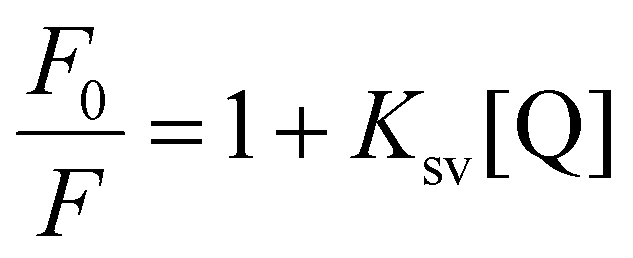DOI:
10.1039/D4DT01914H
(Paper)
Dalton Trans., 2024,
53, 17702-17720
A novel copper(II) complex with a salicylidene carbohydrazide ligand that promotes oxidative stress and apoptosis in triple negative breast cancer cells†
Received
2nd July 2024
, Accepted 4th October 2024
First published on 11th October 2024
Abstract
We report the synthesis, characterization, anti-cancer activity and mechanism of action of a novel water-soluble Cu(II) complex with salicylidene carbohydrazide as the ligand and o-phenanthroline as the co-ligand. The synthesized complex (1) was characterized by FT-IR, EPR, and electronic spectroscopy, as well as single crystal X-ray diffraction. This compound was found to be paramagnetic from EPR spectra and X-ray crystallography revealed that the molecule crystallized in an orthorhombic crystal system. The crystal lattice was asymmetric containing two distinct binuclear copper complexes containing the Schiff base as the major ligand, o-phenanthroline as a co-ligand, two nitrate anions, and two water molecules. The Cu(II) in the first site coordinated with the enolised ligand comprising enolate O−, phenolate O−, and the imine N and N,N from o-phen. The major part of this complex exists as Cu(II) coordinated with two H2O molecules at the second site with nitrate acting as the counter anion. However, a smaller portion of the complex exists where Cu(II) is coordinated with NO3− and H2O, and the remaining water molecule acts as lattice water. It was tested for DNA binding and cleavage properties which revealed that it binds in an intercalative mode to CT-DNA with Kb value of 1.25 × 104 M−1. Furthermore, cleavage studies reveal that the complex has potential for efficient DNA cleavage under both oxidative and hydrolytic conditions. It was able to enhance the rate of cleavage by 2.8 × 108 times. The complex shows good cytotoxicity to breast cancer monolayer (2D) as well as spheroid (3D) systems. The IC50 values for MDA-MB-231 and MCF-7 monolayer culture was calculated as 1.86 ± 0.17 μM and 2.22 ± 0.08 μM, respectively, and in (3D) spheroids of MDA-MB-231 cells, the IC50 value was calculated to be 1.51 ± 0.29 μM. It was observed that the complex outperformed cisplatin in both breast cancer cell lines. The cells treated with complex 1 underwent severe DNA damage, increased oxidative stress and cell cycle arrest which finally led to programmed cell death or apoptosis in triple negative breast cancer cells through an intrinsic pathway.
Introduction
One of the leading causes of death worldwide is cancer and amongst women, breast cancer has the highest mortality rate, making it a major contributor to global mortality statistics.1,2 There are several sub types of breast cancer out of which triple negative breast cancer (TNBC) is the most aggressive subtype. TNBC does not express any of the receptors for hormones like oestrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER 2) that are commonly expressed in other breast cancer sub types. This complexity makes hormone therapy ineffective and leaves chemotherapy as the only treatment option.3–6 Chemotherapy involves the use of mostly platinum-based complexes, of which cisplatin is the most widely used anti-cancer drug but its use is limited by the development of both inherent and acquired resistance and severe side effects.7–10 Therefore, there is a need for improved chemotherapeutic drugs.
Copper is an important micronutrient in humans and is involved in the smooth functioning of various biochemical reactions of the body11,12 making it potentially less toxic than platinum. In addition, it has been reported that copper is absorbed more by cancer tissues of the breast, prostate and brain compared to non-cancerous tissues.13 Therefore, copper-based complexes are expected to have the ability to serve as effective anti-cancer agents. There are several reports which support the anti-cancer potential of copper-based complexes.7,14–16 It is observed that copper complexes have the potential to interact with DNA through binding and cleavage mechanisms, thus affecting the proliferation of cancer cells.17–19 Additionally, it is reported that copper coordinated with Schiff base ligands can lead to cellular apoptosis in cancer cells.20–23 Furthermore, Cu complexes can also increase the intracellular ROS content. In fact, the overproduction of ROS adds to its cytotoxic potential towards cancer cells.18,24–26 Hence, copper complexes are gaining significant attention as they can combat the progression of cancers by targeting their growth, metastatic properties and angiogenesis.27 Interestingly, the cytotoxic potential of a ligand system alone is not as high as that of the metal coordinated complexes.28 This highlights the importance of metal coordination. The choice of ligand also affects the properties of the synthesized complex. Chelating ligands which have N,S/O donors are known to have biological relevance as they mimic bio-active molecule donor environment. Hence, many Cu(II) complexes with such donor groups are reported as having anti-tumour potential due to their ability to form strong interaction and nuclease activity on DNA, ROS production and apoptosis.22,29
In consideration of the above we have synthesized Cu(II) complex 1 with salicylidene carbohydrazide as the ligand and ortho-phenanthroline as the co-ligand. The structure of complex 1 has been characterized by single crystal XRD analysis, IR, UV-VIS and EPR spectroscopy. We have studied the effect of treatment of 1 on the cell viability of triple negative breast cancer cells MDA-MB-231 and MCF-7. We further studied the effect of 1 on ROS generation and the resultant effects on cell cycle progression, DNA cleavage and apoptosis. Western blot analysis was carried out to detect the proteins involved in apoptotic signalling. In addition, we have investigated the anti-tumour potential of complex 1 in a 3D breast cancer model using spheroids.
Experimental
Materials
CT-DNA was purchased from Sigma, plasmid pUC19 DNA was obtained from Thermo Fisher Scientific. Cisplatin was purchased from Sigma. MCF-7 and MDA-MB-231 cell lines were obtained from the National Centre for Cell Science (NCCS), Pune, India. Breast normal cell line MCF-10A was obtained from ATCC, CRL-1034. MDA-MB-231 cell line used in the TUNEL assay and PI uptake was also obtained from ATCC. TUNEL assay was carried out using DeadEnd™ Fluorometric TUNEL System Promega G3250. For propidium iodide (PI) uptake, PI was obtained from Sigma (P4170). All other chemicals were of reagent grade.
Synthesis of the Schiff base ligand 1,5-bis(salicylidene)carbohydrazide (H3L)
In the synthesis of the Schiff base ligand 1,5-bis(salicylidene)carbohydrazide (H3L), the modified method of Bustos et al.30,31 was followed. Briefly, salicylaldehyde (20 mmol) was added directly to a solution of carbohydrazide (10 mmol) dissolved in 50 mL of ethanol. The resulting reaction mixture was stirred at room temperature for 2 h. The mixture was then filtered through a G-3 crucible, and the pale white solid obtained was washed well with ethanol and air-dried. The compound was recrystallized from a methanol–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture at room temperature. The yield of the compound was 70%, and its melting point was 226 °C. The calculated percentages of carbon, hydrogen, and nitrogen for compound C15H14N4O3 were found to be 60.40%, 4.73%, and 18.78%, respectively. The actual percentages found experimentally were 60.91% for carbon, 4.69% for hydrogen, and 18.71% for nitrogen.
:1) mixture at room temperature. The yield of the compound was 70%, and its melting point was 226 °C. The calculated percentages of carbon, hydrogen, and nitrogen for compound C15H14N4O3 were found to be 60.40%, 4.73%, and 18.78%, respectively. The actual percentages found experimentally were 60.91% for carbon, 4.69% for hydrogen, and 18.71% for nitrogen.
Synthesis of the dinuclear Cu(II) complex [(o-phen)Cu(L)Cu(H2O)2](NO3) and its hydrate isomer [(o-phen)Cu(L)Cu(H2O)(NO3)]·H2O
A solution of 0.2 g (0.001 mol) of 1,10-phenanthroline monohydrate in 10 mL of methanol was slowly added to a solution of 0.199 g (0.001 mol) of Cu(OAc)2·H2O in 15 mL of methanol over the course of 30 minutes, with stirring below room temperature (15 °C). The resulting solution turned dark blue. The dark blue solution was then slowly added to a solution of 0.298 g (0.001 mol) of the Schiff base ligand 1,5-bis(salicylidene)carbohydrazide in 50 mL of methanol over the course of 20 minutes. The reaction mixture turned dark green. The mixture was stirred at room temperature for 6 h. Afterward, 0.2415 g (0.001 mol) of Cu(NO3)2·3H2O in 15 mL of methanol was added to the dark green reaction mixture. The dark green solid gradually disappeared as stirring continued for another 6 hours. The solution was then allowed to stand at room temperature overnight. The resulting solution was filtered through a G-4 crucible, and the green filtrate was collected in a 250 mL beaker. The solution was left at room temperature for slow evaporation. A dark green compound was obtained upon concentrating the solution. This compound was collected by filtration, washed with acetonitrile (CH3CN), and dried under vacuum. The compound was recrystallised from methanol at 4 °C. Yield, 60%. Elemental analysis: C27H23N7O8Cu2: Calculated (found): C 46.29 (46.47); H 3.31 (3.40); N 13.99 (14.20) %. ESI-MS in CH3OH: m/z 540.58 [(o-phen)Cu(HL) + H]+ corresponding to [(C12H8N2)Cu(C15H12N4O3) + H]+; m/z 711.60 [(o-phen)Cu2(HL)(H2O)2NO3 + 0.5H2O + H]+ corresponding to [C27H24N7O8Cu2 + 0.5H2O + H]+; and at m/z 739.62 [M + K]+ corresponding to [(o-phen)Cu2L(H2O)2NO3 + K]+ or [C27H23N7O8Cu2 + K]+.
A small fraction of the separated compound was dissolved in a mixture of methanol and water, with a ratio of 9 parts methanol to 1 part water. This solution was filtered through a G4 crucible and collected in a 20 mL test tube. Upon allowing this solution to gradually evaporate, small crystals were formed. These crystals were well-suited for X-ray crystallography and were utilized to determine the crystal structure of the compound.
X-ray crystallography
Single crystal X-ray diffraction data collection was performed on a Bruker D8 VENTURE diffractometer at room temperature (296 K) using Mo Kα radiation of 0.71073 Å wavelength. The data were integrated using Apex3-SAINT.32 A multi-scan absorption correction was applied to the data using the SADABS33 program. The structure was solved using SHELXT34 and refined by full-matrix least squares techniques using the SHELXL35 computer program incorporated into the WinGX36 program package. Hydrogen atoms on aromatic carbons and nitrogens were fixed at calculated positions and refined as the riding model with respect to its parent atom with Uiso(H) = 1.2Ueq(C). Whereas, the hydrogens on water oxygens (O4, O8 and O9) were located from difference electron density peaks and refined freely to have a target O–H distance of 0.85 Å. The hydrogen atom on water oxygen O16 and O17 could not be located due to poor quality data and hence its atom counts were added to final formula units. It should be noted that due to the poor quality of crystals, the higher angle reflections were not observed or were very weak. This contributed to the high values of Rint as well as R1. However, the data could be resolved to a satisfactory level and it is true to the chemical structure of the molecule. The structure presented here is for representation purposes, to show that the desired product has been formed. After several repeated failed attempts of preparation and recrystallization in different solvents, this was the only crystal specimen that could give satisfactory data to resolve a meaningful structure. Moreover, the data showed a twining effect on the crystal lattice. The twin law [−0.483 0 −0.517, 0 −1 0, −1.483 0 0483], which is a 180.0 degree rotation about the [−1 0 1] direct lattice direction was identified using the ROTAX tool incorporated in WinGX. Crystallographic parameters are listed in Table 1.
Table 1 Crystallographic parameters for 1a
|
Crystallographic data for the structural analysis has been deposited with the Cambridge Crystallographic Data Center, CCDC number is 2365897 for compound 1.†
|
| Identification code |
K16 |
| Empirical formula |
C54H48Cu4N14O17 |
| Formula weight |
1419.22 |
| Temperature |
296(2) K |
| Wavelength |
0.71073 Å |
| Crystal system |
Orthorhombic |
| Space group |
Pbcn
|
| Unit cell dimensions |
a = 19.498(4) Å; a = 90° |
|
b = 17.355(3) Å; b = 90° |
|
c = 33.032(6) Å; g = 90° |
| Volume |
11178(3) Å3 |
|
Z
|
8 |
| Density (calculated) |
1.687 Mg m−3 |
| Absorption coefficient |
1.589 mm−1 |
|
F(000) |
5776 |
| Crystal size |
0.150 × 0.150 × 0.100 mm3 |
| Theta range for data collection |
2.850 to 20.896° |
| Index ranges |
−19 ≤ h ≤ 19, −17 ≤ k ≤ 17, −33 ≤ l ≤ 33 |
| Reflections collected |
128317 |
| Independent reflections |
128317 [R(int) = ?] |
| Completeness to theta = 20.896° |
99.1% |
| Absorption correction |
Semi-empirical from equivalents |
| Max. and min. transmission |
0.7440 and 0.5799 |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
128317/1030/918 |
| Goodness-of-fit on F2 |
1.031 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.1184, wR2 = 0.1553 |
| R indices (all data) |
R
1 = 0.2722, wR2 = 0.2023 |
| Extinction coefficient |
n/a |
| Largest diff. peak and hole |
1.407 and −0.839 e Å−3 |
DNA binding
The DNA binding potential of synthesized metal complex 1 was analysed by using UV-Vis spectroscopy on a Jasco V-570 UV/VIS/NIR spectrophotometer with the help of calf thymus DNA (CT-DNA). The DNA stock was prepared by soaking CT-DNA in Tris–HCl buffer, pH 7.4 overnight. The purity of this DNA was tested by taking the ratio of absorbance readings A260/A280 = 1.84 which indicates that the DNA is free of protein. The concentration for CT-DNA was calculated by dividing the UV absorbance at 260 nm with the known molar extinction coefficient22ε260 = 6600 L mol−1 cm−1. The electronic absorbance titration experiment was carried out by adding an increasing amount of CT-DNA, at concentrations ranging from 0 to 40 μM to a fixed concentration of complex 1 [7.14 × 10−5 M]. Using the Wolfe–Shimer equation, the binding constant, Kb, was ultimately determined.
Viscosity measurement
In order to study the effect of complex 1 on the viscosity of CT-DNA, an Ostwald viscometer was used. The CT-DNA concentration was 100 μM and the concentration of complex 1 was varied from 0 to 100 μM. The flow time was recorded thrice with the help of a stopwatch. The viscosity was calculated using (η = (t − t0)/t0), where t0 is the viscosity of the buffer while t is the viscosity of the buffer with complex 1. Data were presented as a plot of (η/η0)1/3 against [complex]/[DNA], where η0 is the viscosity of CT-DNA and η is the viscosity of DNA with the complex.
Competitive binding
A solution of ethidium bromide [EB] = 2 μM and [CT-DNA] = 50 μM was allowed to incubate for 15 min to allow for the binding of EtBr to DNA and their fluorescence spectra were measured. Next an incremental amount of complex 1 (0–55 μM) was added to this solution and spectra were measured using a Jasco spectrofluorometer FP-8500. The incubation time of 2 min was kept constant each time to see the effect of competitive binding of the metal complex with EB. The excitation wavelength λex was 510 nm.
DNA cleavage
DNA cleavage potential of the Cu(II) complex was analysed by employing gel electrophoresis of complex 1-treated pUC19 plasmid DNA. For oxidative conditions a reaction mixture was prepared containing 200 ng of plasmid DNA (50 mM Tris HCl with NaCl), 1 mM H2O2, the indicated concentration of complex 1 and 50 mM Tris HCl, to make a final volume of 10 μL.
Additionally, free radical scavengers like DMSO (2 μL) and NaN3 (500 μM) were added to figure out the involvement of hydroxyl radicals and singlet oxygen respectively in oxidative cleavage. This reaction mixture was incubated for 2 hours at 37 °C. In the case of hydrolytic cleavage, the reaction mixture was prepared with the same concentration of plasmid DNA and the indicated concentration of complex 1 and the volume was adjusted to 10 μL each with the help of Tris HCl buffer. The incubation time for hydrolytic conditions was 3 h at 37 °C. Gel electrophoresis was carried out in 1% agarose gel for 1.5 h in 1× Tris acetic acid EDTA buffer, pH 8.4 at 60 V, and then UV light was used to visualize 0.5 μg mL−1 ethidium bromide (EB)-stained plasmid DNA.
Cell viability assay
The MTT assay was performed to establish the cytotoxic effect of the synthesized Cu(II) complex on breast cancer cell lines such as MDA-MB-231 and MCF-7 and on human mammary epithelial cell line MCF-10A. In the case of breast cancer cells, a cell suspension of 100 μL containing 1 × 104 cells was prepared and seeded in a 96 well plate. Upon overnight incubation, the cells were treated with the mentioned concentration of complex 1 for 24 h. While MCF-10A cells (ATCC, CRL-10317) were cultured in a humidified incubator with 5% CO2 at 37 °C in DMEM/F12 media supplemented with 5% horse serum, epidermal growth factor (20 ng ml−1), insulin (10 μg mL−1), hydrocortisone (0.5 mg ml−1), cholera toxin (100 ng mL−1), and penicillin/streptomycin. All cell viability experiments were conducted between 4–6 passages. Briefly, 3000 cells were seeded per well in 96 well plates (Nunc) and incubated at 37 °C either with DMSO or various concentrations of complex 1 for 24 h. After this the media was changed and the treated cells along with non-treated cells (control) were exposed to MTT for 4 hours in a humidified CO2 incubator. The formazan crystals formed after 4 hours were dissolved in 100 μL DMSO. The mitochondrial dehydrogenase activity in the cells that converts the yellow tetrazolium to purple formazan crystals reflects cell viability. It was measured with the help of an absorbance reading at 570 nm. IC50 values were calculated and a graph was plotted using graph pad prism. In the case of MCF-10A, absorbance was taken at 540 nm (FLUOstar Omega, BMG-Lifetech, Germany) and percent cell viability was calculated.
Colony forming assay
The effect of complex 1 on the cell colony formation ability was analysed by a colony assay as described.37 Briefly, 800 cells were seeded per well in a 6-well plate. The next day these cells were treated with the indicated concentration of complex 1 and left in a humidified CO2 incubator for 7 days without disturbing them. On the 7th day, the cells were fixed with 100% methanol for 20 min and then stained with 0.1% crystal violet dye for 10 min at RT. Finally, they were washed with water, dried and images were taken.
Hoechst imaging
For confocal imaging of the nuclear region, MDA-MB-231 cells were seeded at a density of 5 × 105 cells per dish in 35 mm confocal dishes. Once the cells were attached, the medium was changed and these cells were treated with the indicated concentration of 1 for 24 h. Following the incubation period, the cells were rinsed with 1× PBS, treated with 4% paraformaldehyde for fixation, and permeabilized with 0.1% Triton X-100. Subsequently, the cells were exposed to Hoechst-33342 (1 μg mL−1) in a dark environment for 30 minutes. After rinsing with 1× PBS once more, the confocal dishes were observed using a fluorescence microscope (OLYMPUS CKX53).
ROS analysis
About 5 × 105 cells per dish were seeded in confocal dishes to perform ROS analysis through imaging. TNBC cells were treated with 1 at an IC50 dose and 25 μM t-BHP (tert-butyl hydrogen peroxide), which was used as the positive control. The cells were incubated for 6 h in a CO2 incubator followed by the addition of 10 μM DCFDA to each dish for 30 min and incubation in the dark in the CO2 incubator. Next, a final washing step was performed by using 1× PBS and the cells were observed under a confocal microscope for imaging.
Apoptosis analysis
The experiment was conducted according to the manufacturer's instructions (Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit V13241, Thermo Fisher Scientific). The cells (5 × 105) were seeded in a 6-well plate and treated for 24 hours with the specified concentrations of compound 1. After incubation, the cells were harvested by centrifugation at 3000 rpm for 10 minutes. The collected cells were then resuspended in 100 μL of 1× binding buffer. To this cell suspension, 5 μL of Annexin V and 1 μL of PI (stock of 100 μg mL−1) were added. The cells were incubated at room temperature in the dark for 15 minutes, after which 400 μL of binding buffer was added. The samples were then kept on ice until analyzed using the BD FACS MELODY instrument.
Western blot
The seeding density was 1 × 106 cells per dish in a 90 mm culture plate. They were treated with 1 for 24 h. After this the cells were harvested through centrifugation and lysed with RIPA lysis buffer containing 1× protease inhibitor and centrifuged at 14000 rpm for 14 min. The supernatant protein concentration was analysed using the Bradford method and protein samples were prepared with 45 μg protein per sample. These samples were run in a polyacrylamide gel electrophoresis (PAGE) at 60 V in 1× running buffer. After this the protein was transferred to a PVDF membrane at 55 V at 4 °C for 2 h. The membranes were blocked with 5% BSA in TBST for 1 h and then incubated with the primary antibody overnight at 4 °C. Afterward, the membranes were washed with 1× TBST and incubated with the secondary antibody for 2 h at room temperature. Next, there was a washing step and then the images were developed on an X-ray film using a developer and fixer in a dark room. The loading control used was GAPDH.
Cell cycle arrest
Cells were initially seeded in a 6-well plate at a density of 5 × 105 cells per well. Subsequently, they were treated with varying doses (1, 2, and 3 μM) of compound 1 for a duration of 24 hours. After this incubation period, the cells were harvested by centrifuging them at 3000 rpm for 10 minutes. To obtain the cell pellet, 70% chilled ethanol was added and the mixture was centrifuged again. The resulting pellet was then re-suspended in 1× phosphate buffer and incubated with 10 μg of RNase at 37 °C for 0.5 h. Following this, the cells were stained with (PI) propidium iodide in the dark for 0.5 h at 4 °C. The BD FACS MELODY instrument was used to analyse the DNA content, allowing for the determination of the cell cycle distribution.
TUNEL assay
The TUNEL (TdT-mediated dUTP Nick-End Labeling) assay was performed as per the previously standardized protocol with slight modifications using DeadEnd™ Fluorometric TUNEL System Promega.38 Briefly after complex 1 treatment for 24 h, the MDA-MB-231 cells were washed with PBS, fixed with 4% formaldehyde and equilibrated for 5–10 min. The cells were labelled with fluorescein-12-dUTP using the rTdT reaction mix for 60 min at 37 °C. Then the sample was terminated using the stop solution and counterstained with the DAPI mounting media (Sigma). Slides were visualized under a confocal microscope with excitation and emission at 490 nm and 520 nm, respectively.
Propidium iodide uptake assay
The propidium iodide (PI) uptake experiment was carried out according to a previously standardized protocol with slight modification39 (R&D system). MDA-MB-231 cells were treated with compound 1 for 24 hours. The cells were collected with PBS and incubated with PI (2 mg mL−1) in the dark at 37 °C for 20 min. The stained cells were analysed by flow cytometer (BD FACS Lyric™).
3D studies: multicellular tumour spheroid formation
The spheroids were developed by the liquid overlay method as described.40 Briefly, a 96 well plate was coated with 80 μL of 1.6% Agarose solution in 1× PBS and left to dry under sterile conditions for 1 h. After which the dried wells were covered with 100 μl of 1× PBS. These plates can be stored in a humidified incubator overnight. Next, the cell suspension of MDA-MB-231 cells containing 5000 cells per 100 μL was prepared and transferred to each agarose coated well after removing 1× PBS. The plate was incubated for 1 h in a humidified CO2 incubator, followed by centrifugation of the plate at 3000 rpm for 10 min at 25 °C. After this the plate was incubated for 72 h such that spheroids can be developed.
Multicellular spheroid viability assay (MTT)
Cell viability of multicellular spheroids was assessed following treatment with Cu(II) complex 1 at three concentrations, which are 1, 2, and 3 μM in DMEM for a duration of 48 hours. The MTT assay was employed to evaluate cell viability using the method described in detail for 2D monolayer cultures. Briefly, the spheroids were grown on a 96 well plate which contains 1.6% agarose. 5000 cells per 100 μL were taken and transferred to each agarose coated well and placed in an incubator for 72 h. After this the media was changed and 3 concentrations of complex 1 treatment was given to spheroids in triplicates for 48 h. Furthermore, the spheroids were exposed to MTT for 4 hours in a humidified CO2 incubator. The formazan crystal formed after 4 hours were dissolved in 100 μL DMSO and absorbance reading was measured at 570 nm. The IC50 concentration was subsequently determined.
Multicellular spheroid live/dead fluorescence imaging
Multicellular spheroids grown in 96-well plates were treated with complex 1 at 3 concentrations (1–3 μM) for 72 h. The media was gently removed, followed by washing with 1× PBS. The spheroids were stained with 40 μM each of acridine orange and propidium iodide for a period of 20 min at a constant temperature of 37 °C in the dark. Finally, the fluorescence images were taken with the help of a fluorescence microscope (Olympus).
Results and discussion
Synthesis and characterization
The Schiff base 1,5-bis(salicylidene)carbohydrazide exists in its keto form (A) in the solid as reflected from its IR spectrum (vide infra), however, in solution the keto form (A) may coexist with its enol form (syn or anti) as shown in B or B′ (Scheme 1). Depending on the reaction condition it may coordinate with the metal ion either in its keto form or enol form. If the ligand coordinates in the enol form, then the keto form is converted to the enol form usually resulting in a complex containing only the enolised ligand. The enolisation of the ligand is found to be favoured in the presence of a base such as acetate anion. Thus the dark blue reaction product of Cu(OAc)2·H2O and 1,10-phenanthroline in CH3OH was used to carefully react with the Schiff base in a 1:1:1 ratio such that Cu(II) coordinates with the first site of the enolised ligand as shown in the anti form (B′) comprising enolate O−, phenolate O− and imine N. Enolisation of the Schiff base ligand followed by its deprotonation is facilitated in the presence of the acetate anions coming out during its reaction with the starting Cu(OAc)2-o-phen complex. The fourth and the fifth positions of the Cu(II) ion incorporated into the first site are occupied by the two N donor atoms of the o-phen coligand forming a five-coordinate neutral Cu(II) complex. Then this five-coordinate green compound was reacted in situ with Cu(NO3)2·3H2O to incorporate the metal ion in the available second site comprising N of NH, N of imine and the remaining phenolate O− of the coordinated ligand in the five-coordinate green complex, thus behaving as a monoanionic tridentate ligand for the second Cu(II) ion. The remaining positive charge of the metal ion is compensated by NO3− which may act as a counter anion or as a monodentate anionic ligand. In fact, in the majority of the complexes formed, two water molecules are found to be coordinated with the Cu(II) ion in the second site while NO3− is found to be acting as the counter anion, except a small portion of the product where one aqua ligand and NO3− are found to coordinate with the second Cu(II) ion while the other water molecule is found to be outside the coordination site thus forming a hydrate isomer. This fact is confirmed by X-ray crystallography (vide infra). However, when dissolved in methanol, the coordinated NO3− of the hydrate isomer is found to dissociate from the coordination site and acts as the counter anion, as confirmed by the conductivity measurement of the compound in methanol (Λ = 90.7 Ohm−1 cm2 mol−1) where it behaves as a 1:1 electrolyte,41 suggesting that in solution the compound behave as a uniform mono-cationic species where NO3− acts as the counter anion.
 |
| | Scheme 1 Schiff base ligand 1,5-bis(salicylidene)carbohydrazide in its keto- and enol-forms. | |
HR-MS spectrum
The HR-MS spectrum (in the positive-ion mode) for complex 1 has been recorded in CH3OH and the result is presented in Fig. S1 (ESI).† Complex 1 showed peaks at m/z 540.58 [(o-phen)Cu(HL) + H]+ corresponding to [(C12H8N2)Cu(C15H12N4O3) + H]+; m/z 711.60 [(o-phen)Cu2(HL)(H2O)2NO3 + 0.5H2O + H]+ corresponding to [C27H24N7O8Cu2 + 0.5H2O + H]+; and at m/z 739.62 [M + K]+ corresponding to [(o-phen)Cu2L(H2O)2NO3 + K]+ or [C27H23N7O8Cu2 + K]+.
IR spectra
The free Schiff base ligand 1,5-bis(salicylidene)carbohydrazide exhibits a medium intensity IR band at around 3220 cm−1 with a shoulder near 3325 cm−1 (Fig. S2A†) arising due to ν(N–H) and ν(O–H), respectively.30 The strong IR band at 1682 cm−1 arising due to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) suggests that the free ligand is in its keto form in the solid. The ν(CN) band appears as a strong band at 1620 cm−1 in the IR spectrum of the free Schiff base ligand (Fig. S2A†). The ν(CO) band at 1682 cm−1 and the ν(O–H) shoulder near 3325 cm−1 of the free ligand are found to be absent in the IR spectrum (Fig. S2B†) of complex 1 while the ν(CN) band is found to be shifted to 1595 cm−1 suggesting the coordination of imine nitrogen (CN) to the metal ion (Cu2+) in complex 1. The absence of the ν(CO) band in the complex strongly suggests that the ligand is coordinated with the metal ion in its enol form while the absence of the ν(O–H) in the IR spectrum of the complex suggests the deprotonation of the O–H groups and the coordination of the two phenolate O− in the complex. Thus, the Schiff base ligand behaves as a tri-anionic ligand overall possessing two Cu2+ ions, one in each site, thereby making it a positively charged species with the scope of accommodating one NO3− ion either as a coordinating ligand or as a counter anion (since Cu(NO3)2·3H2O was used in the synthesis of the complex). The appearance of a band at 1409 cm−1 in the IR spectrum of the complex indicates the presence of the NO3− moiety, this band is absent in the IR spectrum of the free Schiff base (Fig. S2C†). It is to be noted that the IR spectrum of cupric nitrate42,43 shows a very strong band centered around 1370 cm−1. The presence of the NO3− moiety is further supported by the X-ray crystal structure of the complex (vide infra).
O) suggests that the free ligand is in its keto form in the solid. The ν(CN) band appears as a strong band at 1620 cm−1 in the IR spectrum of the free Schiff base ligand (Fig. S2A†). The ν(CO) band at 1682 cm−1 and the ν(O–H) shoulder near 3325 cm−1 of the free ligand are found to be absent in the IR spectrum (Fig. S2B†) of complex 1 while the ν(CN) band is found to be shifted to 1595 cm−1 suggesting the coordination of imine nitrogen (CN) to the metal ion (Cu2+) in complex 1. The absence of the ν(CO) band in the complex strongly suggests that the ligand is coordinated with the metal ion in its enol form while the absence of the ν(O–H) in the IR spectrum of the complex suggests the deprotonation of the O–H groups and the coordination of the two phenolate O− in the complex. Thus, the Schiff base ligand behaves as a tri-anionic ligand overall possessing two Cu2+ ions, one in each site, thereby making it a positively charged species with the scope of accommodating one NO3− ion either as a coordinating ligand or as a counter anion (since Cu(NO3)2·3H2O was used in the synthesis of the complex). The appearance of a band at 1409 cm−1 in the IR spectrum of the complex indicates the presence of the NO3− moiety, this band is absent in the IR spectrum of the free Schiff base (Fig. S2C†). It is to be noted that the IR spectrum of cupric nitrate42,43 shows a very strong band centered around 1370 cm−1. The presence of the NO3− moiety is further supported by the X-ray crystal structure of the complex (vide infra).
Electronic spectra
The compound is soluble in methanol as well as in water producing a green solution in both solvents. Since complex 1 is readily soluble in water, water was used as a solvent to record the electronic spectra. The green coloured complex showed a broad band (Fig. S3† inset) centred around 643 nm (ε = 74 mol−1 cm−1). This may be attributed to the ligand field transitions. Additionally, three distinct peaks were observed at 380 nm, 270 nm and 205 nm due to charge transfer transitions. Also the electronic spectrum of complex 1 in DMEM was monitored with time for 48 h to check its stability (Fig. S4†). Overall, there was very little change in the electronic spectra of 1 with time suggesting its stability in DMEM. However, fresh solutions were prepared each time for experimental purposes.
EPR spectra
Compound 1 showed a strong X-band EPR signal (Fig. S5A†) in its powder state confirming that it is paramagnetic. The g values calculated from the powder spectra are g∥ = 2.196 and g⊥ = 2.089 at RT. The methanol solution of the compound also displayed a strong signal at RT which is neither a four-line spectrum (63/65Cu nucleus, I = 3/2) nor a seven-line spectrum (Fig. S5B†). However, the EPR spectrum of 1 in CH3OH at LNT (Fig. S5C†) showed the presence of two Cu(II) centres suggesting that they are very weakly interacting.
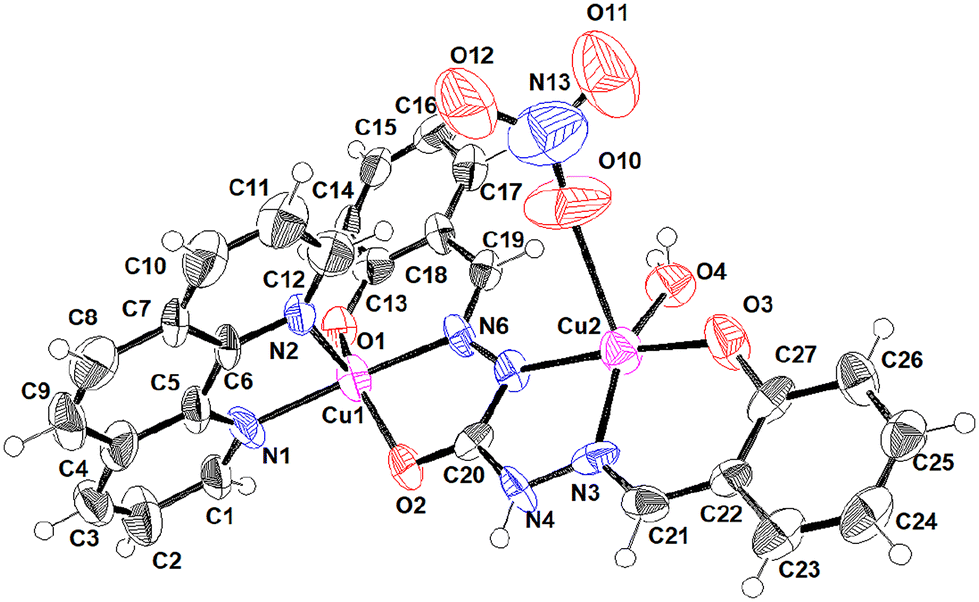
X-ray crystal structure
The molecule crystallized in the orthorhombic crystal system with space group Pbcn. The asymmetric unit of the crystal lattice consists of two different types of binuclear copper complexes, two nitrate anions and two water molecules. The moiety formula of the complexes in the asymmetric unit of the crystal lattice can be given as ‘1.25 [C27H23Cu2N6O5, (NO3)] and 0.75 [C27H21Cu2N7O7, (H2O)]’. The nitrate anion, N14, O13, O14 O15 is disordered in the ratio of 45:55. The nitrate anion, N13, O10, O11 O12, and the water molecule O17 are disordered with respect to each other in the ratio of 75:25 which resulted in different coordination to the central metal Cu2. The positions of disordered atoms were identified from difference electron density peaks. Distance restraints SADI and DFIX were used to achieve meaningful geometry for the disordered atoms. Anisotropic displacement parameters of atoms in the disordered groups were restrained or constrained using SIMU or EADP respectively to converge the anisotropic refinement.
The central copper atom shows a distorted square pyramidal geometry with a geometry index (tau(τ)) ranges of 0.19–0.5. The imine ligand connects the two copper metal centres through its unique hexadentate property. For Cu1 and Cu4, the penta coordination sphere is formed with the bidentate phenanthroline moiety and 3-coordination sites of the ligand. For, Cu2 and Cu3, the coordination is satisfied by the remaining 3-coordination sites of the ligand and water molecules. However, for Cu2, one of the coordination sites is shared between water and nitrate anion. The bond distances and angles of these atoms to central copper are listed in Table 2. In the crystal lattice, the molecules are held together by N–H⋯O and O–H⋯O hydrogen bond interactions. Table 3 gives a list of H-bond interactions in the lattice. In addition to this, the crystal lattice is stabilized by CH⋯O CH⋯π and π⋯π interactions mediated through the aromatic rings. Two different types of Cu complexes are depicted in Fig. 1 and 2.
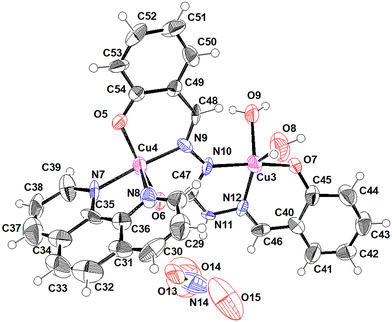 |
| | Fig. 1 ORTEP (50% probability ellipsoids) of the major component (62.5%) where Cu is coordinated with two H2O molecules in the second site. | |
 |
| | Fig. 2 ORTEP (50% probability ellipsoids) of the minor component (37.5%) where Cu is coordinated with NO3− and H2O molecules in the second site. | |
Table 2 Selected bond distances and bond angles for the copper complex
| Cu(1)–O(1) |
1.907(16) |
|
|
| Cu(1)–N(6) |
1.909(19) |
|
|
| Cu(1)–N(1) |
1.98(2) |
|
|
| Cu(1)–O(2) |
2.004(16) |
|
|
| Cu(1)–N(2) |
2.22(2) |
|
|
| Cu(2)–O(3) |
1.894(17) |
|
|
| Cu(2)–N(3) |
1.933(18) |
|
|
| Cu(2)–N(5) |
1.966(18) |
|
|
| Cu(2)–O(4) |
1.974(15) |
|
|
| Cu(2)–O(17′) or Cu(2)–O10 |
2.07(7) or 2.65(6) |
|
|
| Cu(3)–N(12) |
1.94(2) |
|
|
| Cu(3)–O(7) |
1.94(6) |
|
|
| Cu(3)–N(10) |
1.968(17) |
|
|
| Cu(3)–O(9) |
2.069(18) |
|
|
| Cu(3)–O(8) |
2.17(2) |
|
|
| Cu(4)–N(9) |
1.922(18) |
|
|
| Cu(4)–O(5) |
1.927(16) |
|
|
| Cu(4)–O(6) |
1.968(16) |
|
|
| Cu(4)–N(7) |
1.98(2) |
|
|
| Cu(4)–N(8) |
2.23(2) |
|
|
| |
| O(1)–Cu(1)–N(6) |
92.9(9) |
N(12)–Cu(3)–O(7) |
88.5(17) |
| O(1)–Cu(1)–N(1) |
96.5(8) |
N(12)–Cu(3)–N(10) |
81.2(9) |
| N(6)–Cu(1)–N(1) |
170.3(10) |
O(7)–Cu(3)–N(10) |
166(2) |
| O(1)–Cu(1)–O(2) |
149.7(6) |
N(12)–Cu(3)–O(9) |
136.8(8) |
| N(6)–Cu(1)–O(2) |
79.8(9) |
O(7)–Cu(3)–O(9) |
84.6(17) |
| N(1)–Cu(1)–O(2) |
93.0(8) |
N(10)–Cu(3)–O(9) |
96.1(8) |
| O(1)–Cu(1)–N(2) |
116.3(7) |
N(12)–Cu(3)–O(8) |
132.8(9) |
| N(6)–Cu(1)–N(2) |
96.0(8) |
O(7)–Cu(3)–O(8) |
97(2) |
| N(1)–Cu(1)–N(2) |
77.9(10) |
N(10)–Cu(3)–O(8) |
97.4(8) |
| O(2)–Cu(1)–N(2) |
93.8(7) |
O(9)–Cu(3)–O(8) |
90.4(9) |
| O(3)–Cu(2)–N(3) |
92.9(10) |
N(9)–Cu(4)–O(5) |
93.6(9) |
| O(3)–Cu(2)–N(5) |
172.5(8) |
N(9)–Cu(4)–O(6) |
80.9(8) |
| N(3)–Cu(2)–N(5) |
79.8(10) |
O(5)–Cu(4)–O(6) |
162.6(7) |
| O(3)–Cu(2)–O(4) |
86.3(7) |
N(9)–Cu(4)–N(7) |
174.2(10) |
| N(3)–Cu(2)–O(4) |
152.1(8) |
O(5)–Cu(4)–N(7) |
91.9(8) |
| N(5)–Cu(2)–O(4) |
100.9(8) |
O(6)–Cu(4)–N(7) |
93.3(8) |
| O(3)–Cu(2)–O(17′) |
83(3) |
N(9)–Cu(4)–N(8) |
101.8(9) |
| N(3)–Cu(2)–O(17′) |
130(4) |
O(5)–Cu(4)–N(8) |
103.2(7) |
| N(5)–Cu(2)–O(17′) |
100(3) |
O(6)–Cu(4)–N(8) |
94.0(7) |
| O(4)–Cu(2)–O(17′) |
77(4) |
N(7)–Cu(4)–N(8) |
78.5(11) |
Table 3 List of H-bonds
| Symmetry transformations used to generate equivalent atoms: #1 −x + 3/2, y − 1/2, z #2 −x + 3/2, y + 1/2, z #3 −x + 2, −y + 1, −z + 1. |
| N(4)–H(4)⋯O(17)#1 |
0.86 |
1.95 |
2.79(6) |
165.9 |
| N(4)–H(4)⋯O(12′)#1 |
0.86 |
2.08 |
2.92(15) |
166.9 |
| O(4)–H(4B)⋯O(10) |
0.96(10) |
2.4(2) |
3.10(4) |
128(18) |
| O(4)–H(4B)⋯O(17′) |
0.96(10) |
1.9(2) |
2.53(11) |
123(12) |
| O(8)–H(8A)⋯O(15)#2 |
0.92(8) |
2.02(14) |
2.73(9) |
133(14) |
| O(8)–H(8A)⋯O(15′)#2 |
0.92(8) |
2.60(12) |
3.50(10) |
170(22) |
| O(8)–H(8B)⋯O(11) |
0.87(8) |
2.19(15) |
2.85(4) |
133(15) |
| O(8)–H(8B)⋯O(17′) |
0.87(8) |
2.4(2) |
3.02(11) |
129(22) |
| O(9)–H(9A)⋯O(16) |
0.89(8) |
1.88(12) |
2.71(3) |
154(13) |
| O(9)–H(9B)⋯O(5)#3 |
0.92(9) |
1.81(10) |
2.69(2) |
158(21) |
| N(11)–H(11A)⋯N(14) |
0.83(9) |
2.67(15) |
3.36(6) |
143(17) |
| N(11)–H(11A)⋯O(13) |
0.83(9) |
2.10(12) |
2.87(4) |
153(18) |
| N(11)–H(11A)⋯O(13′) |
0.83(9) |
2.49(18) |
3.02(6) |
122(16) |
DNA binding studies using CT-DNA
Absorption spectroscopic studies.
Recording of the electronic absorption spectra is a universally applied technique to study DNA binding behaviour with complexes.44 The electronic absorption spectra of complex 1 was recorded in the absence of CT DNA and with incremental addition of CT-DNA (Fig. 3). It can be observed that there is a decrease in absorbance intensity of 1 upon incremental addition of CT-DNA [0–40 μM]. This phenomenon of hypochromism is associated with intercalative mode of DNA binding. This intercalation is a result of stacking of the planar aromatic group of the complex between DNA base pairs.45 Furthermore, the intrinsic binding constant (Kb) was calculated with the help of equation (eqn (1)).| |  | (1) |
where [DNA] is the concentration of CT-DNA used and εf, εa, and εb denote the free, apparent, and bound metal complex extinction coefficients, respectively. The slope/intercept value from the plots of [DNA]/(εa − εf) versus [DNA] gave the Kb value, which is 1.25 × 104 M−1. This value is 10-fold less than that for the classical DNA intercalator (EB). Hence, it can be inferred that the complex shows moderate binding with CT-DNA.46
 |
| | Fig. 3 (A) Hypochromic effect in the absorbance spectra of 1 (7.14 × 10−5 M) with increasing concentration of CT-DNA (0–40 μM) dissolved in Tris HCl buffer (pH 7.4), a plot of [DNA]/(εa − εf) versus [DNA] is shown in inset (R2 = 0.98031) for 8 points. (B) Plot of relative specific viscosity of CT DNA dissolved in 10 mM Tris–HCl buffer (pH 7.4) with increasing concentration of [1] = 0 to 100 μM at RT, [CT-DNA] = 100 μM. | |
Viscosity measurements.
To better understand the interaction between complex 1 and DNA, we conducted viscosity measurements. These measurements are sensitive to changes in the DNA length and are considered to be the most reliable in determining binding interactions. In the case of intercalative binding, the DNA helix needs to elongate as the base pairs separate to accommodate the binding substrate. However, groove binders behave differently, they have less pronounced or no effect on DNA viscocity.47 Thus, the viscosity experiment can give a better understanding of type or mode of the binding interaction. As observed from Fig. 3B, the addition of copper complex 1 to CT-DNA increases the relative viscosity, suggesting that the Cu(II) complex binds to CT-DNA through intercalation causing lengthening of the DNA double helix. These results are consistent with the electronic spectral data.
Competitive DNA binding by fluorescence studies
Competitive DNA binding experiment is a frequently used effective technique to study the interaction between small molecules and DNA, it utilizes the displacement of typical intercalator ethidium bromide (EB) from the DNA–EB adduct to provide information about binding affinity and binding mode of small molecules to DNA. Ethidium bromide is planar and non-emissive, but gives abundant fluorescence upon binding to DNA. When complex 1 was introduced into a mixture of EB and CT-DNA the fluorescence intensity significantly reduced (Fig. 4). This indicates that the Cu(II) complex is able to displace EB from the DNA–EB adduct thus it competitively binds to CT-DNA, reducing the fluorescence intensity.22,48 The fluorescence quenching data are in good agreement with the linear Stern Volmer equation (eqn (2)).| |  | (2) |
 |
| | Fig. 4 Competitive DNA binding in Tris–HCl buffer, pH 7.4 with incremental addition of 1 (0–55 μM) to an EB-CT-DNA adduct. Inset shows plot of F0/F vs. [Complex]. R2 for linear plot is 0.99295 for 12 points. | |
In the equation mentioned above, F0 represents the fluorescence intensity of the DNA–EB adduct when no quencher is present, while F represents the fluorescence intensity when complex 1 acts as the quencher. The concentration of complex 1 is denoted by [Q], and the Stern–Volmer quenching constant (Ksv) and the apparent binding constant (Kapp) are also included in eqn (2) and (3), respectively. The value of Ksv for complex 1 was determined from the slope of the F0/F versus [Q] plot, which was found to be 6.3 × 104 M−1 (R2 = 0.99295 for 12 points). This indicates a strong affinity between complex 1 and CT-DNA. The Kapp value for complex 1 was estimated using eqn (3), where [complex]50 represents the concentration of complex 1 required for 50% reduction in fluorescence intensity. With KEB = 1.0 × 107 M−1, the calculated Kapp value for complex 1 was 1.41 × 106 M−1, which is similar to that of the classical intercalator, ethidium bromide.49,50
| | | KEB × [EB] = Kapp × [complex]50 | (3) |
DNA cleavage
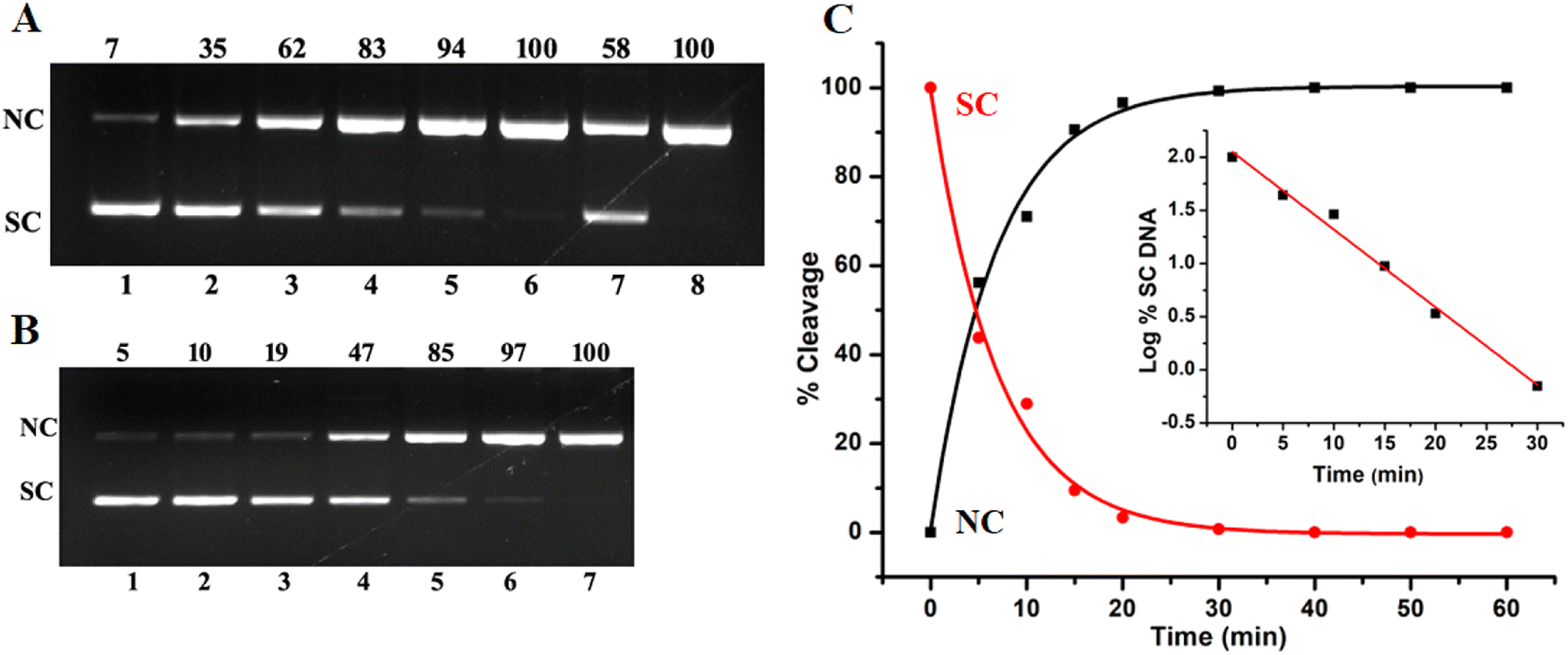
To investigate the cleavage potential of complex 1 on pUC19 plasmid DNA, gel electrophoresis was employed. Plasmid DNA like pUC19, typically adopts a compact supercoiled conformation called form I. When an ss or single-strand scission occurs in supercoiled DNA, it transforms into a nicked circular form called form II, while a ds scission or double-strand scission results in the linear form called form III. In gel electrophoresis, form I usually migrates quickly toward the anode followed by form III and form II is the slowest.47,51–53 DNA cleavage follows two primary pathways: an oxidative pathway, which leads to oxidative damage to the DNA sugar or nitrogen base, and a hydrolytic pathway, which causes hydrolysis of phosphodiester linkages.22,54,55Fig. 5 demonstrates the pUC19 DNA cleavage induced by complex 1 under both oxidative (Fig. 5A) and hydrolytic (Fig. 5B) conditions. As seen in Fig. 5, at just 2.5 μM concentration there is complete conversion to the nicked form under oxidative conditions. A clear suppression of DNA cleavage can be observed in the presence of DMSO, which is a scavenger of hydroxyl radicals. Hence, it can be inferred that hydroxyl radicals are primarily responsible for oxidative cleavage of DNA.
 |
| | Fig. 5 Gel electrophoresis of 200 ng pUC19 DNA by complex 1 at 37 °C in 50 mM Tris HCl having pH 8 showing oxidative cleavage (A) Lane 1, control: pUC19 DNA; lane 2–6 [1] = 0.5, 1, 1.5, 2, 2.5 μM + 1 mM of H2O2; Lane 7, 2.5 μM 1 + 2 μL DMSO; Lane 8, 2.5 μM 1 + 500 μM NaN3. (B) Hydrolytic cleavage Lane 1, pUC19 DNA control; Lane 2–7 [1] = 2.5, 5, 10, 20, 30, and 40 μM. (C) Time course measurement of pUC19 DNA cleavage activity of 1 with the disappearance of SC DNA in red and formation of NC DNA in black with increasing incubation time; the inset shows the plot of log(% SC DNA) vs time. | |
Hydrolytic cleavage involves the breaking of phosphodiester bonds, which are extremely stable bonds (k = 3.6 × 10−8 h−1), it requires enormous enhancement of the hydrolysis rate which is a major challenge. As seen from Fig. 5B under hydrolytic conditions at 40 μM concentration complex 1 has efficiently cleaved 100% of plasmid DNA. This nuclease activity occurred without the presence of any external agent or light. Since this cleavage involves the breaking of the phosphodiester bond which is a very stable bond the cleavage effectiveness of the complex is quite evident.51,56 Furthermore, the kinetics of hydrolytic DNA cleavage was studied. The cleavage of plasmid DNA by complex 1 was analysed by measuring the conversion of SC DNA (form I) to NC DNA (form II) with respect to time. As seen from Fig. 5C, there is a decrease in SC DNA with time and fits well into a single exponential decay curve (Fig. 5C), the plot of log(% SC) against time showed a linear fit (Fig. 5C, inset). The hydrolytic rate constant (kobs) was determined from the slope of the linear fit, it was found to be 10.10 h−1. The obtained high hydrolysis rate constant for 1 indicates that it shows very high nuclease activity and is capable of 2.8 × 108 fold enhancement of the hydrolysis rate compared to unhydrolyzed ds DNA.22,57–59 The results indicate that complex 1 is highly effective in bringing out DNA cleavage oxidatively as well as hydrolytically.
Cell viability assay
As complex 1 under study is highly effective in cleaving DNA under physiological conditions in the absence of any external agent it may have the potential to serve as an anticancer agent. The in vitro cytotoxicity of Cu(II) complex 1 was analysed on breast cancer cell lines such as MDA-MB-231 and MCF-7 using the MTT assay (Fig. 6). Cisplatin is one of the most widely used drugs for solid tumours so it was tested under similar conditions for reference (Fig. 7). Fig. 6 shows that complex 1 is able to significantly reduce the viability of both breast cancer cell lines. At just 1 μM concentration of 1, the cell viability dropped to 60% for MDA-MB-231 and MCF-7. The effect of compound 1 was also tested on human mammary epithelial cell (MCF-10A). The cell viability assay results at doses of 1,10, 25 and 50 μM in MCF-10A cells showed no significant toxicity (p > 0.001) as seen in Fig. S6,† thereby indicating that the compound has a robust safety profile. Notably, the cell viability for MCF-10A at even 50 μM dose of complex 1 was 98% emphasizing its cytotoxic selectivity towards cancer cells. The IC50 values calculated were 1.86 ± 0.17 μM for MDA-MB-231 and 2.22 ± 0.08 μM for MCF-7 (Table 4). Hence, both the IC50 values in breast cancer cells fall in the low micromolar range of concentration (1–5 μM) after 24 h of incubation. It is reported that such complexes can be considered as remarkable cytotoxic agents.14 It is important to highlight that complex 1 has outperformed cisplatin for both the tested breast cancer cell lines as seen in Fig. 7. Lucia M. Balsa et al.1 have reported similar IC50 values (4.0 ± 0.2 μM and 5.2 ± 1.8 μM) of Cu(trp)2 in MDA-MB-231 and MCF-7 cells. Along with this there are several other Cu(II) complexes reported for sub-micromolar or nanomolar cytotoxicity on different tumour cell lines.14 The results establish that Cu(II) complexes are promising candidates for their anti-cancer activity and the complex 1 has significant cytotoxic potential towards breast cancer cells.
 |
| | Fig. 6 The effect of the Cu(II) complex 1 on cell viability of (A) MCF-7 and (B) MDA-MB-231 cell lines. Statistical analysis was performed using GraphPad Prism, and **** indicates a significant difference with p < 0.0001. | |
 |
| | Fig. 7 The viability of (A) MCF-7 and (B) MDA-MB-231 cells was assessed in response to treatment with cisplatin. | |
Table 4 IC50 values calculated for MDA-MB-231 and MCF-7 cells against complex 1
| Cell line |
IC50 for 1 in (μM) |
| MDA-MB-231 |
1.86 ± 0.17 |
| MCF-7 |
2.22 ± 0.08 |
Colony forming assay
The anti-cancer activity of 1 was further analysed with the help of the colony formation assay. Fig. 8 illustrates the impact of the increase in the concentration of 1 on colony formation. It can be observed there is a marked reduction in the number of colonies formed with increasing concentration as compared to the control. The colonies in the control are dense and overpopulated which have decreased drastically upon treatment with 1. At 2 μM dose the colony population has become negligible. The results are in line with MTT data and the cytotoxic potential of 1 is evident.60
 |
| | Fig. 8 MDA-MB-231 cells were treated with indicated concentrations (0, 1 and 2 μM) of complex 1 fixed and stained with crystal violet. | |
Hoechst imaging
Hoechst is a bright blue coloured fluorescent dye that stains the nuclear region. The nucleus of a healthy cell is usually spherical and stains evenly, while for cells undergoing apoptosis the nucleus shrinks, becomes fragmented and condenses.61 This chromatin condensation leads to the formation of bright blue patches when stained with Hoechst.62 In order to see the effects of complex 1 on DNA, MDA-MB-231 cells treated with 1 were stained with Hoechst. Fig. 9 clearly shows all characteristic features of apoptosis like chromatin condensation, nuclear shrinkage and disintegration/fragmentation in all complex 1 treated nuclei in a dose dependent manner. There is a reduction in the number of cells and an increase in the appearance of the late apoptotic feature of the dot like chromatin material at a 3 μM dose as compared to control cells where the nucleus is spherical and evenly stained. All of these signs suggest that complex 1 is causing apoptosis in MDA-MB-231 cells.
 |
| | Fig. 9 Morphological changes in the nuclear region (indicated by arrows) of MDA-MB-231 cells upon treatment with 1 (1, 2 and 3 μM), visualized using Hoechst-33342. Scale bar: 50 μm. | |
Reactive oxygen species (ROS) analysis
The generation of ROS plays a vital role in the activation of mitochondrial-initiated events resulting in apoptosis. Some amount of ROS is produced in our bodies, which is essential for normal biological functions but when it exceeds a threshold level it can cause oxidative stress leading to apoptosis.18 Copper based complexes are redox active and lead to the production of reactive oxygen species which can cause DNA damage.63 In order to measure the intracellular ROS production due to complex 1, DCF-DA was used. In the presence of intracellular ROS this DCF-DA, which is non–fluorescent, gets oxidized to fluorescent DCF. In this experiment a positive control, t-BHP (tert-butyl hydrogen peroxide) was used as it can generate ROS and is more stable. In Fig. 10, we see very less or negligible green fluorescence in the control while there is a bright and large amount of fluorescence visible in complex 1-treated cells. It is also comparable to the positive control treated cells. These results indicate that complex 1 has induced a significant amount of ROS that probably causes oxidative stress in the cells, leading to DNA damage as evidenced by pUC 19 plasmid DNA cleavage experiments.
 |
| | Fig. 10 Intracellular ROS was measured by DCFDA staining of MDA-MB-231 cells after treatment with complex 1 (0, 2 μM) and 25 μM tert-butyl hydrogen peroxide. | |
Apoptosis analysis
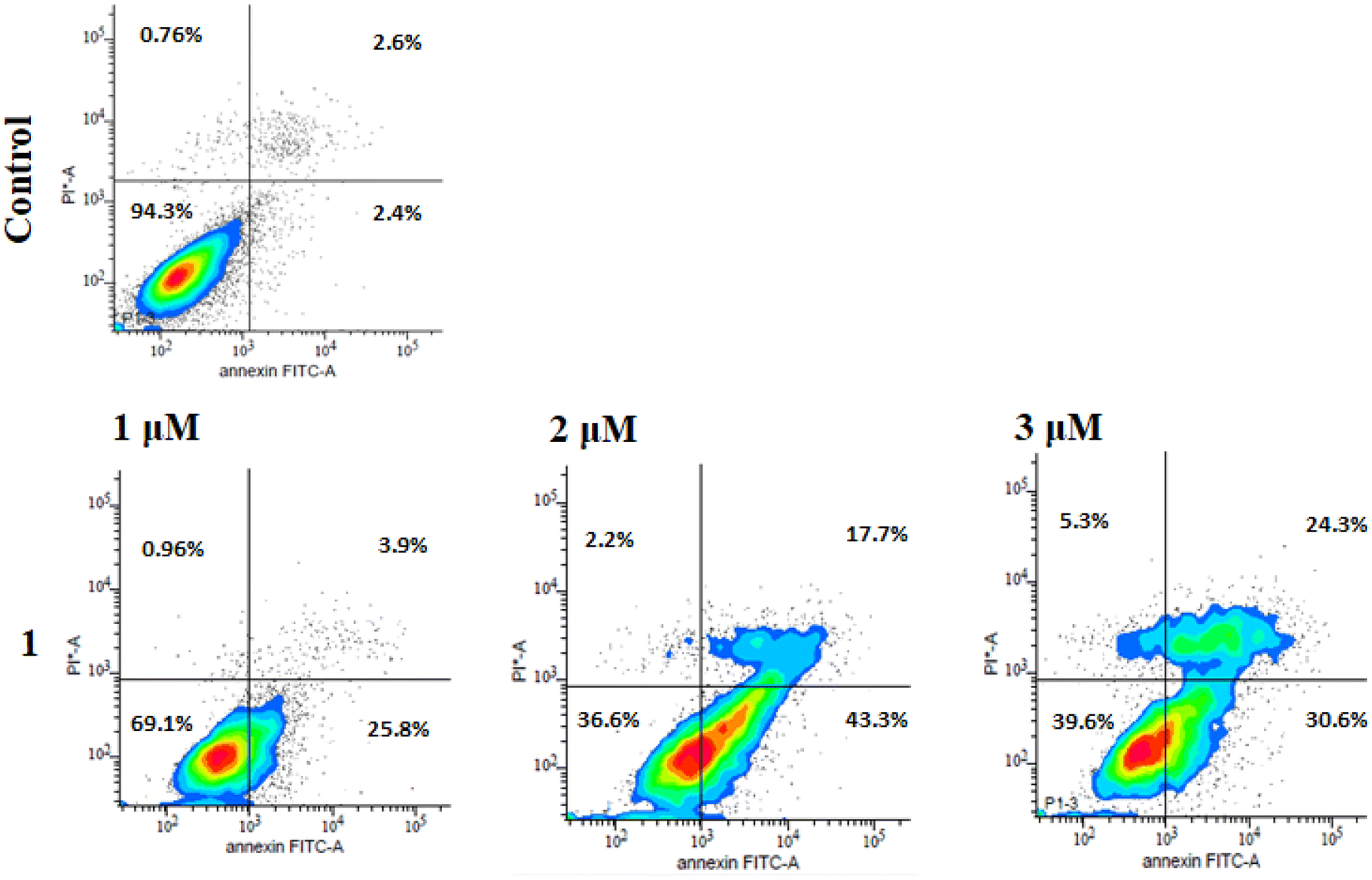
Annexin V-FITC/PI dual staining kits are specifically designed to identify and detect cell apoptosis. During early apoptosis, phosphatidyl serine (PS) translocates to the outer leaflet of the cell membrane. As Annexin V is a phospholipid-binding protein having high affinity for PS, it is able to bind to this exposed phosphatidyl serine and leads to the detection of early apoptotic cells. Propidium iodide helps to detect the later stage of apoptosis as it enters cells when the cell membrane integrity is compromised.64 In order to confirm apoptosis as the mode of cell death, the Annexin V-FITC/PI kit was used and samples were analysed through FACS. Fig. 11 shows an increase in both early and late apoptosis cells in the presence of complex 1. The number of early apoptotic cells is the highest for the IC50 dose which could be due to severe DNA damage and cell cycle arrest at this concentration. Furthermore, there is a sharp increase in the percentage of late apoptotic cells from 3.9% to 24% in a dose dependent manner.
 |
| | Fig. 11 Flow cytometry analysis of MDA-MB-231 breast cancer cells treated with indicated concentrations of 1 (1, 2 and 3 μM), for 24 hours. Viable cells are represented in the lower left quadrant (Annexin V−/PI−), early apoptotic cells in the lower right quadrant (Annexin V+/PI−), and late apoptotic cells in the upper quadrant (Annexin V+/PI+). | |
Western blot
It has been successfully established that complex 1 leads to elevated ROS production, DNA damage and apoptosis. In order to understand the underlying molecular mechanism of action, western blot analysis was performed using various antibodies as indicated in Fig. S7.† It is known from the literature that at the sites of DNA double-strand breaks (DSB) H2AX is phosphorylated to γ-H2AX therefore this phosphorylated histone is considered to be a biomarker for DSB.65 Also in response to DNA damage p53 accumulates in the cell nucleus and it regulates cell cycle arrest. If the DNA damage is severe and beyond repair, it promotes apoptosis of such cells.66 Fig. S7† shows an elevated level of both phosphorylated H2AX (γ-H2AX) as well as p53 at the IC50 dose, clearly indicating DNA damage, this damage may further lead to apoptosis. It is seen from Fig. S7† that there is increasing expression of Cytochrome c, which is known to lead to caspase activation.67 Also there is an increase in the expression of both caspase 3 and cleaved caspase 9 clearly demonstrating the activation of intrinsic apoptosis promoted by severe DNA damage. Furthermore, another characteristic feature of apoptosis is proteolytic cleavage of PARP by caspase 3. PARP is involved in DNA repair and stability, upon cleavage, it cannot recruit repair enzymes to the DNA damage site, thus preventing DNA repair and promoting apoptosis.68 We observe a clear increase in the expression of cleaved PARP with increasing concentration of 1 prominent at 2 μM dose, further confirming that complex 1 has the potential to cause critical DNA damage where cancer cells have to undergo programmed cell death through the intrinsic pathway.
Cell cycle arrest
It has been reported that cancer cells have a higher ROS level compared to normal cells which is further increased due to therapies. This oxidative stress causes DNA damage and triggers a DNA damage response in cancer cells. This response leads to cell cycle arrest and ultimately apoptosis for most cancer cells.69 The cell cycle consists of G1, S, G2 and M phases. Tumour proliferation can be blocked by arrest in different phases of the cell cycle. As evidenced from DCF-DA and Hoechst staining, it is clear that 1 is able to cause oxidative stress and lead to DNA damage. We further evaluated the effect of 1 on cell cycle progression and the phases involved (Fig. 12). It can be observed from Fig. 12 that there is a definite increase in the cell population in both S and G2/M phases of the cell cycle. However, in the case of 1 μM dose of complex 1, there is a reduction in the cell population in S and G2/M phases with an increase in the cell population in G0/G1 suggesting a possible temporary arrest at the G0/G1 phase at 1 μM dose, such that the cell cycle arrest in both S and G2/M phases is limited to just 2 μM (near IC50) and 3 μM dose. The arrest in the S phase is more prominent for the near IC50 dose than G2/M. This could be because the S phase is involved in DNA replication and at the IC50 dose there is more ROS production and DNA damage. Also, as seen in the western blot results, there is more p53 and γ-H2AX expression at this concentration. H2AX is phosphorylated to γ-H2AX upon double stranded DNA damage and p53 responds to DNA breaks by arresting the cell cycle,65,66 suggesting that all the results are consistent with each other.
 |
| | Fig. 12 Cell cycle analysis using FACS indicating the cell population in percentage at different phases of the cell cycle upon treatment with complex 1 (1, 2 and 3 μM) at the indicated concentrations. | |
TUNEL assay
The TUNEL assay is used to detect DNA fragmentation and in situ apoptosis. Apoptotic DNA fragmentation is a characteristic of programmed cell death, or apoptosis.61 So TUNEL assay was performed to further confirm single cell DNA damage by complex 1. The DeadEnd™ Fluorometric TUNEL System used for the experiment can measure these fragmented DNA strands by labelling them with fluorescein-12-dUTP(a) at 3′-OH DNA ends of the fragmented DNA using an enzyme called Terminal Deoxynucleotidyl Transferase, Recombinant, enzyme (rTdT). The fluorescence labelled DNA fragments can be visualized with the help of fluorescence microscopy.
MDA-MB-231 cells treated with complex 1, labelled with fluorescein-12-dUTP and counterstained with DAPI are shown in Fig. 13. It can be observed that the green fluorescent TUNEL positive cells increase with an increase in the complex 1 concentration pointing to an increase in DNA fragmentation due to complex 1 activity. Furthermore, it can be inferred that the extensive DNA fragmentations as seen in Fig. 13 is a characteristic feature of the late apoptotic stage which leads to an increased number of 3′-hydroxyl (3′-OH) ends. The TUNEL assay involves incorporating fluorescein-12-dUTP at these ends such that the fluorescence from labelled DUTP's is directly related to fragmented DNA. Based on these observations and results, it can be concluded that complex 1 induces extensive DNA fragmentation in MDA-MB-231 treated cells. Moreover, the extent of DNA fragmentation increases in a dose-dependent manner, indicating that higher concentrations of complex 1 lead to more significant DNA damage which leads to apoptosis of cells.
 |
| | Fig. 13 (A) The effect of 1 on DNA damage using TUNEL assay, the representative image of MDA-MB-231 cells after treatments with 1 (0.5, 1 and 2 μM), with TUNEL-DAPI co-staining. (B) Bar graph showing the percentage of TUNEL positive cells with respect to the concentration of 1. Data are shown as the mean ± SD. (*) p < 0.05 and (***) p < 0.001. | |
Propidium iodide uptake assay
Propidium iodide (PI) is a DNA intercalating dye that can only enter cells with damaged cell membranes. This cell membrane damage is a result of apoptosis and thus PI helps to detect later stages of apoptosis.64 The PI uptake flow cytometry analysis was performed to analyse cell viability, membrane integrity, changes in DNA content and apoptosis in complex 1 treated cells. After compound 1 treatment, PI staining indicates that MDA-MB-231 cells lose membrane integrity with genomic instability. Fig. 14 shows that there is an overall increasing trend in the percentage of PI positive cells with respect to the control.
 |
| | Fig. 14 (A) The flow cytometry images of propidium iodide (PI) uptake by MDA-MB-231 cells treated with complex 1 (0.5, 1 and 2 μM). (B) Bar graph showing percentage of PI positive cells with respect to different concentrations of 1. Data are shown as the mean ± SD. (****) p <0.0001. | |
Multicellular spheroid viability study using the MTT assay
3D tumours or spheroids can more closely resemble the physiological tissue like morphology of cancer tumour. It has the cell to cell contact that is a more accurate representation of cancer tumour with respect to nutrient and oxygen absorption as well as drug penetration,16 thus multicellular spheroids can serve as models to mimic the tumour microenvironment (TME).40
To verify the anti-cancer potential of complex 1, MTT assay on multicellular spheroid treated with 1 was performed (Fig. 15) and the IC50 value for the treated spheroid was 1.51 ± 0.29 μM which is close to the IC50 value in the monolayer culture of 1.86 ± 0.17 μM. This establishes the potential of synthesized complex 1 as a potent anti-cancer agent.28
 |
| | Fig. 15 Cell viability of 3D multicellular spheroid of MDA-MB-231 against 1 (1, 2 and 3 μM). Statistical analyses were done using GraphPad prism. **** indicates p < 0.0001. | |
Multicellular spheroid live/dead fluorescence imaging
Since, there is a decrease in tumour cell viability due to treatment with complex 1, the morphological changes caused were examined with the help of acridine orange and propidium iodide dual staining. AO and PI both bind to nucleic acids and help visualize them. AO is able to infiltrate live cells and fluoresces green while PI can only enter cells with damaged cell membranes which is a result of apoptosis, and fluoresces orange-red.46
Fig. 16 shows the increase in PI-stained cells in a dose dependent manner. There is evident loss of cell density in complex 1 treated spheroid with respect to the control. We observed that at a 3 μM dose there is ample red fluorescence in the core of the spheroid, which implies that complex 1 was able to penetrate deep inside the spheroid and cause cell death.16 Furthermore, the control spheroid shows signs of cell migration which is reduced in the case of treated spheroid, hence complex 1 may lead to the loss of migration and invasion in cancer tumour.
 |
| | Fig. 16 Fluorescent images of acridine orange/propidium iodide dual staining in multicellular spheroid treated with 1 (1, 2 and 3 μM). | |
Conclusions
In this work, a novel water-soluble Cu(II) Schiff base complex (1) was synthesized and characterized using X-ray crystallography, IR and UV-VIS spectroscopy. The complex was able to bind to CT-DNA by an intercalative mode and cause efficient DNA cleavage by both oxidative and hydrolytic pathways. Furthermore, antitumor properties of the synthesized complex were analysed to establish its therapeutic potential. It was found that this complex is able to inhibit triple negative breast cancer, MDA-MB-231and MCF-7 monolayer cell lines, as well as 3D tumour proliferation. This was studied through IC50 calculation, colony forming assay and fluorescence imaging of spheroids. Furthermore, the safety profile of complex 1 was also evaluated by the MTT assay on the human mammary epithelial cell line (MCF-10A) and it was found that the complex was significantly non-toxic to this cell line. The IC50 value for breast cancer cells falls in the low molar range (1.86 ± 0.17 μM for MDA-MB-231, 2.22 ± 0.08 μM for MCF-7 in 2D monolayer and 1.51 ± 0.29 μM in 3D spheroids). The mode of cell death was inferred to be apoptosis through Hoechst imaging and Annexin V/PI dual staining assay. Additionally, the complex led to an increase in ROS production which is possibly responsible for DNA damage as evident from DNA cleavage results as well as TUNEL assay which showed increase in DNA fragmentation and subsequent increase in PI uptake with respect to control. The cancer cells were arrested in the S and G2/M phases of the cell cycle, as observed in cell cycle analysis, however, when DNA damage was severe and could not be repaired, cells went through apoptosis. Western blot results indicate that complex 1 caused apoptosis by caspase activation via the intrinsic mitochondrial pathway. Taken together our results indicate that the synthesized complex 1 has significant anti-cancer potential.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank SAIF, IIT-Madras for XRD and EPR spectra, and the National Institute of Technology (NIT)-Rourkela for high resolution mass spectrum of the compound. The authors also thank Dr Babu Varghese, SAIF, IIT-Madras for help in solving the structure of the compound and Prof. Rupam Dinda, Department of Chemistry, NIT-Rourkela for help with HRMS results. Facilities provided by the Department of Biological Sciences, BITS Pilani K. K. Birla Goa Campus and the CSIF of our Institute are gratefully acknowledged.
References
- L. M. Balsa, M. C. Ruiz, L. Santa Maria de la Parra, E. J. Baran and I. E. León, J. Inorg. Biochem., 2020, 204, 110975 CrossRef PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, CA Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- S. Karpagam, A. Mamindla, V. K. Sali, R. S. Niranjana, V. S. Periasamy, A. A. Alshatwi, M. A. Akbarsha and V. Rajendiran, Inorg. Chim. Acta, 2022, 531, 120729 CrossRef.
- W. D. Foulkes, I. E. Smith and J. S. Reis-Filho, N. Engl. J. Med., 2010, 363, 1938–1948 CrossRef PubMed.
- S. J. Isakoff, Cancer J., 2010, 16, 53–61 CrossRef PubMed.
- C. Omarini, G. Guaitoli, S. Pipitone, L. Moscetti, L. Cortesi, S. Cascinu and F. Piacentini, Cancer Manage. Res., 2018, 10, 91–103 CrossRef PubMed.
- P. L. Zhang, X. X. Hou, M. R. Liu, F. P. Huang and X. Y. Qin, Dalton Trans., 2020, 49, 6043–6055 RSC.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197 CrossRef PubMed.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef PubMed.
- J. Gu, C. Guo, J. Ruan, K. Li, Y. Zhou, X. Gong and H. Shi, Apoptosis, 2024, 1–19 Search PubMed.
- J. K. Kim, C. Lee, S. W. Lim, A. Adhikari, J. T. Andring and R. McKenna, Nat. Commun., 2020, 11, 4557 CrossRef PubMed.
- Q. Mo, J. Deng, Y. Liu, G. Huang, Z. Li, P. Yu, Y. Gou and F. Yang, Eur. J. Med. Chem., 2018, 156, 368–380 CrossRef PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef PubMed.
- S. Tardito, A. Barilli, I. Bassanetti, M. Tegoni, O. Bussolati, R. Franchi-Gazzola and C. Mucchino, J. Med. Chem., 2012, 55, 10448–10459 CrossRef PubMed.
- S. M. de la Parra, A. I. Romo, J. Rodríguez-López, O. R. Nascimento, G. A. Echeverría, O. E. Piro and I. E. León, Inorg. Chem., 2024, 63, 4925–4938 CrossRef PubMed.
- D. S. Sigman, D. R. Graham and V. D'Aurora, J. Biol. Chem., 1979, 254, 12269–12272 CrossRef PubMed.
- Y. P. Zhang, Q. He, X. H. Zhou, G. H. Liu and A. Q. Yue, J. Mol. Struct., 2023, 1292, 136090 CrossRef.
- D. A. da Silva, A. De Luca, R. Squitti, M. Rongioletti, L. Rossi, C. M. L. Machado and G. Cerchiaro, J. Inorg. Biochem., 2022, 226, 111634 CrossRef PubMed.
- Y. Xia, X. Liu, L. Zhang, J. Zhang, C. Li and N. Zhang, Cancer Cell Int., 2019, 19, 1–11 CrossRef PubMed.
- S. M. Zhang, H. Y. Zhang, Q. P. Qin, J. W. Fei and S. H. Zhang, J. Inorg. Biochem., 2019, 193, 52–59 CrossRef PubMed.
- K. Paliwal, P. Haldar, P. K. S. Antharjanam and M. Kumar, ACS Omega, 2022, 7, 21961–21977 CrossRef PubMed.
- X. Qiao, Z. Y. Ma, C. Z. Xie, F. Xue, Y. W. Zhang, J. Y. Xu and S. P. Yan, J. Inorg. Biochem., 2011, 105, 728–737 CrossRef PubMed.
- Y. Gou, G. J. Huang, J. Li, F. Yang and H. Liang, Coord. Chem. Rev., 2021, 441, 213975 CrossRef.
- X. C. Shi, H. B. Fang, Y. Guo, H. Yuan, Z. J. Guo and X. Y. Wang, J. Inorg. Biochem., 2019, 190, 38–44 CrossRef PubMed.
- S. Gu, P. Yu, J. Hu, Y. Liu, Z. Li, Y. Qian, Y. Wang, Y. Gou and F. Yang, Eur. J. Med. Chem., 2019, 164, 654–664 CrossRef PubMed.
- D. Denoyer, S. A. S. Clatworthy and M. A. Cater, Met. Ions Life Sci., 2018, 18, 469–506 Search PubMed.
- L. M. Balsa, V. Ferraresi-Curotto, M. J. Lavecchia, G. A. Echeverría, O. E. Piro and J. García-Tojal, Dalton Trans., 2021, 50, 9812–9826 RSC.
- M. Hazra, T. Dolai, A. Pandey, S. K. Dey and A. Patra, Bioinorg. Chem. Appl., 2014, 2014, 104046–104059 Search PubMed.
- C. Bustos, O. Burckhardt, R. Schrebler, D. Carrillo, A. M. Arif, A. H. Cowley and C. M. Nunn, Inorg. Chem., 1990, 29, 3996–4001 CrossRef.
- M. K. Koley, N. Duraipandy, M. S. Kiran, B. Varghese, P. T. Manoharan and A. P. Koley, Inorg. Chim. Acta, 2017, 466, 538–550 CrossRef.
-
Bruker (2016) – APEX3-SAINT, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, SADABS-2016/2 – Bruker AXS area detector scaling and absorption Correction, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef PubMed.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, WinGX for Windows, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- L. C. Crowley, M. E. Christensen and N. J. Waterhouse, Cold Spring Harb. Protoc., 2016, 8, pdb-rot087171 CrossRef PubMed.
- R. Thakur, R. Trivedi and N. Rastogi, Sci. Rep., 2015, 5, 10194 CrossRef PubMed.
- L. C. Crowley, A. P. Scott, B. J. Marfell, J. A. Boughaba, G. Chojnowski and N. J. Waterhouse, Cold Spring Harb. Protoc., 2016, 7, 87163 Search PubMed.
- H. Dhandapani, A. Siddiqui, S. Karadkar and P. Tayalia, Adv. Healthcare Mater., 2023, 12, 2300164 CrossRef PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef.
-
P. J. Linstrom, NIST Chemistry WebBook-SRD 69, National Institute of Standards and Technology, 2023 Search PubMed.
-
K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, part B: applications
in coordination, organometallic, and bioinorganic chemistry, John Wiley & Sons, 2009 Search PubMed.
- J. Liu, T. Zhang, T. Lu, L. Qu, H. Zhou, Q. Zhang and L. Ji, J. Inorg. Biochem., 2002, 91, 269–276 CrossRef PubMed.
- B. Selvakumar, V. Rajendiran, P. U. Maheswari, H. Stoeckli-Evans and M. Palaniandavar, J. Inorg. Biochem., 2006, 100, 316–330 CrossRef PubMed.
- K. Paliwal, P. Haldar, P. S. Antharjanam and M. Kumar, ACS Omega, 2023, 8, 21948–21968 CrossRef PubMed.
- P. R. Reddy and A. Shilpa, Chem. Biodiversity, 2011, 8, 1245–1265 CrossRef PubMed.
- J. M. Kelly, A. B. Tossi, D. J. McConnell and C. A. OhUigin, Nucleic Acids Res., 1985, 13, 6017–6603 CrossRef PubMed.
- J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4161–4170 CrossRef PubMed.
- V. Rajendiran, R. Karthik, M. Palaniandavar, H. Stoeckli-Evans, V. S. Periasamy, M. A. Akbarsha, B. S. Srinag and H. Krishnamurthy, Inorg. Chem., 2007, 46, 8208–8221 CrossRef PubMed.
- S. Dhar, P. A. Reddy and A. R. Chakravarty, Dalton Trans., 2004, 697–698 RSC.
- S. Rajalakshmi, T. Weyhermüller, A. J. Freddy, H.R Vasanthi and B.U. Nair, Eur. J. Med. Chem., 2011, 46, 608–617 CrossRef PubMed.
- A. Jayamani, V. Thamilarasan, V. Ganesan and N. Sengottuvelan, Bull. Korean Chem. Soc., 2013, 34, 3695–3702 CrossRef.
- T. Hemnani and M. S. Parihar, Indian J. Physiol. Pharmacol., 1998, 42, 440–452 Search PubMed.
- Y. Zhao, J. Zhu, W. He, Z. Yang, Y. Zhu, Y. Li, J. Zhang and Z. Guo, Chem. – Eur. J., 2006, 12, 6621–6629 CrossRef PubMed.
- J. N. Burstyn and K. A. Deal, Inorg. Chem., 1993, 32, 3585–3586 CrossRef.
- K. A. Deal, A. C. Hengge and J. N. Burstyn, J. Am. Chem. Soc., 1996, 118, 1713–1718 CrossRef.
- P. A. N. Reddy, M. Nethaji and A. R. Chakravarty, Eur. J. Inorg. Chem., 2004, 1440–1446 CrossRef.
- F. Z. Li, J. Q. Xie and F. M. Feng, New J. Chem., 2015, 39, 5654–5660 RSC.
- M. Yavuz and T. Demircan, Mol. Biol. Rep., 2023, 50, 2611–2621 CrossRef PubMed.
- D. P. Dorairaj, J. Haribabu, D. Mahendiran, R. E. Malekshah, S. C. Hsu and R. Karvembu, Appl. Organomet. Chem., 2023, 37, 7087 CrossRef.
- S. Mandal, R. Naskar, R. Mukherjee, A. S. Mondal, A. Das, N. Murmu and T. K. Mondal, New J. Chem., 2023, 47, 15267–15282 RSC.
- S. U. Parsekar, K. Paliwal, P. Haldar, P. S. Antharjanam and M. Kumar, ACS Omega, 2022, 7, 2881–2896 CrossRef PubMed.
- I. Lakshmanan and S. K. Batra, Bio-Protoc., 2013, 3, e374 Search PubMed.
- M. Podhorecka, A. Skladanowski and P. Bozko, J. Nucleic Acids, 2010, 1, 920161 CrossRef.
- T. Ozaki and A. Nakagawara, Cancers, 2011, 3, 994–1013 CrossRef PubMed.
- M. Schuler, E. Bossy-Wetzel, J. C. Goldstein, P. Fitzgerald and D. R. Green, J. Biol. Chem., 2000, 275, 7337–7342 CrossRef PubMed.
- M. Los, M. Mozoluk, D. Ferrari, A. Stepczynska, C. Stroh, A. Renz, Z. Herceg, Z. Q. Wang and K. Schulze-Osthoff, Mol. Biol. Cell, 2002, 13, 978–988 CrossRef PubMed.
- M. D. Kuczler, A. M. Olseen, K. J. Pienta and S. R. Amend, Prog. Biophys. Mol. Biol., 2021, 165, 3–7 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: HR-MS spectra, Fig. S2: IR spectra, and Fig. S3: electronic spectra of 1 in water. Fig. S4: Electronic spectral change with time to check stability of 1 in DMEM for 48 h. Fig. S5: X-band EPR spectra of 1. Fig. S6: Cell viability of MCF-10A against Cu(II) complex 1. Fig. S7: Western blot detection of apoptotic protein expression after treatment with complex 1. CCDC 2365897 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01914h |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a