
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal- versus ligand-centered reactivity of a cobalt-phenylenediamide complex with electrophiles†
Minzhu
Zou
 ,
Sewwandi
Kuruppu
,
Thomas J.
Emge
and
Kate M.
Waldie
*
,
Sewwandi
Kuruppu
,
Thomas J.
Emge
and
Kate M.
Waldie
*
Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA. E-mail: kate.waldie@rutgers.edu
First published on 17th July 2024
Abstract
A new series of [CoIII–CF3]n+ complexes supported by a bidentate redox-active ligand is presented. The cationic [Co–CF3]+ complex was first obtained by reacting [CpCo(tBuUreaopda)] (Cp = cyclopentadienyl, opda = o-phenylenediamide) with an electrophilic trifluoromethyl source, for which the redox-active phenylenediamide ligand serves as a 2e− reservoir to generate [CpCp(tBuUreabqdi)(CF3)]+ (bqdi = benzoquinonediimine). Electrochemical studies of [Co–CF3]+ revealed two reversible 1e− reductions. Chemical reduction with 1 or 2 equiv. reducing agent enabled isolation of the neutral and anionic complexes, respectively, where the [CoIII–CF3] bond remains intact in all three oxidation states (n = +1, 0, −1). Structural analysis shows systematic changes to the redox-active ligand backbone upon reduction, consistent with sequential ligand-centered electron transfer in the series [bqdi]0 to [s-bqdi]˙− to [opda]2−. In contrast, the reaction of [CpCo(tBuUreaopda)] with alkyl triflates resulted in ligand-centered alkylation at the ureayl groups instead of the targeted Co–alkyl bond formation, suggesting less favorable bond formation at cobalt and greater nucleophilic accessibility of the ligand compared to the metal center.
Introduction
The installation of trifluoromethyl substituents in organic molecules is widely prevalent in pharmaceuticals and agrochemicals.1–6 The trifluoromethyl (CF3) group substantially enhances various molecular properties, including lipophilicity, metabolic stability, and bioavailability. While a host of trifluoromethylation reagents have been developed by Tyrra (AgCF3),7 Ruppert and Prakash (TMS–CF3),8–10 Togni,11 Umemoto,12,13 and Ritter,14 organic trifluoromethylation is often achieved via transition metal catalysis (e.g., Cu) to promote C–CF3 bond formation.15–21 However, metal–CF3 bonds, particularly with low-valent late transition metals, are considerably more robust and less reactive compared to the corresponding metal–alkyl analogues.22,23 Thus, forcing conditions (e.g., high temperatures) are generally required to activate and cleave the M–CF3 bond and regenerate the active catalyst, which limits the scope of compatible substrates. Alternatively, two other successful strategies to activate M–CF3 bonds have been demonstrated: (a) oxidation at the metal to favor C–CF3 reductive elimination at ambient temperatures,24–26 and (b) photo-induced homolysis of the M–CF3 bond to afford a reactive CF3˙ radical species.27,28By the latter approach, Soper and co-workers reported the reaction of a CoII-redox-active ligand complex with a trifluoromethyl electrophile, generating a CoIII–CF3 bond with concomitant 1e− oxidation of the ligand.28 The redox activity of the ligand was also proposed to facilitate Co–CF3 bond homolysis by enabling access to a CoII redox isomer upon visible light irradiation. In a related copper system, formation of a CuII–CF3 complex is enabled by the redox-active ligands, where 1e− transfer from two redox-active ligands occurs without changing the oxidation state of the metal.29 These examples highlight the utility of redox-active ligands. Since high formal oxidation states are not always stable nor accessible at first-row late transition metal centers, the installation of redox-active ligands can provide greater flexibility to the electronic structure of the complex by serving as an electron reservoir during redox events,30,31 and their redox potentials can be tuned to match the driving force required for specific transformations.32
Our studies with redox-active ligands have focused on cobalt complexes bearing the phenylenediamide ligand, which has three accessible oxidation states: phenylenediamide (opda2−), semi-benzoquinonediimine (s-bqdi˙−), and benzoquinonediimine (bqdi0) (Scheme 1a).33,34 In the [CpCo(Ropda)] architecture (Cp = cyclopentadienyl), the opda ligand substituents have a significant effect on the redox potentials of the complex, but in all cases, a reversible 2e− oxidation featured is observed.33 Recently, we showed that the isopropyl derivative (R = iPr) reacts with electrophilic trifluoromethyl sources, leading to CoIII–CF3 bond formation via opda ligand-to-substrate 2e− transfer.34 This Co–CF3 bond is robust and does not participate in radical trifluoromethylation, although electrochemical studies suggested that Co–CF3 bond activation may occur under reducing conditions. While the first 1e− reduction of [iPrCo–CF3]+ is reversible and yielded an isolable paramagnetic species [iPrCo–CF3]0, the second 1e− reduction is chemically irreversible (Scheme 1b).
 | ||
Scheme 1 (a) Sequential 1e− redox activity of metal-phenylenediamide complexes. Reductive stability of [CoIII–CF3]+ complexes determined by the (b) isopropyl34 or (c) t-butyl ureayl substituents (this work) on the redox-active ligand. R = (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)NHtBu. (d) Electrophile addition to 1 at the metal or ligand. O)NHtBu. (d) Electrophile addition to 1 at the metal or ligand. | ||
Herein, we expand our investigations to establish how the presence of electron-withdrawing ureayl substituents on the opda ligand affects the reactivity of [Co–CF3]n+ complexes. In contrast to the isopropyl derivative, the cationic [tBuUreaCo–CF3]+ complex exhibits two reversible 1e− reduction features, which allows the neutral and anionic complexes to be isolated (Scheme 1c). Structural characterization of both reduced complexes demonstrates that the Co–CF3 bond is fully maintained upon reduction and confirms that both electron transfers are localized to the redox-active ligand core. In contrast, the analogous CoIII–alkyl bond formation is not observed between 1 and alkyl triflate reagents, instead resulting in O-alkylation at the carbonyl groups of the ureayl substituents (Scheme 1d).
Results and discussion
Synthesis and characterization of [Co–CF3] complex 2
The synthesis and characterization of the neutral Co-phenylenediamine complex 1 was previously reported.33 In brief, the tBuUreaopdaH2 ligand precursor was deprotonated with 2 equiv. potassium hydride, followed by salt metathesis with CpCo(CO)I2 in THF. Complex 1 is diamagnetic and exhibits a two-legged piano-stool coordination geometry with the electron-rich tBuUreaopda ligand and η5-Cp ligand. Treatment of 1 with Umemoto's reagent ([DBT–CF3]+) in anhydrous MeCN at 25 °C results in a gradual color change from deep purple to dark red (Scheme 2). After 2 days, the red product was collected and purified by column chromatography and recrystallization. Complex 2 was isolated in excellent yield (92%) and was characterized by NMR spectroscopy, mass spectrometry, and single crystal X-ray crystallography. The 1H NMR spectrum shows the signal for the Cp ring protons at δH 5.81 ppm, which is more downfield than the Cp signal for the neutral complex 1 (δH 5.01 ppm in CD3CN; Fig. S8†).33 The 1H NMR resonances appear sharper with higher resolution in C6D6, and the urea NH signal for 2 is clearly observed in this solvent (δH 8.25 ppm; Fig. S9†). The trifluoromethyl ligand appears at δF −11.45 ppm by 19F NMR (Fig. S11†), which falls within the typical range observed for other CoIII–CF3 systems.27,34–36 The high-resolution mass analysis of 1 shows the signal corresponding to the [CpCo(tBuUreabqdi)(CF3)]+ cation (Fig. S29†), further confirming the successful installation of the trifluoromethyl ligand into this complex. | ||
| Scheme 2 Synthesis of complex 2. | ||
Dark red block single crystals of 2 suitable for X-ray diffraction were obtained by layering hexanes upon a saturated cobalt solution in THF at −35 °C. As shown in Fig. 1, the structure of 2 exhibits a clear change in the coordination geometry at the metal, transitioning from the two-legged piano stool geometry in 1 to the three-legged piano stool geometry in 2 due to formation of the new Co–trifluoromethyl bond. The Co–CF3 bond lengths are 1.953(3) and 1.964(3) Å for the two molecules in the unit cell, which lie within the typical bond lengths observed for other CoIII–CF3 complexes in the CCDC database (1.88–1.98 Å). In this structure, the ureayl substituents are oriented such that the NH groups are directed on the same side of the Co-metallocycle plane, with both NH bonds forming a hydrogen bonding interaction with one triflate anion (Fig. S1†). Looking at the phenylene ligand backbone, the C–C bond lengths alternate between short (ca. 1.34 Å) and long (ca. 1.44 Å), consistent with the fully-oxidized bqdi state of the redox-active ligand.37 This assignment is further supported by the N–Cphenylene bond lengths (1.306–1.313 Å), which are consistent with nitrogen–carbon double bonds and indicate an increase in the oxidation state of the ligand compared to 1. Thus, complex 2 is formulated as a coordinatively saturated complex with a low-spin CoIII center with neutral bqdi ligand. This result mirrors the reactivity seen for [CpCo(iPropda)] with [DBT–CF3]+, where the redox-active ligand provides two electrons for Co–CF3 bond formation via ligand-to substrate 2e− transfer.34
 | ||
| Fig. 1 Structure of 2. Hydrogen atoms and OTf− counterions omitted for clarity. tert-Butyl groups shown as capped sticks for clarity. Ellipsoids shown at 30% probability. | ||
In our previous work with [CpCo(iPropda)], we found that the rate of trifluoromethyl addition to the metal correlated with the difference in the redox potentials of the metal complex and the electrophilic CF3 source.34 Here, we note that the more positive reduction potential for 1, due to the electron withdrawing properties of the ureayl substituents, results in a larger potential difference with Umemoto's reagent (ΔE = E1/2(1) − Ep,c([DBT–CF3]+) = 0.76 V). Consequently, the trifluoromethylation reaction with 1 is slow: it takes 24 h to reach 93% conversion in the presence of 2 equiv. [DBT–CF3]+, or 98 h to reach the same conversion with 1.1 equiv. [DBT–CF3]+ (Fig. S12–S15†). Nonetheless, this reaction proceeds with high yield, resulting in a stable [CoIII–CF3]+ product that has been isolated and fully characterized.
The redox properties of 2 were studied by cyclic voltammetry (CV) in MeCN with 0.1 M [nBu4N][PF6] (Fig. 2). This complex exhibits two reversible reductions at E1/2 = –0.18 and −0.68 V vs. Fc+/0, both of which are 1e− processes. A further chemically-irreversible reduction is observed when the working electrode potential is scanned further negative – the peak potential for this feature is Ep,c = −1.99 V vs. Fc+/0 at 100 mV s−1 (Fig. S42†). For the reversible redox features, the oxidative and reductive peak currents show a linear dependence on the square root of the scan rate (ν1/2), confirming freely diffusing species in solution (Fig. S43†). Several previous reports have demonstrated the electrochemical properties of half-sandwich [CpCo] complexes, which typically show reversible or quasi-reversible features associated with the CoIII/CoII and CoII/CoI redox couples.38–47 However, the redox potentials for 2 are more positive by comparison, despite the presence of the formally anionic CF3 ligand; for example, [CpCo(bpy)(MeCN)]2+ (bpy = 2,2′-bipyridine) undergoes reduction at −0.40 and −1.15 V vs. Fc+/0 in MeCN.38,43 This deviation suggests that one or both reduction processes for 2 may not be localized to the metal center. We recently reported that the isopropyl analogue [CpCo(iPrbqdi)(CF3)]+ undergoes 1e− reduction at the redox-active bqdi ligand, but the second 1e− reduction is not reversible.34 The high reversibility of both reduction processes for 2 suggests that [Co–CF3]n+ may be isolable in three different oxidation states as the cationic, neutral, and anionic complexes (n = +1, 0, −1). This consideration prompted our further studies to explore the behavior of this complex under reducing conditions (vide infra).
 | ||
| Fig. 2 CV of 2 in MeCN (1 mM [2] and 0.1 M [nBu4N][PF6]; scan rate 100 mV s−1). | ||
Synthesis and characterization of [Co–CF3] complexes 3 and 4
To probe the structure and stability of 2 upon reduction by one or two electrons, we selected suitable reducing agents based on the observed redox potentials to isolate the singly- and doubled-reduced complexes. Chemical reduction of 2 with 1 equiv. decamethylferrocene (Cp*2Fe, E1/2 = −0.51 V vs. Fc+/0 in MeCN)48 results in a rapid reaction, generating the neutral complex 3 in high yield within 10 min at 25 °C (Scheme 3, step B). Complex 3 is easily purified by extraction into pentane and filtration to remove the decamethylferrocenium triflate salt, which is insoluble in nonpolar solvents. High-resolution mass spectrometry analysis confirms that the Co–CF3 bond remains intact in this product (Fig. S30†). As expected, the 1e− reduction of 2 results in a paramagnetic species that could not be characterized by NMR spectroscopy. Evans NMR method was employed to determine the magnetic moment of 3 in solution, which was found to be 1.75 μB (Fig. S16†). This value is consistent with an S = ½ system.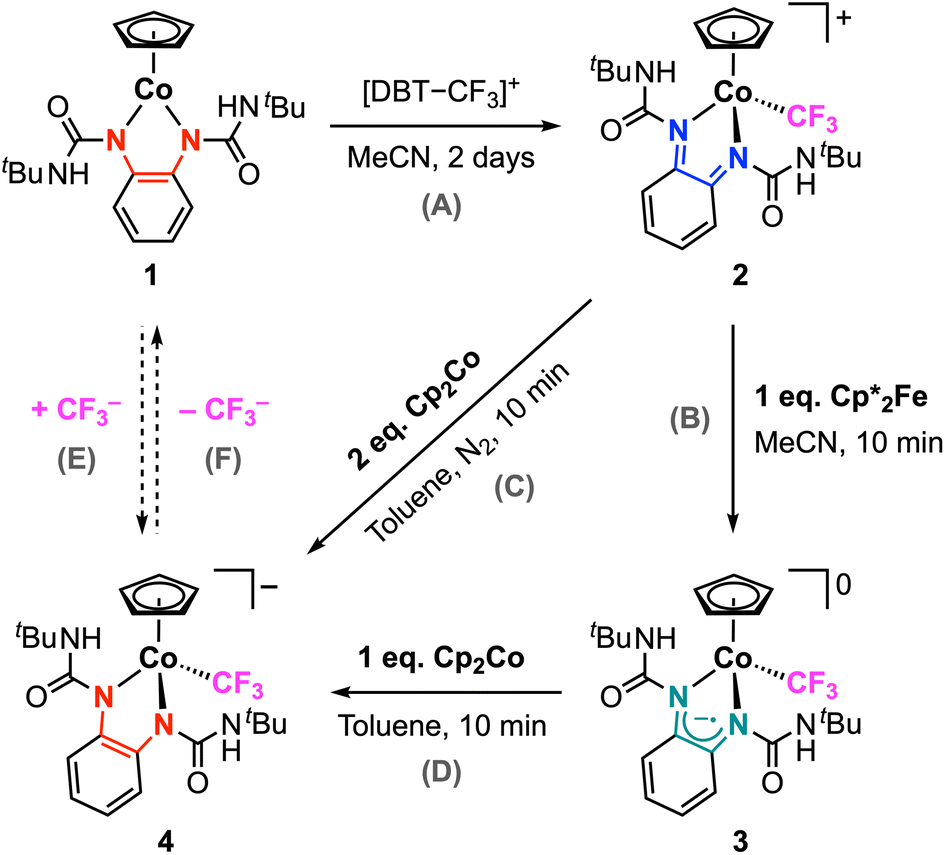 | ||
| Scheme 3 Synthesis of 3 and 4via CF3+ addition to 1 and chemical reduction of 2. | ||
The slow evaporation of a saturated cobalt solution of 3 in pentane at −25 °C yielded dark plate single crystals suitable for X-ray diffraction. The structure of 3 is shown in Fig. 3a and selected structural metrics are presented in Table 1, which highlights the key differences between 3 and 2. It is evident that the 3-legged piano stool geometry is maintained in 3, indicating that 1e− reduction of 2 does not change the coordination number at the metal. The Co–CF3 bond length exhibits a small contraction from 1.953(3) to 1.924(3) Å for 2 and 3, respectively, suggesting increased covalency upon reduction. There are minor lengthenings of the Co–N bonds (< 0.03 Å). The most significant structural changes are observed in the redox-active ligand. The N–Cphenylene bond lengths are much longer (1.352(3) and 1.357(3) Å) for 3, indicating an increase in the ligand oxidation state and suggesting that 1e− reduction of 2 occurs at the bqdi ligand to generate the semi-benzoquinonediimine radical anion (s-bqdi˙−).37 Furthermore, the phenylene ring backbone shows a smaller degree of alternation in the C–C bond lengths compared to 2, which is also consistent with an intermediate s-bqdi˙− oxidation state assignment. Overall, this structural analysis indicates that 3 is best described as a low-spin CoIII with the s-bqdi˙− ligand radical anion. This formulation is also supported by the EPR analysis of 3, which shows characteristic features for a ligand-centered radical and closely resembles the EPR metrics for the previously reported isopropyl analogue [CpCo(iPrs-bqdi)(CF3)]0 (Fig. 4).34
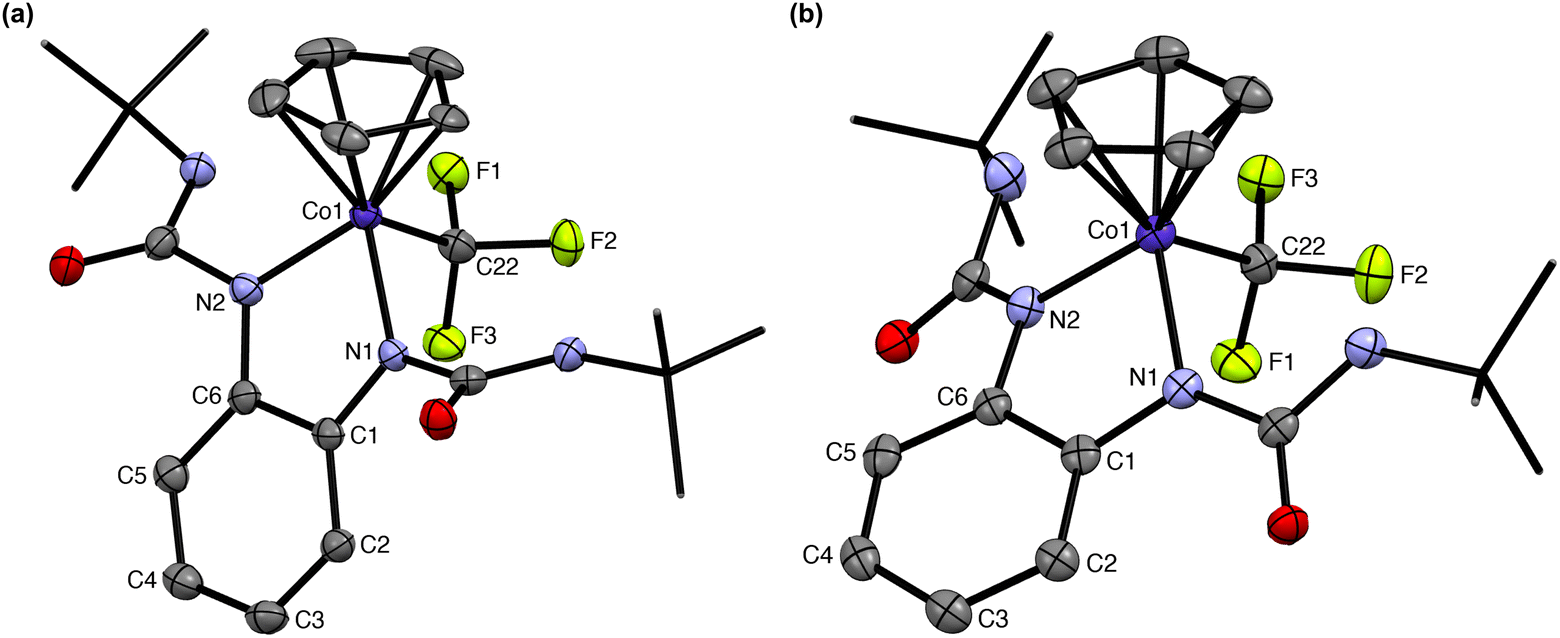 | ||
| Fig. 3 Structures of (a) 3 and (b) 4. Hydrogen atoms and counterion omitted for clarity. tert-Butyl groups shown as capped sticks for clarity. Ellipsoids shown at 50% and 30% probability, respectively. | ||
 | ||
| Fig. 4 EPR spectrum of 3 in toluene at room temperature. giso = 1.9926. A(14N) = 17.3 MHz, A(14N′) = 16.9 MHz, A(59Co) = 56.2 MHz. Linewidth = 0.71 mT. | ||
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
3 | 4 | |
|---|---|---|---|
| a The bond lengths and angle of 2 are listed for one molecule in the crystal lattice, and the second molecule showed similar parameters. | |||
| Co1–N1 | 1.907(2) | 1.916(2) | 1.964(2) |
| Co1–N2 | 1.893(2) | 1.931(2) | 1.932(2) |
| Co1–CF3 | 1.953(3) | 1.924(3) | 1.940(3) |
| Co1–Cpcentroid | 1.686 | 1.716 | 1.721 |
| N1–C1 | 1.309(4) | 1.352(3) | 1.409(4) |
| N2–C6 | 1.313(4) | 1.357(3) | 1.405(4) |
| N1–Co1–N2 | 82.15(10) | 82.60(9) | 83.83(10) |
This electronic structure assignment was further corroborated using DFT calculations (see ESI† for details). The calculations were performed using the BP86 functional with the def2-TZVP basis set (and def2-TZVPP for Co), which we have previously utilized to probe the electronic structure of this family of complexes.33,34 We find that these methods are able to accurately capture the structural metrics of complex 3. Using this approach, the doublet ground state of this complex was confirmed, with the spin density being localized on the redox-active ligand (80%), primarily on the nitrogen atoms (46% total). As shown in Fig. 5, only 10% of the spin density is localized on the cobalt center.
 | ||
| Fig. 5 Mulliken spin density plot (isovalue = 0.005) of [CpCo(tBuUreas-bqdi)(CF3)] 3 using BP86/def2TZVP(C,H,N,O,F)/def2TZVPP(Co). | ||
Chemical reduction of 2 with 2 equiv. cobaltocene (Cp2Co, E1/2 = −1.33 V vs. Fc+/0 in CH2Cl2)49 yields the anionic complex 4, which can also be obtained via chemical reduction of 3 with 1 equiv. cobaltocene (Scheme 3, steps C and D). The red-brown complex 4 is diamagnetic and displays well-resolved NMR data. This analysis was performed in C6D6, which is surprisingly able to solubilize this product despite it being a metal complex salt. The Cp signal appears at δH 4.59 ppm, which is the most upfield chemical shift in this series of [CpCo] complexes. This trend is consistent with the highly electron-rich, anionic character of 4 compared to the neutral (1 and 3) and cationic (2) complexes. Notably, the 19F NMR spectrum still shows a signal at δF −2.52 ppm consistent with the presence of the trifluoromethyl ligand coordinated to cobalt. This is further supported by the high-resolution mass spectrum that shows the signal corresponding to 4 (detected as 4+; Fig. S31†). Complex 4 exhibits reasonable stability in solution, with no decomposition observed after 24 h (Fig. S20†). This behavior contrasts with [iPrCo–CF3]n+, which could only be isolated in the cationic (n = 1) and neutral (n = 0) states as stable species.34 Thus, complexes 2–4 provide a remarkable demonstration of three isolable oxidation states where the [Co–CF3] bond remain intact.
Single crystals of 4 were obtained by vapor diffusion of pentane into a saturated cobalt solution in THF. For 4, the metal complex is anionic and thus the crystal structure shows the presence of the cobaltocenium cation in the lattice (Fig. S3†). The structure of the anion is highlighted in Fig. 3b. Clearly, the Co–CF3 bond is still present in this complex, with the length of this bond being 1.940(3) Å. The C–Nphenylene bond lengths, 1.405(4) and 1.409(4) Å, are typical of carbon–nitrogen single bonds in the dianionic opda2− ligand.37 The C–C bonds in the phenylene backbone have distances between 1.39–1.40 Å, consistent with the aromatic benzene ring in the fully reduced ligand. Given these structural metrics, we assign 4 as a low-spin CoIII center with three anionic ligands: Cp−, CF3−, and the dianionic opda2− ligand. There is an increase in the Co–N bond lengths upon each 1e− reduction (i.e., 2 < 3 < 4), suggesting a decrease in the Co-redox-active ligand bond strength as the ligand gets reduced but the coordination number at cobalt remains unchanged. This trend is consistent with the frontier molecular orbitals for this series (vide infra): the SOMO of 3 and HOMO of 4 show π*-antibonding character between the cobalt d and ligand π* orbitals, correlating with a decrease in metal–ligand bonding interactions upon reduction. In addition, we note that the ureayl substituents in 4 are approaching coplanarity with the phenylenediamide core – the angles between the urea groups and the central N–C–C–N plane are only 14–25°, whereas the corresponding angles in 2 and 3 are significantly larger (47–60° and 66–82°, respectively). This suggests some degree of delocalization of electron density from the electron rich opda2− onto the electron-withdrawing ureayl arms. We also see that the Co–N bond lengths are noticeably different, with one bond showing significant lengthening (1.932(2) vs. 1.964(2) Å), causing a slight twisting distortion in the phenylenediamide backbone. These structural changes may contribute to the small increase in Co–Cpcentroid bond distance upon each 1e− reduction due to differences in the steric profile of the coordinated ligand.
In principle, complex 4 could be obtained via the direct nucleophilic addition of the trifluoromethyl anion at the cobalt center in 1 (Scheme 3, step E). To probe this possible route to 4, complex 1 was treated with 2 equiv. TMS–CF3 and 2 equiv. CsF in THF. No reaction was observed after 18 h. We note that 1 and the other neutral cobalt complexes in this family do not exhibit reactivity with other common anionic ligands (e.g., chloride, iodide, etc.). Thus, the direct addition of nucleophile sources to 1 is not accessible, but we are still able to generate 4via our alternative stepwise procedure. In this novel route, we take advantage of the nucleophilic nature of 1 to achieve Co–CF3 bond formation via ligand-to-substrate 2e− transfer, followed by the stepwise addition of 2e− electrons via electrochemical or chemical reduction (Scheme 3, steps A–D). Overall, this process is equivalent to the net addition of CF3− to 1, and thus may be considered formal umpolung of the original electrophilic CF3+ source to the anionic CF3− ligand. Despite 4 being a very electron-rich anionic complex, the Co–CF3 bond is robust. Desage-El Murr, Fensterbank, and co-workers reported a CuII–CF3 complex that, upon heating, underwent intramolecular nucleophilic transfer to the ligand, demonstrating the formal umpolung of the starting CF3+ reagent.29 Similarly, we examined the stability of 4 by heating a solution in CD3CN at 50 °C for several hours. After one day, no significant changes were observed in the 1H and 19F NMR spectra, indicating that the transfer of CF3− from this complex is not readily accessible under thermal conditions (Scheme 3, step F).
Absorption spectra comparison
The UV-vis-NIR absorption behavior of complexes 2–4 was investigated in MeCN solution (Fig. S34–S36†). The electronic absorption spectrum of 2 displays two dominant absorbances at 440 and 536 nm in the visible range, with extinction coefficients of 6000 and 4100 M−1 cm−1, respectively. These features are assigned as metal–ligand and intra-ligand charge transfer transitions observed for diiminoquinone-type complexes.50,51 The neutral complex 3 shows significantly different absorbance features, with strong UV-vis absorbances between 300–400 and 500–600 nm and the appearance of near-IR bands between 800–1000 nm, typical of metal complexes containing ligand-radical species.52 The electronic absorption spectrum of 4 exhibits a strong band at 290 nm (extinction coefficient = 17 000 M−1 cm−1), but otherwise shows only weak transitions in the visible region.The frontier molecular orbitals for 2–4 are presented in Fig. 6. For each complex, there is clearly little contribution from the trifluoromethyl ligand to either the HOMO or LUMO. The HOMO of 2 is π*-antibonding with contributions from both the cobalt dxy and ligand π orbitals, in agreement with the DFT calculations for the related acetonitrile complex [CpCo(tBubqdi)(MeCN)]2+.33 The LUMO of 2 is π*-antibonding with respect to the cobalt dyz and ligand π* orbitals, but this orbital has much more pronounced ligand character. The SOMO of 3 is very similar to the LUMO of 2 and shows major ligand π* contributions, as does the HOMO for the fully reduced complex 4. This trend is consistent with both reduction processes being centered on the redox-active ligand without any change in the cobalt oxidation state: for each 1e− reaction, the electron addition primarily occurs to the π* orbitals of bqdi and s-bqdi for 2 and 3, respectively. The HOMO of 4 shows some π delocalization onto the ureayl arms thanks to the electron-withdrawing properties of these substituents and the extreme electron richness of this anionic complex. The ability of the ureayl groups to accept electron density in this fashion likely contributes to its stability. The LUMO of 4 shows no contributions from the phenylenediamide ligand, as expected for the fully reduced opda2− dianionic ligand.
 | ||
| Fig. 6 Calculated frontier molecular orbitals (isovalue = 0.04) for (a) 2, (b) 3, and (c) 4. | ||
The electrochemical reduction of 2 was also followed in situ by UV-vis-NIR spectroelectrochemistry (SEC). Initial electrochemical reduction of 2 by SEC results in clear isosbestic points at 400 and 565 nm, accompanied with absorbance changes in the visible range and the appearance of weak transitions in the near-IR range that are assigned to 3 (Fig. 7a). Scanning the working electrode potential further negative causes the reduction of 3 to in situ generate 4, which is evidenced by the growth of the absorbance bands at 292 nm and decay of the absorbances at 382, 498, and 575 nm (Fig. 7b). Remarkably, following the production of 4 in the SEC cell, the application of an oxidative potential to the working electrode results in the full recovery of the electronic transitions associated with 3 and then 2 (Fig. S37†), consistent with the high reversibility of these redox processes observed in our CV studies.
 | ||
| Fig. 7 UV–vis SEC reductions in MeCN in 0.2 M [nBu4N][PF6]. (a) One-electron reduction of 2 (orange trace) to 3 (blue trace). (b) One-electron reduction of 3 (blue trace) to 4 (cyan trace). | ||
Mid-IR spectroscopy was also employed to probe the CO and N–H stretching vibrations of the ureayl substituents in 1–4 (Fig. S38–S41 and Table S4†). The CO stretching frequencies in 1 and 3 are very similar, suggesting comparable carbonyl bond strengths despite their different geometries and oxidation state assignments. Complex 2 shows the highest CO stretching frequency, consistent with the least electron density on the electron-withdrawing ureayl arms in this cationic complex. The carbonyl stretching frequency decreases with each 1e− reduction process (i.e., 2 > 3 > 4) as the electron density in the redox-active ligand backbone and delocalization onto the ureayl groups increases. The N–H stretching frequencies in 2–4 show the opposite trend, with this frequency increasing as the electron richness of the complex increases.
Reactivity of complex 1 with alkyl triflates
Given the great success of electrophilic trifluoromethyl addition at 1, we were inspired to explore the analogous reaction with electrophilic alkyl sources. We note that M–CF3 and M–CH3 (or M–alkyl) bonds are generally considered to have very different properties, and M–alkyl complexes are typically more reactive than the analogous perfluorinated derivatives.22 As an initial NMR scale investigation, 1 was treated with excess methyl triflate in CDCl3, resulting in a gradual color change from dark purple to dark blue. This reaction was repeated in diethyl ether with stirring overnight at 25 °C. The blue product was successfully isolated, but its structure was wholly inconsistent with the anticipated alkyl addition product [CpCo(tBuUreabqdi)(CH3)]+. Instead, characterization of this product by NMR spectroscopy, mass spectrometry, and structural analysis confirms its formulation as complex 5, where electrophilic methyl addition has occurred at both carbonyl groups in the ligand scaffold (Scheme 4a). We note that the use of methyl triflate is critical here, as no reaction is observed when 1 is treated with methyl iodide.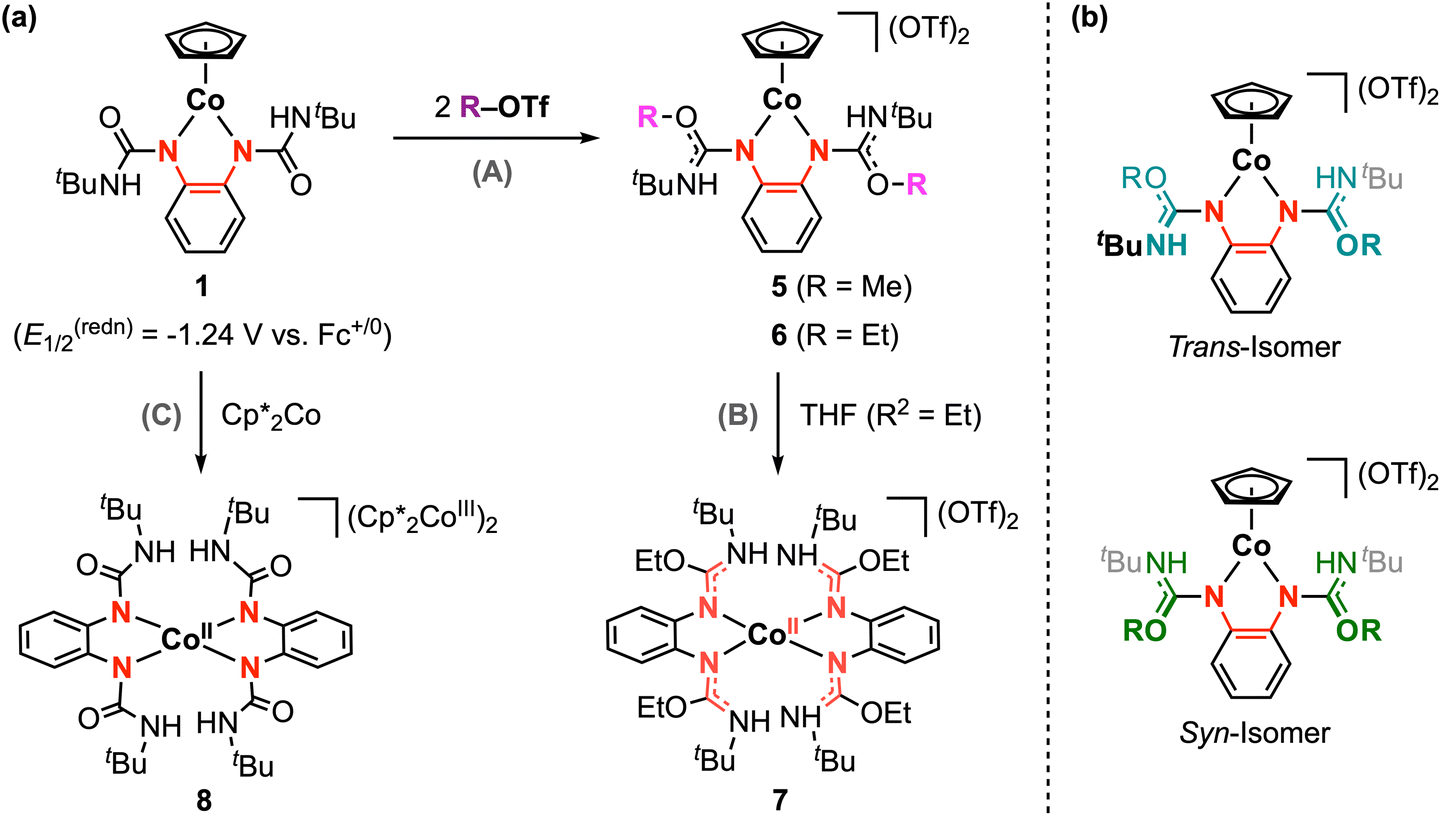 | ||
| Scheme 4 (a) Reactivity of 1 with alkyl triflates and observed reductive decomposition pathways. (b) trans and syn isomers of O-alkylated complexes. | ||
The 1H NMR of 5 shows the Cp signal at δH 5.59 ppm, which is shifted downfield by 0.57 ppm compared to the starting complex 1 (Fig. S21†). This trend is consistent with the increased charge of the dicationic complex 5, but this signal is more upfield that that of other dicationic [CpCo] half-sandwich complexes with neutral nitrogen-based ligands,43 likely due to the lack of through-bond communication between the [CpCo] fragment and the ureayl substituents (vide infra). The 1H and 13C NMR spectra of 5 also reveal the presence of two structural isomers in solution that differ with respect to the relative position of the two methoxy groups with respect to the Co-phenylenediamide plane. These species are assigned as the trans and syn isomers, which are related via rotation of the C–N single bond on the ureayl arms (Scheme 4b). The isomers exhibit distinct chemical shifts for their respective ligand NH and methoxy signals, as well as a small difference in the chemical shift of the Cp signal. Lowering the temperature of the NMR solution from 25 °C to −25 °C increases the sharpness of the signals, but overall, the ratio of the two isomers remains roughly unchanged at 82:18 (Fig. S26†). We have previously observed similar behavior in solution for [CpCo(iPropda)] where two conformations that differ with respect to the orientation of the isopropyl substituents are resolved at lower temperatures.33 DFT calculations for 5 predict that the trans isomer is slightly more stable by −0.24 kcal mol−1, but this energy difference between the trans and syn isomers should be considered negligible within the expected accuracy of these methods (Table S6†). Thus, we are not able to confidently assign which isomer of 5 is favored in solution.
Complex 5 is not stable in solution at room temperature over several hours, and recrystallization attempts at low temperature largely resulted in high-defect crystals as both isomers compete for crystallization. Luckily, a high-quality single crystal of the trans isomer was obtained by vapor diffusion of pentane into a saturated THF solution at −35 °C. The structure of 5 and select structural metrics are provided in the ESI.† The C–Nphenylene bond lengths have increased to 1.377(6) and 1.391(5) Å, indicating a slight decrease in bond order compared to 1. The bond lengths in the phenylene ring are very similar for 1 and 5, suggesting this complex is best described as a CoIII center and an electron-rich opda-type ligand with some degree of delocalization across the metallocycle. The C–O urea bond lengths are significantly longer in 5 (1.298(6) and 1.304(6) Å) compared to 1 (1.219(3) and 1.227(2) Å), while the C–NtBu bond lengths are noticeably shorter (1.288(6) and 1.295(5) Å) compared to 1.341(3) and 1.343(3) Å for 5 and 1, respectively. These bond lengths changes indicate that methylation of the carbonyl groups in 1 results in delocalization of the positive charge and partial double-bond character across the O–C–NtBu fragment in 5. In this structure, the ureayl substituents are nearly perpendicular to the phenylenediamide backbone: the angles between the planes defined by the urea groups and the plane defined by the Co-phenylenediamide fragment are 78° and 81°. Thus, no substantial delocalization or electronic communication is expected between the inner coordination sphere of the complex and the ureayl substituents, likely accounting for the two-legged piano stool geometry of 5. Typically, cationic [CpCoIII] complexes prefer a coordinatively-saturated, three-legged piano stool geometry, and the two-legged geometry is reserved for [CpCoI(L)2]0 complexes (L = neutral) or [CpCoIII(X)2]0 systems with formally anionic X− ligands. To the best of our knowledge, no examples of [CpCoIII(L)2]2+ have been reported to date. In our case, the bidentate ligand in 5 is formally neutral and the three-legged geometry may be expected. However, a zwitterionic description for the ligand is more reasonable – methylation at the carbonyl groups installs the positive charge on the ligand periphery, and the lack of extended conjugation means the electron-rich phenylenediamide core remains largely unaffected. Thus, the ligand-centered reactivity at 1 increases the charge of the complex, while the unique structure of the resulting methylated ligand in 5 enables the two-legged geometry to be maintained.
This ligand-centered reactivity is also observed from the reaction of ethyl triflate with 1, yielding complex 6 following the same procedure described above (Scheme 4a). High-resolution mass spectrometry confirms the incorporation of two ethyl groups in this product. The 1H and 13C NMR analysis for 6 shows similar diagnostic signals as 5. Again, two species are clearly observed in this data, which are analogously assigned as the trans and syn isomers of 6. The ratio of the isomers in solution is identical within error for 5 and 6. As shown in Fig. S27,† the 1H NMR resonances for 6 are sharper and the J-coupling patterns for ethyl substituents are readily resolved. This contrasts with the broader signals observed for 5, which is reasonably attributed to the increased steric bulk of the ethoxy substituents causing greater restriction to the rotation of the C–N bonds and less facile interconversion between isomers. Complex 6 is unstable in solution and decomposes at ambient conditions; however, we were able to confirm the structure of the trans isomer by single-crystal X-ray diffraction (Fig. S5†). The urea C–O and C–NtBu bond lengths are within 0.01 Å of the corresponding values for 5. The phenylenediamide structural metrics are also very similar to 5, leading us to assign 6 as a CoIII complex with a zwitterionic bidentate ligand.
The reaction of 1 with R–OTf (R = Me, Et) stands in stark contrast to the anticipated Co–R bond formation, in which ligand-to-substrate 2e− transfer would be promoted by bond formation at the cobalt center. As discussed above, we have observed a correlation between the accessibility of Co–CF3 bond formation and the redox potential difference between the metal complex and the electrophilic reagent.33 Considering the very negative reduction potentials of electrophilic alkylation reagents53 and the rather positive potential for oxidation of 1, the potential difference between 1 and the alkyl triflate may be too large to be effectively compensated for by Co–R bond formation. This effect is compounded by the anticipated weaker strength of Co–alkyl vs. Co–perfluoroalkyl bonds, providing less driving force for cobalt–carbon bond formation with methyl or ethyl triflate.22 Furthermore, the divergent ligand-centered reactivity with alkyl triflates also suggests that in the absence of accessible electrophile addition at the metal, the ureayl groups have sufficient nucleophilicity to undergo facile and rapid alkylation at the oxygen centers. Thus, the redox-active ligand substituents have a significant impact on directing substrate reactivity and selectivity, and the design of future systems should carefully consider these factors to achieve selective bond formation at the metal as opposed to ligand functionalization.
As briefly mentioned above, complexes 5 and 6 slowly decompose in solution at room temperature. It is not always feasible to determine the product(s) of decomposition; however, we obtained single crystals for one decomposition product (complex 7, Fig. S6†) while attempting to recrystallize complex 6 from a THF/pentane solution at −35 °C. The X-ray structure of 7 shows a distorted tetrahedral geometry with two opda-type ligands at the cobalt center. The C–NtBu and C–Nphenylene bond lengths are both ca. 1.32 Å, indicating delocalization across these bonds and intermediate carbon–nitrogen double bond character. In addition, two triflate counterions were found in the crystal lattice. Thus, complex 7 is best formulated as [CoII(EtL0)2](OTf)2. In a related experiment, chemical reduction of 1 with decamethylcobaltocene (Cp*2Co, E1/2 = −1.91 V vs. Fc+/0 in MeCN)49 also resulted in Cp ligand loss to afford complex 8, [CoII(tBuUreaopda)2]2−, where both redox-active ligands are in the dianionic opda2− form (Fig. S7†). The mechanism by which these reductive decomposition reactions occur has not been elucidated and may warrant further investigation in the future.
Conclusions
We have demonstrated that the reactivity of [CpCo(Ropda)] complexes with electrophilic reagents can be directed to occur either at the cobalt center accompanied with two-electron transfer from the redox-active ligand or entirely at the ligand scaffold where the cobalt center merely serves as the supporting core. The well-defined [Co–CF3]+ complex 2 was obtained through the reaction of the neutral Co-phenylenediamide complex 1 with an electrophilic CF3 source, achieved via ligand-to-substrate two-electron transfer. Detailed studies showed that 1 can be reversibly reduced to the corresponding neutral and anionic species, enabling isolation of [Co–CF3]n+ in three different oxidation states, where the oxidation state of cobalt is unchanged. This reactivity scheme provides a novel stepwise route to achieve the net addition of CF3− to cobalt, which is not accessible via direct addition. In contrast, we have shown that complex 2 undergoes selective reaction at the ligand ureayl groups in the presence of alkyl electrophiles. Comparing this divergent reactivity, we arrive at two general requirements to achieve Co–R bond formation through redox-active ligand promoted reactivity: (1) The difference between the redox potentials for the neutral cobalt complex and electrophilic substrate (R+) should be sufficiently small to maintain the requisite thermodynamic driving force for the overall ligand-to-substrate 2e− transfer and Co–R bond formation. This requirement is more limiting for alkyl electrophiles that generally form weaker Co–C bonds compared to the trifluoromethyl ligand. (2) The redox-active ligand substituents must be carefully selected to tune the redox properties of the complex while also not introducing possible sites for off-target, ligand-centered electrophile addition. This study thus provides insights for the design of redox-active ligand scaffolds to promote selective bond forming reactions.Author contributions
Conceptualization, M. Z. and K. M. W.; methodology, M. Z. and K. M. W.; investigation, M. Z., S. K., and T. J. E.; writing – original draft, M. Z. and K. M. W.; writing – review and editing, M. Z., S. K. and K. M. W.; supervision, K. M. W.Data availability
Crystallographic data for new compounds 2–8 have been deposited at the CCDC under 2294060–2294065 and 2294086.† All other data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ACS Petroleum Research Fund (65171-DNI3) and Rutgers, The State University of New Jersey. The Rigaku SYNERGY-S X-ray diffractometer was partially funded by an NSF MRI Award (CHE-2117792). We acknowledge the Office of Advanced Research Computing (OARC) at Rutgers University for providing access to the Amarel Cluster and associated research computing resources.References
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- Q. Wang, H. Song and Q. Wang, Chin. Chem. Lett., 2022, 33, 626–642 CrossRef CAS.
- W. E. Tyrra, J. Fluorine Chem., 2001, 112, 149–152 CrossRef CAS.
- I. Ruppert, K. Schlich and W. Volbach, Tetrahedron Lett., 1984, 25, 2195–2198 CrossRef CAS.
- G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757–786 CrossRef CAS PubMed.
- X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- T. Umemoto and S. Ishihara, Tetrahedron Lett., 1990, 31, 3579–3582 CrossRef CAS.
- T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156–2164 CrossRef CAS.
- H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed.
- A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120–9123 CrossRef CAS PubMed.
- X. Wang, Y. Ye, S. Zhang, J. Feng, Y. Xu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 16410–16413 CrossRef CAS PubMed.
- H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793–3798 CrossRef CAS PubMed.
- N. D. Litvinas, P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 536–539 CrossRef CAS PubMed.
- P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882–2885 CrossRef CAS PubMed.
- Y. Li, L. Wu, H. Neumann and M. Beller, Chem. Commun., 2013, 49, 2628–2630 RSC.
- J. Jacquet, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Chem. Sci., 2016, 7, 2030–2036 RSC.
- O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- Y. Ye and M. S. Sanford, Synlett, 2012, 2005–2013 CAS.
- S. Liu, H. Liu, S. Liu, Z. Lu, C. Lu, X. Leng, Y. Lan and Q. Shen, J. Am. Chem. Soc., 2020, 142, 9785–9791 CrossRef CAS PubMed.
- J. R. Bour, N. M. Camasso and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 8034–8037 CrossRef CAS PubMed.
- C. F. Harris, C. S. Kuehner, J. Bacsa and J. D. Soper, Angew. Chem., Int. Ed., 2018, 57, 1311–1315 CrossRef CAS PubMed.
- C. S. Kuehner, A. G. Hill, C. F. Harris, C. A. Owens, J. Bacsa and J. D. Soper, ACS Catal., 2023, 13607–13617, DOI:10.1021/acscatal.3c03832.
- J. Jacquet, E. Salanouve, M. Orio, H. Vezin, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Chem. Commun., 2014, 50, 10394–10397 RSC.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- M. Zou, T. J. Emge and K. M. Waldie, Inorg. Chem., 2023, 62, 10397–10407 CrossRef CAS PubMed.
- M. Zou and K. M. Waldie, Chem. Commun., 2023, 59, 14693–14696 RSC.
- M. C. Leclerc, J. M. Bayne, G. M. Lee, S. I. Gorelsky, M. Vasiliu, I. Korobkov, D. J. Harrison, D. A. Dixon and R. T. Baker, J. Am. Chem. Soc., 2015, 137, 16064–16073 CrossRef CAS PubMed.
- P. Liebing, F. Oehler, M. Wagner, P. F. Tripet and A. Togni, Organometallics, 2018, 37, 570–583 CrossRef CAS.
- K. Chlopek, E. Bothe, F. Neese, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2006, 45, 6298–6307 CrossRef CAS PubMed.
- U. Koelle, J. Organomet. Chem., 1980, 184, 379–383 CrossRef CAS.
- U. Koelle and S. Ohst, Inorg. Chem., 1986, 25, 2689–2694 CrossRef CAS.
- T. Nagasawa and T. Nagata, Biochim. Biophys. Acta, 2007, 1767, 666–670 CrossRef CAS PubMed.
- M. Fang, E. S. Wiedner, W. G. Dougherty, W. S. Kassel, T. Liu, D. L. DuBois and R. M. Bullock, Organometallics, 2014, 33, 5820–5833 CrossRef CAS.
- M. van der Meer, E. Glais, I. Siewert and B. Sarkar, Angew. Chem., Int. Ed., 2015, 54, 13792–13795 CrossRef CAS PubMed.
- K. M. Waldie, S.-K. Kim, A. J. Ingram and R. M. Waymouth, Eur. J. Inorg. Chem., 2017, 2017, 2755–2761 CrossRef CAS.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- N. Elgrishi, D. A. Kurtz and J. L. Dempsey, J. Am. Chem. Soc., 2017, 2017, 239–244 CrossRef PubMed.
- D. A. Kurtz, D. Dhar, N. Elgrishi, B. Kandemir, S. F. McWilliams, W. C. Howland, C.-H. Chen and J. L. Dempsey, J. Am. Chem. Soc., 2021, 143, 3393–3406 CrossRef CAS PubMed.
- S. K. Mandal, C. Sunil and J. Choudhury, ACS Catal., 2024, 14, 2058–2070 CrossRef CAS.
- J. R. Aranzaes, M.-C. Daniel and D. Astruc, Can. J. Chem., 2006, 84, 288–299 CrossRef.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- M. van der Meer, S. Manck, S. Sobottka, S. Plebst and B. Sarkar, Organometallics, 2015, 34, 5393–5400 CrossRef CAS.
- S. Suhr, R. Walter, J. Beerhues, U. Albold and B. Sarkar, Chem. Sci., 2022, 13, 10532–10545 RSC.
- W. Kaim, Coord. Chem. Rev., 2011, 255, 2503–2513 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 714–723 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2294060–2294065 and 2294086. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01655f |
| This journal is © The Royal Society of Chemistry 2024 |