
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunability of triplet excited states and photophysical behaviour of bis-cyclometalated iridium(III) complexes with imidazo[4,5-f][1,10]phenanthroline†
Katarzyna
Choroba
 *a,
Joanna
Palion-Gazda
a,
Mateusz
Penkala
a,
Patrycja
Rawicka
b and
Barbara
Machura
*a
*a,
Joanna
Palion-Gazda
a,
Mateusz
Penkala
a,
Patrycja
Rawicka
b and
Barbara
Machura
*a
aInstitute of Chemistry, Faculty of Science and Technology, University of Silesia, Szkolna 9, 40-006 Katowice, Poland. E-mail: katarzyna.choroba@us.edu.pl; barbara.machura@us.edu.pl
bInstitute of Physics, Faculty of Science and Technology, University of Silesia, 75 Pułku Piechoty 1a, 41-500 Chorzów, Poland
First published on 17th October 2024
Abstract
This is a comprehensive study of the photophysical behaviour of heteroleptic iridium(III) complexes with imidazo[4,5-f][1,10]phenanthroline (imphen) as an ancillary ligand, represented by the general formula [Ir(N∩C)2(imphen)]PF6. As cyclometalating ligands, 2-phenylpyridine (Hppy), 2-phenylquinoline (Hpquin), 2-phenylbenzothiazole (Hpbztz), and 2-(2-pyridyl)benzothiophene (pybzthH) were used. The impact of structural modifications of cyclometalating ligands was widely explored by a combination of steady-state and time-resolved optical techniques accompanied by theoretical calculations. We evidenced that the cyclometalating ligands induce essential changes in the nature of the emissive excited state and the emission characteristics of [Ir(N∩C)2(imphen)]PF6. While the complex [Ir(ppy)2(imphen)]PF6 (1) is a typical 3MLLCT emitter, the lowest triplet states of [Ir(pquin)2(imphen)]PF6 (2), [Ir(pbztz)2(imphen)]PF6 (3) and [Ir(pybzth)2(imphen)]PF6 (4) have a predominant 3LCN∩C character. The phosphorescence colour of the investigated Ir(III) complexes changes from greenish-yellow to red, their quantum yields vary from 56 to 2%, and their triplet excited-state lifetimes fall in the 743–3840 ns range. The highest photoluminescence quantum yield was revealed for 2 in CH2Cl2, while complex 3 in MeCN shows the most pronounced increase in the lifetime. Both complexes 2 and 3 show an increased efficiency of singlet oxygen generation. The herein discussed structure–property relationships are of high significance for controlling photoinduced processes in heteroleptic iridium(III) complexes with the imphen-based ancillary ligand, and making further progress in effectively tuning the emission energies, quantum yields and excited-state lifetimes of these systems by structural modifications of cyclometalating ligands, especially the π-conjugation, the position of the N-donor and the presence of sulfur heteroatoms.
Introduction
Heteroleptic iridium(III) complexes [Ir(N∩C)2(N∩N)]X (N∩C – cyclometalating ligand, N∩N – diimine, and X – counter anion) are among the most attractive targets for the design of photoluminescent materials for employment in optoelectronics,1–15 photocatalysis2,16–23 and life science.2,7,19,24–34 The great advantages of these systems are good photo- and thermal stability, high phosphorescence quantum yields and long-lived triplet excited states due to the large ligand-field splitting and spin–orbit coupling constant of the Ir(III) ion, as well as their wide colour emission tunability achieved by an appropriate combination of cyclometalating and ancillary ligands.5,6,35–46 In general, the highest occupied molecular orbital (HOMO) of [Ir(N∩C)2(N∩N)]+ resides on the d orbitals of the Ir(III) ion and the π orbitals of cyclometalating ligands, while the lowest unoccupied molecular orbital (LUMO) is contributed by the π* orbitals of an ancillary ligand. The photoexcitation of [Ir(N∩C)2(N∩N)]+ may generate electronic transitions of metal-to-ligand charge transfer (MLCT), ligand-to-ligand charge transfer (LLCT), and ligand-centered (LC) nature, or lead to their superposition. A combination of MLCT and LLCT, frequently observed in these systems, is generally denoted as metal–ligand-to-ligand charge transfer (MLLCT). Due to the heavy atom effect, the photoinduced excited singlet states undergo ultrafast intersystem crossing (ISC), forming 3MLLCT, 3LC, or mixed 3MLLCT/3LC.6,7,36,42,47–51Each of these triplet excited states in [Ir(N∩C)2(N∩N)]+ brings different photophysical parameters of the resulting complexes. Structureless emission profiles with high quantum yields but shorter lifetimes are expected for 3MLLCT excited states, while triplet excited states with a predominant ligand character are characterised by emission bands with a well-resolved vibronic structure and prolonged excited state lifetimes but lower quantum yields.6,13,42,52,53 The photophysical parameters of 3MLLCT emitters are generally sufficient for employment as OLED components, but they are commonly detrimental for applications in photodynamic therapy (PDT), time-resolved bioimaging, or triplet–triplet annihilation up-conversion (TTA UC), where systems with much longer excited state lifetimes are required.2,26,31,54
Although the excited state properties of cyclometalated Ir(III) complexes have been in the scientific interest for over 50 years, precise control of excited states in [Ir(N∩C)2(N∩N)]+ still remains challenging and requires further systematic studies to identify reliable structure–property relationships, and make progress in the further improvement of phosphorescent materials and potent chemotherapeutic drugs.
In the present work, we have investigated the role of structurally different cyclometalating ligands in determining the photophysical characteristics of heteroleptic iridium(III) complexes with imidazo[4,5-f][1,10]phenanthroline (imphen) as an ancillary ligand (Scheme 1).
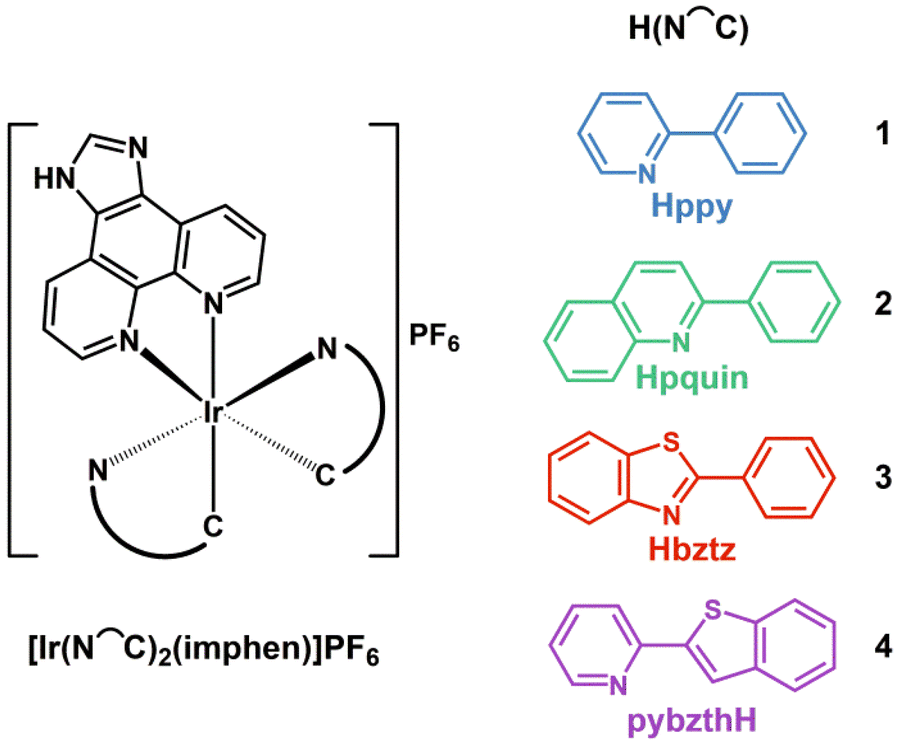 | ||
| Scheme 1 Discussed iridium(III) complexes 1–4. H(N∩C) stands for different cyclometalating ligands: Hppy – 2-phenylpyridine, Hpquin – 2-phenylquinoline, Hpbztz – 2-phenylbenzothiazole, and pybzthH – 2-(2-pyridyl)benzothiophene. | ||
The employment of these ligand combinations constitutes the novelty of the current contribution. Over the last two decades, derivatives of imidazo[4,5-f][1,10]phenanthroline have been predominately combined with the [Ir(ppy)2]+ core (Hppy – 2-phenylpyridine), and what is really important, the resulting complexes [Ir(ppy)2(R1,R2-imphen)]X have been successfully applied in anticancer therapy, bioimaging, conversion of solar energy, organic light-emitting diodes, photocatalysis, and luminescence sensing.28,30,55–57 In contrast, heteroleptic iridium(III) complexes [Ir(N∩C)2(R1,R2-imphen)]+ bearing 2-phenylquinoline (Hpquin), 2-phenylbenzothiazole (Hpbztz), and 2-(2-pyridyl)benzothiophene (pybzthH) as cyclometalating ligands are extremely rare.58–61 The survey of the Reaxys database (date of search: 26-06-2024) revealed 111 records for [Ir(ppy)2(R1,R2-imphen)]X, 2 for [Ir(pquin)2(R1,R2-imphen)]X, 2 for [Ir(pbztz)2(R1,R2-imphen)]X and 0 for [Ir(pybzth)2(R1,R2-imphen)]X. The employment of a new ligand combination leads to novel excited-state and physical properties of the resulting Ir(III) complexes, and thus facilitates the novel use of these systems in biology and optoelectronics. To the best of our knowledge, the photobehaviours of [Ir(ppy)2(imphen)]PF6 (1) [Ir(pquin)2(imphen)]PF6 (2), [Ir(pbztz)2(imphen)]PF6 (3) and [Ir(pybzth)2(imphen)]PF6 (4) have been firstly discussed herein. Considering the fact that the imphen platform can be widely modified at the 1H- and C2-positions,62,63 the herein presented structure–property relationships seem to be crucial for making further progress in tuning the photophysical properties of heteroleptic iridium(III) complexes with the imphen-based ancillary ligand by structural modifications of cyclometalating ligands.
Results and discussion
Synthesis and structural characterization of heteroleptic Ir(III) complexes
The heteroleptic iridium(III) complexes [Ir(ppy)2(imphen)](PF6) (1), [Ir(pquin)2(imphen)](PF6) (2), [Ir(pbztz)2(imphen)](PF6) (3) and [Ir(pybzth)2(imphen)](PF6) (4) were prepared using the standard synthetic methodology of this type of Ir(III) complex,64–67 that is a bridge-splitting reaction of the appropriate dinuclear precursor [Ir(μ-Cl)2(N∩C)4] with imidazo[4,5-f][1,10]phenanthroline (imphen), followed by anion exchange of the chloride salt with NH4PF6 (Scheme S1†).The formation of the target complexes was confirmed by elemental analysis, 1H, 13C{1H} and 31P{1H} NMR spectroscopy (Fig. S1–S4†), HRMS (Fig. S5†), and the FT-IR technique (Fig. S6†). For all investigated systems, the signals in the 1H and 13C{1H} NMR spectra were fully assigned using multidimensional techniques 1H–1H COSY, 1H–13C{1H} HMQC, and 1H–13C{1H} HMBC (Fig. S1–S4†). The aromatic hydrogen protons of the ancillary ligand are generally downfield in relation to those of cyclometalating ligands. On analyzing the correlations between the structure of the cyclometalating ligand and the imphen proton signals, it can be noticed that the extending conjugation of the N-part of the cyclometalating ligands (2 and 4) leads to small upfield shifts of the H-2 proton signals in the imidazole ring compared to those of complexes 1 and 4, with pyridyl as the N-part. An opposite trend is observed for the H-4 and H-11 proton signals of the 1,10-phenanthroline framework, which experience a downfield shift for 2 and 4 relative to those of 1 and 4. The signal originating from the H-1 proton attached to the nitrogen atom in the imidazole ring appears above 14 ppm in the 1H NMR spectra of compounds 1 and 3, while it completely disappears in the case of 2 and 4 owing to the replacement of the N–H protons by deuterium atoms from the solvent. In agreement with the σ-donating abilities, the 13C{1H} NMR signals of the carbon coordinated to the Ir(III) center show a noticeable downfield shift compared to other aromatic carbons. The analysis of the 31P{1H} NMR spectra confirms that the signals recorded for the PF6− ion practically do not differ in their shifts. In each case, we observed a septet signal resulting from phosphorus–fluorine couplings. High-resolution mass spectrometry (HRMS) analysis was performed on four Ir(III) compounds in both positive and negative ionization modes (Fig. S5†). In positive ion mode, the HRMS spectra of compounds 1–4 display signals for the corresponding [Ir(N∩C)2(imphen)]+ cations, along with their characteristic isotope patterns. In negative ion mode, all compounds exhibit strong signals for the PF6− ions. The presence of the counter anion in the investigated systems is also supported by intense bands appearing at ∼845 cm−1 and ∼555 cm−1 in the FT-IR spectra68 (Fig. S6†).
The molecular structure of 2 was further characterized by single-crystal X-ray crystallography and is presented in Fig. 1.
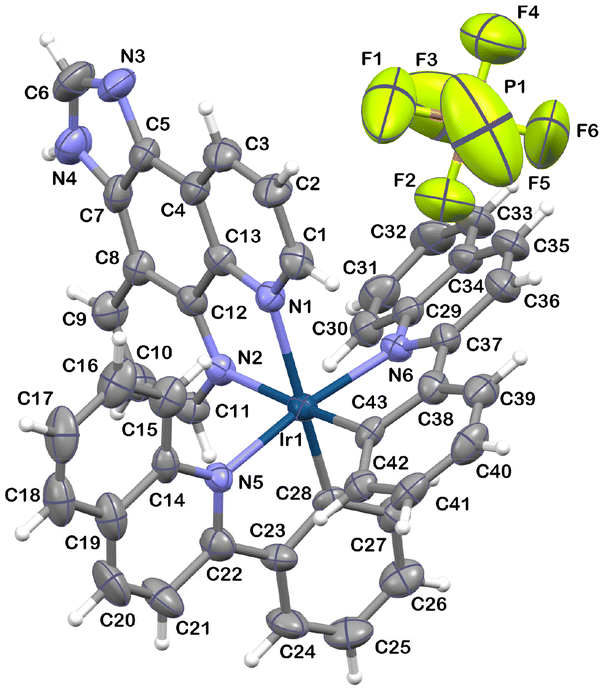 | ||
| Fig. 1 Molecular structure of 2 with atom numbering. Selected bond lengths [Å] and angles [°]: Ir(1)–N(1) 2.195(7); Ir(1)–N(2) 2.166(7); Ir(1)–N(5) 2.081(7); Ir(1)–N(6) 2.078(7); Ir(1)–C(28) 1.998(7); Ir(1)–C(43) 1.996(8) N(2)–Ir(1)–N(1) 75.7(3); N(5)–Ir(1)–N(1) 104.9(3); N(5)–Ir(1)–N(2) 84.1(3); N(6)–Ir(1)–N(1) 80.6(3); N(6)–Ir(1)–N(2) 102.8(3); N(6)–Ir(1)–N(5) 172.2(3); C(28)–Ir(1)–N(1) 170.5(3); C(28)–Ir(1)–N(2) 96.7(3); C(28)–Ir(1)–N(5) 79.7(3); C(28)–Ir(1)–N(6) 95.7(3); C(43)–Ir(1)–N(1) 97.0(3); C(43)–Ir(1)–N(2) 171.6(3); C(43)–Ir(1)–N(5) 93.9(3); C(43)–Ir(1)–N(6) 79.8(3); C(43)–Ir(1)–C(28) 90.9(3); C(1)–N(1)–Ir(1) 127.8(6). Thermal ellipsoids are drawn at the 50% probability level. | ||
As is typical of this class of compounds,69 the Ir(III) ion of 2 shows a distorted octahedral coordination environment, with the imphen ligand coordinated trans to the cyclometalated phenyl rings. The cyclometalating ligands remain in the cis-C,C and trans-N,N arrangement, as in the chloro-bridged precursor [Ir(μ-Cl)2(pquin)4]. Consistent with the trans-effect of the carbon donors of pquin ligands, the bond lengths of Ir–Nimphen become slightly elongated [2.166(7) Å and 2.195(7) Å] relative to Ir–Npquin [2.078(7) Å and 2.081(7) Å]. A noticeable angular distortion from the octahedral geometry is reflected in the N–Ir–N and N–Ir–C bite angles due to the formation of five-membered metallocycles upon coordination [75.7(3)°, 79.7(3)° and 79.8(3)°]. The full structural data, including the analysis of short intra- and intermolecular contacts detected in structure 2, are provided in the ESI (Tables S1–S3).†
Computational investigations
To gain a better understanding of how structural modifications of cyclometalating ligands affect the photophysical behaviour of [Ir(N∩C)2(imphen)]+ systems, DFT and DT-DFT calculations were performed using Gaussian-16 software70 at the PCM/PBE0/SDD/def2-TZVP level,71–80 and the relevant theoretical findings are briefly presented prior to the discussion of the optical properties of 1–4. Selected optimized structural parameters in ground-state geometries (S0) are presented in Tables S4 and S5,† and compared to the available structural data obtained from X-ray analysis of 2, demonstrating a good agreement between the experimental and theoretical values.The partial molecular orbital energy level diagram with plots of the electron density distribution in the HOMO and LUMO of 1–4 is depicted in Fig. 2. Complexes 1 and 2 show very similar HOMO and LUMO electron density distributions. Their HOMOs dominantly reside on the d-orbital of the Ir(III) center and the phenyl rings of the ppy and pquin ligands, while their LUMOs are largely contributed by the π* imphen ligand orbitals (Fig. 2 and Fig. S7–S11†). The extending conjugation of the N-part of the cyclometalating ligand (pyridine in ppy and quinoline in pquin) leads to a slight destabilization of both the HOMO and LUMO levels of 2 relative to 1, resulting in an insignificant increase of the HOMO–LUMO energy gap of 2, that is 3.61 eV for 1 and 3.63 eV for 2.
 | ||
| Fig. 2 Partial molecular orbital energy level diagram with the plots of electron density distributions in the HOMO (below) and the LUMO (above) of 1–4 (see also Fig. S7–S10†). | ||
More noticeable variations in the HOMO and LUMO energies and electron distributions are induced by isomeric pbztz and pybzth cyclometalating ligands. The LUMO energy levels of both 3 and 4 are lowered compared to complex 1, while opposite effects on the HOMO energy level are observed. While pbztz lowers the HOMO level and complex 3 shows the highest calculated energy gap (3.70 eV), pybzth with an electron-rich benzothiophene C-part increases the HOMO level, which results in a significantly reduced HOMO–LUMO energy gap for 4 (3.20 eV).
A striking difference between 3 and 4 can also be noticed in the HOMO and LUMO electron density distributions. By analogy to 1 and 2, the electron density in the HOMO of 3 is dominated by the orbitals of the Ir(III) center and the phenyl rings of pbztz. Conversely, the HOMO of 4 shows a decrease in metal character. Compared to 3, the metal contribution to the HOMO of 4 is decreased twice. The HOMO of complex 4 is dominantly localized on pybzth, with the contribution of the benzothiophene core exceeding over 50%. The LUMOs of both 3 and 4 show a reduced contribution of the π* orbitals of imphen compared to 1 and 2. They are almost equally contributed by the π* orbitals of imphen and the cyclometalating ligand.
Time-dependent (TD-DFT) calculations estimated the S0 → S1 excitation to occur at 450 nm for 1, 446 nm for 2, 435 nm for 3 and 508 nm for 4. For all complexes 1–4, this lowest-energy excitation originates from an almost pure HOMO to LUMO orbital, so it can be assigned to an electronic transition of the mixed character: 1MLLCT for 1–3, and 1ILCT/1MLCT for 4. The contribution of 1ILCT in 4 can be assigned to the donor–acceptor nature of pybzth, constituting the electron-rich benzothiophene and electron-deficient pyridine units.52,81,82 The characteristics of other calculated electronic transitions of 1–4 are presented in Tables S6–S9,† and the electron density distributions of MOs involved in this excitation are presented in Fig. S11.†
The phosphorescence energies of the investigated complexes were theoretically calculated as the vertical energy difference between the ground singlet and triplet excited states ΔET1–S0. The solvent environment was taken into account by PCM equilibrium solvation with the linear response approach. The triplet emissions are predicted at 583 nm for 1, 608 nm for 2, 548 nm for 3 and 662 nm for 4 (Table S10†), and the spin density surfaces indicate that the phosphorescence of these systems is contributed by the 3MLLCT and 3LC excited states (Fig. S12†).
Electrochemistry
To provide more insight into the HOMO and LUMO energy levels and their characteristics, complexes 1–4 and the free ligands were studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in MeCN on a glassy carbon working electrode and 0.1 M Bu4NPF6 as the supporting electrolyte. All CV and DPV voltammograms are presented in the ESI (Table S11 and Fig. S13),† and the most relevant data are presented in Fig. 3 and Table 1.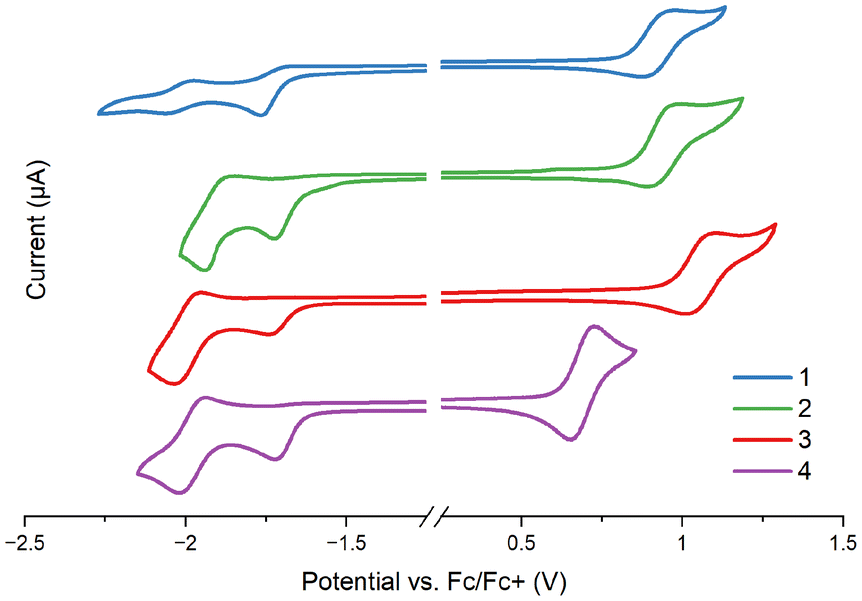 | ||
| Fig. 3 Cyclic voltammetry of complexes 1–4. CG working electrode; electrolyte: MeCN/Bu4NPF6; scan rate: 0.1 V s−1. | ||
| Complex | E ox1pa [V] | E ox1pc [V] | ΔEox1 [V] | E ox11/2 [V] | E red1pa [V] | E ox1onset [V] | E redonset 1 [V] | IP [eV] | EA [eV] |
|---|---|---|---|---|---|---|---|---|---|
| E pa and Epc – anodic and cathodic potentials vs. Fc/Fc+; ΔE – peak potential difference estimated from the equation ΔE = Epa − Epc; E1/2 – redox potentials estimated from the equation E1/2 = (Epa + Epc)/2; Eox1onset – oxidation onset potential; Ered1onset – reduction onset potential; IP – ionization potential estimated from the equation IP = |e−|(5.1 + Eox1onset); EA – electron affinity estimated from the equation EA = |e−|(5.1 + Ered1onset); CG working electrode; electrolyte: MeCN/Bu4NPF6; scan rate: 0.1 V s−1. | |||||||||
| 1 | 0.98 | 0.88 | 0.10 | 0.93 | −1.77 | 0.83 | −1.68 | 5.93 | 3.42 |
| 2 | 0.99 | 0.89 | 0.10 | 0.94 | −1.72 | 0.85 | −1.62 | 5.95 | 3.48 |
| 3 | 1.11 | 1.01 | 0.10 | 1.06 | −1.74 | 0.97 | −1.64 | 6.07 | 3.46 |
| 4 | 0.73 | 0.65 | 0.07 | 0.69 | −1.72 | 0.62 | −1.63 | 5.72 | 3.47 |
All complexes show multi-stage redox characteristics (Fig. S13†). The first irreversible reduction peak of 1–4, associated with the LUMO level, appears in a narrow potential range from −1.72 to −1.77 V, and remains almost unperturbed by structural modifications of the cyclometalating ligand. With reference to the electrochemical data of the free ligands, DFT calculations of 1–4, and previous findings for [Ir(N∩C)2(N∩N)]PF6,83–85 it can be safely assigned to the reduction of the imphen ancillary ligand; the cyclometalating ligands are reduced at noticeably lower potentials (Table S11†).
The first reversible oxidation wave appears between 0.69 and 1.06 V versus the ferrocene/ferrocenium redox couple, and it is noticeably affected by the cyclometalating ligand. Striking differences in Eox1 are observed upon the replacement of ppy by pbztz and pybzth. In agreement with the DFT calculations which show the stabilization of the HOMO for 3 and its destabilization for 4 relative to the HOMO of 2, the oxidation potentials of 3 and 4 are anodically and cathodically shifted in relation to complex 2, respectively. In turn, the extending conjugation of the N-part of the cyclometalating ligand upon the replacement of ppy by pquin leads to small variations in Eox1 values, in agreement with the DFT calculations. Such findings are indicative of the oxidation process occurring on the metal-centered orbitals with a contribution of the C-part of the cyclometalating ligand, as previously reported for related systems.83,86–88
Photophysical properties
The UV-Vis absorption spectra of 1–4 were recorded in dichloromethane and acetonitrile (Fig. 4 and Fig. S14†). The absorption maxima and molar extinction coefficients are listed in Table S12.† All examined complexes display absorption profiles typical of bis-cyclometalated Ir(III) systems. Very intense high-energy bands are accompanied by broad and noticeably weaker absorption bands in a lower-energy region. Spin-forbidden 3MLCT transitions, facilitated by the heavy Ir atom, are represented by very weak bands appearing above 500 nm.39,41,89–91 | ||
| Fig. 4 UV-Vis spectra of 1–4 in acetonitrile (c = 10−5 M and c = 5 × 10−4 M for the curves in the inset). | ||
A detailed inspection of the UV-Vis data of 1–4 shows that structural modifications of cyclometalating ligands have a noticeable impact on the absorption intensity of [Ir(N∩C)2(imphen)](PF6). In general, complexes bearing cyclometalating ligands with a higher degree of π-conjugation (2–4vs.1) have larger molar extinction coefficients, especially in the 300–500 nm region. Additionally, the visible-light-absorption abilities of 4 are enhanced relative to those of 1–3, as a result of the replacement of phenyl in ppy, pquin and pbztz by the more electron-rich benzothiophene core.52,81,82
In accordance with the previous findings39,41,89–91 and TD-DFT calculations (Fig. S7–S11 and Tables S6–S9†), intense high-energy absorption bands in the 225–340 nm range are dominantly assigned to the spin-allowed 1π → π* (1LC) transition occurring within the cyclometalating and ancillary ligands, while absorptions at lower energies mainly originate from spin-allowed charge-transfer transitions 1MLLCT for 1–3 and 1ILCT/1MLCT for 4. Before investigating the excited-state properties of 1–4, their stability and photostability in solution were proved by UV-Vis spectroscopy, as shown in Fig. S15–S20.†
The photoluminescence studies revealed that all the obtained Ir(III) complexes are emissive in solutions at room temperature (RT), in the solid state, and in a frozen matrix of EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (4:1 v/v) at 77 K, as summarized in Fig. 5 and 6. More detailed photophysical characteristics of 1–4 are presented in the ESI (Table S13 and Fig. S21–S24).†
:MeOH (4:1 v/v) at 77 K, as summarized in Fig. 5 and 6. More detailed photophysical characteristics of 1–4 are presented in the ESI (Table S13 and Fig. S21–S24).†
 | ||
| Fig. 5 Emission spectra of compounds 1–4 in CH2Cl2, MeCN, and an EtOH:MeOH (4:1 v/v) rigid matrix at 77 K, along with their respective photoluminescence lifetimes and quantum yields. For samples with decay profiles fitted to biexponential functions, average lifetime values τav were given (see also Fig. S21–S24†). | ||
 | ||
| Fig. 6 Solid-state emission spectra of complexes 1–4 (a). Photographs of CH2Cl2 solutions and the solid samples of compounds 1–4 under UV light (b), along with their CIE 1931 chromaticity plots (c). | ||
For all complexes 1–4, their lifetimes are in the microsecond domain, and their photoluminescence intensities and lifetimes are substantially decreased in the presence of molecular oxygen, which indicates that the emission originates from a triplet excited state (Fig. S25†).
The cyclometalating ligand induces noticeable changes in the emission characteristics of the resulting [Ir(N∩C)2(imphen)](PF6). In solutions at RT, the phosphorescence colour ranges from greenish-yellow to red (Fig. 6), the quantum yields vary from 56 to 2%, and the triplet excited-state lifetimes fall in the 740–3840 ns range (Table S13†). In CH2Cl2, the luminescence energies of [Ir(N∩C)2(imphen)](PF6) follow the order: 3 > 2 > 1 > 4. The highest photoluminescence quantum yield was found for 2 in CH2Cl2, while complex 3 in MeCN shows the most pronounced increase in the lifetime. The radiative constants of 1 and 2 are one order higher than that of complex 4.
All the obtained Ir(III) complexes also exhibit photoluminescence in the solid state, with the emission maxima and quantum yields falling in the ranges of 560–695 nm and 8–0.7%, respectively (Fig. 6).
Regarding the emission energies and profiles in different media, the Ir(III) compounds can be divided into three groups: {1}, {2} and {3 and 4}. Complex 1 shows excitation-independent, broad and structureless bands in both solutions at RT and the EtOH:MeOH matrix at 77 K. The frozen-state emission of 1 occurs in a higher energy region and shows a prolonged lifetime in relation to the RT emission in agreement with the rigidochromic effect.92,93 The increased environment polarity produces a bathochromic shift of the emission maximum of 1 (591 nm in CH2Cl2 and 601 nm in MeCN). All these findings are indicative of the lowest triplet state of 3MLLCT character.6,7,36,42,47–51
For other complexes 2–4, the room-temperature emission appears almost in the same range as its frozen-state emission band. The frozen-state phosphorescence spectra of these systems exhibit sharp vibronic structures, and they are largely superimposed with the phosphorescence of the corresponding free ligand, confirming the contribution of 3LCN∩C to the emission at 77 K. As shown in Fig. 5, the frozen emission profile of 4 is noticeably red-shifted relative to those of 2 and 3.
The striking differences between 2 and 3–4 concern the room-temperature emission profiles. While complex 2 shows very poorly structured emission bands in MeCN and CH2Cl2, the emission bands of 3 and 4 in solution at RT display a well-resolved vibronic progression. With reference to the previous findings,6,13,42,52,53 the loss of vibronic progression in the PL solution spectra of 2 at RT may be associated with the increased 3MLCT character in the triplet emissive state.
To get a better insight into the nature of the triplet emitting excited state of 2–4, their solvent-dependent emission spectra at room temperature and variable-temperature emission spectra (Fig. 7) have been recorded. As shown in Fig. 7, the emission of 2–4 shows totally different solvent and temperature dependencies in relation to 1. While the 3MLLCT emission band of 1 is noticeably affected by the solvent polarity and temperature, the phosphorescence energies of 3 and 4 remain insensitive to the environment and do not exhibit a rigidochromic effect, which is typical of 3LCN∩C emission. The changes in the emission intensities of 4, observed on the low energy side of the emission band, may be attributed to the contribution of the 3ILCTN∩C component, in agreement with the donor–acceptor nature of pybzth.
 | ||
| Fig. 7 Variable-temperature emission spectra of 1–4 in 2-methyltetrahydrofuran (a) and normalized solvent-dependent emission spectra of 1–4 at room temperature (b). | ||
For complex 2, cooling results in a much sharper vibronic progression of the emission profile, and the emission energy undergoes only a slight blue shift below 120 K. Also, the room temperature emission of 2 is independent of the solvent polarity. These findings are supportive of the predominant 3LCN∩C character in the emitting excited state of complex 2.
The decreased contribution of the MLLCT character in the triplet excited states of 2–4 in relation to complex 1 is further supported by the thermally induced shifts ΔEs = E0–0(77 K) − E0–0(298 K),94 equal to 2342 cm−1 for 1, 700 cm−1 for 2, 370 cm−1 for 3 and 278 cm−1 for 4. The variations in the emission profiles of 1–4 can be rationalized by the lowering of the triplet excited state localized on the cyclometalating ligand in the order: ppy > pquin > pbztz > pybzth (Fig. S26†). The triplet emissions of the ligands were induced by addition of 10% ethyl iodide, and 3LCH(N∩C) energies were estimated by taking the tangent line on the high-energy side of the frozen-state phosphorescence band, and its intersection with the wavelength axis (Fig. S27†).
The inspection of the photoluminescence lifetimes and quantum yields (Table S13†) shows a striking difference between complexes 3 and 4, both emitted from the triplet excited state of the predominant 3LCN∩C character. Relative to 3, complex 4 with markedly red-shifted emission is characterized by shorter photoluminescence lifetimes and lower quantum yields. This can be rationalized by the fact that the red phosphor (4) easily decays to the ground state through the non-radiative channels.95,96
Femto- and nanosecond transient absorption
To determine further characteristics of the excited states of 1–4, femtosecond transient absorption (fs-TA) spectroscopy and laser flash photolysis were used, and the studies were performed in acetonitrile solution upon excitation at 355 nm. The photostability of 1–4 in TA experiments was confirmed by comparison of the UV-Vis spectra of the Ir(III) complexes recorded before and after irradiation (Fig. S28†). For all systems 1–4, the ultrafast intersystem crossing cannot be determined with our experimental setup, as it occurs in a time scale shorter than the instrument response. The results of fs-TA measurements and global fitting analyses are presented in Fig. 8 and also provided in the ESI (Fig. S29–S32).† A global analysis of the TA maps was carried out using a linear unidirectional sequential model implemented in Optimus™ software. This analysis allowed us to perform the deconvolution of the transient spectra into evolution-associated spectra (EAS), providing the decay-associated spectra (DAS) as a linear combination of the EAS. The laser flash photolysis studies are summarized in Fig. S33–S36.† | ||
| Fig. 8 The results of fs-TA measurements and global fitting analyses of 1–4: (a) 2D time–wavelength plots; (b) fs-TA spectra at selected time delays (ps); and (c) decay-associated spectra DASi with appropriate time constants ti (see also Fig. S29–S32†). | ||
The laser pulse excitation of 1 in MeCN at 355 nm results in an instant formation of negative and positive bands in the 360–400 nm and 405–615 nm regions, respectively. The negative feature corresponds to a ground-state bleaching (GSB), falling in the wavelength region of the lowest energy charge-transfer absorption in the UV-Vis spectrum of 1 (Fig. 8). The initially formed broad excited-state absorption (ESA) band in the visible region undergoes rapid evolution. Within 3 ps, it evolves into two overlapping bands covering the 405–490 and 490–615 nm ranges. At longer time delays, up to 300 ps, the intensity enhancement of the TA band at longer wavelengths is accompanied by a decrease in the intensity of the higher energy ESA band. The well-defined isosbestic point at 490 nm allows us to assign these absorptions to two different transient species, undergoing interconversion. The weaker TA signal (405–490 nm) can be attributed to the transitions of the 3MLCTppy excited state, while the stronger TA band (490–615 nm) represents the transitions of the 3MLCTimphen excited state. Such observations are indicative of the occurrence of 3LLCT from ppy to imphen in the excited state. The LLCT transitions shift the electron density from the cyclometalating ligand to the ancillary one.90,97–100 Accordingly, the lowest triplet excited state of 1 can be safely defined as 3MLLCT. The given interpretation is fully supported by the global fit analysis, resulting in three components for the best fitting of fs-TA data of 1. The components with time constants of 0.79 ps (DAS1) and 42.76 ps (DAS2) represent the formation of the relaxed 3MLCT excited state and the 3LLCT process occurring from ppy to imphen, respectively. The slowest component with the infinite lifetime is attributed to the relaxation from the lowest triplet state (T1) to the ground state (S0). Both the negative and positive TA signals remain visible up to the end of the delay stage (7 ns), and the relaxed fs-TA profiles are spectrally identical to those recorded with the laser flash photolysis technique. It allows us to assume that the excited state formed in the fs-regime is the final triplet excited state (Fig. 8).
Contrary to 1, the fs-TA and ns-TA spectra of 2–4 display only positive bands, both in the UV and UV-Vis regions. This indicates that the Ir(III) complexes bearing cyclometalating ligands with a higher degree of π-conjugation (2–4vs.1) exhibit stronger excited-state absorption than ground-state absorption in the visible range, which makes them interesting as potential reverse saturable absorbers.66,101–106 For complexes 2–4, the ground-state bleaching is manifested by a dip in the 420–460 nm range, covering the wavelength region of the lowest energy charge-transfer absorption of these systems (Fig. S37†). As shown in Fig. S33–S36,† the ns-TA spectra consist of two ESA bands, with maxima at 415 and 505 nm for 2, 390 and 485 nm for 3, and 385 and 475 nm for 4, and separated by ground-state bleaching. Such findings are indicative of the triplet excited state localized on the cyclometalating ligand.90 The assignment of the ns-TA spectral features of 2–4 to the 3LCN∩C excited state is further supported by the high similarity between the TA spectra of 2–4 and free cyclometalating ligands (Fig. S33–S36†). Also, the triplet excited-state lifetimes determined on the basis of the triplet state kinetic decay curves of ns-TA signals (Fig. S34–S36 and Table S13†) resemble the lifetimes obtained from the decay of the 3LCN∩C emission of 2–4 in acetonitrile (Fig. S22–S24 and Table S13†), implying that the transient absorbing excited state is the same as the emitting triplet excited state.
Some differences between 2–3 and 4 can be noticed when their fs-TA and ns-TA results are compared (Fig. 9). The relaxed fs-TA profile of 4 is spectrally identical to those recorded with the laser flash photolysis technique, supporting that the relaxed excited state formed in the fs-regime is the final 3LCN∩C excited state of 4. In contrast, the fs-TA spectra of 2 and 3 show considerably stronger absorption in the 550–560 nm range for 2 and 520–620 nm for 3 compared to the signals in the ns-TA spectra (Fig. 9). By analogy to 1, complex 2 exhibits a well-defined isosbestic point at 537 nm which can be safely assigned to the interconversion of 3MLCTpquin into 3MLCTimphen, and thus the occurrence of 3LLCT transitions from pquin to imphen. In agreement with the extending conjugation of the N-part of the cyclometalating ligand (pyridine in ppy and quinoline in pquin), the ESAMLLCT of 2 is noticeably red-shifted relative to 1 (Fig. 8). At the longer delay time, that is in the ns-TA spectra, the ESA of 2 in the 550–560 nm range disappeared. Similar variations, but without the presence of a well-defined isosbestic point in the fs-TA spectra, are observed for 3.
 | ||
| Fig. 9 TA spectral profiles of 1–4 at a 6 ns delay time (fs-TA studies) versus the first delay time profiles measured using the laser flash photolysis technique. | ||
On the basis of the TA results, it can be assumed that the population of the 3LCN∩C excited state in systems 2–4 occurs in a few steps. The optically populated 1MLCT excited state undergoes a rapid ISC process and then vibrational relaxation to the relaxed 3MLCT excited state. In the next stage, LLCT transitions shift the electron density from the cyclometalating ligand to the ancillary one.90,97–100 Finally, the relaxed 3MLCT/3LLCT excited state undergoes conversion into the lower energy 3LCN∩C one. The utilization of pybzth as the cyclometalating ligand in [Ir(N∩C)2(imphen)]PF6 significantly facilitates the population of 3LCN∩C.
Singlet oxygen generation
All investigated Ir(III) complexes show a noticeable decrease in the phosphorescence intensity and lifetime under the air-equilibrated conditions (Fig. S25†), which may indicate the ability of these systems to transfer energy from the triplet excited state to molecular oxygen (3O2), and the formation of singlet oxygen (1O2). Singlet oxygen, one of the reactive oxygen species (ROS), is known for acting as an efficient cytotoxic agent in photodynamic therapy (PDT).27,30,34,107,108To evaluate the singlet oxygen generation abilities of 1–4, we utilized an indirect strategy based on the oxidation reaction of diphenylisobenzofuran (DPBF) with Ir(III) complexes as photosensitizers, upon irradiation at 420 nm. Complexes 1–4 in DMSO are photostable, showing no absorbance decrease upon excitation at 420 nm (Fig. S20†). DPBF acts as an efficient singlet oxygen scavenger, and undergoes a rapid conversion into 1,2-dibenzoylbenzene.54,109–111 Therefore, the progress of DPBF photooxidation can be monitored by the gradual decrease of the lowest energy absorption maximum of DPBF (at 417 nm), and quantitatively compared by plotting A/A0 against the irradiation time (Fig. 10). The singlet oxygen quantum yields (ΦΔ) of 1–4 were obtained by comparison with [Ru(bipy)3](PF6)2 as a standard photosensitizer (ΦΔ = 0.656)112 using the following equation:

 | ||
| Fig. 10 Plots of A/A0 against the irradiation time for the photooxidation reaction of DPBF using Ir(III) complexes (1–4) and [Ru(bipy)3](PF6)2 as sensitizers. A0 and A stand for absorbance at t = 0 s and after the given irradiation time, respectively. | ||
The investigated Ir(III) complexes show moderate 1O2 quantum yields,54,113,114 decreasing in the order: 2 (76.4%) > 3 (64.1%) > 4 (60.6%) > 1 (58.3%). The obtained findings indicate the increased 1O2 efficiency in the case of Ir(III) complexes with a more extended π-conjugated N-part of the cyclometalating ligand. However, there is no clear correlation between the ΦΔ and the excited state lifetime or emission quantum yield. Nevertheless, it can be assumed that the contribution of 3LC to the excited state of [Ir(N∩C)2(imphen)]PF6 is beneficial for the ability to generate 1O2via energy transfer.
Conclusions
To investigate the impact of cyclometalating ligands on the nature of the triplet excited state and emission characteristics of heteroleptic iridium(III) complexes with imidazo[4,5-f][1,10]phenanthroline (imphen) as an ancillary ligand, a series of [Ir(N∩C)2(imphen)]PF6 complexes with four different cyclometalating ligands were prepared and widely explored by a combination of steady-state and time-resolved optical techniques in combination with theoretical calculations. The complex [Ir(ppy)2(imphen)]PF6, with 2-phenylpyridine as the cyclometalating ligand, was found to be a typical 3MLLCT emitter. The extending conjugation of the N- and C-parts of the cyclometalating ligand resulted in a predominant 3LCN∩C character of the triplet excited state of the investigated Ir(III) complexes. Consequently, structural modifications of the cyclometalating ligands induce essential changes in the emission characteristics. The phosphorescence colour of these systems changes from greenish-yellow to red, the quantum yields vary from 56 to 2%, and the triplet excited-state lifetimes fall in the 743–3840 ns range. Complexes 2 and 3 are characterized by noticeably prolonged lifetimes, and show an increased 1O2 efficiency. Since the imphen framework can be easily further modified by incorporation of both electron-withdrawing and electron-donating groups into the 1H- and C2-positions, the herein presented structure–property relationships are of high importance for making further progress in the development of more efficient imidazo[4,5-f][1,10]phenanthroline-based Ir(III) luminophores for optoelectronics, photocatalysis and life science.Experimental section
Iridium(III) chloride, ammonium hexafluorophosphate, and appropriate H(N∩C) ligands were all commercially available, and they were used without further purification. Solvents used for synthesis were of reagent grade, while those for spectroscopic measurements were of HPLC grade. The ligand imphen was prepared employing the method reported previously,115 by mixing 1,10-phenanthroline-5,6-dione and formaldehyde in glacial acetic acid, with the addition of ammonium acetate. Iridium(III) compounds 1–4 were obtained according to a modified literature method.64–67 All reactions were carried out under an inert argon atmosphere.Synthesis of [Ir(μ-Cl)2(N∩C)4]
0.15 g of IrCl3·3H2O (0.5 mmol) and 1.25 mmol of appropriate H(N∩C) ligand (0.2 g of Hppy, 0.25 g of Hpquin, and 0.26 g of Hpbztz or pybzthH) were dissolved in a 2-methoxyethanol:water mixture (3:1 v/v). The resulting solution was heated in an autoclave for 12 hours to 120 °C, maintained at the temperature for 24 hours and cooled down to RT for another 24 hours. The resulting precipitate was filtered and washed with ethanol and diethyl ether.
Synthesis of [Ir(N∩C)2(imphen)]PF6 complexes
0.25 mmol of appropriate [Ir(μ-Cl)2(N∩C)4] dimer (0.25 g for Hppy and 0.32 g for Hpquin, Hpbztz, or pybzthH as H(N∩C)) mixed with 0.5 mmol (0.11 g) of imphen ligand was dissolved in a 30 mL chloroform:methanol mixture (3:1 v/v) and heated under reflux for 24 h. The solution was evaporated using a rotary evaporator to approx. 3 mL and after that, 5 mL of saturated aqueous solution of NH4PF6 was added dropwise and mixed for 12 h, resulting in the precipitation of the crude product. The precipitate was filtered, washed with diethyl ether and recrystallized from acetonitrile or methanol.
![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) F6−) ppm.
F6−) ppm.
F6−) ppm.
F6−) ppm.
F6−) ppm.
F6−) ppm.
F6−) ppm.
F6−) ppm.
General techniques
NMR spectra were recorded with a Bruker Avance 500 NMR spectrometer at 298 K using DMSO-d6 as the solvent. HRMS analyses were performed using a Waters Xevo G2 Q-TOF mass spectrometer with an ESI ion source in positive and negative modes. FT-IR spectra were recorded using a Nicolet iS5 FTIR spectrophotometer with the KBr pellet method. Elemental analyses were performed using an Elementar Vario EL Cube for C, H and N contents. The X-ray structural data of compound 2 were collected using a four-circle diffractometer Gemini A Ultra (Oxford Diffraction) with a CCD Atlas detector using graphite monochromated MoKα radiation (λ = 0.71073 Å). The crystallographic data in CIF format were deposited in the Cambridge Crystallographic Data Center, CCDC 2369644. Electrochemical measurements were performed with an Autolab PGSTAT 128N potentiostat using GC, Ag, and Pt as the working, reference, and counter electrodes, respectively; supporting electrolyte: 0.1 M Bu4NPF6 in MeCN, scan rate: 0.1 V s−1, and referenced against Fc/Fc+. UV-Vis spectra were recorded using a Thermo Scientific Evolution 220 spectrometer. The singlet oxygen generation efficiency was measured using an Evolution 220 UV–vis spectrometer in DMSO with 1,3-diphenylisobenzofuran as a probe. The DFT and TD-DFT calculations were performed using Gaussian-1670 software, using the PBE0 functional74,75 with the Stuttgart/Dresden Relativistic Small Core ECP basis set76,77 for Ir and the def2-TZVP basis set78–80 for other elements, and PCM71–73 corresponding to acetonitrile. The photoluminescence measurements were performed using an FLS-980 fluorescence spectrophotometer (Edinburgh Instruments). The femtosecond transient absorption spectra were recorded using a pump–probe transient absorption system (Ultrafast Systems, Helios) described previously.116–118 The triplet state lifetimes of the compounds were measured using an LKS 60 laser flash photolysis spectrometer (Applied Photophysics). Further details concerning the experimental setup and methodology are presented in the ESI.†Author contributions
K. C.: methodology, investigation, visualization, formal analysis, data curation, writing – original draft, and writing – review and editing; J. P-G.: investigation, visualization, formal analysis, and writing – review and editing; M. P.: investigation, visualization, and data curation; P. R.: investigation, visualization, and data curation; B. M.: conceptualization, methodology, investigation, formal analysis, funding acquisition, project administration, supervision, validation, writing – original draft, and writing – review and editing. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI at https://doi.org/10.1039/D4DT01996B. Additional experimental details; NMR, HRMS, and FT-IR spectra; crystal data and structure refinement, short intra- and intermolecular contacts; DFT calculations: comparison of theoretical singlet, triplet, and experimental bond lengths and angles, selected molecular orbitals, percentage contribution of selected molecular fragments to the frontier molecular orbitals, calculated electronic transitions, comparison of the experimental and calculated luminescence properties, spin density maps; electrochemical measurements: DPV and CV voltammograms; UV-Vis spectra, absorption maxima and molar extinction coefficients, UV-Vis stability and photostability; summary of the luminescence properties, emission spectra of Ir(III) complexes in argon-saturated and air-equilibrated solutions, emission spectra of Ir(III) complexes and their respective H(N∩C) ligands at 77 K; photodamage tests, summary of the fs-TA and ns-TA measurements, comparison of decay associated spectra and UV-Vis spectra.Crystallographic data for 2 were deposited with the Cambridge Crystallographic Data Center, CCDC 2369644.†
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was cofinanced by funds allocated to the European City of Science Katowice 2024 and the Research Excellence Initiative of the University of Silesia in Katowice. The authors would like to thank dr inż. M. Siwy for the elemental analyses and dr inż. K. Erfurt for the HRMS measurements.References
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- X. Cui, J. Zhao, Z. Mohmood and C. Zhang, Chem. Rec., 2016, 16, 173–188 CrossRef CAS PubMed.
- D. Ma, T. Tsuboi, Y. Qiu and L. Duan, Adv. Mater., 2017, 29, 1603253 CrossRef PubMed.
- E. Matteucci, A. Baschieri, A. Mazzanti, L. Sambri, J. Ávila, A. Pertegás, H. J. Bolink, F. Monti, E. Leoni and N. Armaroli, Inorg. Chem., 2017, 56, 10584–10595 CrossRef CAS PubMed.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- S. Lee and W.-S. Han, Inorg. Chem. Front., 2020, 7, 2396–2422 RSC.
- Y. Zhang and J. Qiao, iScience, 2021, 24, 102858 CrossRef CAS PubMed.
- J. Jayabharathi, V. Thanikachalam and S. Thilagavathy, Coord. Chem. Rev., 2023, 483, 215100 CrossRef CAS.
- Y. Lan, D. Liu, J. Li, H. Wan and Y. Mei, Dyes Pigm., 2023, 210, 111032 CrossRef CAS.
- T. Yang, A. Zhou, Y. He, Z. Yao, X. Song, X. Tao and Y. Tao, Mater. Adv., 2023, 4, 631–640 RSC.
- P. L. dos Santos, P. Stachelek, Y. Takeda and P. Pander, Mater. Chem. Front., 2024, 8, 1731–1766 RSC.
- Z.-P. Yan, M.-X. Mao, Q.-M. Liu, L. Yuan, X.-F. Luo, X.-J. Liao, W. Cai and Y.-X. Zheng, Adv. Funct. Mater., 2024, 34, 2402906 CrossRef CAS.
- Z. Yan, Z. Wang, X. Zhuang, Z. Li, C. Kai, X. Song, J. Liang, H. Bi and Y. Wang, Adv. Opt. Mater., 2024, 12, 2303214 CrossRef CAS.
- J. Yan, Y. Pan, I.-C. Peng, W.-Y. Hung, B. Hu, G. Ni, S.-M. Yiu, Y. Chi and K. C. Lau, Inorg. Chem. Front., 2024, 11, 2413–2426 RSC.
- J. Yan, M. Song, D.-Y. Zhou, G. Ni, M. Gu, S.-M. Yiu, X. Zhou, L.-S. Liao and Y. Chi, Adv. Opt. Mater., 2024, 12, 2301739 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed.
- F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- X. Zhang, Y. Hou, X. Xiao, X. Chen, M. Hu, X. Geng, Z. Wang and J. Zhao, Coord. Chem. Rev., 2020, 417, 213371 CrossRef CAS.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- D. Kim, V. Q. Dang and T. S. Teets, Chem. Sci., 2023, 15, 77–94 RSC.
- Y.-L. Li, A.-J. Li, S.-L. Huang, J. J. Vittal and G.-Y. Yang, Chem. Soc. Rev., 2023, 52, 4725–4754 RSC.
- L. Qin, X. Xin, R. Wang, H. Lv and G.-Y. Yang, J. Catal., 2023, 417, 89–97 CrossRef CAS.
- C. Caporale and M. Massi, Coord. Chem. Rev., 2018, 363, 71–91 CrossRef CAS.
- A. Zamora, G. Vigueras, V. Rodríguez, M. D. Santana and J. Ruiz, Coord. Chem. Rev., 2018, 360, 34–76 CrossRef CAS.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- P.-Y. Ho, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2020, 413, 213267 CrossRef CAS.
- X. Tian, C. De Pace, L. Ruiz-Perez, B. Chen, R. Su, M. Zhang, R. Zhang, Q. Zhang, Q. Wang, H. Zhou, J. Wu, Z. Zhang, Y. Tian and G. Battaglia, Adv. Mater., 2020, 32, 2003901 CrossRef CAS PubMed.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- B. H. Jhun, D. Song, S. Y. Park and Y. You, Top. Curr. Chem., 2022, 380, 35 CrossRef CAS PubMed.
- T. Yang, M. Zhu, M. Jiang, F. Yang and Z. Zhang, Front. Pharmacol., 2022, 13, 1025544 CrossRef CAS PubMed.
- J. Zhou, J. Li, K. Y. Zhang, S. Liu and Q. Zhao, Coord. Chem. Rev., 2022, 453, 214334 CrossRef CAS.
- B. Kar, U. Das, N. Roy and P. Paira, Coord. Chem. Rev., 2023, 474, 214860 CrossRef CAS.
- L. M. Groves, C. Schotten, J. Beames, J. A. Platts, S. J. Coles, P. N. Horton, D. L. Browne and S. J. A. Pope, Chem. – Eur. J., 2017, 23, 9407–9418 CrossRef CAS PubMed.
- A. F. Henwood and E. Zysman-Colman, Chem. Commun., 2017, 53, 807–826 RSC.
- A. J. Huckaba and M. K. Nazeeruddin, Comments Inorg. Chem., 2017, 37, 117–145 CrossRef CAS.
- C.-J. Li, S.-Y. Yin, H.-P. Wang, Z.-W. Wei and M. Pan, J. Photochem. Photobiol., A, 2019, 379, 99–104 CrossRef CAS.
- B. Liu, L. Lystrom, S. Kilina and W. Sun, Inorg. Chem., 2019, 58, 476–488 CrossRef CAS PubMed.
- E. Kabir, Y. Wu, S. Sittel, B.-L. Nguyen and T. S. Teets, Inorg. Chem. Front., 2020, 7, 1362–1373 RSC.
- T. M. Stonelake, K. A. Phillips, H. Y. Otaif, Z. C. Edwardson, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Inorg. Chem., 2020, 59, 2266–2277 CrossRef CAS PubMed.
- S. DiLuzio, V. Mdluli, T. U. Connell, J. Lewis, V. VanBenschoten and S. Bernhard, J. Am. Chem. Soc., 2021, 143, 1179–1194 CrossRef CAS PubMed.
- S. Yoon and T. S. Teets, Chem. Commun., 2021, 57, 1975–1988 RSC.
- S. A. Fitzgerald, E. N. Payce, P. N. Horton, S. J. Coles and S. J. A. Pope, Dalton Trans., 2023, 52, 16480–16491 RSC.
- Z. Hendi, D. O. Kozina, V. V. Porsev, K. S. Kisel, J. R. Shakirova and S. P. Tunik, Molecules, 2023, 28, 2740 CrossRef CAS PubMed.
- E. Martínez-Vollbert, C. Philouze, T. Cavignac, C. Latouche, F. Loiseau and P.-H. Lanoë, Dalton Trans., 2024, 53, 4705–4718 RSC.
- K.-C. Tang, K. L. Liu and I.-C. Chen, Chem. Phys. Lett., 2004, 386, 437–441 CrossRef CAS.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, J. Phys. Chem. A, 2009, 113, 2–4 CrossRef CAS PubMed.
- F. Messina, E. Pomarico, M. Silatani, E. Baranoff and M. Chergui, J. Phys. Chem. Lett., 2015, 6, 4475–4480 CrossRef CAS PubMed.
- Z. Kuang, X. Wang, Z. Wang, G. He, Q. Guo, L. He and A. Xia, Chin. J. Chem. Phys., 2017, 30, 259–267 CrossRef CAS.
- F. Monti, A. Baschieri, L. Sambri and N. Armaroli, Acc. Chem. Res., 2021, 54, 1492–1505 CrossRef CAS PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- A. Kapturkiewicz and A. Kamecka, RSC Adv., 2021, 11, 29308–29322 RSC.
- S. K. Seth and P. Purkayastha, Eur. J. Inorg. Chem., 2020, 2020, 2990–2997 CrossRef CAS.
- C. Jin, J. Liu, Y. Chen, G. Li, R. Guan, P. Zhang, L. Ji and H. Chao, Dalton Trans., 2015, 44, 7538–7547 RSC.
- L. Bai, W.-D. Fei, Y.-Y. Gu, M. He, F. Du, W.-Y. Zhang, L.-L. Yang and Y.-J. Liu, J. Inorg. Biochem., 2020, 205, 111014 CrossRef CAS PubMed.
- J. Chen, W. Li, G. Li, X. Liu, C. Huang, H. Nie, L. Liang, Y. Wang and Y. Liu, Eur. J. Med. Chem., 2024, 265, 116078 CrossRef CAS PubMed.
- J. Liu, C. Jin, B. Yuan, X. Liu, Y. Chen, L. Ji and H. Chao, Chem. Commun., 2017, 53, 2052–2055 RSC.
- Q.-Z. Yuan, Q. Fan, H. Lv, W.-W. Chen, X.-X. Yang, D.-K. Cao and J. Wen, Inorg. Chem., 2020, 59, 17071–17076 CrossRef CAS PubMed.
- Yu. P. Parshina, T. A. Kovylina, A. N. Konev, A. A. Belikov, P. O. Baber, A. D. Komarova, E. A. Romaeva and L. N. Bochkarev, Russ. J. Gen. Chem., 2022, 92, 2666–2675 CrossRef CAS.
- J. R. Shakirova, V. A. Baigildin, A. I. Solomatina, R. B. Aghakhanpour, V. V. Pavlovskiy, V. V. Porsev and S. P. Tunik, Adv. Funct. Mater., 2023, 33, 2212390 CrossRef CAS.
- S. Kumar, S. Singh, A. Kumar, K. S. R. Murthy and A. K. Singh, Coord. Chem. Rev., 2022, 452, 214272 CrossRef CAS.
- S. Naithani, T. Goswami, F. Thetiot and S. Kumar, Coord. Chem. Rev., 2023, 475, 214894 CrossRef CAS.
- H. Woo, S. Cho, Y. Han, W.-S. Chae, D.-R. Ahn, Y. You and W. Nam, J. Am. Chem. Soc., 2013, 135, 4771–4787 CrossRef CAS PubMed.
- T. Li, M. Cui, G. Ran and Q. Song, Dyes Pigm., 2015, 112, 1–7 CrossRef CAS.
- L. Wang, P. Cui, S. Kilina and W. Sun, J. Phys. Chem. C, 2017, 121, 5719–5730 CrossRef CAS.
- B. Joshi and M. Shivashankar, ACS Omega, 2023, 8, 43408–43432 CrossRef CAS PubMed.
- A. M. Heyns and G. J. van Schalkwyk, Spectrochim. Acta, Part A, 1973, 29, 1163–1175 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408–3420 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- H. Patil, A. Gupta, A. Bilic, S. L. Jackson, K. Latham and S. V. Bhosale, J. Electron. Mater., 2014, 43, 3243–3254 CrossRef CAS.
- Y. Jeon, T.-M. Kim, J.-J. Kim and J.-I. Hong, New J. Chem., 2015, 39, 9591–9595 RSC.
- K. K.-W. Lo, C.-K. Chung, T. K.-M. Lee, L.-H. Lui, K. H.-K. Tsang and N. Zhu, Inorg. Chem., 2003, 42, 6886–6897 CrossRef CAS PubMed.
- Q. Zhao, S. Liu, M. Shi, F. Li, H. Jing, T. Yi and C. Huang, Organometallics, 2007, 26, 5922–5930 CrossRef CAS.
- D. Tordera, A. Pertegás, N. M. Shavaleev, R. Scopelliti, E. Ortí, H. J. Bolink, E. Baranoff, M. Grätzel and M. K. Nazeeruddin, J. Mater. Chem., 2012, 22, 19264–19268 RSC.
- C. Dragonetti, L. Falciola, P. Mussini, S. Righetto, D. Roberto, R. Ugo, A. Valore, F. De Angelis, S. Fantacci, A. Sgamellotti, M. Ramon and M. Muccini, Inorg. Chem., 2007, 46, 8533–8547 CrossRef CAS PubMed.
- K. Y. Zhang, S. P.-Y. Li, N. Zhu, I. W.-S. Or, M. S.-H. Cheung, Y.-W. Lam and K. K.-W. Lo, Inorg. Chem., 2010, 49, 2530–2540 CrossRef CAS PubMed.
- I. González, M. Natali, A. R. Cabrera, B. Loeb, J. Maze and P. Dreyse, New J. Chem., 2018, 42, 6644–6654 RSC.
- J.-H. Kim, S.-Y. Kim, S. Jang, S. Yi, D. W. Cho, H.-J. Son and S. O. Kang, Inorg. Chem., 2019, 58, 16112–16125 CrossRef CAS PubMed.
- P. A. Scattergood, A. M. Ranieri, L. Charalambou, A. Comia, D. A. W. Ross, C. R. Rice, S. J. O. Hardman, J.-L. Heully, I. M. Dixon, M. Massi, F. Alary and P. I. P. Elliott, Inorg. Chem., 2020, 59, 1785–1803 CrossRef CAS PubMed.
- C. E. Elgar, H. Y. Otaif, X. Zhang, J. Zhao, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Chem. – Eur. J., 2021, 27, 3427–3439 CrossRef CAS PubMed.
- A. J. Lees, Chem. Rev., 1987, 87, 711–743 CrossRef CAS.
- A. J. Lees, Comments Inorg. Chem., 1995, 17, 319–346 CrossRef CAS.
- J. M. Favale, C. E. Hauke, E. O. Danilov, J. E. Yarnell and F. N. Castellano, Dalton Trans., 2020, 49, 9995–10002 RSC.
- Z. Chen, H. Zhang, D. Wen, W. Wu, Q. Zeng, S. Chen and W.-Y. Wong, Chem. Sci., 2020, 11, 2342–2349 RSC.
- S. Chen, H. Bi, W. Tian and Y. Liu, Molecules, 2022, 27, 286 CrossRef CAS PubMed.
- Y.-J. Cho, S.-Y. Kim, M. Cho, K.-R. Wee, H.-J. Son, W.-S. Han, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 15162–15169 RSC.
- S. Tschierlei, A. Neubauer, N. Rockstroh, M. Karnahl, P. Schwarzbach, H. Junge, M. Beller and S. Lochbrunner, Phys. Chem. Chem. Phys., 2016, 18, 10682–10687 RSC.
- Y.-J. Cho, S.-Y. Kim, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 8778–8786 RSC.
- R. Bevernaegie, L. Marcélis, A. Moreno-Betancourt, B. Laramée-Milette, G. S. Hanan, F. Loiseau, M. Sliwa and B. Elias, Phys. Chem. Chem. Phys., 2018, 20, 27256–27260 RSC.
- C. Wang, L. Lystrom, H. Yin, M. Hetu, S. Kilina, S. A. McFarland and W. Sun, Dalton Trans., 2016, 45, 16366–16378 RSC.
- B. Liu, L. Lystrom, S. L. Brown, E. K. Hobbie, S. Kilina and W. Sun, Inorg. Chem., 2019, 58, 5483–5493 CrossRef CAS PubMed.
- T. Lu, C. Lu, P. Cui, S. Kilina and W. Sun, J. Mater. Chem. C, 2021, 9, 15932–15941 RSC.
- J. Lu, Q. Pan, S. Zhu, R. Liu and H. Zhu, Inorg. Chem., 2021, 60, 12835–12846 CrossRef CAS PubMed.
- M. E. Olumba, R. M. O'Donnell, T. N. Rohrabaugh Jr. and T. S. Teets, Inorg. Chem., 2022, 61, 19344–19353 CrossRef CAS PubMed.
- G. Li, Z. Jiang, M. Tang, X. Jiang, H. Tu, S. Zhu, R. Liu and H. Zhu, Molecules, 2023, 28, 566 CrossRef CAS PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- M. A. Munegowda, A. Manalac, M. Weersink, S. A. McFarland and L. Lilge, Coord. Chem. Rev., 2022, 470, 214712 CrossRef PubMed.
- B. Sanasam, M. K. Raza, D. Musib, M. Pal, M. Pal and M. Roy, ChemistrySelect, 2020, 5, 13824–13830 CrossRef CAS.
- B. Sanasam, M. K. Raza, D. Musib and M. Roy, J. Chem. Sci., 2021, 133, 42 CrossRef CAS.
- Z. Yuan, J. He, Z. Mahmood, L. Xing, S. Ji, Y. Huo and H.-L. Zhang, Dyes Pigm., 2022, 199, 110049 CrossRef CAS.
- L. V. Lutkus, S. S. Rickenbach and T. M. McCormick, J. Photochem. Photobiol., A, 2019, 378, 131–135 CrossRef CAS.
- D. Ashen-Garry and M. Selke, Photochem. Photobiol., 2014, 90, 257–274 CrossRef CAS PubMed.
- J. Liu, X. Yang, S. Wu, P. Gong, F. Pan, P. Zhang, C.-S. Lee, C. Liu and K. M.-C. Wong, J. Mater. Chem. B, 2024, 12, 3710–3718 RSC.
- K. Choroba, M. Penkala, J. Palion-Gazda, E. Malicka and B. Machura, Inorg. Chem., 2023, 62, 19256–19269 CrossRef CAS PubMed.
- K. Choroba, S. Kula, A. Maroń, B. Machura, J. Małecki, A. Szłapa-Kula, M. Siwy, J. Grzelak, S. Maćkowski and E. Schab-Balcerzak, Dyes Pigm., 2019, 169, 89–104 CrossRef CAS.
- K. Choroba, S. Kotowicz, A. Maroń, A. Świtlicka, A. Szłapa-Kula, M. Siwy, J. Grzelak, K. Sulowska, S. Maćkowski, E. Schab-Balcerzak and B. Machura, Dyes Pigm., 2021, 192, 109472 CrossRef CAS.
- A. Szlapa-Kula, M. Małecka, A. M. Maroń, H. Janeczek, M. Siwy, E. Schab-Balcerzak, M. Szalkowski, S. Maćkowski, T. Pedzinski, K. Erfurt and B. Machura, Inorg. Chem., 2021, 60, 18726–18738 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details; NMR, HRMS, and FT-IR spectra; crystal data and structure refinement, short intra- and intermolecular contacts; DFT calculations: comparison of theoretical singlet, triplet, and experimental bond lengths and angles, selected molecular orbitals, percentage contribution of selected molecular fragments to the frontier molecular orbitals, calculated electronic transitions, comparison of the experimental and calculated luminescence properties, spin density maps; electrochemical measurements: DPV and CV voltammograms; UV-Vis spectra, absorption maxima and molar extinction coefficients, UV-Vis stability and photostability; summary of the luminescence properties, emission spectra of Ir(III) complexes in argon-saturated and air-equilibrated solutions, emission spectra of Ir(III) complexes and their respective H(N∩C) ligands at 77 K; photodamage tests, summary of the fs-TA and ns-TA measurements, comparison of decay associated spectra and UV-Vis spectra (PDF). CCDC 2369644. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01996b |
| This journal is © The Royal Society of Chemistry 2024 |