
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mix and match – controlling the functionality of spin-crossover materials through solid solutions and molecular alloys†
Malcolm A.
Halcrow

School of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: m.a.halcrow@leeds.ac.uk
First published on 6th August 2024
Abstract
The influence of dopant molecules on the structure and functionality of spin-crossover (SCO) materials is surveyed. Two aspects of the topic are well established. Firstly, isomorphous inert metal ion dopants in SCO lattices are a useful probe of the energetics of SCO processes. Secondly, molecular alloys of iron(II)/triazole coordination polymers containing mixtures of ligands were used to tune their spin-transitions towards room temperature. More recent examples of these and related materials are discussed that reveal new insights into these questions. Complexes which are not isomorphous can also be co-crystallised, either as solid solutions of the precursor molecules or as a random distribution of homo- and hetero-leptic centres in a molecular alloy. This could be a powerful method to manipulate SCO functionality. Published molecular alloys show different SCO behaviours, which may or may not include allosteric switching of their chemically distinct metal sites.
Introduction
Spin-crossover (SCO) compounds undergo a transition between high-spin and low-spin electronic states under a physical stimulus (Fig. 1).1–3 SCO is usually found in compounds of first row transition ions from the middle of the d-block,4 and is particularly common in six-coordinate iron(II)5–15 and cobalt(II)4,16 compounds with N-donor ligands; and, in iron(III) compounds with mixed N/O ligation.17 It occurs equally in molecular compounds, and in coordination polymers and framework materials containing those metal/ligand combinations.12–15 SCO in such materials is usually effected by cooling or heating, but it can also be induced by applying pressure to the sample;18 with visible light irradiation;19–21 and with more specialised stimuli such as intense magnetic fields22 or synchrotron X-rays.20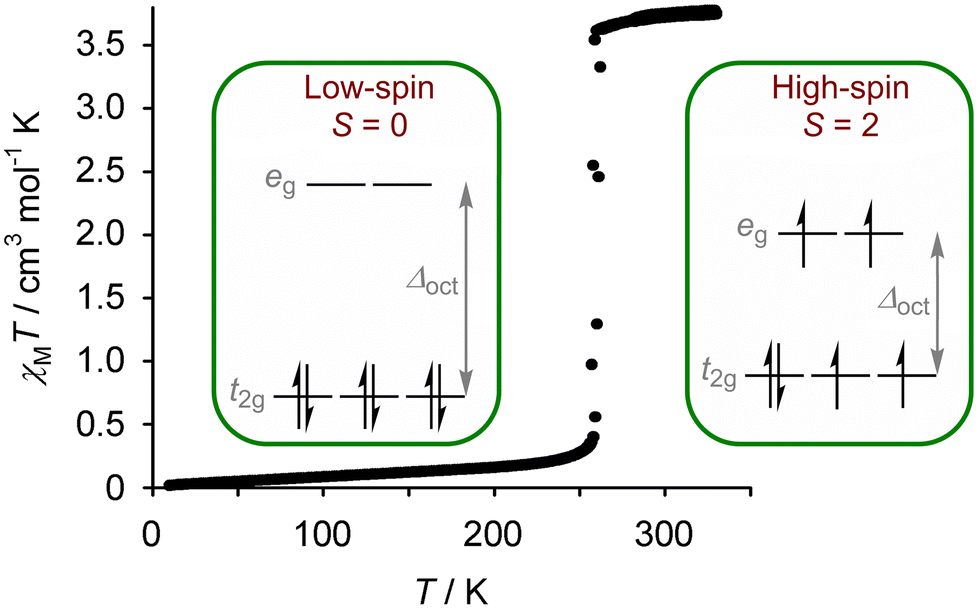 | ||
| Fig. 1 Magnetic susceptibility data from an iron(II) spin-crossover complex, showing the change in magnetic moment associated with an abrupt transition between its low-spin and high-spin d-electron configurations at 260 K. Data are taken from ref. 28. | ||
A spin transition at a metal ion significantly rearranges its coordination sphere, which induces wider structural changes in the bulk material.23 These changes propagate through a sample via elastic interactions between the switching centres, and the form of an SCO transition reflects the strength and dimensionality of these interactions.24 That is, the magnitude of the structural changes at the individual switching sites, and their mutual disposition in the lattice (i.e. the crystal packing).25 SCO in different compounds can occur gradually or abruptly, with or without thermal hysteresis, and in one or more discrete steps.26,27 Cooperative (abrupt, hysteretic) transitions near room temperature are desirable for most practical applications. Such cooperative SCO is most common in iron(II) chemistry, and most of the examples cited below are iron(II) compounds.
As well as these structural changes, a spin transition changes the magnetic moment of a material, and often its colour (Fig. 1).29 Modulating permittivity,30 conductivity,31 fluorescence32 and bulk moduli33 by SCO switching is also established, while the thermodynamic and structural consequences of SCO are also relevant to certain applications.34,35 Moreover, some molecular and coordination polymer SCO materials are processable at the micro and nano scales,36–38 and SCO switching in single molecules has been demonstrated inside an STM.37–39
These aspects make SCO compounds attractive as switchable components in nanoelectronic devices.36,40 SCO materials have also been used in prototype macroscopic applications as diverse as mechanical actuation;34,41 solid state refrigeration;35,42 impact sensors;43 switchable optical waveguides44 and microwave absorbers;45 and, for thermochromic 3D printing.46 There is a continued drive to produce new single-component or hybrid SCO materials with useful materials properties and switching characteristics.
Introducing new functionality into SCO compounds is usually achieved by molecular design and chemical synthesis. Appending fluorophores or other functional substituents to the periphery of SCO molecules, affords single-component compounds combining both properties.30–32 Multifunctional salts of an SCO cation and redox-active or protic anion(s) have also produced crystals with switchable semiconducting or dielectric properties.30,31,47 Lastly decorating SCO centres with alkyl chains, or incorporating them into hybrid polymers, has yielded switchable amphiphiles48 and other soft materials.49
This Perspective explores an alternative approach to manipulating SCO functionality by combining SCO centres with inert, or functional, auxiliary molecules in homogeneous solid solutions. Controlling crystal polymorphism and morphology by incorporating foreign dopants is established in the crystal engineering community.50,51 Using molecular cocrystals52 or mixed-component frameworks53 to change the properties of other families of metal–organic materials is also well known. There is great scope to produce SCO materials with controlled switching properties and embedded functionality using these principles, without extended chemical syntheses.
Isomorphous dopants
The simplest SCO solid solutions have the form [FezM1−zLn]m+, where SCO-active [FeIILn]m+ and the inert dopant [MIILn]m+ (M = Mn, Co, Ni, Zn, Cd etc.) are cocrystallised in different fractions z. The [FeLn]m+ and [MLn]m+ precursors and [FezM1−zLn]m+ cocrystal are crystallographically isomorphous, and the bulk composition of the doped material usually mirrors the stoichiometry of the crystallisation solution. Such isomorphous doping of SCO compounds has been studied since the 1970s, as a probe of the interplay between SCO and the elastic interactions in different lattices (Tables S1 and S2†).26 From that work, the effects of inert dopants on SCO materials are largely predictable based on the identity of the dopant ion and its concentration in the sample. SCO in a typical series of isomorphous FezZn1−z materials is shown in Fig. 2.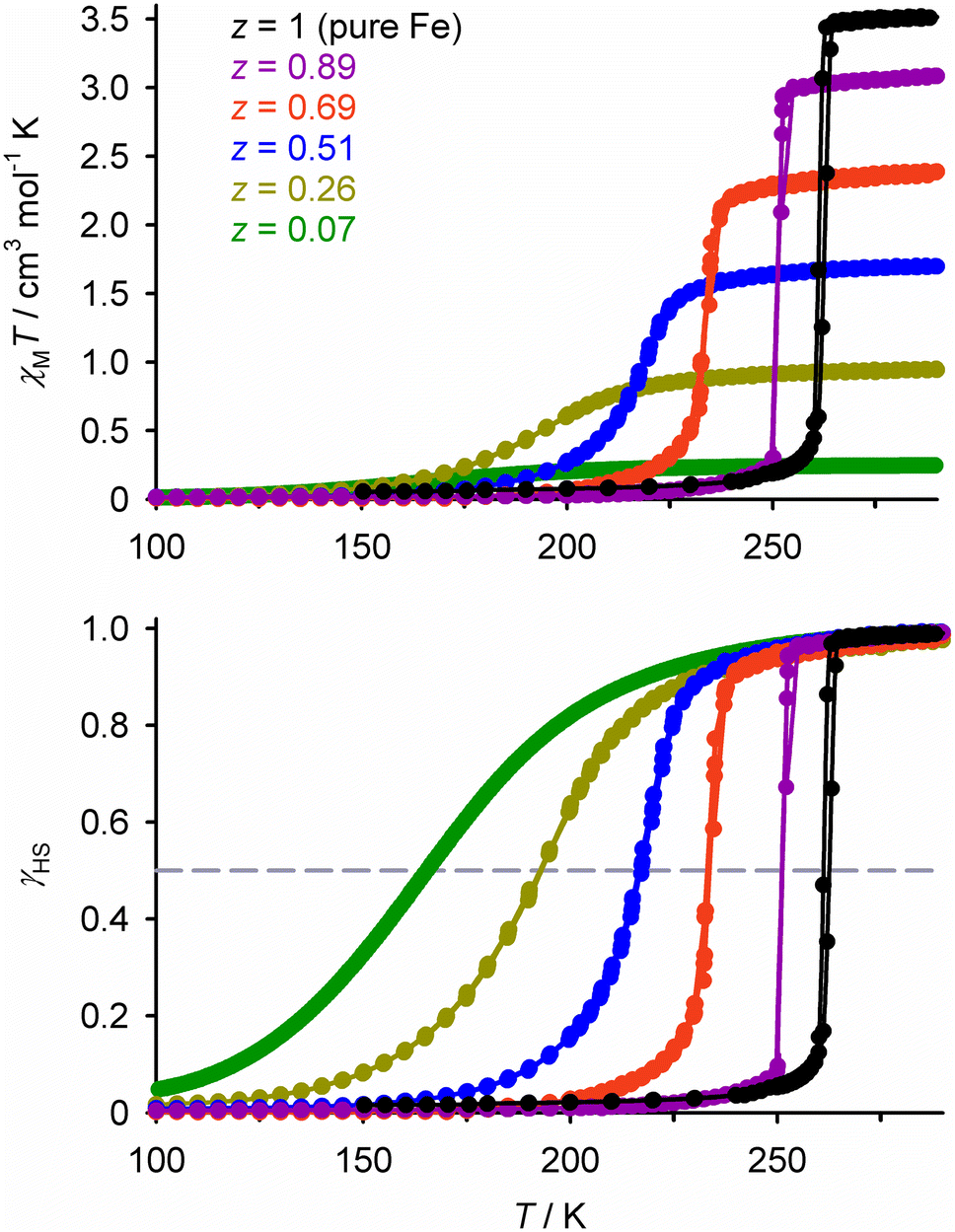 | ||
| Fig. 2 SCO in different compositions of [FezZn1−z(1-bpp)2][BF4]2 (Scheme 1). Top, the measured magnetic susceptibility data. Bottom, the same data expressed as the high-spin fraction in the iron content of the samples (γHS). Data points are linked by spline curves for clarity and the dashed line corresponds to γHS = 0.5, indicating the SCO midpoint at T½. This figure has been adapted from ref. 63 with permission from the Royal Society of Chemistry, copyright 2023. | ||
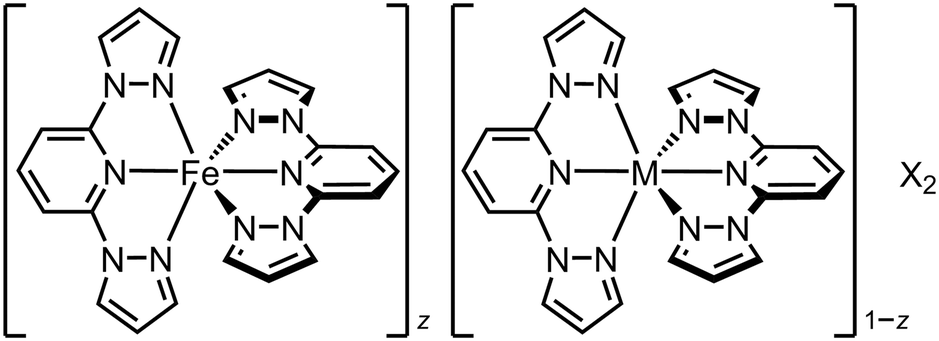 | ||
| Scheme 1 The solid solutions [FezM1−z(1-bpp)2][BF4]2 (1-bpp = 2,6-bis{pyrazol-1-yl}pyridine; M2+ = Co2+, Ni2+, Zn2+ or Ru2+; X− = BF4− or ClO4−).63 | ||
Doping with most common transition ions lowers the SCO transition temperature, as expressed by its midpoint temperature T½ (Fig. 2). That correlates with the ionic radius of the dopant ion M. Larger dopant ions expand and rigidify the lattice, reducing the chemical pressure exerted on the SCO switching centres by their nearest neighbour lattice sites. Thus, since the ionic radius of high-spin iron(II) (78 pm) is larger than its low-spin state (61 pm), a lattice containing larger dopant ions stabilises the high-spin form of an iron(II) complex. The ionic radii of common six-coordinate dopant ions are:54
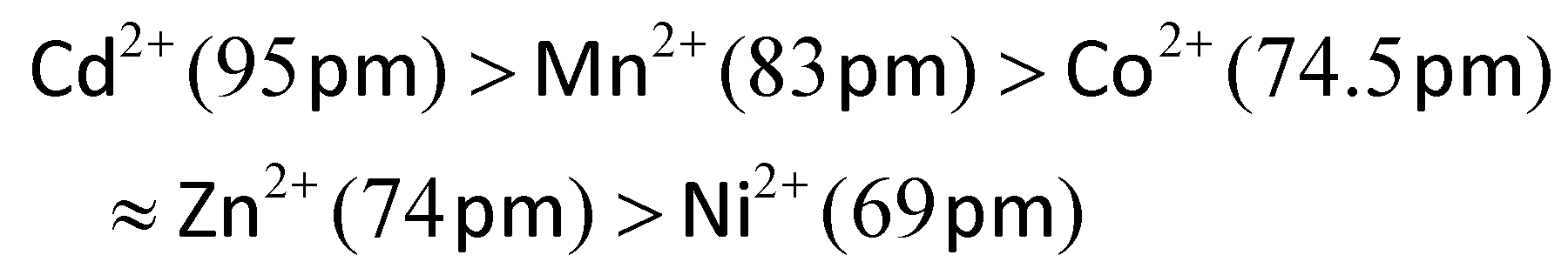
The influence of those dopant ions on T½ follows the same trend, when different dopants are compared in the same SCO host material (Fig. S1 and S2†).55–63 Doping with nickel(II), whose ionic radius lies between the iron(II) spin states, has a roughly neutral effect on the internal lattice chemical pressure. Thus, nickel dopants induce much smaller changes to T½ than the other dopants listed (Fig. 3).
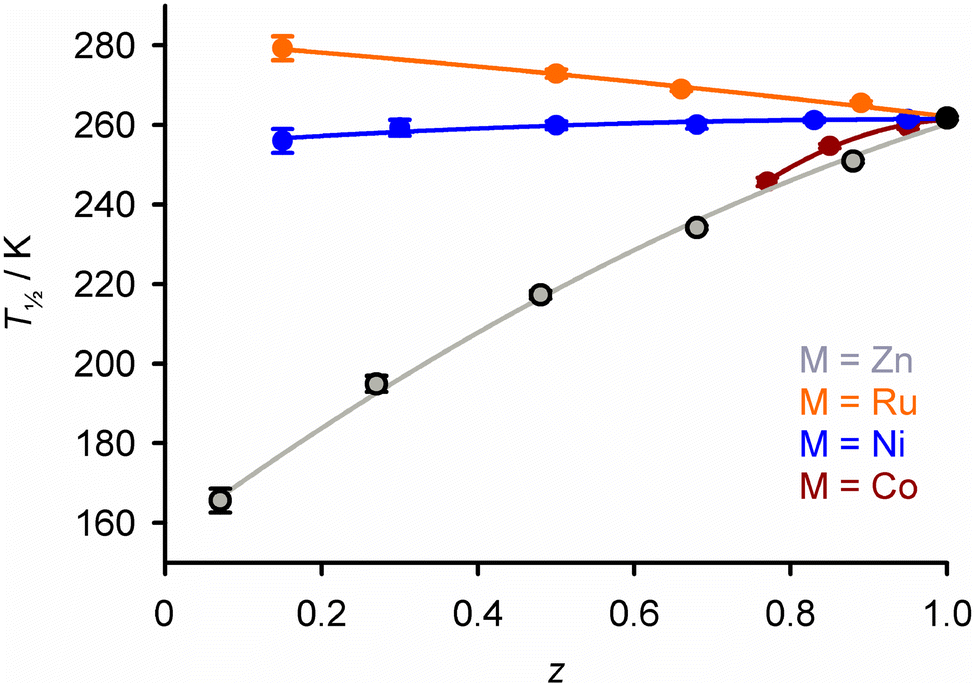 | ||
| Fig. 3 Variation of T½ with composition in [FezM1−z(1-bpp)2][BF4]2 (Scheme 1) with different dopant ions. The FezZn1−z data correspond to those in Fig. 2. Analogous data for other doped SCO compounds are in Fig. S1 and S2.† This figure has been adapted from ref. 63 with permission from the Royal Society of Chemistry, copyright 2023. | ||
An exception to that generalisation occurs in two sets of [FezRu1−zLn]m+ materials, where doping with ruthenium(II) leads to increasing T½ (Fig. 3).63,64 That cannot be explained by the ionic radius of Ru2+, which is similar to Ni2+. Rather, it may reflect the stereochemical rigidity of the [RuLn]m+ dopant molecules, which influences the molecular geometry of the [FeLn]m+ switching centres as the dopant concentration increases.63
The second trend in Fig. 2 is that larger dopant concentrations reduce the cooperativity of the spin-transition. That reflects attenuation of the elastic interactions between the remaining switching centres, as more lattice sites are occupied by inert dopant spacers.26 As a result, doping broadens the temperature range of the transition as shown in Fig. 2. If this broadening extends the SCO below 100 K the transition may become incomplete, as a fraction of the sample becomes kinetically trapped in its high-spin form at such low temperatures.65–67 For the same reason, doping hysteretic SCO materials progressively narrows the hysteresis width, so the hysteresis may be lost above a particular dopant concentration.56–58,62,65 Moreover some SCO transitions occur in stepwise fashion, reflecting the presence of multiple unique iron sites in the lattice or involvement of crystallographic phase changes in the SCO. Such structured SCO becomes continuous, and any intermediate phase transitions are quenched, above a threshold concentration of inert dopants.68–70 This weakened cooperativity is reflected in calorimetric data, where ΔH and ΔS of SCO (per mole of iron) decrease steadily on doping as the transition broadens.58,63,71,72
The first computational treatments of these effects employed a mean field model describing the macroscopic thermodynamics of SCO transitions. This worked well for data like Fig. 2, but could not treat more complicated behaviours.26,73 More recent approaches to the problem include Ising Hamiltonians that couple spin state changes to structural transformations; and, mechanoelastic models considering lattices as spheres linked by pairwise elastic interactions (i.e. springs). These approaches can model hysteretic and multi-step thermal SCO, by manipulating the coupling matrix or elastic interactions between lattice sites in each spin state.74 The current state-of-the-art employs Monte Carlo simulations of mechanoelastic models to describe the nucleation and progression of SCO through model lattices.24,75 Such simulations have rationalised phenomena such as the fact that SCO in single crystals propagates from lattice defects; and, surface effects on SCO in very small nanoobjects.76
While many of the studies in this section have involved molecule-based materials, doping inert metal ions into coordination polymers or frameworks during the crystallisation process has a similar impact on their SCO T½ and cooperativity (Table S2†).77–80 The isomorphous 3D frameworks [Fe(μ3-ta)2] and [Cu(μ3-ta)2] (Hta = 1H-1,2,3-triazole) cannot be co-crystallised from solution, but homogeneous solid solutions were achieved by grinding them in a ball mill.81 Pure [Fe(μ3-ta)2] exhibits a high-temperature spin-transition with very wide thermal hysteresis,82,83 but SCO in the Cu-doped samples was not measured.81
High dilutions of iron(II) inside isomorphous host crystals are used to probe the photophysics of isolated iron(II) centres in rigid lattices.84 Such samples typically contain <1% of the iron compound doped into an optically transparent manganese(II) or zinc(II) host. These studies are outside the scope of this article, but examples are collected in Table S3.†
Probing slow SCO kinetics with isomorphous dopants
The above principles have recently been applied to probe kinetically inhibited SCO in two very different materials. [Fe(NCBH3)2(2MeL)] (2MeL = N,N′-dimethyl-N,N′-bis-{pyrid-2-ylmethyl}-1,2-diaminoethane; Scheme 2) is apparently high-spin, but it can be reversibly converted to its low-spin state by irradiation with red light at 10 K (a reverse-LIESST effect20,21). That implies its low-spin form is thermodynamically accessible, despite it not being observed under normal conditions.85 This was confirmed by doping the compound with nickel(II), which lowers the SCO activation energy by attenuating lattice cooperativity, while only slightly influencing T½ (Fig. 3). Although [FezNi1−z(NCBH3)2(2MeL)] remained high-spin when z = 0.52, when z = 0.48 and 0.41 a cooperative but incomplete spin-transition was revealed at T½ = 72–73 K. Doping with ca. 50% zinc did not have the same effect, since the reduced SCO activation energy was offset by its shifting T½ to even lower temperature.86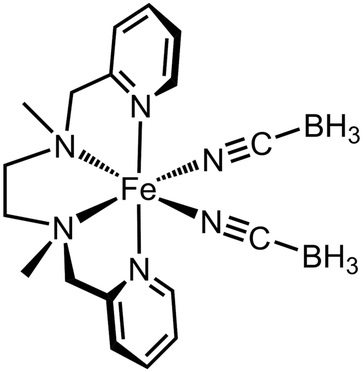 | ||
| Scheme 2 [Fe(NCBH3)2(2MeL)].85,86 | ||
Unique behaviour was found recently in [Me2NH2]6[Fe3(μ-trzs)6(H2O)6] ([trzs]2− = 2-{1,2,4-triazol-4-yl}ethane-1,1-disulfonate; Scheme 3). The central iron atom in the trinuclear anion exhibits gradual SCO upon slow cooling, which is centred at 355 K and shows a 90 K thermal hysteresis width.87 Cooling the sample more rapidly traps it in its high-spin state which is metastable at low temperatures (a TIESST effect88), and it can only relax to its thermodynamic low-spin form on rewarming to ca. 250 K. That relaxation temperature is at least 100 K higher than for any other spin-state trapping phenomenon to date. A different salt of the same anion exhibits similar SCO and TIESST properties, but with a narrower thermal hysteresis of 50 K.89
 | ||
| Scheme 3 [Me2NH2]6[Fe3(μ-trzs)6(H2O)6].87,90 | ||
Exceptionally, doping the dimethylammonium complex salt with zinc(II) leads to widening of the thermal hysteresis, as its high → low-spin T½ shifts to lower temperature while the low → high-spin transition is unchanged. The high-temperature TIESST properties are also retained in the doped materials.90 Thus the hysteretic SCO and thermal trapping in [Me2NH2]6[Fe3(μ-trzs)6(H2O)6] are not a function of lattice cooperativity. Rather, they reflect slow relaxation of the individual molecules in the material during thermal cycling. The concentrated negative charge around the periphery of the complex may contribute to these novel properties.90
Isomorphous dopants in reverse spin-crossover materials
Reverse-SCO, a low → high-spin transition on cooling and high → low-spin on warming, is rare because of its unfavourable electronic and vibrational entropy. However that may be offset by the entropy associated with an order![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :disorder transition elsewhere in the lattice, which is coupled to the spin-state change. Hence, reverse-SCO materials often contain molecules with flexible alkyl chain substituents.91
:disorder transition elsewhere in the lattice, which is coupled to the spin-state change. Hence, reverse-SCO materials often contain molecules with flexible alkyl chain substituents.91
[Co(terpyOC16H33)2][BF4]2 (terpyOC16H33 = 4′-hexadecoxy-2,2′:6′,2′′-terpyridine) exhibits an abrupt reverse-SCO transition at T½ = 239 K, with a 43 K thermal hysteresis loop.92 Doping the compound with up to 70% zinc(II) gradually increases the reverse-SCO T½ as z decreases.93 Conversely doping the same complex with 20% iron lowers T½ by 20 K, and induces a “reverse-TIESST” effect when the sample is rapidly cooled.93,94 These are opposite trends to those in conventional SCO materials (see above), but they can again be understood from the dopants’ ionic radii. The ionic radius of zinc(II) is similar to high-spin cobalt(II),54 so zinc doping increases T½ by stabilising the low-temperature, high-spin form of the sample. The iron(II) dopant is low-spin in this system, with a small ionic radius. That stabilises the low-spin, high-temperature state of the reverse-SCO host lattice and lowers T½.
Another unusual aspect is that the cooperativity of the hysteretic reverse-SCO in [CozM1−z(terpyOC16H33)2][BF4]2 (M = Zn or Fe) is not affected by metal doping.93 That may be because the transition is driven by ligand conformational changes, which are not influenced by the introduction of inert metal centres.
Isomorphous dopants as functional centres
The low-spin state of iron(II) is diamagnetic while, as a non-Kramers ion, its high-spin state is also EPR-silent above helium temperatures in a typical X-band or Q-band spectrometer.95 However, SCO in iron(II) complexes can be monitored by EPR if the sample is doped with small quantities of an EPR-active metal ion. Manganese(II) and copper(II) dopants are usually used for this purpose, and typically ≤3% of the dopant ion is required (i.e. FezM1−z with z ≥ 0.97). In one case, good EPR data were obtained with a sample prepared from a commercial Fe(II) salt containing trace Mn(II) impurities, without any further enrichment.96The anisotropy of Cu(II) EPR spectra97 and the zero-field splitting of Mn(II) ions98 are sensitive to the symmetry and local geometry of the dopant lattice sites. This can be a useful probe of structural changes during SCO, for example to detect crystallographic phase changes in the absence of structural data (Table S4†).99–101 EPR can also monitor the response of dopant species to SCO in the host lattice (see below).102
A few SCO iron(II) compounds are single ion magnets (SIMs) below 5 K, in their photogenerated high-spin form.103–105 However cobalt(II) complexes are often better SIMs than their iron(II) analogues, because of their larger zero-field splitting.‡106 There is one report of a cobalt(II) centre exhibiting SIM properties when doped into an iron(II) SCO lattice, although the effect of spin-state switching on that SIM dopant could not be probed.107
Doping anions and lattice solvent
Many SCO compounds are charged, and preparing their salts with different anions can be facile. Different salts of the same SCO complex cations are not always isomorphous, and can have very different spin state properties.47 However, where isomorphous salts of the same SCO complex can be compared, T½ shows a (usually inverse) correlation with anion size.23Non-stoichiometric mixed-anion SCO salts [FeLn]X2zY2−2z (X−, Y− = anion; 0 ≤ z ≤ 1) mostly show switching properties intermediate between pure [FeLn]X2 and [FeLn]Y2 (Table S5†).108–111 However an exception is the coordination polymers [Fe(μ-atrz)3][BF4]2z[SiF6]1−z (atrz = 4-amino-1,2,4-triazole), whose SCO occurs more abruptly and at 60–70 K higher temperature than in the pure BF4− and SiF62− salts.112,113 Such anion doping can control T½ without attenuating the cooperativity of an SCO transition, as occurs with metal doping. It has also been used to probe the kinetics of SCO in highly cooperative lattices, which can be sensitive to changes in anion size.110,111
Different lattice solvents also impact SCO in solvate materials, especially when they are not isomorphous.26 That is particularly evident in materials undergoing single-crystal-to-single-crystal solvent exchange, which allows a range of SCO behaviours to be accessed from the same crystal precursor.14,114 However, the effect of mixed lattice solvent has only been studied systematically in one case. Crystals of [Fe(pic)3]Cl2·(EtOH)0.74·(iPrOH)0.26 (pic = 2-{aminomethyl}-pyridine) undergo more gradual thermal SCO than the pure, isomorphous ethanol or 2-propanol solvates.115 That should reflect local heterogeneities in the crystal lattice induced by a random distribution of differently shaped solvent molecules.
Non-isomorphous dopants
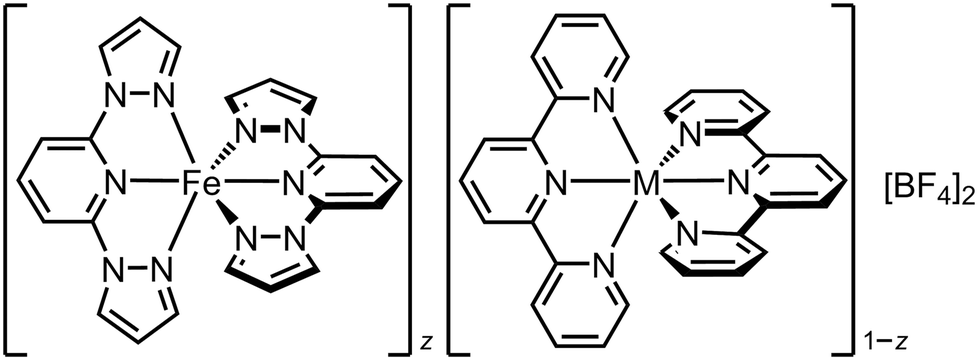
Chemically analogous [FeIILn]m+ and [MIILn]m+ compounds are not always structurally isomorphous, but that need not prevent them from co-crystallising. We have exploited that to activate a high-spin iron(II) compound to undergo SCO. The perchlorate salt of the iron complex in Fig. 2, [Fe(1-bpp)2][ClO4]2, is high-spin and adopts a different crystal phase from [Ni(1-bpp)2][ClO4]2.108 Polycrystalline [FezNi1−z(1-bpp)2][ClO4]2 (z = 0.53 and 0.74; Scheme 1) adopt the nickel complex structure and exhibit complete SCO at 250 K, which is abrupt with narrow hysteresis for z = 0.74 (Fig. 4).116 Compositions containing less nickel form mixed-phase solids exhibiting incomplete abrupt SCO, which implies z = 0.74 is close to the maximum iron content the SCO-active phase can accommodate. This could be a powerful new way to induce SCO in otherwise inactive materials.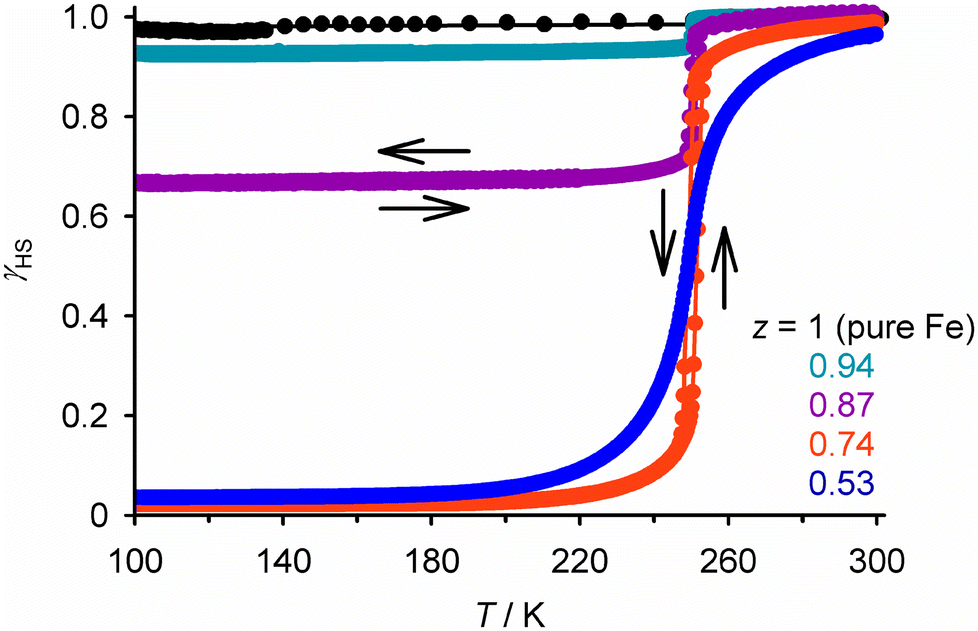 | ||
| Fig. 4 SCO in the non-isomorphous doped materials [FezNi1−z(1-bpp)2][ClO4]2 (Scheme 1). Data for z < 1 were measured in cooling and warming modes. Other details as for Fig. 2. This figure has been adapted from ref. 116 with permission from the Royal Society of Chemistry, copyright 2024. | ||
The iron(II) and cobalt(II) perchlorate complexes of [2,6-bis{pyrazol-1-yl}pyrid-4-yl]carboxylic acid (1-bppCOOH) are not isomorphous,117,118 but can be combined in a rapidly precipitated powder showing gradual SCO above room temperature. While the pure cobalt complex is a field-induced SIM at 3.5 K, [Fe0.92Co0.08(1-bppCOOH)2][ClO4]2 did not show SIM properties.118
Completely different complex molecules may also be co-crystallised, as long as they have compatible molecular symmetry or shape.52 We investigated this through the solid solutions [Fe(1-bpp)2]z[M(terpy)2]1−z[BF4]2 (terpy = 2,2′:6′,2′′-terpyridine; M = Co, Cu or Ru; Scheme 4).119–121 The precursors [Fe(1-bpp)2][BF4]2 and [M(terpy)2][BF4]2 are not isomorphous but adopt the same crystal packing motif, which probably helps them cocrystallise. Five ruthenium-containing samples were obtained with 0.95 ≥ z ≥ 0.28.119 These predominantly adopted one or the other of the precursor crystal phases, although a fraction of the other phase was also present when z ≈ 0.5. The unit cell volumes of the major phase in each sample show a linear correlation with z, confirming they are genuine solid solutions (Fig. 5).122
 | ||
| Scheme 4 The solid solutions [Fe(1-bpp)2]z[M(terpy)2]1−z[BF4]2 (M2+ = Co2+, Cu2+ or Ru2+).102,119–122 | ||
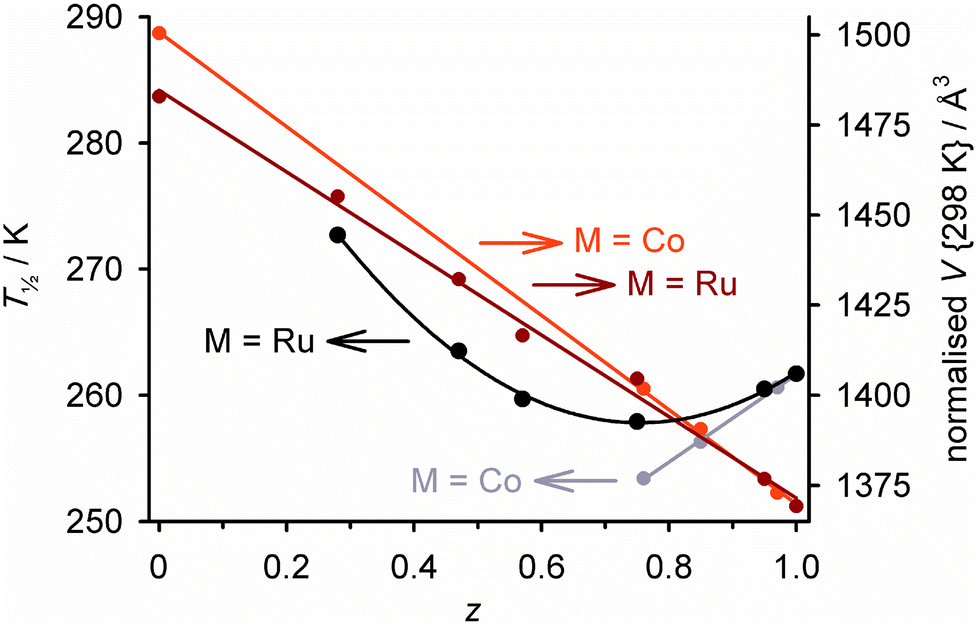 | ||
| Fig. 5 Composition dependence of T½ (black/grey) and the normalised unit cell volume at room temperature (dark/pale red) in [Fe(1-bpp)2]z[M(terpy)2]1−z[BF4]2 (M2+ = Co2+ or Ru2+; Scheme 4).123 Each plot is connected by a regression line for clarity. The materials adopt purely or predominantly the [Fe(1-bpp)2][BF4]2 crystal phase for z > ca. 0.5, and the [M(terpy)2][BF4]2 phase at other compositions. The data are replotted from ref. 122. | ||
Thermal SCO in [Fe(1-bpp)2]z[Ru(terpy)2]1−z[BF4]2 becomes more gradual with increased ruthenium doping, as expected. However, T½ follows a more complicated dependence on z than for the isomorphous solid solutions in Fig. 2 and 3, reaching a minimum near z ≈ 0.7 before increasing again with greater ruthenium doping (Fig. 5).119 That could reflect changes to the phase composition of the materials as z varies, and/or the effect of ruthenium dopants raising T½ through their rigid molecular structures (Fig. 3).63 Attempts to cocrystallise [Fe(1-bpp)2][BF4]2 with two other ruthenium(II) complexes also yielded homogeneous SCO-active solid solutions with one of the dopants, but not with the other.124
[Fe(1-bpp)2]z[Co(terpy)2]1−z[BF4]2 (0.97 ≥ z ≥ 0.76) are cocrystals of two different SCO compounds.125,126 Magnetic measurements implied the iron host lattice and cobalt dopant complex undergo thermal SCO simultaneously, but that was difficult to confirm.120 Light-Induced Excited Spin State Trapping (LIESST) measurements at low temperature, which measure the high → low state transition under kinetic control,20,21,26 were more informative (Fig. 6).122 EPR spectroscopy, which selectively probes the low-spin state of the [Co(terpy)2]2+ sites, showed the low-spin dopant population mirrors the spin state of the host lattice as the LIESST experiment proceeds.102
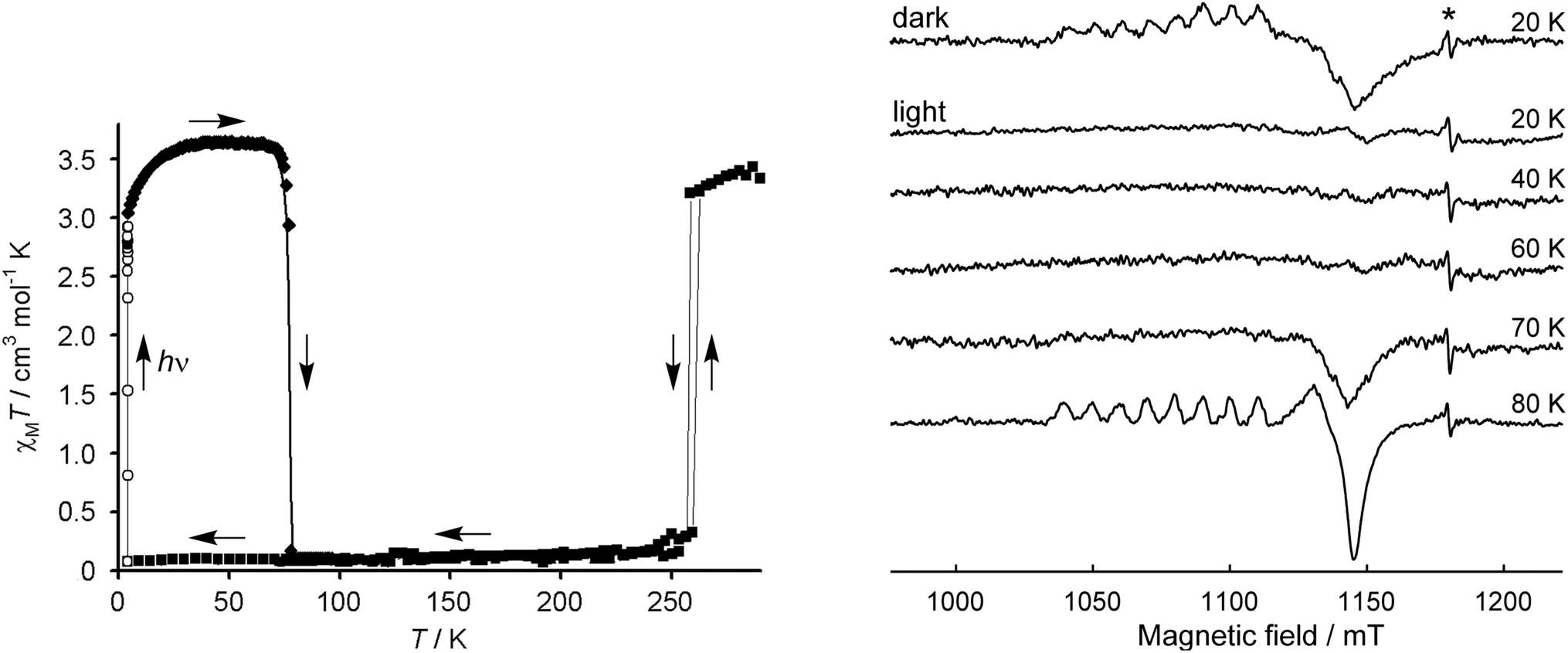 | ||
| Fig. 6 Left: LIESST experiment on the [Fe(1-bpp)2][BF4]2 host lattice. The sample was cooled to 10 K then irradiated with green light, causing photoexcitation to its high-spin state which is metastable at those temperatures.20,21,26 Rewarming to 80 K leads to thermal high → low spin relaxation. Right: Q-band EPR spectra of [Fe(1-bpp)2]0.97[Co(terpy)2]0.03[BF4]2 under LIESST conditions. The sample was measured prior to illumination (20 K, dark); irradiated and remeasured (20 K, light); then warmed at 10 K intervals up to its high → low relaxation temperature. The asterisk marks a radical impurity. This figure has been adapted from ref. 28 with permission from the Royal Society of Chemistry, copyright 2006; and from ref. 102 with permission from the American Chemical Society, copyright 2018. | ||
That is, allosteric spin state switching of the iron and cobalt sites in the material occurs under these conditions (Fig. 6).102 Since cobalt(II) compounds do not themselves exhibit LIESST effects,127 the spin state of [Co(terpy)2]2+ in this system is controlled by thermal expansion or contraction of the [Fe(1-bpp)2][BF4]2 host lattice as its spin state changes.
The materials in Scheme 4 were prepared by cocrystallising preformed [Fe(1-bpp)2][BF4]2 and [M(terpy)2][BF4]2 from a weakly associating solvent. Minimal ligand exchange between the metal centres was detected under those conditions, so the solid solutions are not contaminated by heteroleptic species like [Fe(1-bpp)(terpy)]2+.119,120 In contrast ligand redistribution was observed in other cocrystals of two SCO molecules, which are described in the Molecular Alloys section below.128,129
Stoichiometric salts of cationic and anionic SCO centres
Cations and anions exhibiting SCO and other functionalities have been combined to form multifunctional salts.30,31,47,52 There is one such compound that combines two SCO centres, namely [FeII(tpm)(tpm′)][FeIII(azp)2][ClO4]·2MeCN (tpm = tris{pyrazol-1-yl}methane, tpm′ = tris{3,5-dimethylpyrazol-1-yl}methane, H2azp = 2,2′-azodiphenol; Scheme 5). The compound undergoes very gradual thermal SCO with a small hysteresis, involving both its cation and anion sites. However Mössbauer spectroscopy showed these do not undergo SCO simultaneously; T½ for the cations is near room temperature, when the anions are still predominantly low-spin.130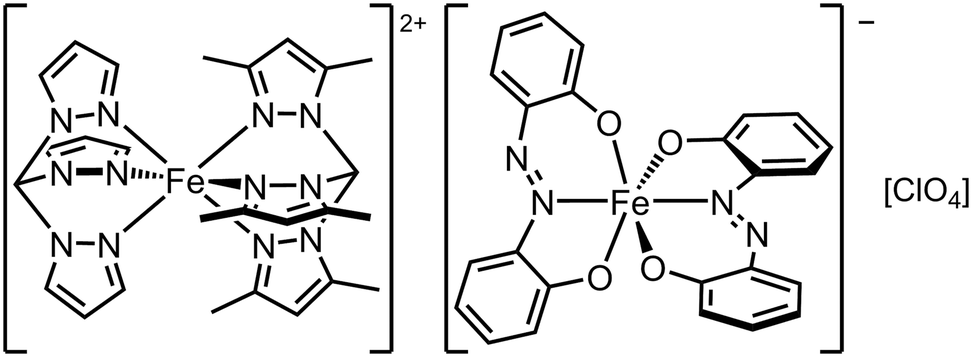 | ||
| Scheme 5 The [FeII(tpm)(tpm′)][FeIII(azp)2]ClO4 SCO double salt.130 | ||
Molecular alloys
Solid solutions of different ligands within the same SCO material were first investigated in the iron/triazole coordination polymers12 [Fe(μ-atrz)3n(μ-Htrz)3−3n][ClO4]2·H2O (atrz = 4-amino-1,2,4-triazole, Htrz = 4H-1,2,4-triazole; 0 < n < 1).131 Homoleptic [Fe(Htrz)3][ClO4]2·H2O and [Fe(atrz)3][ClO4]2·H2O both exhibit SCO with moderate thermal hysteresis, at T½ = 311 K for the Htrz polymer and 188 K for the atrz material. Varying the ratio of the ligands in the synthesis mixture afforded molecular alloys, whose SCO properties evolved continuously with the ligand ratio n between those two limits (Fig. 7). That allowed fine control of T½ using the appropriate ligand stoichiometries while retaining SCO hysteresis, which would not have been possible using inert metal dopants for example.132,133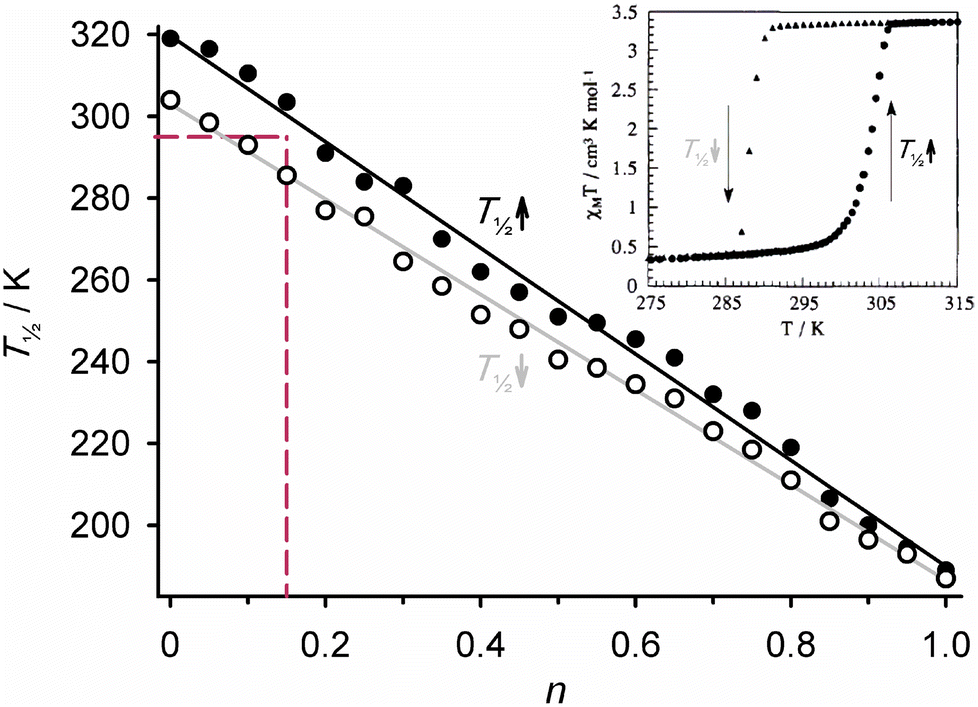 | ||
| Fig. 7 Measured (circles) and calculated (lines) dependence of T½ on composition in [Fe(μ-atz)3n(μ-Htz)3−3n][ClO4]2·H2O. The dashed lines indicate the composition z that is thermally bistable at room temperature (magnetic data shown in inset). The data were replotted from ref. 132, while the inset is adapted from ref. 134 with permission from Springer/Nature, copyright 2009. | ||
The composition [Fe(μ-Htrz)2.55(μ-atrz)0.45][ClO4]2·H2O exhibits a hysteretic spin-transition spanning room temperature (Fig. 7),131 which was employed in a prototype display device based on pixels of a thermochromic SCO material.133 Ligand alloys from the same family were used to produce nanoparticles undergoing SCO just above, or at, room temperature.135,136 The principle has since been extended to the new alloy formulations [Fe(Htrz)1+n−m(trz)2−n(atrz)m][BF4]n·xH2O, where the ratios of the triazole/triazolate ligands and the polymer cationic charge can be continuously varied. These have found use in hybrid polymer materials showing thermally switchable piezoelectric and semiconducting properties.137–139
The Hofmann networks [Fe(μ-pyz){M(CN)4}] (pyz = pyrazine, M = Ni, Pd or Pt) contain [Fe{μ4-M(CN)4}] 44 sheets linked by ditopic pyz ligands, bridging through the axial coordination sites of the iron atoms.14,15 They each undergo a hysteretic thermal spin-transition, which spans room temperature in the platinum-containing material.140 Molecular alloys [Fe(μ-pyz)n(μ-pyzNH2)1−n{M(CN)4}] (pyzNH2 = aminopyrazine) have been prepared, for both M = Pd and Pt.141 All the compositions were isomorphous by powder diffraction. As the pyzNH2 fraction increases T½ shifts to lower temperature, the hysteresis width narrows and the transition becomes less complete, reflecting that pure [Fe(μ-pyzNH2){Pt(CN)4}] is a high-spin material. The plot of T½vs. n in this system is also approximately linear.141
The 1D coordination polymer [Fe(NCEt)2(μ-ebtz)2][BF4]2 (ebtz = 1,2-bis{tetrazol-2-yl}ethane) shows a slow, hysteretic spin-transition at T½ = 101 K that is only revealed by measuring at very slow thermal scan rates. The kinetic barrier to SCO is associated with a reorientation of the EtCN ligands between the spin states.142 In [Fe(NCEt)2n(NCPr)2−2n(μ-ebtz)2][BF4]2 (n = 0.85 and 0.72) the transition hysteresis narrows and T½ increases with larger butyronitrile content. That was attributed to the larger PrCN ligand fraction sterically restricting the capacity of the remaining EtCN ligands to undergo their conformational rearrangement.143 While the structural chemistry is different, the consequences of ligand-doping on SCO in this system resemble nickel-doping in [Fe(NCBH3)2(2MeL)] (Scheme 2).86
The above molecular alloys all exhibit continuous, cooperative SCO at temperatures varying with their chemical composition. In contrast, some other coordination polymer molecular alloys show quite different behaviours. An example is [Fe(μ-btix)3n(μ-btzx)3−3n][ClO4]2 (Scheme 6; btix = 1,4-bis{1,2,4-triazol-1-ylmethyl}benzene, btzx = 1,4-bis{tetrazol-1-ylmethyl}benzene). The precursor compounds are 1D coordination polymers with the same chemical connectivity, but which are not crystallographically isomorphous. Moreover [Fe(μ-btzx)3][ClO4]2 exhibits an abrupt spin-transition at T½ = 200 K,144 while [Fe(μ-btix)3][ClO4]2 is high-spin.145
 | ||
| Scheme 6 The ditopic bis-(azolylmethyl)benzene ligands employed in ref. 144–146, and a schematic of their 1D iron(II) coordination polymers. | ||
Up to 20% btix can be introduced into the [Fe(μ-btzx)3][ClO4]2 structure (i.e. 0.05 ≤ n ≤ 0.2) without affecting its crystallinity by powder diffraction.146 All those compositions show broad, incomplete SCO which is resolvable into two or three steps at T½ ≈ 190, 140 and 60 K (Fig. 8). The lower temperature features increase in intensity with respect to the 190 K step as n increases, but their temperatures do not change significantly. The 190, 140 and 60 K steps were attributed to resolved SCO in [Fe(N-tetrazolyl)6]2+, [Fe(N-tetrazolyl)5(N-triazolyl)]2+ and [Fe(N-tetrazolyl)4(N-triazolyl)2]2+ centres in the heteroleptic polymer chains.146
 | ||
| Fig. 8 Magnetic data for [Fe(μ-btix)0.6(μ-btzx)2.4][ClO4]2 (Scheme 6), and (inset) the first derivative of the data highlighting the three components in its SCO. This figure has been adapted from ref. 146 with permission from the American Chemical Society, copyright 2014. | ||
Similar behaviour occurs in [Fe(NCS)2n(NCBH3)2−2n(μ-bpe)2]·solvent (bpe = 1,2-bis{pyrid-4-yl}ethane; 0 < n < 1). These materials can be crystallised as 1D coordination polymers, or as interpenetrated 2D sheet structures, from different solvents. Two of the 1D compounds exhibited gradual thermal SCO below 150 K, which again occurred in two discrete steps. The height of the steps correlated reasonably with the expected number of Fe(NCS)2 and Fe(NCS)(NCBH3) centres in each sample.147
Ligand alloys based on two molecular SCO systems have been explored. Isomorphous [Fe(LF)(HLF)]·H2O and [Fe(LCl)(HLCl)]·H2O (H2LX = a {5-halosalicyl}thiosemicarbazone; Scheme 7) exhibit abrupt spin-transitions at T½ = 317 and 230 K respectively. Cocrystallisation of H2LF, H2LCl and an iron(III) salt in different ratios yields [Fe(H0.5LF)2n(H0.5LCl)2−2n]·H2O (Scheme 7; 0 ≤ n ≤ 1).128 These contain a random distribution of LF and LCl ligands, with statistical ratios of homoleptic and heteroleptic iron centres. Consistent with that, their crystallographic unit cell volume changes linearly with composition (Fig. 9). Their SCO becomes broadened as n increases from n = 0 to 0.5, then progressively regains its abruptness from n = 0.5 to 1. In contrast to Fig. 7, T½ does not vary linearly with temperature across the whole composition range (Fig. 9). However, the graph shows T½ can be fine-tuned about room temperature by controlling the composition between 0.90 ≤ n ≤ 1.128
 | ||
| Scheme 7 The [Fe(H0.5LF)2n(H0.5LCl)2−2n]·H2O molecule-based molecular alloys.128 | ||
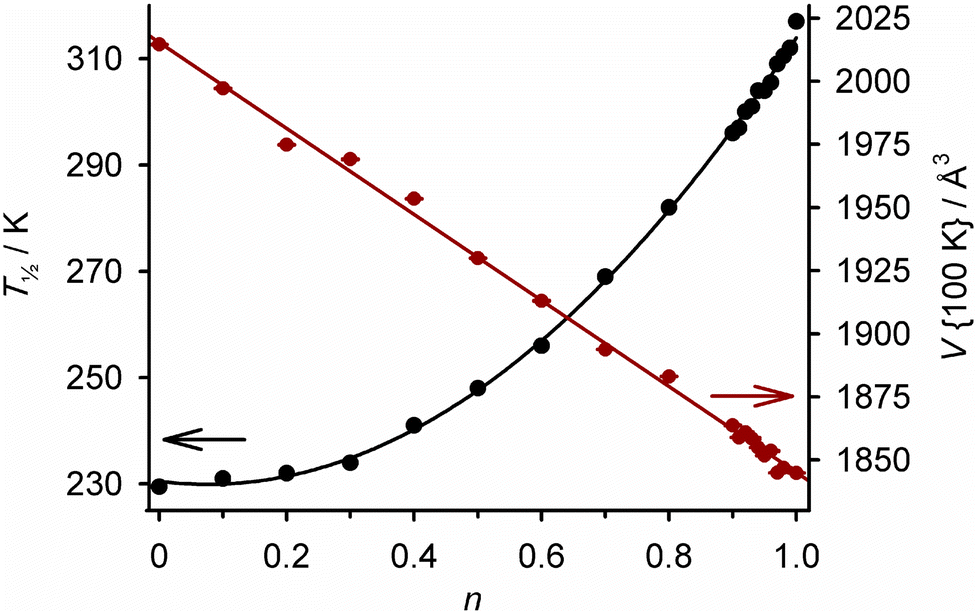 | ||
| Fig. 9 Composition dependence of T½ (black) and the crystallographic unit cell volume at 100 K (red) in [Fe(H0.5LF)2n(H0.5LCl)2−2n]·H2O (Scheme 7). Both plots are connected by regression lines for clarity. The data are replotted from ref. 128. | ||
Crystals of [Fe(Me21,3-bpp)][ClO4]2 (Scheme 8; T½ = 378 K) and [Fe(Me1,3-bpp)][ClO4]2 (T½ = 184 K) are not isomorphous.148 None-the-less, homogeneous solid solutions [Fe(Me21,3-bpp)2n(Me1,3-bpp)2−2n][ClO4]2 with n ≤ 0.5 were achieved by treating Fe[ClO4]2·6H2O with different ratios of those ligands.129 The materials are isomorphous with the pure Me1,3-bpp complex, and their unit cell volumes increased linearly with n which again confirms their homogeneity (Fig. 10). Their SCO becomes more gradual as n increases over this range, while T½ shifts to higher temperature with a reasonably linear dependence on composition (Fig. 10). However T½ for [Fe(Me21,3-bpp)][ClO4]2 (i.e. n = 1.0), which is not isomorphous with the alloy materials, is underestimated by this trend by ca. 100 K. Evidently crystal packing significantly influences the spin state properties of these compounds.
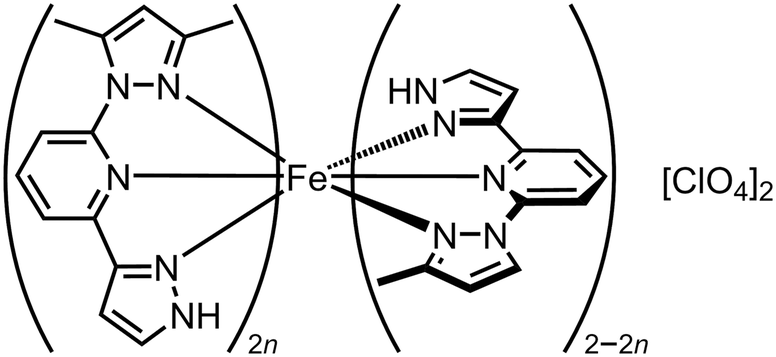 | ||
| Scheme 8 The [Fe(Me21,3-bpp)2n(Me1,3-bpp)2−2n][ClO4]2 molecular alloys.129 | ||
 | ||
| Fig. 10 Composition dependence of T½ (black) and the normalised unit cell volume at 100 K (red) in [Fe(Me21,3-bpp)2n(Me1,3-bpp)2−2n][ClO4]2 (Scheme 8).123 Both plots are connected by regression lines for clarity. The data are replotted from ref. 129 and 148. | ||
To summarise, most known molecular alloys contain chemically heterogeneous switching sites that undergo SCO simultaneously, in one transition step.128,129,132,141,143 Their SCO temperatures and cooperativity vary smoothly with their chemical composition, between the limiting behaviours shown by their pure component compounds (Fig. 7, 9 and 10). However there are also examples of the opposite behaviour, with chemically distinct iron centres undergoing SCO independently of each other at different temperatures (Fig. 8).146,147 This latter observation implies reduced structural cooperativity within those alloy materials, but more detailed explanation of their differences will require further study.
Emissive SCO nanoparticles have been produced by adding an organic fluorophore dopant to the synthetic emulsion. However there the fluorophore is noncovalently included in the surfactant nanoparticle coating, rather than in the bulk of the SCO nanomaterial.149,150
Ligand alloys by post-synthetic modification
Another route to molecular alloys of SCO materials is by post-synthetic modification of a preformed coordination polymer (Table S6†). This was achieved through heterogeneous Schiff base condensation reactions of insoluble [Fe(μ-atrz)3]X2 (X− = anion) with aldehyde reagents in refluxing solvents (Scheme 9). This method was first used to append fluorophores to the coordination polymer scaffold (R = anthryl, pyrenyl, rhodaminyl etc.), which led to 10–15% functionalisation yields (i.e. n = 0.10–0.15; Scheme 9).151,152 The products exhibit complete thermal SCO, at different temperatures from the [Fe(μ-atrz)3][ClO4]2 starting material but with similar cooperativity. The modified polymers were fluorescent, and showed maxima in their temperature-dependent emission intensities at the SCO T½.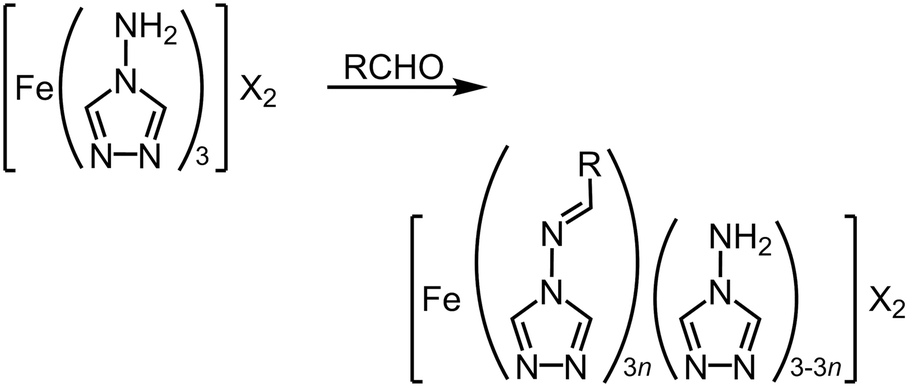 | ||
| Scheme 9 Post-synthetic modification of [Fe(μ-atrz)3]X2 coordination polymers.151–156 | ||
A protocol for the reaction in Scheme 9 has since been published that allows the degree of functionalisation to be controlled over the entire composition range in refluxing ethanol (i.e. 0 < n ≤ 1).153,154 That implies swelling of the material during the heterogeneous reaction, to allow the aldehyde to access the interior of the insoluble polymer particles rather than simply reacting at their surface. Consistent with that, the success of the procedure depended on the reaction solvent used.155 When R = 4-methoxyphenyl and X− = NO3−, the hysteretic spin transition of the starting material became overlaid with a gradual SCO, which grew in at the expense of the abrupt transition as n increased. That suggests a heterogeneous distribution of imine groups at intermediate compositions, with domains of reacted and unreacted material within the sample.153
The transformation in Scheme 9 has also been achieved as a solid-gas reaction, by treating [Fe(μ-atrz)3][MeC6H4SO3]2 with volatile aldehyde or ketone vapours. Again, complete reaction of the amino functions in the solid was observed. Formaldehyde vapour transformed the purple SCO-active starting material to a white high-spin product, while the other vapours studied had a smaller effect on the spin state of the material.156
A different form of post-synthetic ligand modification was performed by treating [Fe(μ-pyz){Pt(CN)4}]140 with controlled quantities of elemental halogens, which undergo oxidative addition to the Pt centres in the material.157 The porosity of the Hofmann framework allows the halogen molecules to access the interior of the solid, so complete conversion can be achieved. Intermediate compositions of [Fe(μ-pyz){PtIVI2(CN)4}n{PtII(CN)4}1−n] (0 < n < 1) retain the hysteretic spin-transition showed by the parent framework, while T½ increases linearly from 295 K at n = 0 to 391 K when n = 1 (Fig. 11). Triggering the spin-transition at elevated temperatures activates the iodine atoms to migrate between Pt centres.158
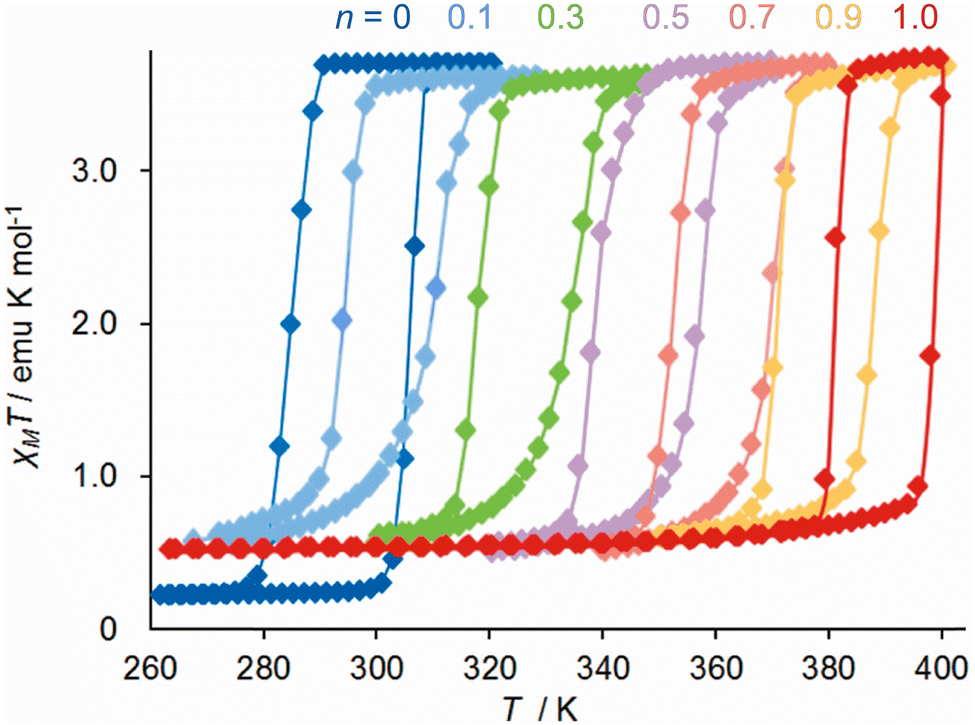 | ||
| Fig. 11 Magnetic data from six compositions of [Fe(μ-pyz){PtI2(CN)4}n{Pt(CN)4}1−n]. This figure has been adapted from ref. 158 with permission from the American Chemical Society, copyright 2011. | ||
This concept has recently been generalised by treating other Pt(II)- or Au(I)-containing SCO Hofmann frameworks with Br2 or I2. Those heterogeneous oxidative additions also proceed to completion and can be reversed by treating the halogenated products with ascorbic acid, a mild reducing agent. However partially halogenated compositions of those materials were not yet reported.159–161
The chemical structure of preformed SCO materials has also been modified through other heterogeneous reactions,162–167 photochemical transformations19,168,169 and chemical or electrochemical redox.170,171 Guest exchange within the pores of an SCO framework material,14,15 or lattice solvent exchange in a solvate crystal,114,172 can achieve similar ends through supramolecular chemistry. Few of these have yet been realised with the stoichiometric control shown in Fig. 11, but there is great scope for further investigations.
Conclusions
The concept of co-crystallising SCO materials with inert dopants was introduced nearly 50 years ago, to probe lattice contributions to the thermodynamics and kinetics of cooperative spin-transitions.26 Studies of this type continue to be an important testbed for models of SCO in the solid state (Tables S1–S3†).24 However, solid solutions and molecular alloys can also be used to tune SCO switching properties for bespoke applications.Different types of solid solution have complementary advantages, as a means of controlling T½. The effects of metal ion doping are best understood. This gives the most predictable stoichiometric control of T½, but at the cost of reduced switching cooperativity at higher dopant levels (Fig. 2). Anion doped SCO materials can also behave predictably, if the mixed-anion materials are isomorphous with their individual component salts.108,109,116 In that case T½ can be controlled without affecting cooperativity, although the accessible range of transition temperatures varies for different materials.
Some ligand-doped molecular alloys show the best performance to date, in controllably accessing a range of T½ values spanning 80–100 K while retaining their cooperative switching properties (Fig. 7, 9 and 11).128,132,158 Indeed, molecular alloys of iron/1,2,4-triazole coordination polymers have been used to fine-tune their performance for particular applications.133,137,139 However, while individual molecular alloy systems can behave predictably, different molecular alloy families show quite different relationships between SCO and composition (see below). To date, there are insufficient data available to predict the performance of a new molecular alloy system.
As well as controlling T½, solid solutions can introduce new functionality into SCO lattices. Adding small quantities of dopants as spectroscopic probes (Table S4†), or perturbing lattice dynamics using mixed-anion compositions (Table S5†), can shed light on the structural chemistry of SCO. That has been especially useful for cooperative SCO materials without crystallographic data.100,101,110 Functional metal ion or ligand dopants have introduced switchable fluorescence151,152 and single ion magnetism107 into existing SCO materials. An otherwise inert dopant has also been used as a structural auxiliary, to activate a high-spin compound to undergo SCO.116 Some of these have been achieved simply using appropriate metal ion dopants, but that is not always necessary. Different molecules with desired functionalities may be combined in a homogeneous solid solution, if their shapes and symmetry are compatible.119–122,124
This survey also highlights different SCO behaviours in solid solutions containing heterogeneous switching sites. Multiple SCO centers combined in the same material may undergo SCO simultaneously, in allosteric fashion (see e.g.Fig. 7 and 11).102,128,129,132,141,143 The relationship between T½ and composition in these systems is often linear (Fig. 7 and 10),129,132,141 but is non-linear in at least one example (Fig. 9).128 Alternatively, chemically distinct switching sites in the same lattice may undergo SCO independently of each other, at different temperatures (Fig. 8).130,146,147 While the intensity of each SCO step changes with the composition of the sample, their temperatures stay roughly constant.§146,147
While diffraction data are not always available, most allosteric SCO has been shown to involve different switching centres disordered within the same lattice site.102,128,129,141,143 Moreover, all known allosteric SCO switching is based on cooperative spin-transitions, although that cooperativity can be reduced at intermediate compositions.102,128,129,132,141,143 Conversely, mixed materials showing independent switching sites have shown very gradual SCO equilibria, with weak cooperativity.130,146,147 Thus, allosteric switching appears to be more likely in cooperative lattices. That makes intuitive sense, but more data are required to establish if that is a general trend.
As a final comment, all the solid solutions in this survey were assumed to be homogeneous materials, unless evidence was found to the contrary. However, it was shown that while the bulk stoichiometries of [MzM′1−z(phen)3][PF6]2 (M, M′ = Fe, Ni or Ru; phen = 1,10-phenanthroline) and related co-crystals could be controlled predictably, individual crystallites within those samples had quite variable compositions by EDX analysis.173,174 Those compounds are not SCO-active, and there has been no comparable investigation of doped SCO crystals. However, if present, sample heterogeneity could contribute to the broadening of SCO in doped and molecular alloy materials at increased dopant levels.
Data availability
No primary research results, software or code have been included in this Perspective submission, and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our own work in this area was performed by Drs C. A. Tovee, T. D. Roberts, L. J. Kershaw Cook, S. Greatorex and I. Capel Berdiell, as well as by the author. Contributions from collaborators listed in the references are also gratefully acknowledged.References
- Spin Crossover in Transition Metal Compounds I–III: Topics in Current Chemistry, ed. P. Gütlich and H. A. Goodwin, Springer-Verlag, Berlin, 2004, vol. 233–235 Search PubMed.
- Spin-crossover materials – properties and applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013, p. 568 Search PubMed.
- J. Zarembowitch, F. Varret, A. Hauser, J. A. Real and K. Boukheddaden, C. R. Chim., 2018, 21, 1056 CrossRef.
- J. Olguín, Coord. Chem. Rev., 2020, 407, 213148 CrossRef.
- M. A. Halcrow, Polyhedron, 2007, 26, 3523–3576 CrossRef.
- G. A. Craig, O. Roubeau and G. Aromí, Coord. Chem. Rev., 2014, 269, 13–31 CrossRef.
- L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef CAS.
- H. L. C. Feltham, A. S. Barltrop and S. Brooker, Coord. Chem. Rev., 2017, 344, 26–53 CrossRef CAS.
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303–7338 RSC.
- H. S. Scott, R. W. Staniland and P. E. Kruger, Coord. Chem. Rev., 2018, 362, 24–43 CrossRef CAS.
- K. Senthil Kumar, Y. Bayeh, T. Gebretsadik, F. Elemo, M. Gebrezgiabher, M. Thomas and M. Ruben, Dalton Trans., 2019, 48, 15321–15337 RSC.
- O. Roubeau, Chem. – Eur. J., 2012, 18, 15230–15244 CrossRef.
- N. F. Sciortino and S. M. Neville, Aust. J. Chem., 2014, 67, 1553–1562 CrossRef.
- Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef.
- B. Kumar, A. Paul, D. J. Mondal, P. Paliwal and S. Konar, Chem. Rec., 2022, 22, e202200135 CrossRef PubMed.
- R. G. Miller, S. Narayanaswamy, J. L. Tallon and S. Brooker, New J. Chem., 2014, 38, 1932–1941 RSC.
- D. J. Harding, P. Harding and W. Phonsri, Coord. Chem. Rev., 2016, 313, 38–61 CrossRef CAS.
- A. B. Gaspar, G. Molnár, A. Rotaru and H. J. Shepherd, C. R. Chim., 2018, 21, 1095–1120 CrossRef CAS.
- M. M. Khusniyarov, Chem. – Eur. J., 2016, 22, 15178–15191 CrossRef.
- D. Unruh, P. Homenya, M. Kumar, R. Sindelar, Y. Garcia and F. Renz, Dalton Trans., 2016, 45, 14008–14018 RSC.
- G. Chastanet, C. Desplanches, C. Baldé, P. Rosa, M. Marchivie and P. Guionneau, Chem. Sq., 2018, 2–2 Search PubMed.
- M. Owczarek, M. Lee, S. Liu, E. R. Blake, C. S. Taylor, G. A. Newman, J. C. Eckert, J. H. Leal, T. A. Semelsberger, H.-P. Cheng, W. Nie and V. S. Zapf, Angew. Chem., Int. Ed., 2022, 61, e202214335 CrossRef PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- A.-I. Popa, L. Stoleriu and C. Enachescu, J. Appl. Phys., 2021, 129, 131101 CrossRef CAS.
- P. Guionneau, M. Marchivie and G. Chastanet, Chem. – Eur. J., 2021, 27, 1483–1486 CrossRef CAS.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- Y. Fang, Y.-S. Meng, H. Oshio and T. Liu, Coord. Chem. Rev., 2024, 500, 215483 CrossRef CAS.
- C. Carbonera, J. S. Costa, V. A. Money, J. Elhaïk, J. A. K. Howard, M. A. Halcrow and J.-F. Létard, Dalton Trans., 2006, 3058–3066 RSC.
- O. Kahn, J. Kröber and C. Jay, Adv. Mater., 1992, 4, 718–728 CrossRef CAS.
- Y. Sekine, R. Akiyoshi and S. Hayami, Coord. Chem. Rev., 2022, 469, 214663 CrossRef CAS.
- M. Wang, Z.-Y. Li, R. Ishikawa and M. Yamashita, Coord. Chem. Rev., 2021, 435, 213819 CrossRef CAS.
- M. K. Javed, A. Sulaiman, M. Yamashita and Z.-Y. Li, Coord. Chem. Rev., 2022, 467, 214625 CrossRef CAS.
- M. Mikolasek, M. D. Manrique-Juarez, H. J. Shepherd, K. Ridier, S. Rat, V. Shalabaeva, A.-C. Bas, I. E. Collings, F. Mathieu, J. Cacheux, T. Leichle, L. Nicu, W. Nicolazzi, L. Salmon, G. Molnár and A. Bousseksou, J. Am. Chem. Soc., 2018, 140, 8970–8979 CrossRef.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef.
- K. G. Sandeman, APL Mater., 2016, 4, 111102 CrossRef.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef.
- K. Senthil Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef.
- L. Kipgen, M. Bernien, F. Tuczek and W. Kuch, Adv. Mater., 2021, 33, 2008141 CrossRef CAS and 2021, 33, 2170354 (correction).
- S. Johannsen, S. Ossinger, J. Grunwald, A. Herman, H. Wende, F. Tuczek, M. Gruber and R. Berndt, Angew. Chem., Int. Ed., 2022, 61, e202115892 CrossRef CAS PubMed.
- R. Torres-Cavanillas, M. Gavara-Edo and E. Coronado, Adv. Mater., 2024, 36, 2307718 CrossRef CAS.
- M. Piedrahita-Bello, J. E. Angulo-Cervera, A. Enriquez-Cabrera, G. Molnár, B. Tondu, L. Salmon and A. Bousseksou, Mater. Horiz., 2021, 8, 3055–3062 RSC.
- M. S. Reis, Coord. Chem. Rev., 2020, 417, 213357 CrossRef CAS.
- K. Boukheddaden, M. H. Ritti, G. Bouchez, M. Sy, M. M. Dîrtu, M. Parlier, J. Linares and Y. Garcia, J. Phys. Chem. C, 2018, 122, 7597–7604 CrossRef CAS.
- L. Zhang, J. Anorld, C. Chichi, S. Calvez, Y. Zhang, L. Salmon, G. Molnár, K. Ridier and A. Bousseksou, Adv. Opt. Mater., 2024, 12, 2303252 CrossRef.
- O. I. Kucheriv, V. V. Oliynyk, V. V. Zagorodnii, V. L. Launets and I. A. Gural'skiy, Sci. Rep., 2016, 6, 38334 CrossRef.
- M. Piedrahita-Bello, J. E. Angulo-Cervera, R. Courson, G. Molnár, L. Malaquin, C. Thibault, B. Tondu, L. Salmon and A. Bousseksou, J. Mater. Chem. C, 2020, 8, 6001–6005 RSC.
- S.-Z. Zhao, H.-W. Zhou, C.-Y. Qin, H.-Z. Zhang, Y.-H. Li, M. Yamashita and S. Wang, Chem. – Eur. J., 2023, 29, e202300554 CrossRef PubMed.
- K. Kuroiwa, Inorganics, 2017, 5, 45 CrossRef.
- A. Enriquez-Cabrera, A. Rapakousiou, M. Piedrahita Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 CrossRef CAS.
- W. Kras, A. Carletta, R. Montis, R. A. Sullivan and A. J. Cruz-Cabeza, Commun. Chem., 2021, 4, 38 CrossRef CAS PubMed.
- S. Pu and K. Hadinoto, Chem. Eng. Res. Des., 2024, 201, 45–66 CrossRef CAS.
- R. Tsunashima, CrystEngComm, 2022, 24, 1309–1318 RSC.
- J. Bitzer and W. Kleist, Chem. – Eur. J., 2019, 25, 1866–1882 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Ganguli, P. Gütlich and E. W. Müller, Inorg. Chem., 1982, 21, 3429–3433 CrossRef CAS.
- J.-P. Martin, J. Zarembowitch, A. Dworkin, J. G. Haasnoot and E. Codjovi, Inorg. Chem., 1994, 33, 2617–2623 CrossRef CAS.
- J.-P. Martin, J. Zarembowitch, A. Bousseksou, A. Dworkin, J. G. Haasnoot and F. Varret, Inorg. Chem., 1994, 33, 6325–6333 CrossRef CAS.
- T. Tayagaki, A. Galet, G. Molnár, M. C. Muñoz, A. Zwick, K. Tanaka, J.-A. Real and A. Bousseksou, J. Phys. Chem. B, 2005, 109, 14859–14867 CrossRef PubMed.
- Z. Yu, T. Kuroda-Sowa, H. Kume, T. Okubo, M. Maekawa and M. Munakata, Bull. Chem. Soc. Jpn., 2009, 82, 333–337 CrossRef.
- C. Baldé, C. Desplanches, J.-F. Létard and G. Chastanet, Polyhedron, 2017, 123, 138–144 CrossRef.
- M. S. Sylla, C. Baldé, N. Daro, C. Desplanches, M. Marchivie and G. Chastanet, Eur. J. Inorg. Chem., 2018, 297–304 CrossRef CAS.
- C. Das, S. Dey, A. Adak, C. Enachescu and P. Chakraborty, Cryst. Growth Des., 2023, 23, 3496–3508 CrossRef CAS.
- P. Ghosh, C. M. Pask, H. B. Vasili, N. Yoshinari, T. Konno, O. Cespedes, C. Enachescu, P. Chakraborty and M. A. Halcrow, J. Mater. Chem. C, 2023, 11, 12570–12582 RSC.
- P. Chakraborty, M. Sy, H. Fourati, T. Delgado, M. Dutta, C. Das, C. Besnard, A. Hauser, C. Enachescu and K. Boukheddaden, Phys. Chem. Chem. Phys., 2022, 24, 982–994 RSC.
- J. Kusz, R. Bronisz, M. Zubko and G. Bednarek, Chem. – Eur. J., 2011, 17, 6807–6820 CrossRef CAS PubMed.
- N. Paradis, G. Chastanet, F. Varret and J.-F. Létard, Eur. J. Inorg. Chem., 2013, 968–974 CrossRef CAS.
- N. Paradis, G. Chastanet, T. Palamarciuc, P. Rosa, F. Varret, K. Boukheddaden and J.-F. Létard, J. Phys. Chem. C, 2015, 119, 20039–20050 CrossRef CAS.
- T. Kohlhaas, H. Spiering and P. Gütlich, Z. Phys. B: Condens. Matter, 1997, 102, 455–459 CrossRef CAS.
- A. B. Gaspar, V. Ksenofontov, S. Reiman, P. Gütlich, A. L. Thompson, A. E. Goeta, M. C. Muñoz and J. A. Real, Chem. – Eur. J., 2006, 12, 9289–9298 CrossRef CAS.
- S. Zheng, M. A. Siegler, J. S. Costa, W.-T. Fu and S. Bonnet, Eur. J. Inorg. Chem., 2013, 1033–1042 CrossRef CAS.
- R. Jakobi, H. Spiering and P. Gütlich, J. Phys. Chem. Solids, 1992, 53, 267–275 CrossRef CAS.
- A. Rotaru, M. M. Dîrtu, C. Enachescu, R. Tanasa, J. Linares, A. Stancu and Y. Garcia, Polyhedron, 2009, 28, 2531–2536 CrossRef CAS.
- A. Hauser, J. Jeftić, H. Romstedt, R. Hinek and H. Spiering, Coord. Chem. Rev., 1999, 190–192, 471–491 CrossRef CAS.
- J. Pavlik and R. Boča, Eur. J. Inorg. Chem., 2013, 697–709 CrossRef CAS.
- J. Pavlik and J. Linares, C. R. Chim., 2018, 21, 1170–1178 CrossRef CAS.
- C. Enachescu and W. Nicolazzi, C. R. Chim., 2018, 21, 1179–1195 CrossRef CAS.
- T. Tayagaki, A. Galet, G. Molnár, M. C. Muñoz, A. Zwick, K. Tanaka, J.-A. Real and A. Bousseksou, J. Phys. Chem. B, 2005, 109, 14859–14867 CrossRef CAS.
- C. Lefter, S. Tricard, H. Peng, G. Molnár, L. Salmon, P. Demont, A. Rotaru and A. Bousseksou, J. Phys. Chem. C, 2015, 119, 8522–8529 CrossRef CAS.
- B. R. Mullaney, L. Goux-Capes, D. J. Price, G. Chastanet, J.-F. Létard and C. J. Kepert, Nat. Commun., 2017, 8, 1053 CrossRef PubMed.
- C. Das, S. Dey, A. Adak, C. Enachescu and P. Chakraborty, Cryst. Growth Des., 2023, 23, 3496–3508 CrossRef CAS.
- J.-S. M. Lee, Y. Fujiwara, S. Kitagawa and S. Horike, Chem. Mater., 2019, 31, 4205–4212 CrossRef CAS.
- M. Grzywa, R. Röβ-Ohlenroth, C. Muschielok, H. Oberhofer, A. Błachowski, J. Żukrowski, D. Vieweg, H.-A. Krug von Nidda and D. Volkmer, Inorg. Chem., 2020, 59, 10501–10511 CrossRef CAS.
- A. B. Andreeva, K. N. Le, K. Kadota, S. Horike, C. H. Hendon and C. K. Brozek, Chem. Mater., 2021, 33, 8534–8545 CrossRef CAS.
- I. Krivokapic, P. Chakraborty, R. Bronisz, C. Enachescu and A. Hauser, Angew. Chem., Int. Ed., 2010, 49, 8509–8512 CrossRef CAS.
- Y. S. Ye, X. Q. Chen, Y. D. Cai, B. Fei, P. Dechambenoit, M. Rouzières, C. Mathonière, R. Clérac and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 18888–18891 CrossRef CAS PubMed.
- X. Li, D. Zhang, Y. Qian, W. Liu, C. Mathonière, R. Clérac and X. Bao, J. Am. Chem. Soc., 2023, 145, 9564–9570 CrossRef CAS PubMed.
- V. Gómez, C. Sáenz de Pipaón, P. Maldonado-Illescas, J. C. Waerenborgh, E. Martin, J. Benet-Buchholz and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2015, 137, 11924–11927 CrossRef PubMed.
- G. A. Craig, J. S. Costa, S. J. Teat, O. Roubeau, D. S. Yufit, J. A. K. Howard and G. Aromí, Inorg. Chem., 2013, 52, 7203–7209 CrossRef.
- A. Moneo-Corcuera, D. Nieto-Castro, C. Sáenz de Pipaón, V. Gómez, P. Maldonado-Illescas and J. R. Galán-Mascarós, Dalton Trans., 2018, 47, 11895–11902 RSC.
- A. Moneo-Corcuera, D. Nieto-Castro, J. Cirera, V. Gómez, J. Sanjosé-Orduna, C. Casadevall, G. Molnár, A. Bousseksou, T. Parella, J. M. Martínez-Agudo, J. Lloret-Fillol, M. H. Pérez-Temprano, E. Ruiz and J. R. Galán-Mascarós, Chem, 2023, 9, 377–393 Search PubMed.
- S. Hayami, M. R. Karim and Y. H. Lee, Eur. J. Inorg. Chem., 2013, 683–696 CrossRef.
- S. Hayami, Y. Shigeyoshi, M. Akita, K. Inoue, K. Kato, K. Osaka, M. Takata, R. Kawajiri, T. Mitani and Y. Maeda, Angew. Chem., Int. Ed., 2005, 44, 4899–4903 CrossRef.
- R. Ohtani, S. Egawa, M. Nakaya, H. Ohmagari, M. Nakamura, L. F. Lindoy and S. Hayami, Inorg. Chem., 2016, 55, 3332–3337 CrossRef PubMed.
- S. Hayami, D. Urakami, Y. Kojima, H. Yoshizaki, Y. Yamamoto, K. Kato, A. Fuyuhiro, S. Kawata and K. Inoue, Inorg. Chem., 2010, 49, 1428–1432 CrossRef.
- A. Ozarowski, S. A. Zvyagin, W. M. Reiff, J. Telser, L.-C. Brunel and J. Krzystek, J. Am. Chem. Soc., 2004, 126, 6574–6575 CrossRef.
- H. Daubric, J. Kliava, P. Guionneau, D. Chasseau, J.-F. Létard and O. Kahn, J. Phys.: Condens. Matter, 2000, 12, 5481–5494 CrossRef.
- B. J. Hathaway, Struct. Bonding, 1984, 57, 56–118 CrossRef.
- C. Duboc, Chem. Soc. Rev., 2016, 45, 5834–5847 RSC.
- A. Ozarowski and B. R. McGarvey, Inorg. Chem., 1989, 28, 2262–2266 CrossRef.
- R. C. W. Sung and B. R. McGarvey, Inorg. Chem., 1999, 38, 3644–3650 CrossRef.
- C. M. Pask, S. Greatorex, R. Kulmaczewski, A. Baldansuren, E. J. L. McInnes, F. Bamiduro, M. Yamada, N. Yoshinari, T. Konno and M. A. Halcrow, Chem. – Eur. J., 2020, 26, 4833–4841 CrossRef.
- S. V. Tumanov, S. L. Veber, S. Greatorex, M. A. Halcrow and M. V. Fedin, Inorg. Chem., 2018, 57, 8709–8713 CrossRef PubMed.
- X. Feng, C. Mathonière, I.-R. Jeon, M. Rouzières, A. Ozarowski, M. L. Aubrey, M. I. Gonzalez, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2013, 135, 15880–15884 CrossRef PubMed.
- C. Mathonière, H.-J. Lin, D. Siretanu, R. Clérac and J. M. Smith, J. Am. Chem. Soc., 2013, 135, 19083–19086 CrossRef.
- A. Urtizberea and O. Roubeau, Chem. Sci., 2017, 8, 2290–2295 RSC.
- A. Sarkar, S. Dey and G. Rajaraman, Chem. – Eur. J., 2020, 26, 14036–14058 CrossRef CAS.
- B. Drahoš, I. Šalitroš, I. Císařová and R. Herchel, Dalton Trans., 2021, 50, 11147–11157 RSC.
- C. Carbonera, C. A. Kilner, J.-F. Létard and M. A. Halcrow, Dalton Trans., 2007, 1284–1292 RSC.
- I. Capel Berdiell, R. Kulmaczewski, N. Shahid, O. Cespedes and M. A. Halcrow, Chem. Commun., 2021, 57, 6566–6569 RSC.
- T. D. Roberts, C. M. Pask, I. Capel Berdiell, F. Tuna and M. A. Halcrow, J. Mater. Chem. C, 2022, 10, 16353–16362 RSC.
- M. Książek, M. Weselski, M. Kaźmierczak, A. Półrolniczak, A. Katrusiak, D. Paliwoda, J. Kusz and R. Bronisz, Chem. – Eur. J., 2024, 30, e202302887 CrossRef.
- X. Yang, A. Enriquez-Cabrera, D. Toha, Y. Coppel, L. Salmon and A. Bousseksou, Dalton Trans., 2023, 52, 10828–10834 RSC.
- X. Yang, A. Enriquez-Cabrera, K. Jacob, Y. Coppel, L. Salmon and A. Bousseksou, Dalton Trans., 2024, 53, 6830–6838 RSC.
- S. Xue, Y. Guo and Y. Garcia, CrystEngComm, 2021, 23, 7899–7915 RSC.
- D. Chernyshov, B. Vangdal, K. W. Törnroos and H.-B. Bürgi, New J. Chem., 2009, 33, 1277–1282 RSC.
- M. A. Halcrow, H. B. Vasili, C. M. Pask, A. N. Kulak and O. Cespedes, Dalton Trans., 2024, 53, 6983–6992 RSC.
- A. Abhervé, M. Clemente-León, E. Coronado, C. J. Gómez-García and M. López-Jordà, Dalton Trans., 2014, 43, 9406–9409 RSC.
- V. García-López, F. J. Orts-Mula, M. Palacios-Corella, J. M. Clemente-Juan, M. Clemente-León and E. Coronado, Polyhedron, 2018, 150, 54–60 CrossRef.
- C. A. Tovee, C. A. Kilner, J. A. Thomas and M. A. Halcrow, CrystEngComm, 2009, 11, 2069–2077 RSC.
- M. A. Halcrow, Chem. Commun., 2010, 46, 4761–4763 RSC.
- R. Docherty, F. Tuna, C. A. Kilner, E. J. L. McInnes and M. A. Halcrow, Chem. Commun., 2012, 48, 4055–4057 RSC.
- G. Chastanet, C. A. Tovee, G. Hyett, M. A. Halcrow and J.-F. Létard, Dalton Trans., 2012, 41, 4896–4902 RSC.
- The crystal structures of the two precursor compounds in these figures have different numbers of molecules in their asymmetric unit (Z = 2 and 4 for Fig. 5, and Z = 4 and 8 for Fig. 10). For consistency, the unit cell data in both plots are normalised to the same value of Z.
- L. J. Kershaw Cook and M. A. Halcrow, Polyhedron, 2015, 87, 91–97 CrossRef CAS.
- J. M. Holland, J. A. McAllister, Z. Lu, C. A. Kilner, M. Thornton-Pett and M. A. Halcrow, Chem. Commun., 2001, 577–578 RSC.
- C. A. Kilner and M. A. Halcrow, Dalton Trans., 2010, 39, 9008–9012 RSC.
- C. Enachescu, I. Krivokapic, M. Zerara, J. A. Real, N. Amstutz and A. Hauser, Inorg. Chim. Acta, 2007, 360, 3945–3950 CrossRef CAS.
- Y.-Y. Wu, Z.-Y. Li, S. Peng, Z.-Y. Zhang, H.-M. Cheng, H. Su, W.-Q. Hou, F.-L. Yang, S.-Q. Wu, O. Sato, J.-W. Dai, W. Li and X.-H. Bu, J. Am. Chem. Soc., 2024, 146, 8206–8215 CrossRef CAS PubMed.
- C. Bartual-Murgui, C. Pérez-Padilla, S. J. Teat, O. Roubeau and G. Aromí, Inorg. Chem., 2020, 59, 12132–12142 CrossRef CAS PubMed.
- W. Phonsri, B. A. I. Lewis, G. N. L. Jameson and K. S. Murray, Chem. Commun., 2019, 55, 14031–14034 RSC.
- J. Kröber, E. Codjovi, O. Kahn, F. Grolibre and C. Jay, J. Am. Chem. Soc., 1993, 115, 9810–9811 CrossRef.
- O. Kahn, L. Sommier and E. Codjovi, Chem. Mater., 1997, 9, 3199–3205 CrossRef CAS.
- O. Kahn and C. Jay Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- I. Šalitroš, N. T. Madhu, R. Boča, J. Pavlik and M. Ruben, Monatsh. Chem., 2009, 140, 695–733 CrossRef.
- J. R. Galán-Mascarós, E. Coronado, A. Forment-Aliaga, M. Monrabal-Capilla, E. Pinilla-Cienfuegos and M. Ceolin, Inorg. Chem., 2010, 49, 5706–5714 CrossRef.
- M. Giménez-Marqués, M. L. García-Sanz de Larrea and E. Coronado, J. Mater. Chem. C, 2015, 3, 7946–7953 RSC.
- S. Rat, M. Piedrahita-Bello, L. Salmon, G. Molnár, P. Demont and A. Bousseksou, Adv. Mater., 2018, 30, 1705275 CrossRef.
- M. Piedrahita-Bello, K. Ridier, M. Mikolasek, G. Molnár, W. Nicolazzi, L. Salmon and A. Bousseksou, Chem. Commun., 2019, 55, 4769–4772 RSC.
- M. Piedrahita-Bello, B. Martin, L. Salmon, G. Molnár, P. Demont and A. Bousseksou, J. Mater. Chem. C, 2020, 8, 6042–6051 RSC.
- S. Bonhommeau, G. Molnár, A. Galet, A. Zwick, J.-A. Real, J. J. McGarvey and A. Bousseksou, Angew. Chem., Int. Ed., 2005, 44, 4069–4073 CrossRef CAS.
- Y. Gong, Z.-H. Li, X. Yan, Y.-Q. Wang, C.-Y. Zhao, W.-K. Han, Q.-T. Hu, H.-S. Lu and Z.-G. Gu, Chem. – Eur. J., 2020, 26, 12472–12480 CrossRef CAS PubMed.
- A. Białońska and R. Bronisz, Inorg. Chem., 2012, 51, 12630–12637 CrossRef.
- M. Książek, M. Weselski, M. Kaźmierczak, A. Tołoczko, M. Siczek, P. Durlak, J. A. Wolny, V. Schünemann, J. Kusz and R. Bronisz, Chem. – Eur. J., 2020, 26, 14419–14434 CrossRef.
- E. Coronado, M. Giménez-Marqués, G. M. Espallargas, F. Rey and I. J. Vitórica-Yrezábal, J. Am. Chem. Soc., 2013, 135, 15986–15989 CrossRef CAS PubMed.
- Y. Chen, S.-Y. Zhang, X.-Q. Zhao, J.-J. Zhang, W. Shi and P. Cheng, Inorg. Chem. Commun., 2010, 13, 699–702 CrossRef CAS.
- N. C. Galve, E. Coronado, M. Giménez-Marqués and G. M. Espallargas, Inorg. Chem., 2014, 53, 4482–4490 CrossRef PubMed.
- H. Dote, M. Kaneko, K. Inoue and S. Nakashima, Bull. Chem. Soc. Jpn., 2018, 91, 71–81 CrossRef CAS.
- C. Bartual-Murgui, S. Vela, M. Darawsheh, R. Diego, S. J. Teat, O. Roubeau and G. Aromí, Inorg. Chem. Front., 2017, 4, 1374–1383 RSC.
- L. Salmon, G. Molnár, D. Zitouni, C. Quintero, C. Bergaud, J.-C. Micheaud and A. Bousseksou, J. Mater. Chem., 2010, 20, 5499–5503 RSC.
- C. M. Quintero, I. A. Gural'skiy, L. Salmon, G. Molnár, C. Bergaud and A. Bousseksou, J. Mater. Chem., 2012, 22, 3745–3751 RSC.
- C.-F. Wang, R.-F. Li, X.-Y. Chen, R.-J. Wei, L.-S. Zheng and J. Tao, Angew. Chem., Int. Ed., 2015, 54, 1574–1577 CrossRef CAS.
- C.-F. Wang, G.-Y. Yang, Z.-S. Yao and J. Tao, Chem. – Eur. J., 2018, 24, 3218–3224 CrossRef CAS.
- A. Enríquez-Cabrera, L. Routaboul, L. Salmon and A. Bousseksou, Dalton Trans., 2019, 48, 16853–16856 RSC.
- A. Enríquez-Cabrera, K. Ridier, L. Salmon, L. Routaboul and A. Bousseksou, Eur. J. Inorg. Chem., 2021, 2000–2016 CrossRef.
- A. Enríquez-Cabrera, L. Getzner, L. Salmon, L. Routaboul and A. Bousseksou, New J. Chem., 2022, 46, 22004–22012 RSC.
- E. Resines-Urien, L. Piñeiro-López, E. Fernandez-Bartolome, A. Gamonal, M. Garcia-Hernandez and J. S. Costa, Dalton Trans., 2020, 49, 7315–7318 RSC.
- G. Agustí, R. Ohtani, K. Yoneda, A. B. Gaspar, M. Ohba, J. F. Sánchez-Royo, M. C. Muñoz, S. Kitagawa and J. A. Real, Angew. Chem., Int. Ed., 2009, 48, 8944–8947 CrossRef.
- R. Ohtani, K. Yoneda, S. Furukawa, N. Horike, S. Kitagawa, A. B. Gaspar, M. C. Muñoz, J. A. Real and M. Ohba, J. Am. Chem. Soc., 2011, 133, 8600–8605 CrossRef CAS PubMed.
- S.-G. Wu, L.-F. Wang, Z.-Y. Ruan, S.-N. Du, S. Gómez-Coca, Z.-P. Ni, E. Ruiz, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2022, 144, 14888–14896 CrossRef CAS.
- B.-H. Lyu, Z.-Y. Ruan, W. Cui, S.-G. Wu, Z.-P. Ni and M.-L. Tong, Inorg. Chem. Front., 2023, 10, 3577–3583 RSC.
- L.-F. Wang, S.-G. Wu, Z.-Y. Ruan, A.-Q. Jian, W. Cui, Z.-P. Ni and M.-L. Tong, Sci. China: Chem., 2023, 66, 1744–1749 CrossRef CAS.
- J. E. Clements, J. R. Price, S. M. Neville and C. J. Kepert, Angew. Chem., Int. Ed., 2014, 53, 10164–10168 CrossRef PubMed.
- Y. Komatsumaru, M. Nakaya, F. Kobayashi, R. Ohtani, M. Nakamura, L. F. Lindoy and S. Hayami, Z. Anorg. Allg. Chem., 2018, 644, 729–734 CrossRef CAS.
- S. Kawabata, S. Chorazy, J. J. Zakrzewski, K. Imoto, T. Fujimoto, K. Nakabayashi, J. Stanek, B. Sieklucka and S. Ohkoshi, Inorg. Chem., 2019, 58, 6052–6063 CrossRef CAS.
- J. H. Askew and H. J. Shepherd, Dalton Trans., 2020, 49, 2966–2971 RSC.
- Y. Gong, W.-K. Han, H.-S. Lu, Q.-T. Hu, H. Tu, P.-N. Li, X. Yan and Z.-G. Gu, J. Mater. Chem. C, 2021, 9, 5082–5087 RSC.
- G. Yang, S.-G. Wu, Z.-Y. Ruan, Y.-C. Chen, K.-P. Xie, Z.-P. Ni and M.-L. Tong, Angew. Chem., Int. Ed., 2023, 62, e202312685 CrossRef CAS.
- L.-F. Wang, W.-M. Zhuang, G.-Z. Huang, Y.-C. Chen, J.-Z. Qiu, Z.-P. Ni and M.-L. Tong, Chem. Sci., 2019, 10, 7496–7502 RSC.
- L.-F. Wang, B.-H. Lv, F.-T. Wu, G.-Z. Huang, Z.-Y. Ruan, Y.-C. Chen, M. Liu, Z.-P. Ni and M.-L. Tong, Sci. China: Chem., 2022, 65, 120–127 CrossRef CAS.
- H.-Y. Wang, J.-Y. Ge, C. Hua, C.-Q. Jiao, Y. Wu, C. F. Leong, D. M. D'Alessandro, T. Liu and J.-L. Zuo, Angew. Chem., Int. Ed., 2017, 56, 5465–5470 CrossRef CAS.
- M. Palacios-Corella, V. García-López, J. C. Waerenborgh, B. J. C. Vieira, G. M. Espallargas, M. Clemente-León and E. Coronado, Chem. Sci., 2023, 14, 3048–3055 RSC.
- E. Resines-Urien, E. Fernandez-Bartolome, A. Martinez-Martinez, A. Gamonal, L. Piñeiro-López and J. S. Costa, Chem. Soc. Rev., 2023, 52, 705–727 RSC.
- J. Bouzaid, M. Schultz, Z. Lao, J. Bartley, T. Bostrom and J. McMurtrie, Cryst. Growth Des., 2012, 12, 3906–3916 CrossRef CAS.
- J. Bouzaid, M. Schultz, Z. Lao, T. Bostrom and J. McMurtrie, Cryst. Growth Des., 2015, 15, 62–69 CrossRef CAS.
- D. Shao, L. Shi, L. Yin, B.-L. Wang, Z.-X. Wang, Y.-Q. Zhang and X.-Y. Wang, Chem. Sci., 2018, 9, 7986–7991 RSC.
- R. Rabelo, L. Toma, N. Moliner, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, R. Ruiz-García and J. Cano, Chem. Commun., 2020, 56, 12242–12245 RSC.
- H. Zenno, Y. Sekine, Z. Zhang and S. Hayami, Dalton Trans., 2024, 53, 5861–5870 RSC.
- W. Hibbs, P. J. van Koningsbruggen, A. M. Arif, W. W. Shum and J. S. Miller, Inorg. Chem., 2003, 42, 5645–5653 CrossRef CAS PubMed.
- N. Pittala, F. Thétiot, C. Charles, S. Triki, K. Boukheddaden, G. Chastanet and M. Marchivie, Chem. Commun., 2017, 53, 8356–8359 RSC.
- E. Cuza, C. D. Mekuimemba, N. Cosquer, F. Conan, S. Pillet, G. Chastanet and S. Triki, Inorg. Chem., 2021, 60, 6536–6549 CrossRef CAS PubMed.
- V. Jornet-Mollá, C. Giménez-Saiz, D. S. Yufit, J. A. K. Howard and F. M. Romero, Chem. – Eur. J., 2021, 27, 740–750 CrossRef PubMed.
- D. L. Reger, J. R. Gardinier, W. R. Gemmill, M. D. Smith, A. M. Shahin, G. J. Long, L. Rebbouh and F. Grandjean, J. Am. Chem. Soc., 2005, 127, 2303–2316 CrossRef CAS.
- M. S. Shongwe, B. A. Al-Rashdi, H. Adams, M. J. Morris, M. Mikuriya and G. R. Hearne, Inorg. Chem., 2007, 46, 9558–9568 CrossRef CAS PubMed.
- J. Tang, J. S. Costa, S. Smulders, G. Molnár, A. Bousseksou, S. J. Teat, Y. Li, G. A. van Albada, P. Gamez and J. Reedijk, Inorg. Chem., 2009, 48, 2128–2135 CrossRef CAS PubMed.
- A. Lennartson, A. D. Bond, S. Piligkos and C. J. McKenzie, Angew. Chem., Int. Ed., 2012, 51, 11049–11052 CrossRef CAS.
- W. Phonsri, D. J. Harding, P. Harding, K. S. Murray, B. Moubaraki, I. A. Gass, J. D. Cashion, G. N. L. Jameson and H. Adams, Dalton Trans., 2014, 43, 17509–17518 RSC.
- Y.-Y. Zhu, H.-Q. Li, Z.-Y. Ding, X.-J. Lü, L. Zhao, Y.-S. Meng, T. Liu and S. Gao, Inorg. Chem. Front., 2016, 3, 1624–1636 RSC.
- R. Kulmaczewski, F. Bamiduro, O. Cespedes and M. A. Halcrow, Chem. – Eur. J., 2021, 27, 2082–2092 CrossRef CAS.
- H.-J. Sheng, C.-C. Xia, X.-Y. Zhang, C.-C. Zhang, W.-J. Ji, Y. Zhao and X.-Y. Wang, Inorg. Chem., 2022, 61, 12726–12735 CrossRef CAS PubMed.
- J. Li, X.-P. Sun, S. Bi, M. Xu, S. Jia, Z. Tang, P. Ma, J. Wang, J. Tao and J. Niu, Inorg. Chem., 2022, 61, 17932–17936 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables of metal-doped, anion-doped and molecular alloy SCO materials. See DOI: https://doi.org/10.1039/d4dt01855a |
| ‡ There are examples of the same cobalt(II) complex exhibiting either SCO or SIM properties in different solid forms.175–177 |
| § A similar dichotomy occurs in stoichiometric crystals containing the same molecule in multiple crystallographic lattice sites, which may undergo SCO simultaneously178–181 or separately under different conditions.182–190 |
| This journal is © The Royal Society of Chemistry 2024 |