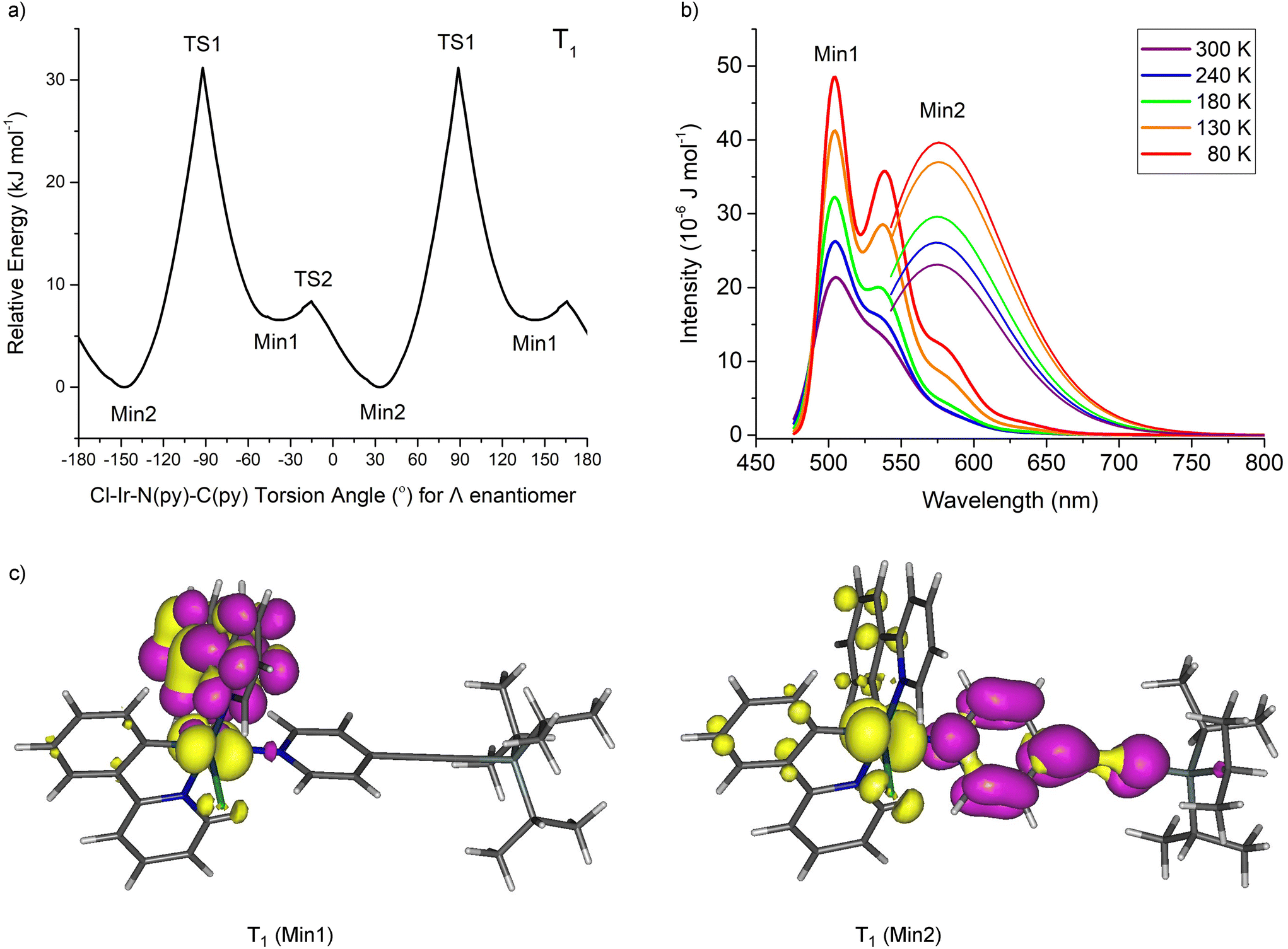 | ||
| Fig. 6 (a) Relative triplet excited state (T1) energies of 1 with respect to fixed Cl–Ir–N–C dihedral angles. (b) Simulated vibronic emission spectra from Min1 and Min2 T1 optimised geometries. (c) Natural transition orbitals (NTO, purple = particle and yellow = hole) for Min1 and Min2 T1 optimised geometries. | ||
Natural transition orbital (NTO) analyses show the vibronic emission to arise from ppy to iridium-ppy (3MLCT state) transition whereas the broad emission is mainly py to iridium charge transfer (from 3MLCT state). The key vibrational modes for the vibronic band observed at 539 nm are the ring breathing modes of the ppy ligands. The different particle (‘acceptor’/‘LUMO’) NTOs are responsible for the different emissions observed in these systems 1–3 and in other iridium complexes elsewhere.21 The broad emission reported for complex A suggests a similar Min2 conformer present at the T1 excited state as found for 1–3 here. Broad emissions are expected in these Ir(N^C)2(py-X)Cl complexes assuming that the particle NTO in the T1 state is located at the pyridine ligand which would be facilitated by a conjugated and electron-withdrawing substituent at X. The T1 Min2 conformer could be located here computationally for complexes A and C but not for B and D which indicates that complex C could generate broad emissions like A and 1–3.
The different emission behaviours for 1–3 observed in DCM and in MeTHF in the 180 to 300 K range suggest different conformer make-ups in the solutions at different temperatures. These varied conformer distributions could be due to solvent viscosities where MeTHF is considerably more viscous than DCM especially at lower temperatures. The broad emissions are dominant in the 180 to 130 K range in MeTHF indicating that the Min2 conformers at the excited states are the major conformers. The sudden change from 130 K to 80 K for 1–3 in MeTHF is related to the reported glass transition temperature for MeTHF at 128 K where only vibronic emissions from the Min1 conformer excited states are present at temperatures below 130 K.
The dual emission process in 1–3 is summarised schematically in Fig. 7. The transition from T1(Min1) to the unchanged geometry S0(Min1)′ results in the vibronic emission at 2.46 eV. The transition of T1(Min2) to the corresponding unchanged ground state geometry S0(Min2)′ affords the broad emission at 2.13 eV. The relative population ratio of T1(Min1) and T1(Min2) states depends on the temperature and solvent/viscosity factors which influence the energy barrier between these excited states.
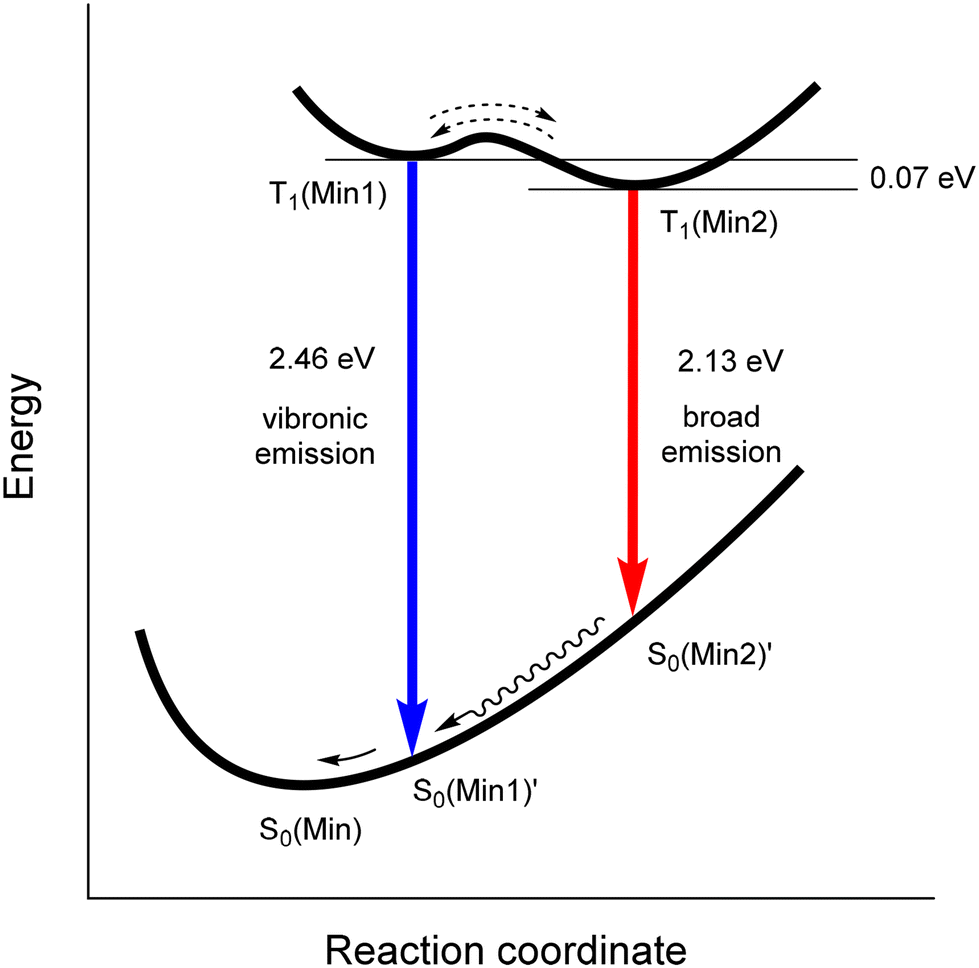 | ||
| Fig. 7 Excited state diagram summarising the observed dual emissions in 1–3. | ||
Conclusion
It is established here for the first time that two distinct conformers are present in iridium complexes as can be observed using variable temperature NMR and emission measurements. These experimental observations where two rotational barriers are present for one iridium rotor are supported by hybrid-DFT computations which reproduced the distinct rotational barrier energies and the different emissions from the two rotational conformers. Dual phosphorescent emissions were present where the vibronic emission is linked to the phenylpyridine (ppy) ligands and the broad emission involves the pyridine ligand. The broad phosphorescent emission exists in these chiral complexes when an electron-withdrawing, conjugated substituent is attached to the pyridine ligand. The dual emissions are remarkably dependent on solvent and temperature due to different viscosities. It is hoped these intriguing emission properties from synthetically facile chiral iridium complexes can be exploited in imaging/sensor applications in future.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
AB and RJD also gratefully acknowledge the EPSRC (EP/K007785/1; EP/K007548/1) for funding this work.References
- W.-P. To, Q. Wan, G. S. M. Tong and C.-M. Che, Trends Chem., 2020, 2, 796–812 CrossRef.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef PubMed.
- P. L. dos Santos, P. Stachelek, Y. Takeda and P. Pander, Mater. Chem. Front., 2024, 8, 1731–1766 RSC.
- (a) P.-Y. Ho, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2020, 413, 213267 CrossRef; (b) H. Shi, Y. Wang, S. Lin, J. Lou and Q. Zhang, Dalton Trans., 2021, 50, 6410–6417 RSC; (c) S. Aoki, K. Yokoi, Y. Hisamatsu, C. Balachandran, Y. Tamura and T. Tanaka, Top. Curr. Chem., 2022, 380, 36 CrossRef CAS; (d) B. Joshi and M. Shivashankar, ACS Omega, 2023, 8, 43408–43432 CrossRef CAS PubMed.
- (a) M. Jin, T. S. Chung, T. Seki, H. Ito and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139, 18115–18121 CrossRef CAS PubMed; (b) M. Jin, S. Yamamoto, T. Seki, H. Ito and M. A. Garcia-Garibay, Angew. Chem., Int. Ed., 2019, 58, 18003–18010 CrossRef CAS PubMed; (c) T. Li, D. Sylvinson, M. Ravinson, R. Haiges, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2020, 142, 6158–6172 CrossRef PubMed.
- R. Gao, D. G. Ho, B. Hernandez, M. Selke, D. Murphy, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 14828–14829 CrossRef CAS.
- E. Baranoff, I. Jung, R. Scopelliti, E. Solari, M. Grätzel and M. K. Nazeeruddin, Dalton Trans., 2011, 40, 6860–6867 RSC.
- M. C. DeRosa, P. J. Mosher, G. P. A. Yap, K. S. Focsaneanu, R. J. Crutchley and C. E. B. Evans, Inorg. Chem., 2003, 42, 4864–4872 CrossRef PubMed.
- G. Mu, C. Jiang and T. S. Teets, Chem. – Eur. J., 2020, 26, 11877–11886 CrossRef PubMed.
- C. Jiang and T. S. Teets, Inorg. Chem., 2022, 61, 8788–8796 CrossRef PubMed.
- G. D. Sutton, C. Jiang, G. Liu and T. S. Teets, Dalton Trans., 2023, 52, 3195–3202 RSC.
- J. Yang, W. T. Wang, Z. D. Shi, R. Yang, X. L. Liao, B. Yang and C. Z. Gao, Appl. Organomet. Chem., 2023, 37, e6964 CrossRef.
- M. C. DeRosa, P. J. Mosher, C. E. B. Evans and R. J. Crutchley, Macromol. Symp., 2003, 196, 235–248 CrossRef.
- E. C. Constable, C. E. Housecroft, G. E. Schneider and J. A. Zampese, Polyhedron, 2013, 52, 530–537 CrossRef CAS.
- F. P. Gasparro and N. H. Kolodny, J. Chem. Educ., 1977, 54, 258–261 CrossRef.
- (a) Z.-J. Chen, H.-F. Lu, C.-W. Chiu, F.-M. Hou, Y. Matsunaga, I. Chao and J.-S. Yang, Org. Lett., 2020, 22, 9158–9162 CrossRef PubMed; (b) S. Pérez-Estrada, B. Rodríguez-Molina, L. Xiao, R. Santillan, G. Jiménez-Osés, K. N. Houk and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2015, 137, 2175–2178 CrossRef PubMed.
- F. Xue, Y. Lu, Z. Zhou, C. Zhang, S. Fang, H. Yang and S. Yang, J. Coord. Chem., 2014, 67, 1353–1360 CrossRef.
- C. Li, X.-Q. Dong, Q. Wang, C.-X. Ren and Y.-Q. Ding, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1205 CrossRef PubMed.
- (a) M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669–1678 RSC; (b) J. Li, Y. Zhang, H. Zhang, X. Xuan, M. Xie, S. Xia, G. Qu and H. Guo, Anal. Chem., 2016, 88, 5554–5560 CrossRef PubMed; (c) A. T. Bui, A. Grichine, A. Duperray, P. Lidon, F. Riobé, C. Andraud and O. Maury, J. Am. Chem. Soc., 2017, 139, 7693–7696 CrossRef; (d) S.-C. Lee, J. Heo, H. C. Woo, J.-A. Lee, Y. H. Seo, C.-L. Lee, S. Kim and O. Kwon, Chem. – Eur. J., 2018, 24, 13706–13718 CrossRef PubMed; (e) K. Daus, S. Tharamak, W. Pluempanupat, P. A. Galie, M. A. Theodoraki, E. A. Theodorakis and M. L. Alpaugh, Sci. Rep., 2023, 13, 20529 CrossRef PubMed; (f) M. Paez-Perez and M. K. Kuimova, Angew. Chem., Int. Ed., 2024, 63, e202311233 CrossRef PubMed.
- C. F. R. Mackenzie, S.-Y. Kwak, S. Kim and E. Zysman-Colman, Dalton Trans., 2023, 52, 4112–4121 RSC.
- (a) S. Sharma, H. Kim, Y. H. Lee, T. Kim, Y. S. Lee and M. H. Lee, Inorg. Chem., 2014, 53, 8672–8680 CrossRef; (b) R. D. Sanner, N. J. Cherepy, H. P. Martinez, H. Q. Pham and V. G. Young Jr., Inorg. Chim. Acta, 2019, 496, 119040 CrossRef; (c) R. Davidson, Y.-T. Hsu, C. Bhagani, D. Yufit and A. Beeby, Organometallics, 2017, 36, 2727–2735 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, crystallographic, electrochemical, photophysical data and computational details. CCDC 2143627. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02321h |
| This journal is © The Royal Society of Chemistry 2024 |