
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and redox properties of single-atom bridged diuranium complexes supported by aryloxides†
Fang-Che
Hsueh
 a,
Luciano
Barluzzi
a,
Thayalan
Rajeshkumar
b,
Rosario
Scopelliti
c,
Ivica
Zivkovic
d,
Laurent
Maron
*b and
Marinella
Mazzanti
*a
a,
Luciano
Barluzzi
a,
Thayalan
Rajeshkumar
b,
Rosario
Scopelliti
c,
Ivica
Zivkovic
d,
Laurent
Maron
*b and
Marinella
Mazzanti
*a
aGroup of Coordination Chemistry, Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLaboratoire de Physique et Chimie des Nano-objets, Institut National des Sciences Appliquées, 31077 Toulouse, Cedex 4, France
cInstitut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland
dLaboratory for Quantum Magnetism, Institute of Physics, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 23rd July 2024
Abstract
Single-atom (group 15 and group 16 anions) bridged dimetallic complexes of low oxidation state uranium provide a convenient route to implement multielectron transfer and promote magnetic communication in uranium chemistry, but remain extremely rare. Here we report the synthesis, redox and magnetic properties of N3−, O2−, and S2− bridged diuranium complexes supported by bulky aryloxide ligands. The U(IV)/U(IV) nitride [Cs(THF)8][(U(OAr)3)2(μ-N)], 1 could be prepared and characterized but could not be reduced. Reduction of the neutral U(IV)/U(IV) complexes [(U(OAr)3)2(μ-X)] A (X = O) and B (X = S) led to the isolation and characterization of the U(IV)/U(III) and U(III)/U(III) analogues. Complexes [(K(THF)4)2(U(OAr)2)2(μ-S)2], 5 and [K(2.2.2-cryptand)]2[(U(OAr)3)2(μ-S)], 6 are the first examples of U(III) sulphide bridged complexes. Computational studies and redox properties allow the reactivity of the dimetallic complexes to be related to their electronic structure.
Introduction
Single-atom (group 15 and group 16 anions) bridged dimetallic complexes of uranium have attracted a large number of studies motivated by the need for a better understanding of uranium–ligand bonding interactions1–18 but also because they provide a convenient route for implementing multielectron transfer in uranium chemistry,16,19–28 as they show unexpected catalytic activity29,30 and promote magnetic communication.2,23,31–35 Most of the reported single-atom bridged uranium complexes contain uranium in high oxidation states (+IV to +VI), with complexes of U(IV) being by far the most studied. Until recently, only one single-atom bridged complex containing uranium in the +III oxidation state, [(U(C5H5)2)2(μ-O)], had been crystallographically characterized,36 but a synthetic route could not be identified. In contrast, oxide, sulphide, or nitride bridged diuranium(IV) complexes were conveniently synthesized by reacting a U(III) precursor with stoichiometric amounts of carefully chosen oxidizing atom transfer reagents.7,13,23 Alternative routes to oxide and sulphide bridged diuranium(IV) complexes include abstraction of sulphide or oxide during the reduction of CO2 or CS2 by U(III) complexes.16,37We reported the synthesis of diuranium(IV) oxide and nitride complexes supported by tris(tert-butoxy)siloxide ligands, which were prepared by the reaction of the U(III) complex [U(OSi(OtBu)3)3]2 with atom transfer reagents.12,23 We showed that these complexes can be reduced with KC8, leading to the rational synthesis of nitride- and oxide-bridged diuranium(III) complexes I and II (Fig. 1).19,23,38–40
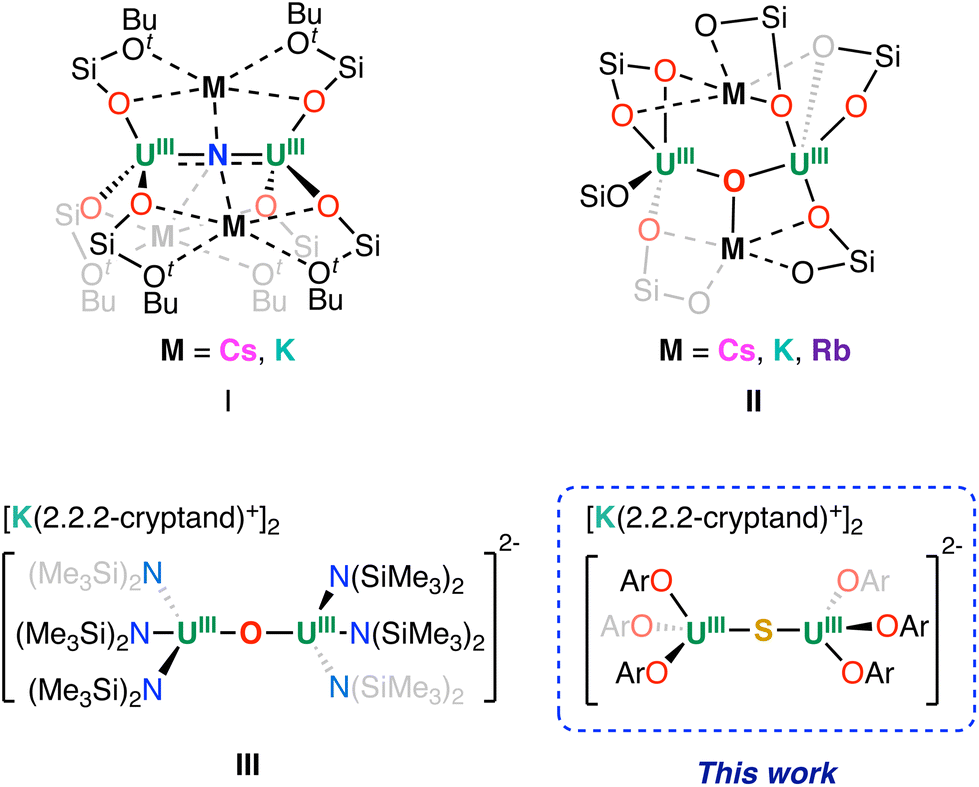 | ||
| Fig. 1 Previously reported U(III) bridging complexes. | ||
Attempts to prepare the analogous sulphide bridged diuranium(III) complex have so far only led to U(IV) decomposition products.41 Complexes I and II showed high reactivity towards N2 resulting in the first examples of four electron reduction and cleavage of N2 effected by a uranium complex in the absence of external reducing agents.19,23,39,40 In contrast, the two- and three-electron reduction of the triphenylsiloxide oxide-bridged diuranium(IV) complex [(U(OSiPh3)3(DME))2(μ-O)] yielded formal “U(II)/U(IV)”, and “U(I)/U(IV)” complexes via ligand migration and formation of uranium–arene δ-bond interactions.27 Remarkably, these complexes can promote the reduction of substrates restoring the original ligand arrangement. Recently, we also investigated the reduction of diuranium(IV) complexes supported by the –N(SiMe3)2 ligand and found that, while the nitride-bridged U(IV) complex [NBu4][(U(N(SiMe3)2)3)2(μ-N)]32 could not be reduced further, reduction of the oxide-bridged complex [(U(N(SiMe3)2)3)2(μ-O)] allowed the isolation of the diuranium(III) analogue (complex III in Fig. 1).21
Surprisingly this complex did not reduce N2, but showed unprecedented reactivity, including four electron reduction of azobenzene by a single metal centre through the delivery of “masked U(II)”.20 Overall, these results highlighted that the attractive and diverse reactivity demonstrated by the single-atom bridged diuranium(III) complexes drastically depend on the steric and electronic properties of the supporting ligand. Hence, we set out to investigate the possibility of accessing U(III)–X–U(III) complexes with X = N, O, S, using the 2,6-di-tert-butylphenoxide supporting ligand. Taking advantage of the reported diuranium(IV) complexes [(U(OAr)3)2(μ-X)], (X = O or S, complex A and B; OAr = 2,6-di-tert-butylphenoxide),7 we were able to prepare the U(III)–O–U(III) and U(III)–O–U(IV) derivatives and compare their redox properties with those of the amide and siloxide analogues and crystallographically characterize the first example of a diuranium(III) sulphide-bridged complex.
Results and discussion
U(IV)/U(IV) complexes
At first, we synthesised the previously reported chalcogenide bridged diuranium(IV) complexes with aryloxide ligands, [(U(OAr)3)2(μ-X)] (X = O or S, complex A and B; OAr = 2,6-di-tert-butylphenoxide).7The complex [(U(OAr)3)2(μ-O)] (A) was prepared in 80% yield using a modified literature procedure7 by reacting [U(OAr)3] in THF at −80 °C with the N2O adduct of the N-heterocyclic carbene 1,3-dimesitylimidazol 2-ylidene (IMes), IMesN2O37,42 (Scheme 1). The sulphide complex [(U(OAr)3)2(μ-S)] (B) was prepared in 91% by reacting [U(OAr)3] with 0.5 equiv. PPh3S according to a slightly modified version of the previously reported procedure7 (Scheme 1).
 | ||
| Scheme 1 Synthesis of the U(IV)/U(IV) bridging complexes. | ||
To compare the redox reactivity and magnetic properties of compounds presenting different linkers we also pursued the synthesis of the nitride-bridged analogous complex that had not been reported previously. The synthesis of the nitride bridged diuranium(IV) complex [Cs(THF)8][(U(OAr)3)2(μ-N)] (1) (Scheme 1) was achieved by reacting [U(OAr)3] with alkali azide, a method that had previously led to the isolation of several nitride-bridged complexes supported by amide or siloxide ligands.5,12,28,32,40,43,44
The addition of 0.5 equiv. of CsN3 to 1.0 equiv. of [U(OAr)3] in THF at −40 °C for 4 days, resulted in the formation of a dark orange solution. Analysis of the reaction mixture by 1H NMR spectroscopy in THF-d8 showed the full consumption of the U(III) precursor and appearance of a new species (Fig. S5†). Single crystals suitable for X-ray diffraction studies were obtained in 88% yield from a concentrated THF/n-hexane solution at −40 °C and were identified as the dimeric complex, [Cs(THF)8][(U(OAr)3)2(μ-N)] (1). The 1H NMR spectrum of isolated complex 1 in THF-d8 shows broad resonances at δ 5.74 and −8.54 ppm at room temperature (Fig. S7†), while at lower temperatures (−40 °C), three resonances at δ 96.95, −19.72, and −39.15 ppm are observed, suggesting fluxional behaviour (Fig. S7†). Complex 1 crystallizes in the space group P21/c, with the full molecule generated by symmetry. The solid-state molecular structure of 1 (Fig. 2) shows an ion pair, consisting of an outer-sphere [Cs(THF)8]+ cation and the [(U(OAr)3)2(μ-N)]− anion. Each uranium centre is tetra-coordinated in a distorted tetrahedral geometry. The two uranium(IV) ions are bridged by a nitride (N3−) anion and are each bound by three –OAr ligands. The U–OAr bond distances (2.178(4)–2.226(4) Å) are elongated compared to the U(III) precursor, [U(OAr)3] (2.149(4)–2.165(3) Å).45 This elongation could be due to reduced tert-butyl interactions between the aryloxides upon the formation of the bridging complex. The OAr–U–OAr and N–U–OAr bond angles are in the range of 94.21(17)–138.43(12)°. The values of the U–N–U bond distances (2.0612(5) Å) and angle (180.0°) are consistent with the previously reported anionic UIV–N–UIV complexes supported by –OSi(OtBu)3 and –N(SiMe3)2 ligands (2.032(12)–2.083(5) Å; 168.4(3)–179(1)°).32,46
 | ||
| Fig. 2 Molecular structure of the anionic species [(U(OAr)3)2(μ-N)]− in (1) with portions of the aryloxide ligands depicted as wire frames for clarity. Thermal ellipsoids are drawn at the 50% probability level. [Cs(THF)8]+ cation, hydrogen atoms, and methyl groups on the –OAr ligands have been omitted for clarity. | ||
U(III)/U(III) complexes
With the bridging U(IV)/U(IV) complexes A, B and 1 in hand, we next investigate their redox reactivity.At first, we explored the reduction of complex 1. Attempts to reduce complex 1 with excess KC8 at −80 °C proved unsuccessful and led to a complex mixture of unreacted 1 and unidentified species (Fig. S8†). The reaction of 1 with 1.0 equiv. of KC8 in THF-d8 at −40 °C resulted in a mixture of 1 and a new species, as observed by 1H NMR spectroscopy (Fig. S9†). The addition of a second equiv. of KC8 to the reaction mixture at −40 °C led to the full consumption of 1 and formation of a new species, as indicated by 1H NMR spectroscopy (Fig. S9†). Unfortunately, multiple attempts to isolate single crystals of this species for X-ray diffraction studies proved unsuccessful.
When the reduction of A was performed with 5.0 equiv. of KC8 in presence of 5.0 equiv. of LiI, a putative U(III)/U(III) was also formed that decomposes over time at −40 °C (Fig. S13†). The product formed from the decomposition of the U(III)/U(III) complex at −40 °C could be crystallographically characterized as the mix-valent diuranium(III)/(IV) complex, [Li(THF)4][U(OAr)3(μ-O)] (2) (Fig. 3). The 1H NMR spectrum of complex 2 is practically the same as that observed for the putative [K(THF)x][(U(OAr)3)2(μ-O)] analogue suggesting that cations are outer-sphere.
 | ||
| Fig. 3 Molecular structure of the anionic species of [(U(OAr)3)2(μ-O)]− (2) with portions of the aryloxide ligands depicted as wire frames for clarity. Thermal ellipsoids are drawn at the 50% probability level. [Li(THF)4]+ cation, hydrogen atoms, solvent molecules, and methyl groups on the –OAr ligands have been omitted for clarity. | ||
Complex 2 crystallizes in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with the full molecule generated by symmetry. The solid-state molecular structure of complex 2 (Fig. 3) displays an anionic dinuclear complex, [(U(OAr)3)2(μ-O)]− where the two uranium ions are bridged by an oxide (O2−), and each uranium is tetra-coordinated by three –OAr ligands and one bridging oxide in a distorted tetrahedral environment. The structure is completed by one outer-sphere [Li(THF)4]+ cation. The U–OAr distances (2.208(3)–2.246(3) Å) are longer compared to the U–OAr lengths in the U(III) precursor, [U(OAr)3] (2.149(4)–2.165(3) Å) and U(IV) complex, [U(OAr)4] (2.135(4) Å).47 This elongation is attributed to the release of steric repulsion upon the formation of the bridging complex. The OAr–U–OAr and Ooxide–U–OAr bond angles (93.16(12)–137.63(12)°) are comparable to those found in 1. The U–Ooxide distances (2.15427(12) Å) are in the range of the previously reported U(III)/U(IV) complexes, [K(2.2.2-cryptand)][(U(N(SiMe3)2)3)2(μ-O)] (2.067(6)–2.273(6) Å)21 and [K(DME)4][(K(DME)U(calix[4]tetrapyrrole))2(μ-O)] (2.017(10)–2.226(10) Å).48
, with the full molecule generated by symmetry. The solid-state molecular structure of complex 2 (Fig. 3) displays an anionic dinuclear complex, [(U(OAr)3)2(μ-O)]− where the two uranium ions are bridged by an oxide (O2−), and each uranium is tetra-coordinated by three –OAr ligands and one bridging oxide in a distorted tetrahedral environment. The structure is completed by one outer-sphere [Li(THF)4]+ cation. The U–OAr distances (2.208(3)–2.246(3) Å) are longer compared to the U–OAr lengths in the U(III) precursor, [U(OAr)3] (2.149(4)–2.165(3) Å) and U(IV) complex, [U(OAr)4] (2.135(4) Å).47 This elongation is attributed to the release of steric repulsion upon the formation of the bridging complex. The OAr–U–OAr and Ooxide–U–OAr bond angles (93.16(12)–137.63(12)°) are comparable to those found in 1. The U–Ooxide distances (2.15427(12) Å) are in the range of the previously reported U(III)/U(IV) complexes, [K(2.2.2-cryptand)][(U(N(SiMe3)2)3)2(μ-O)] (2.067(6)–2.273(6) Å)21 and [K(DME)4][(K(DME)U(calix[4]tetrapyrrole))2(μ-O)] (2.017(10)–2.226(10) Å).48
The U–O–U angle (180.0°) is slightly more linear compared to those found in the U(III)/U(IV) analogues supported by –N(SiMe3)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 and calix[4]tetrapyrrole48 ligands (177.8(4)–179.0(6)°).
21 and calix[4]tetrapyrrole48 ligands (177.8(4)–179.0(6)°).
Lastly, to isolate the U(III)–O–U(III) product, we performed the reduction of A in presence of 2.2.2-cryptand. The addition of 2.0 equiv. of 2.2.2-cryptand and 5.0 equivalents of KC8 to A at −80 °C, resulted in the formation of a new set of resonances in the 1H NMR spectrum (Fig. S15†) recorded at −80 °C that were assigned to the U(III)/U(III) [K(2.2.2-cryptand)]2[(U(OAr)3)2(μ-O)] (3) (Scheme 2). Decomposition of complex 3 was observed by 1H NMR spectroscopy after a few hours at −40 °C (Fig. S17†), but it could be isolated analytically pure in 80% yield from washing with cold toluene and n-hexane at −80 °C.
 | ||
| Scheme 2 Reduction of A and reactivity of 3 with azobenzene. | ||
The formulation of complex 3 was confirmed by the isolation of the U(III)–O–U(IV) analogue from redox reactivity studies. The reaction of complex 3 with PhNNPh resulted in the isolation of the U(III)–O–U(IV) analogue oxide-bridged complex while the reduced azobenzene radical remains outer-sphere. The addition of 1.0 equiv. of PhNNPh to a solution of 3 in THF-d8 at −80 °C, led immediately to a colour change from dark red-brown to dark yellow-brown, full consumption of the starting material and the formation of a new species as indicated by 1H NMR spectroscopy (Fig. S18†). Single crystals suitable for X-ray diffraction studies were obtained from concentrated THF solution at −40 °C, and identified as the complex [K(2.2.2-cryptand)(THF)][(U(OAr)3)2(μ-O)] (4), co-crystallized with singly reduced [K(2.2.2-cryptand)(THF)][PhNNPh] in 52% yield (Scheme 2).
Complex 4 crystallizes in the space group P, with the full molecule generated by symmetry. The solid-state structure of 4 (Fig. 4) was found to be very similar to the lithium analogue (2), with one outer-sphere [K(2.2.2-cryptand)(THF)]+ cations instead of [Li(THF)4]+ cation. The U–OAr distances 2.204(6)–2.223(6) and the U–Ooxide distances 2.1421(7) are comparable to those observed in U(III)/U(IV) complex 2. The OAr–U–OAr and Ooxide–U–OAr bond angles (95.5(2)–137.6(2)°) are comparable to those found in 2. The U–O–U angle (180.0°) remains consistent with the linear arrangement observed in 2. Additionally, the N–N bond length (1.3(1) Å) of singly reduced [K(2.2.2-cryptand)(THF)][PhNNPh] is equivalent to that reported for the anionic compound, [K(2.2.2-cryptand)][PhNNPh] (1.34(3) Å).49 The N–N bond distance, being longer than neutral PhNNPh (1.25 Å) and shorter than the dianionic analogue [K(18-c-6)]2[PhNNPh] (1.40(3) Å),27 is diagnostic of the radical monoanionic nature of the anion [PhNNPh]−.
 | ||
| Fig. 4 Molecular structure of the anionic species [(U(OAr)3)2(μ-O)]− in (4), co-crystallized with [PhNNPh]−, with portions of the aryloxide ligands depicted as wire frames for clarity. Thermal ellipsoids are drawn at the 50% probability level. Two [K(2.2.2-cryptand)(THF)]+ cations, hydrogen atoms, and the methyl groups on the –OAr ligands have been omitted for clarity. | ||
The reactivity of the U(III)–O–U(III) complex 3 is different from that previously reported for the analogous amide complex [K(2.2.2-cryptand)]2[(U(N(SiMe3)2)3)2(μ-O)],20 in which the reaction with azobenzene leads to cleavage of a U–O bond followed by four-electron reduction and cleavage of the N–N bond (yielding a U(VI) bis-imido complex) and the release of a U(IV) terminal oxide complex. Considering the very similar redox properties measured for complex 3 and the amide-supported U(III)–O–U(III) complex (see Electrochemistry section), the difference in reactivity is probably determined by the high stability of the terminal oxide complex [U(O)(N(SiMe3)2)3] compared to the aryloxide analogue.
Additionally, complex 3, similarly to what observed for [K(2.2.2-cryptand)]2[(U(N(SiMe3)2)3)2(μ-O)],20 shows no reactivity towards N2 reduction. This lack of reactivity contrasts with what was previously reported for the analogous oxide-bridged diuranium(III) complex supported by siloxides ligands [(U(OSi(OtBu)3)3)2(μ-O)]23,39,40 and can be explained by the lower reducing ability of 3 compared to the siloxide complex (see Electrochemistry section).
Similar to the reduction of the oxide complex A, the addition of 2.0 equiv. of KC8 to B at −80 °C in THF-d8 resulted in a mixture of U(III)/U(IV), “[K(THF)x][(U(OAr)3)2(μ-S)]” and U(III)/U(III), “[K(THF)x]2[(U(OAr)3)2(μ-S)]” complexes as indicated by 1H NMR spectroscopy (Fig. S20†). The addition of excess 3.0 equiv. of KC8 (5.0 equiv. in total) to B at −80 °C in THF-d8 led to the full consumption of U(III)/U(IV), “[K(THF)x][(U(OAr)3)2(μ-S)], and the concomitant formation of a putative U(III)/U(III), complex “[K(THF)x]2[(U(OAr)3)2(μ-S)]” and KOAr, as observed by 1H NMR spectroscopy (Fig. S20†). Attempts to isolate single crystals of the putative “[K(THF)x]2[(U(OAr)3)2(μ-S)]” for XRD analysis were not successful. In addition, storing the reaction mixture in THF-d8 at −40 °C for 3 weeks resulted in the full decomposition of “[K(THF)x]2[(U(OAr)3)2(μ-S)]” and the formation of [KU(OAr)4], KOAr, and other species, as observed by 1H NMR spectroscopy (Fig. S21†).
Only a few dark crystals suitable for X-ray crystallography analysis of the complex [(K(THF)4)2(U(OAr)2)2(μ-S)2] (5) (Scheme 3) were obtained from a concentrated THF/n-hexane mixture at −40 °C. Attempts to isolate analytically pure complex 5 by changing different recrystallization conditions proved unsuccessful due to the similar solubility of the 5, [KU(OAr)4] and KOAr.
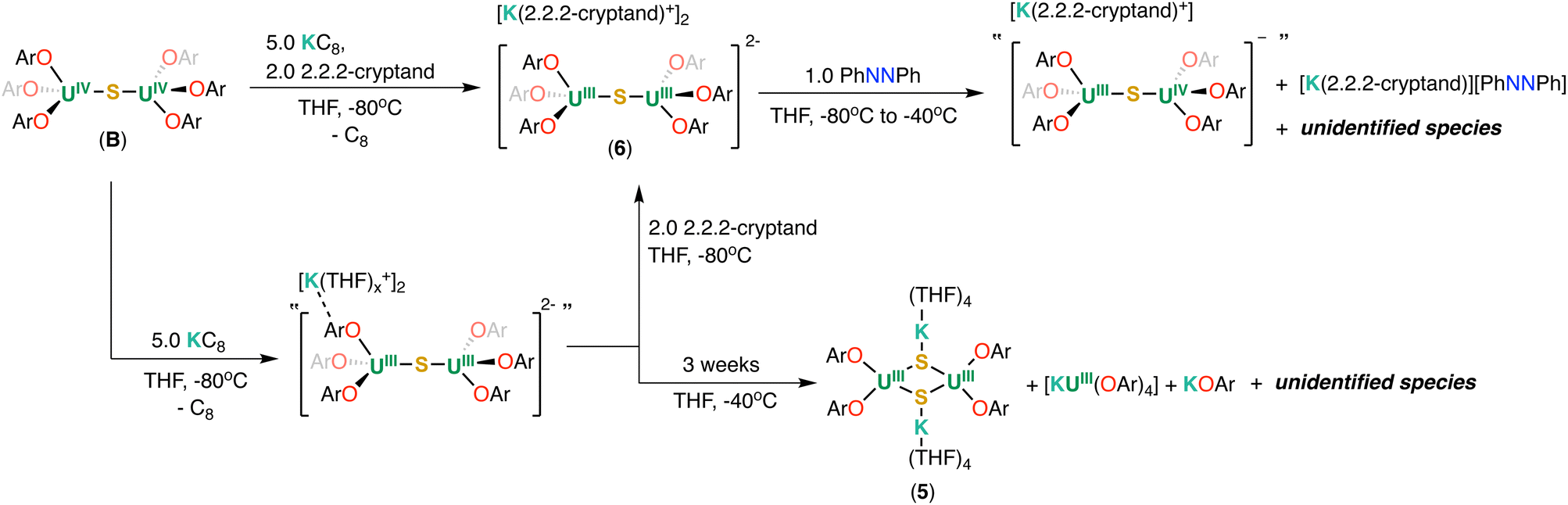 | ||
| Scheme 3 Reduction of B and reactivity of 6 with azobenzene. | ||
Complex 5 crystallizes in the space group P, with the full molecule generated by inversion symmetry. The solid-state structure of 5 (Fig. 5) shows a dinuclear complex consisting of two equivalent U(III) moieties, bridged by two sulphide ligands. Each uranium centre is tetra-coordinated in a distorted tetrahedral geometry and is bound by two –OAr ligands and the two bridging sulphides. Two K+ cations are bound by one sulphide ligand and four THF molecules. Notably, one K+ ion is above the U2S2 plane, while the other one is below the plane. The U–OAr bond distances (2.2191(19)–2.220(2) Å) are longer than those found in the U(IV) precursor, [(U(OAr)3)2(μ-S)] (U–OAr: 2.079(9)–2.125(8) Å).7 The U–S distances (2.6946(7)–2.6988(8) Å) are longer than those reported for uranium(IV) sulphides dimers supported by cyclopentadienyl ligands and –OSi(OtBu)3 ligands, [(η5-1,3-R2C5H3)2U2(μ-S)2] (R = Me3C or Me3Si) (2.605(2)–2.612(1) Å)50,51 and [Cs(THF)2(U(OSi(OtBu)3)3)2(μ-S)2] (2.639(3)–2.677(3) Å).38 The values of the U–S–U angles are equivalent in 5 (91.52(2)°) and are smaller than those reported for the bis-sulphide U(IV) complexes (95.19(7)–98.34(10)°).38,50,51 Moreover, the OAr–U–OAr, S–U–OAr and S–U–S bond angles are in the range of 88.48(2)–119.24(6)°.
 | ||
| Fig. 5 Molecular structure of [(K(THF)4)2(U(OAr)2)2(μ-S)2] (5) with portions of the aryloxide ligands and THF ligands depicted as wire frames for clarity. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms and methyl groups on the –OAr ligands have been omitted for clarity. | ||
As with the oxide system, performing the reduction of B in the presence of 2.2.2-cryptand allowed the isolation of the desired U(III) sulphide complex. Upon addition of 2.0 equiv. of 2.2.2-cryptand and 5.0 equivalents of KC8 to B at −80 °C, a new set of resonances appeared in the 1H NMR spectrum (Fig. S23†) recorded at −80 °C. Eventually, dark single crystals suitable for X-ray diffraction studies were obtained in 81% yield from a concentrated THF/n-hexane mixture at −40 °C, and were identified as the complex [K(2.2.2-cryptand)]2[(U(OAr)3)2(μ-S)] (6) (Scheme 3).
Complex 6 is stable at −40 °C in THF-d8 for up to 1 week as showed by 1H NMR spectroscopy (Fig. S25†), suggesting that the encapsulation of the cations in the cryptand prevents their binding to the ligands and rearrangement processes from occurring.
Complex 6 crystallizes in the triclinic space group P. The solid-state molecular structure of complex 6 (Fig. 6 and Table 1) shows an ion pair consisting of two [K(2.2.2-cryptand)]+ cations and the [(U(OAr)3)2(μ-S)]2− dianion. The two uranium(III) ions in 6 are bridged by a sulphide (S2−) ligand, and each is bound by three –OAr ligands. The U–OAr bond distances (2.236(4)–2.250(4) Å) are longer compared to the U(IV) precursor, [(U(OAr)3)2(μ-S)] (2.079(9)–2.125(8) Å)7 and the previously reported U(IV) [(((t-BuArO)3tacn)U)2(μ-S)] (2.203(4) Å)33 supported by polydentate aryloxide ligand, but are comparable to complex 5 (2.2191(19)–2.220(2) Å). The U–S bond distances (2.6612(16)–2.6668(16) Å) are longer than those found in [(U(OAr)3)2(μ-S)] (2.5881 Å) and [(((t-BuArO)3tacn)U)2(μ-S)] (2.592(6) Å), but slightly shorter than those in complex 5 (2.6946(7)–2.6988(8) Å). The U–S–U bond angle (173.56(7)°) is slightly smaller compared to those found in the precursor (180.0(1)°) and the reported U(IV)[(((t-BuArO)3tacn)U)2(μ-S)] (180.0°). The range of OAr–U–OAr and S–U–OAr bond angles (105.93(16)–117.50(15)°) is smaller than that found in the precursor, B (92.1(2)–156.2(4)°).
| Complex | 1 | 2 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| U–OAr (Å) | 2.178(4)–2.226(4) | 2.208(3)–2.246(3) | 2.204(6)–2.223(6) | 2.2191(19)–2.220(2) | 2.236(4)–2.250(4) |
| U–X (Å) | 2.0612(5) | 2.15427(12) | 2.1421(7) | 2.6946(7)–2.6988(8) | 2.6612(16)–2.6668(16) |
| U–X–U (°) | 180.0 | 180.0 | 180.0 | 91.52(2) | 173.56(7) |
 | ||
| Fig. 6 Molecular structure of the dianionic species [(U(OAr)3)2(μ-S)]2− in (6) with portions of the aryloxide ligands depicted as wire frames for clarity. Thermal ellipsoids are drawn at the 50% probability level. Two [K(2.2.2-cryptand)]+ cations, hydrogen atoms, and methyl groups on the –OAr ligands have been omitted for clarity. | ||
Remarkably, complexes 5 and 6 are the first examples of diuranium(III) bridged by sulphide or bis-sulphide moieties. Previously, only diuranium(IV) bridged by sulphide or bis-sulphide ligands have been reported. Complex 6 showed similar reactivity with azobenzene as 3. The reaction of 6 with 1.0 equiv. of PhNNPh in THF at −80 °C, resulted in a mixture of unreacted 6 and a new species, as observed by 1H NMR spectroscopy at −80 °C (Fig. S26†). The full consumption of the starting material was observed when the reaction mixture was warmed up to −40 °C after 1 hour as indicated by 1H NMR spectroscopy (Fig. S26†). The 1H NMR spectrum of the reaction mixture showed the formation of a major species that was assigned as the mono-reduced U(III)–S–U(IV), species suggesting that complex 6 had transferred one electron to the PhNNPh species as found for the U(III)–O–U(III) complex. No reaction with N2 was observed for complex 6.
Electrochemistry
Cyclic voltammograms were measured for 3 mM THF solutions of complexes A, B, 1, and of previously reported [(U(OSi(OtBu)3)3)2(μ-O)]39 and [(U(N(SiMe3)2)3)2(μ-O)]8 (Fig. 7) complexes (Table 2). | ||
| Fig. 7 Cyclic voltammograms for complexes, A (red), B (blue), 1 (purple), [(U(N(SiMe3)2)3)2(μ-O)] (green) and [(U(OSi(OtBu)3)3)2(μ-O)] (yellow) in THF. Conditions: Pt disk working electrode, referenced to the Fc+/0 couple; 0.1 M [NBu4][BPh4] electrolyte in THF. Scan rate (100 mV s−1). Arrows indicate the scan direction. | ||
| E pc | [(U(OSi(OtBu)3)3)2(μ-O)] | [(U(N(SiMe3)2)3)2(μ-O)] | 1 | A | B |
|---|---|---|---|---|---|
| UIVUIV/UIIIUIV | −3.32 | −1.99 | −3.09 | −2.09 | −1.98 |
| UIIIUIV/UIIIUIII | — | −3.03 | — | −2.96 | −2.60 |
| UIVUV/UIVUIV | — | — | −1.03 | — | — |
| E pa | [(U(OSi(OtBu)3)3)2(μ-O)] | [(U(N(SiMe3)2)3)2(μ-O)] | 1 | A | B |
|---|---|---|---|---|---|
| UIIIUIII/UIIIUIV | −2.48 | −2.81 | — | −2.70 | −2.29 |
| UIIIUIV/UIVUIV | — | −1.78 | −2.90 | −1.85 | −1.66 |
| UIVUIV/UIVUV | — | — | −0.84 | — | — |
The cyclic voltammogram for complex A in THF revealed two quasi-reversible redox events at values of E1/2 = −2.83, and −1.97 V vs. Fc+/0. The two measured waves display reduction events with Epc values of −2.09 V and −2.96 V vs. Fc+/0, respectively. The event at −2.09 V vs. Fc+/0 is assigned to the reduction of the U(IV)–O–U(IV) complex to the U(III)–O–U(IV) analogue, while the more negative event is assigned to the reduction of the mixed valent species to the U(III)–O–U(III) species with both events being in the range of values reported in the literature for the U(IV)/U(III) couple.52 The large difference (0.86 V) between the redox potential measured for the two uranium centres indicate a strong degree of electronic communication between them. Large separation between the U(IV)/U(V), U(V)/U(V), and U(V)/U(VI) redox couples have been previously reported for bis-oxo bridged complexes supported by polydentate tris-aryloxide ligands [(((nP,MeArO)3tacn)UV)2(μ-O)2] and were explained in terms of highly covalent U–O bonds in the bis(μ-oxo) diamond core that supports the electronic coupling.53
The values of reduction potential measured for A are significantly more positive than the value reported for the U(III)/U(IV) (Epc value at −3.32 V vs. Fc+/0) couple in the bridging oxide complexes supported by –OSi(OtBu)3 ligands,39 indicating a lower reducing power of the U(III)–O–U(III) moiety when supported by aryloxide ligands compared to siloxides. However, these values are comparable to the two different U(III)/U(IV) couples (Epc values at −1.99 V and −3.03 V vs. Fc+/0) measured for the bridging oxide complex supported by –N(SiMe3)2 ligands.
Furthermore, the values of the reduction potentials of A (Epc values at −2.09 V and −2.96 vs. Fc+/0) are more negative than those reported for the U(III) monomeric complex, [U(OAr)3](OAr = 2,6-di-tert-butylphenoxide) (Epc value at −1.30 V vs. Fc+/0),7 which, unlike 3, can reduce N2 in non-polar solvents. Unfortunately, the reactivity towards N2 could not be probed for complex 3 in non-polar solvents due to the low solubility. However, the lack of reactivity with N2 may also be due to the presence of aryloxide sterically hindering the U(III)–O–U(III) cavity, which prevents N2 side-on binding.
The voltammogram of B exhibits similar redox events, which can be assigned to the two U(III)/U(IV) couples. The two reduction potentials for B (Epc values at −1.98 and −2.60 V vs. Fc+/0) are more positive than those of A with a larger peak separation of 0.86 V for A compared to B (0.62 V) suggesting that the sulphide (S2−) linker decreases the reducing power of the complex compared to the oxide linker and promotes a weaker electronic communication.
In contrast, the voltammogram of 1 shows two distinct redox processes that are further apart compared to those observed for A and B. The first cathodic redox event (E1/2 = −3.0 V vs. Fc+/0) is assigned to the U(III)/U(IV) couple while the second event (E1/2 = −0.94 V vs. Fc+/0) is assigned to the U(IV)/U(V) couple. Only the one-electron reduction to U(III)/U(IV) is observable for 1 and is significantly shifted to more negative potentials compared to the oxide A and sulphide B complexes. These results indicate that the nature of both supporting ligands and single-atom linker affect significantly the redox potential of single-atom bridged dinuclear uranium complexes. Nitride linkers shift significantly the first reduction potential towards very negative values compared to sulphide and oxide which prevents the isolation of U(III)/U(III) complexes. The presence of siloxide ligands also shifts to very negative potentials the U(IV)/U(III) reduction potential compared to amide and aryloxide ligands, but chemical reduction with KC8 allowed to isolate U(III)–O–U(III) complexes that showed the ability to reduce and even cleave dinitrogen,23,35 suggesting electronic stabilization provided by the multiple binding modes available to the siloxide ligands.
Magnetic properties
To further investigate the electronic structures and bonding in the U(IV) oxide (A), sulphide (B), nitride (1), U(III) oxide (3) and sulphide (6) complexes, variable temperature magnetometry experiments were performed for complexes A, B, 1, 3 and 6 under an applied magnetic field of 1 T (Fig. 8). The measured values of the magnetic moment per ion at 300 K are 3.94μB for A, 3.39μB for B, 3.57 for 1 and decrease smoothly to a low value of 0.77μB for A and B and 0.40μB for 1 at 2 K and tending to zero, which is typical of the magnetic singlet 5f2 uranium(IV) ions subject to modest temperature-independent paramagnetism.54–56 In the case of 6, the magnetic moment per ion is 3.29μB at 300 K. The temperature dependence shows a smooth decrease until 10 K, and a steeper one below that temperature, reaching a value of 1.77μB at 2 K. This trend is characteristic of uranium(III) ions, owing to the 4I9/2 ground state of the f3 ion.55,57 On the other hand, complex 3 possesses a magnetic moment per ion at 300 K of 3.15μB, which decreases to a low value of 0.60μB at 2 K. Similar low values at 2 K have previously been observed even in U(III) complexes.55 | ||
| Fig. 8 Temperature dependent SQUID magnetization data plotted as a function of μeffvs. temperature (top), and χMvs. temperature (bottom) for complexes A (red), B (blue), 1 (purple), 3 (green) and 6 (black) (measured under an applied field of 1 T). | ||
The temperature dependence of the magnetic susceptibility (Fig. 8), for complexes A, B and 6 shows its continuous increase when decreasing the temperature, as expected for isolated uranium(IV) (A, B)33,53,58,59 and uranium(III)55,57 (6) ions. On the other hand, the U(IV) ions in complex 1 are antiferromagnetically coupled, as evidenced by the maximum at 70 K in the χMvs. T plot. Unambiguous antiferromagnetic coupling is very rare for U(IV)–U(IV) complexes2,33 and is usually weaker, but we recently reported a similar behaviour with a maximum at 90 K in the χMvs. T plot for the uranium(IV) nitride-bridged complex [NBu4][(U(N(SiMe3)2)3)2(μ-N)].32 Interestingly, complex 3 also exhibits a maximum at 9 K in the χMvs. T plot (Fig. 8), this maximum and downturn suggest the presence of antiferromagnetic exchange coupling, but the contribution of the single-ion crystal field effect cannot be ruled out.2
Dinuclear complexes of uranium(III) are rare and only a few examples of magnetic communication between U(III) centres have been reported.19,39,60 These include the diuranium(III) bridging nitride and oxide complexes, [K3(UIII(OSi(OtBu)3)3)2(μ-N)] and [Cs2(UIII(OSi(OtBu)3)3)2(μ-O)],19,39 which showed antiferromagnetic coupling with a maximum at 23 K and 20 K, respectively. Stronger antiferromagnetic coupling with the highest value of χM at 110 K was reported for an arene-bridged U(III) dimer.25
Computational studies
To gain some insights into the reduction behaviour of complexes A, B and 1, DFT calculations (B3PW91) including solvent and dispersion corrections were carried out. For the sake of comparison, the tris(tert-butoxy)siloxide equivalent of these three complexes were also computed (labelled SiA, SiB and Si1). The reduction of the complexes with KC8 was computed as two successive single electron reduction with the formation of a mixed valence intermediate complex (Fig. 9). | ||
| Fig. 9 Computed reduction reactions of complexes SiA, SiB, Si1, A, B and 1 by KC8. The energies are given in kcal mol−1 at 298 K. | ||
The two-electron reduction is computed to be thermodynamically favourable for all complexes. However, a closer look at the results of Fig. 9 clearly shows that first the reductions are easier for aryloxide complexes than for the siloxide ones, in line with the experimentally observed higher reducing power of the siloxide complexes. Secondly, it appears that the reduction of the nitride complexes is more complicated than the oxides and sulphides, since the first electron transfer reaction is endothermic (endergonic), which is again in line with experiment. The latter can be easily explained by analysing the boding situation in the complexes A, B and 1 using Natural Bonding Orbital (NBO) analysis. In the starting complexes, NBO indicates presence of two single U–X bonds which are strongly polarized toward X (82 to 91%) so that bonding seems similar at first sight. However, at the second order donor–acceptor, the nitride complex is the only one to display large donation from nitrogen lone pairs to empty uranium orbitals, resulting in electron delocalization in between the nitride and the two uranium. This delocalization strengthens the U–N bonds as evidence by the Wiberg Bond Indexes (WBI) which are around 1 for O and S (0.79 and 1.03) while it grows to 1.22 for N. This multiple bond character of two U–N bonds involves more uranium orbital in bonding and therefore less prompted to accept electrons, making it less reducible. The difference of reducing power between the siloxide and aryloxide ligand was also analysed using bonding analysis. It is noteworthy that the nature of U–OAr and U–OSi are quite different in the different U–O–U complexes. Indeed, as reflected in the Natural Charges (see ESI†), the U–OSi bond appears to much more ionic than the U–OAr ones. Indeed, the oxygen charges in the former is around −1.2 while is it only −0.8 in the latter complexes. Since the charge of uranium centres are similar with the two sets of ligands, it clearly means that the empty f orbitals at uranium are higher in energy in the U(IV)–O–U(IV) siloxide compared to the U(IV)–O–U(IV) aryloxide case making this complex more difficult to reduce as observed experimentally.
Conclusions
In summary, we reported the synthesis, redox and magnetic properties of a series of N3−, O2−, and S2− bridged diuranium complexes supported by bulky aryloxide ligands and compared their properties to analogous complexes supported by siloxides. The U(IV)/U(IV) nitride [Cs(THF)8][(U(OAr)3)2(μ-N)], 1 could be prepared and characterized but could not be chemically reduced. Reduction of the neutral U(IV)/U(IV) complexes [(U(OAr)3)2(μ-X)], A (X = O) and B (X = S) led to the isolation and characterization of rare U(III)/U(IV) and U(III)/U(III) analogues. Notably, complexes [(K(THF)4)2(U(OAr)2)2(μ-S)2], 5 and [K(2.2.2-cryptand)]2[(U(OAr)3)2(μ-S)], 6 are the first examples of U(III) sulphide bridged complexes. The redox properties of diuranium complexes are significantly different for siloxide and aryloxide supported complexes and indicate a significantly stronger reducing power for the siloxide complexes. In contrast, the easily accessible first U(III)/U(IV) couple facilitate one-electron transfer reactions to substrates such as azobenzene for the aryloxide supported diuranium complexes. Computational studies show that the nature of U–OAr and U–OSi are quite different in the U–O–U complexes with the U–OSi bond more ionic than the U–OAr ones. As a result, the empty f uranium orbitals are found higher in energy in the U(IV)–O–U(IV) siloxide compared to the U(IV)–O–U(IV) aryloxide rendering the analogue U(III)–O–U(III) significantly more reducing. The redox behaviour of the oxide- and sulphide-bridged complexes is similar while reduction of the nitride bridged complexes is more complicated due to the significant multiple bond character of the U–N–U bridge. Such different interaction is corroborated by the observed magnetic communication for the U(IV)–N–U(IV) complex compared to the U(IV)–O–U(IV) and U(IV)–S–U(IV) systems, showing a magnetic behaviour typical of isolated U(IV) ions. Finally the data reported here show that both single-atom linkers and supporting ligands can be used to tune magnetic communication and redox reactivity in diuranium complexes.Author contributions
F.-C. H. designed and performed all the experiments, analysed the data, prepared all the figure and wrote the manuscript; L. B. isolated and characterised 1; T. R. and L. M. performed the computational studies, analysed them and wrote the computational section; I. Z. measured and analysed the magnetic data; R. S. measured and analysed the XRD data; M. M. conceived and supervised the project, analysed the data, wrote the manuscript.Data availability
Synthetic details, analytical data including depictions of all spectra and coordinate data of all computationally optimised species, are documented in the ESI.† Crystallographic data is made available via the CCDC. The data that support the findings of this study are openly available in the Zenodo repository at https://doi.org/10.5281/zenodo.12191401.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant numbers 212723, and the École Polytechnique Fédérale de Lausanne (EPFL). We thank Dr. Farzaneh Fadaei Tirani for important contributions to the X-ray single crystal structure analyses. L. M. is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing.References
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- B. M. Gardner, D. M. King, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2017, 8, 6207–6217 RSC.
- D. M. King, B. E. Atkinson, L. Chatelain, M. Gregson, J. A. Seed, A. J. Wooles, N. Kaltsoyannis and S. T. Liddle, Dalton Trans., 2022, 51, 8855–8864 RSC.
- J. Z. Du, D. Hunger, J. A. Seed, J. D. Cryer, D. M. King, A. J. Wooles, J. van Slageren and S. T. Liddle, J. Am. Chem. Soc., 2021, 143, 5343–5348 CrossRef CAS PubMed.
- J. Z. Du, D. M. King, L. Chatelain, E. L. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- T. M. Rookes, B. M. Gardner, G. Balázs, M. Gregson, F. Tuna, A. J. Wooles, M. Scheer and S. T. Liddle, Angew. Chem., Int. Ed., 2017, 56, 10495–10500 CrossRef CAS PubMed.
- L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2013, 32, 1193–1198 CrossRef CAS.
- S. M. Franke, F. W. Heinemann and K. Meyer, Chem. Sci., 2014, 5, 942–950 RSC.
- C. Camp, M. A. Antunes, G. Garcia, I. Ciofini, I. C. Santos, J. Pecaut, M. Almeida, J. Marcalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC.
- C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed.
- M. Keener, L. Maria and M. Mazzanti, Chem. Sci., 2023, 14, 6493–6521 RSC.
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed.
- L. Maria, I. C. Santos, V. R. Sousa and J. Marcalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed.
- P. L. Arnold, C. J. Stevens, N. L. Bell, R. M. Lord, J. M. Goldberg, G. S. Nichol and J. B. Love, Chem. Sci., 2017, 8, 3609–3617 RSC.
- R. J. Ward, S. P. Kelley, W. W. Lukens and J. R. Walensky, Organometallics, 2022, 41, 1579–1585 CrossRef CAS.
- N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed.
- D. K. Modder, C. T. Palumbo, I. Douair, R. Scopelliti, L. Maron and M. Mazzanti, Chem. Sci., 2021, 12, 6153–6158 RSC.
- D. K. Modder, C. T. Palumbo, I. Douair, F. Fadaei-Tirani, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2021, 60, 3737–3744 CrossRef CAS PubMed.
- L. Barluzzi, M. Falcone and M. Mazzanti, Chem. Commun., 2019, 55, 13031–13047 RSC.
- M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed.
- P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS.
- B. Vlaisayljevich, P. L. Diaconescu, W. L. Lukens Jr., L. Gagliardi and C. C. Cummins, Organometallics, 2013, 32, 1341–1352 CrossRef.
- V. Mougel, C. Camp, J. Pecaut, C. Coperet, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed., 2012, 51, 12280–12284 CrossRef CAS PubMed.
- R. A. K. Shivaraam, M. Keener, D. K. Modder, T. Rajeshkumar, I. Zivkovic, R. Scopelliti, L. MaronMaron and M. Mazzanti, Angew. Chem., Int. Ed., 2023, 62, e202304051 CrossRef PubMed.
- M. Keener, F. Fadaei-Tirani, R. Scopelliti, I. Zivkovic and M. Mazzanti, Chem. Sci., 2022, 13, 8025–8035 RSC.
- J. X. Wang, Y. Gurevich, M. Botoshansky and M. S. Eisen, Organometallics, 2008, 27, 4494–4504 CrossRef CAS.
- J. X. Wang, Y. Gurevich, M. Botoshansky and M. S. Eisen, J. Am. Chem. Soc., 2006, 128, 9350–9351 CrossRef CAS PubMed.
- L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC.
- C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC.
- O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC.
- B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689–2694 CrossRef CAS.
- O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC.
- L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed.
- N. Jori, T. Rajeshkumar, R. Scopelliti, I. Zivkovic, A. Sienkiewicz, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 9232–9242 RSC.
- N. Jori, M. Keener, T. Rajeshkumar, R. Scopelliti, L. Maron and M. Mazzanti, Chem. Sci., 2023, 14, 13485–13494 RSC.
- N. Jori, PhD, EPFL, 2023.
- A. G. Tskhovrebov, B. Vuichoud, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2013, 135, 9486–9492 CrossRef CAS PubMed.
- J. Du, D. M. King, L. Chatelain, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS PubMed.
- J. M. Berg, D. L. Clark, J. C. Huffmann, D. E. Morris, A. P. Sattelberger, W. E. Streib, W. G. Van Der Sluys and J. G. Watkin, J. Am. Chem. Soc., 1992, 114, 10811–10821 CrossRef CAS.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433–3436 CrossRef CAS.
- L. Barluzzi, S. R. Giblin, A. Mansikkamaki and R. A. Layfield, J. Am. Chem. Soc., 2022, 144, 18229–18233 CrossRef CAS PubMed.
- D. Q. Wang, G. H. Hou, G. F. Zi and M. D. Walter, Organometallics, 2020, 39, 4085–4101 CrossRef CAS.
- S. C. Wang, T. Y. Li, Y. Heng, G. H. Hou, G. F. Zi and M. D. Walter, Organometallics, 2021, 40, 2149–2165 CrossRef CAS.
- J. Riedhammer, D. P. Halter and K. Meyer, Chem. Rev., 2023, 123, 7761–7781 CrossRef CAS PubMed.
- A. C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- J. A. Seed, L. Birnoschi, E. L. Lu, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem, 2021, 7, 1666–1680 CAS.
- I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC.
- E. J. Schelter, P. Yang, B. L. Scott, J. D. Thompson, R. L. Martin, P. J. Hay, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2007, 46, 7477–7488 CrossRef CAS PubMed.
- G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2008, 47, 3040–3042 CrossRef CAS PubMed.
- M. Tricoire, N. Jori, F. Fadaei-Tirani, R. Scopelliti, I. Zivkovic, L. Natrajan and M. Mazzanti, Chem. Commun., 2024, 60, 55–58 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2362157–2362161. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01819b |
| This journal is © The Royal Society of Chemistry 2024 |