
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co3O4 (111) surfaces in contact with water: molecular dynamics study of the surface chemistry and structure at room temperature†
Tim
Kox
and
Stephane
Kenmoe
 *
*
Department of Theoretical Chemistry, University of Duisburg-Essen, Universitätstrasse 2, D-45141, Essen, Germany. E-mail: stephane.kenmoe@uni-due.de
First published on 16th July 2024
Abstract
In this work, we have used ab initio molecular dynamics at room temperature to study the adsorption and dissociation of a thin water film on Co3O4 (111) surfaces, considering the O-rich and Co-rich terminations named as the A-type and B-type surface terminations, respectively. We investigate the occupation of active sites, the hydrogen bond network at the interface and the structural response of the surfaces to water adsorption. On both terminations, water adsorbs via a partial dissociative mode. The contact layer is populated by molecular water as well as OH groups and surface OH resulting from proton transfer to the surface. The B-termination is more reactive, with a higher degree of dissociation in the contact layer with water (46%). On the B-terminated surface, water barely adsorbs on the Co2+ sites and almost exclusively binds and dissociates on the Co3+ sites. The interaction with the surface consists mostly of Co3+–Ow bonds and proton transfer exclusively to the 3-fold unsaturated surface Os1. Hydrogen bonds between water molecules in the aqueous film dominate the hydrogen bond network and no hydrogen bonds between water and the surface are observed. The A-terminated surface is less reactive. Water binds covalently on Co2+ sites, with a dissociation degree of 13%. Proton transfer occurs mostly on the 3-fold unsaturated surface oxygens Os1. Besides, short-lived surface OH arising from proton transfer to 3-fold unsaturated surface oxygens Os2 is observed. H-bonding to surface Os1 and Os2 constitutes 12.7% and 19.8% of the H-bond network, respectively, and the largest contribution is found among the water molecules (67.4%). On both surfaces, the coordination number of the active sites drives the relaxations of the outermost atom positions to the their bulk counterparts. The occupation of active sites on B-termination could reach up to 3 adsorbates per Co3+ leading to a binding motif in which the Co is octahedrally coordinated and which was observed experimentally.
1. Introduction
Co3O4 is a potential candidate for many catalytic oxidation reactions.1–6 Naturally grown nanoparticles show rhombicuboctahedral grains exposing the (111), (001) and (101) facets, with a percentage of exposure of 47%, 39% and 13% at room temperature, respectively.7 Various recipes have been proposed to selectively promote the exposure of the desired crystal facets on Co3O4 nanoparticles for particular catalytic reactions. The prominent (111) facet is found to be very active in many processes like for example the activation of 2-propanol.8 Co3O4 nano-hexagons exposing the (111) facets can be synthesized using hydrothermal routes.9,10 In such processes, water can play a controversial role as it could destabilize11,12 or stabilize co-adsorbed species.13–15 However, compared to the (001) and (101) facets that were studied at different coverage regimes,16–19 the interaction of water with the Co3O4 (111) surface has not been studied to an extent that would allow a fundamental understanding of its role in the catalytic activity.Recently, the adsorption of water (D2O) on the A-terminated Co3O4 (111) surface was investigated using temperature-programmed and time-resolved IRAS under UHV in combination with isotopic exchange experiments.20 It was found that water interacts strongly with this surface termination. At low adsorption temperature (200 K), within the submonolayer coverage regime at 200 K, OD/D2O networks with various topologies are stabilized following the partial dissociation of D2O. The surface OD groups anchor molecular D2O species. This favors the formation of OD/D2O networks instead of isolated OD species under kinetic control. Temperature destabilizes the OD/D2O network, which breaks up into OD/D2O clusters and weakens the interaction within the clusters and with the surface. This leads to the desorption of molecular D2O from 210 to 470 K. Above 470 K, only isolated OD species remain stable on the surface until they finally desorb at 540 ± 20 K.
In another study, DFT calculations and surface IRAS with isotopically labeled water were combined to study the adsorption and dissociation of water on an atomically defined Co3O4 (111) surface.21 Also in this study, the A-termination was considered and it was also found that D2O interacts strongly with the surface and OD groups were found to be stable up to 500 K and above. At lower temperature, the study confirmed the coexistence of OD groups and molecular D2O adsorbates within extended partially dissociated OD/D2O networks. Interestingly, the study revealed the existence of a new structural motif: a hybrid structure between hexaaquacobalt in solution and a surface cation, with an octahedral coordination. This binding motif consists of a “half-hydrated” surface Co2+ ion binding to three D2O molecules and was predicted to be observed on d6 to d8 transition metal cations on similar oxide surfaces.
In a following work, the structure of the Co3O4 (111) surface in thermodynamic equilibrium with realistic pressures of O2/H2O and H2/H2O was investigated, using density functional theory.22 The study supported the hydroxylation of the surface under oxidative and reductive conditions. It was found that a reductive environment promotes the exposure of surface Co2+ and large structural distortions favor the reduction and stabilization of the Co-rich termination. At 423 K for example, hydroxylation is prominent both on the O-rich and Co-rich surfaces even at water pressures as low as 10−15 bar. Meanwhile, a higher vapor pressure of water (∼10−11 bar) is needed to observe non-dissociated water molecules on the O-rich surface. The study predicted the key role of hydroxyl groups and the structure in shaping the surface's catalytic properties.
Though these contributions provided atomistic insight into the interface, the dynamic description of the interface is missing. In addition, studies have focused on the so-called A-termination (Co2+ terminated) and information on the B termination is missing. Besides, in ref. 23, based on a combined TPD and DFT study, the (001) facet was predicted to be the most attractive for liquid phase catalytic processes. The reason evoked was that on the (111) facet, water molecules may block active sites and inhibit catalytic processes taking place at low temperatures as they do not desorb even at elevated temperatures of more than 450 °C because of the highly unsaturated nature of the surface Co ions. However, in our recent work,7 the hydroxylated Co3O4 (111) surface was found to be more attractive for activation of 2-propanol decomposition compared to the (001) and (101) facets. More interestingly, the presence of surface hydroxyls at room temperature led to interesting surface reconstructions precursing a Mars–van-Krevelen catalytic route for 2-propanol oxidation at the interface.
To elucidate the controversial role of water in catalytic processes, more insight is needed into the properties of interfacial water. In this work, we use ab initio molecular dynamics simulations to investigate the adsorption and dissociation of a thin water film of 24 molecules on the A- and B-terminations of the Co3O4 (111) surface. Such an amount of water could be observed on many oxides under ambient conditions.24 We investigate the structure of interfacial water and the water induced relaxations of the underlying substrates: the magnitude of these relaxations as well as their driving force. Additionally, we investigate the proton transfer to the surface and within the water film as well as the hydrogen bond network at the interface.
2. Computational procedure and material systems
The surfaces were created from the Co3O4 bulk spinel structure described in ref. 25 and 26. Cleaving the bulk along the (111) plane leads to the formation of the so-called A- and B-termination27 displayed in Fig. 1. The surface models are stoichiometric, consisting of slabs of 13 layers followed by a vacuum region of 25 Å. One can distinguish two different layers of cobalt atoms on either side of layers of oxygen atoms. On the one hand, there are layers containing only octahedrally coordinated Co3+ atoms (layers L2, L6, and L10) and on the other hand, there are buckled atomic planes where one-half of Co consists of tetrahedrally coordinated Co2+ and the other half is made up of octahedrally coordinated Co3+ atoms (L0, L4, L8, and L12). | ||
| Fig. 1 Side view of the A- and B-terminations of the Co3O4 (111) surface (A and B). Co3+ ions are displayed in purple, Co2+ ions in green, and oxygen in red. The atomic layers are labeled on the left. Layers L6–L12 are kept frozen in all studies. The corresponding top views with different types of surface oxygens are shown below (C and D). | ||
In each case, the bottom 5 surface layers were frozen at bulk positions while the remaining part of the slab together with the water film adsorbed asymmetrically was allowed to relax. Because of the asymmetry imposed by the one-sided adsorption and freezing part of the slab, a dipole correction was applied to cancel the electric field gradient in the vacuum. An orthorhombic supercell with a (2 × 2) periodicity in the lateral directions x and y with the dimensions 19.81 Å × 11.44 Å × 40.0 Å was used for both terminations to perform molecular dynamics simulations.
Spin polarized Born–Oppenheimer molecular dynamics (MD) simulations were performed at the Γ point using the CP2K/Quickstep package.28 NVT conditions were imposed on the system using a Nosé–Hoover thermostat with a time constant of 1 ps and a target temperature of 300 K. The molecular dynamics trajectories were propagated at a time step of 0.5 fs for a total simulation time of 20 ps. Though most of the systems appear to be reasonably equilibrated from 5 ps (see Fig. S1–S6 in the ESI†), the properties reported in this study were computed considering the last 10 ps of the simulations. Within this time interval, we observed long-lived binding motifs in the contact layer as well as interfacial proton transfer.
Exchange and correlation effects were treated within the PBE formulation29 of the Generalized Gradient Approximation (GGA) plus a Grimme D3 correction30 to efficiently describe dispersion interactions. The Hubbard correction31 term (U = 2 eV) was used for a correct description of Co 3d states. The 3s, 3p, 3d, and 4s electrons of Co atoms and the 2s and 2p electrons of O atoms were treated as valence electrons, and the core electrons were described with Goedecker–Teter–Hutter (GTH) pseudopotentials. Double-ζ quality local basis functions with one set of polarization functions (DZVP) together with plane waves with a cutoff of 500 Ry were used to constitute the basis sets.
3. Results and discussion
3.1. A-termination
State of dissociation of water and active sites occupation. Fig. 2(A) shows the structure of the water film on the A-terminated Co3O4 (111) surface after 20 ps. Water molecules adsorb on this termination via covalent bonding between the top cobalt ions and water oxygens as well as hydrogen bonds to the surface oxygens. Only one water molecule out of the 24 which are present in the water film dissociates, whereby further short-lived dissociation events could be observed during the simulation time.
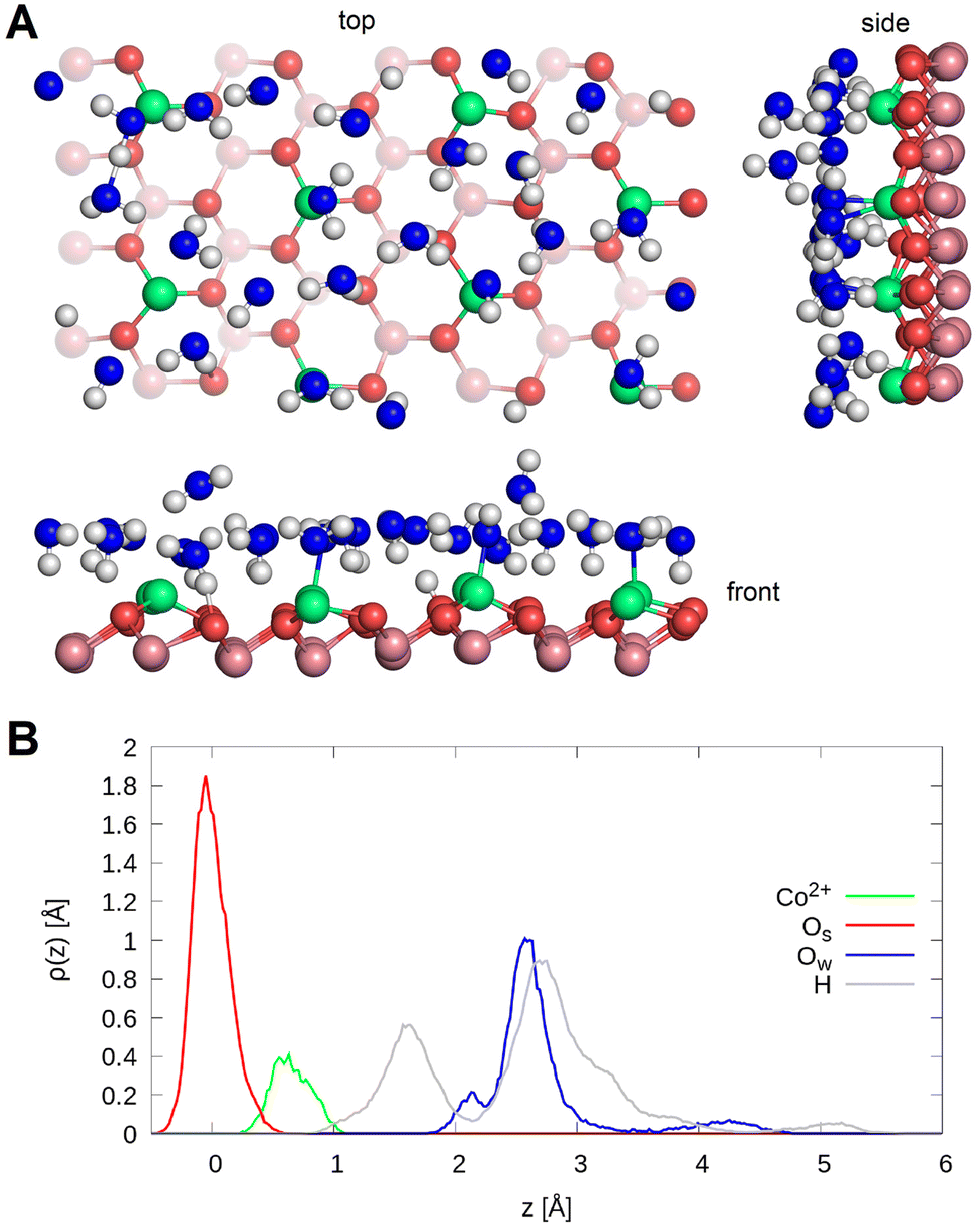 | ||
| Fig. 2 (A) The A-terminated Co3O4 (111) topmost surface layers in contact with a water film of 24 molecules. Top, side and front views of the final equilibrium trajectory. (B) Density profiles of the topmost surface cobalt and oxygens, as well as the oxygens and hydrogens of the water molecule. The center of mass of layer L1 is chosen as the reference. | ||
To investigate this in more detail, the density profiles and the radial and bond angle distributions of the topmost cobalt atoms in layer L0 to the bonded oxygen atoms were calculated. The density profiles of the oxygen atoms of the water molecules are displayed in Fig. 2(B) and show three distinct peaks. The first one located at 2.1 Å can be assigned to the water molecules, which adsorb on Co2+ ions and form a hydrogen bond to a nearby surface oxygen as well. The second peak at 2.6 Å represents water molecules, which either adsorb on a cobalt ion or form a hydrogen bond to the surface. The third peak at 4.2 Å represents the physisorbed water molecules.
Three peaks can also be observed for the density profiles of hydrogen atoms of water. These are located at 1.6 Å, 2.8 Å and 5.1 Å respectively. The peak at 1.6 Å can be assigned to the protons transferred to the surface and the hydrogen of water forming hydrogen bonds to the surface. These peaks overlap because the oxygen atom of the surface to which the proton binds protrudes by about 0.5 Å as the red tail illustrates. The peak at 2.8 Å illustrates the hydrogen atoms involved in hydrogen bonding within the water film, and the one at 5.1 Å represents the hydrogen atoms of the physisorbed water molecules.
Table 1 lists the integrals of the density profile for the different species. It can be seen that only a small proportion of the water molecules (2.6 out of 24) adsorb on the cobalt atoms and at the same time form a hydrogen bond to the oxygen atoms of the surface. The largest proportion of water molecules (19.4 out of 24) exhibit only one of the above-mentioned binding modes and the remaining proportion (2.0 out of 24) is physisorbed. Among the hydrogen atoms, 14.8 out of 48 are either transferred to the surface or form a hydrogen bond to the surface. More precisely, on an average only 1.25 of the hydrogen atoms are transferred to the surface and the rest form hydrogen bonds to the surface. The majority of hydrogen atoms (31.8 out 48) form hydrogen bonds within the water film and a minority (1.5 out of 48) belong to the physisorbed water molecules and point away from the surface.
| Atom type | Integration range (Å) | Integral | Assignment |
|---|---|---|---|
| Ow | 0.0–2.25 | 2.6 | H2O − Co2+ + H–Ow–H–Os |
| Ow | 2.25–3.5 | 19.6 | H2O − Co2+/H–Ow–H–Os |
| Ow | 3.5–6.0 | 2.0 | H2O physisorbed |
| H | 0.0–2.15 | 14.8 | H+ Os/H–Ow–H–Os |
| H | 2.15–4.4 | 31.8 | H–Ow–H–Ow |
| H | 4.4–6.0 | 1.5 | H2O physisorbed |
To investigate the strength of interfacial bonds, the radial distribution function of the topmost cobalt atoms to the oxygen atoms of molecular water and hydroxide molecules is shown in Fig. 3. It is seen that oxygen atoms of molecular water and hydroxide molecules have bonding distances of 1.8 to 2.6 Å to topmost surface cobalt atoms. The two molecules are considered separately, the hydroxide molecules are located on an average at a distance of 1.9 Å while the water molecules have a distance of 2.1 Å. The integral of the radial density distribution gives in total 9.9 water or hydroxide molecules chemisorbed to the Co2+ ions, with an average of 1.2 molecules per site.
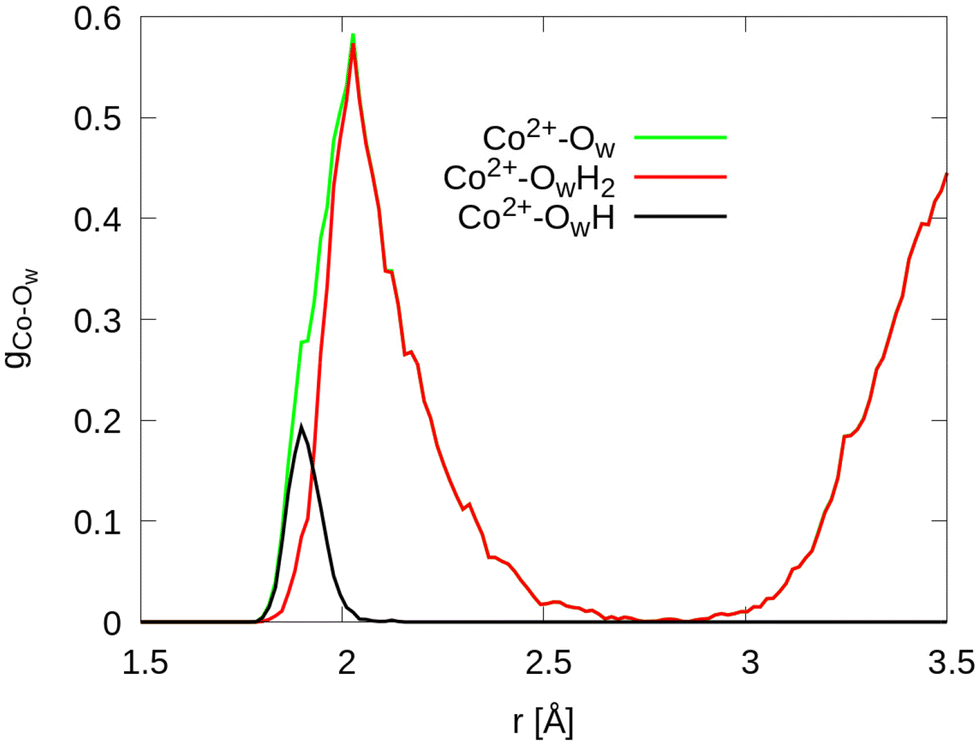 | ||
| Fig. 3 Radial distribution function of topmost Co2+ atoms in layer L0 to the oxygen atoms of the water and hydroxide molecules (Ow) for the A-terminated Co3O4 (111) surface, with projections on the intact water molecules (OwH2) and hydroxide molecules (OwH), respectively. | ||
The hydrogen bond network. For the analysis of the network of hydrogen bonds and the dissociation of the adsorbed water molecules, the radial density distribution of the different oxygens to all hydrogens and the orientation of the water OH vectors were calculated. A distinction is made between, on the one hand, the oxygen atoms of the water and hydroxide molecules denoted by Ow and, on the other hand, the surface oxygen atoms denoted by Os1 and Os2. The latter differ in the following: only Co3+ atoms in layer L2 are bound to the Os2 atoms, whereas the Os1 atoms bind to two Co3+ atoms in L2 as well as to one Co2+ in layer L0 (see Fig. 1, bottom left).
Fig. 4(A) shows the radial density distribution function of the different oxygens to all hydrogens. All the different types of oxygen have a peak between 0.8 Å and 1.3 Å. These denote the hydrogen atoms of molecular water and those of hydroxide molecules or the protons transferred to the surface Os1 and Os2. In addition, for some of the oxygens, a maximum is observed between 1.3 Å and 2.2 Å which indicates hydrogen bonds. From Table 2 that lists the corresponding integrals, it can be observed that out of 48 hydrogen atoms considered in the simulation, one is transferred to a surface oxygen of type Os1 and another one is temporarily transferred to a surface Os2 and it recombines later with a hydroxide molecule to form a water molecule chemisorbed on a Co2+ ion. The rest of the hydrogen atoms constitute the intramolecular bonding within molecular water and hydroxide molecules. The small deviation of 0.02 above 48 shows that some hydrogens are present for a short time within the radius of 1.3 Å of two oxygen atoms. In addition, from the number of hydrogen bonds listed in Table 2, it can be seen that more protons are transferred to the surface Os1, while the Os2 atoms form more hydrogen bonds to the water molecules.
 | ||
| Fig. 4 (A) Radial density distribution of the different oxygen atoms Ow, Os1 and Os2 to the hydrogen atoms. (B) Orientation of the different OH vectors H2Ow, OwH and OsH for the A-terminated Co3O4 (111) surface. | ||
| Atom type | Integration range (0.0–1.3 Å) | Integration range (1.3–2.2 Å) |
|---|---|---|
| H to Ow | 46.77 | 26.09 |
| H to OS1 | 1.0 | 4.94 |
| H to OS2 | 0.25 | 7.67 |
| H to O | 48.02 | 38.7 |
Fig. 4(B) shows the angle distribution of the OH vectors for intact water and hydroxide molecules, as well as for surface OH arising from proton transfer to the surface. Three maxima can be observed for molecular water. They are located at −1.0 (180°), −0.7 (135°) and 0.2 (78°). Most of the OH vectors are found in the vicinity of 78°. This indicates that intact water molecules lie almost parallel to the surface with a slight tilt out of the surface plane. These are the OH vectors forming the hydrogen bonds between the adsorbed water molecules. The maxima at −1.0 and −0.7 correspond to the hydrogen bonds to the surface oxygen atoms. Meanwhile, water molecules adsorbed on the cobalt atoms do not point directly to the surface and can therefore be assigned to the maximum at −0.7. The hydrogen atoms of the hydroxide molecules point away from the surface.
Only one proton is transferred to the oxygen atom Os1 and another short-lived one to the Os2 atom for a duration 1.25 ps on an average after which it recombines to form molecular water. The former occurs ∼2.5 ps after the beginning of the simulation and the resulting surface OH remains until the end of the simulation, while in the latter case, the proton is transferred back and forth and recombines twice, within an ∼5 ps time interval. The corresponding lifetimes read 0.9 ps and 1.5 ps, respectively (see Fig. S2 in the ESI†). Thus, a degree of dissociation in the contact layer of 13% can be calculated from a total of 9.9 chemisorbed water molecules, of which 1.25 are dissociated.
 | ||
| Fig. 5 (A) RDF of the topmost Co2+ ions in layer L0 to the surface oxygens. (B) Cosine of the corresponding bond angles on the A-terminated Co3O4 (111) surface. In the presence of water, the water oxygens are considered as well. The prominent water binding motifs are also displayed (C and D). | ||
| Coverage | Co2+–Os | Co2+–Ow | Co2+–O |
|---|---|---|---|
| 0 | 3.0 | — | 3.0 |
| 24 | 3.0 | 1.2 | 4.2 |
For deeper insight into the water induced relaxations, we further investigated the changes in the bond angles between the topmost Co2+ ions and their neighboring oxygen atoms at the interface, including the surface O as well as the O in the water film in contact with the surface. Their cosine distribution is shown in Fig. 5 (right). A shift in the cosine value is observed from −0.33 to −0.45 after relaxation of the bulk surface. This corresponds to an angle shift from 109.5° to 117° and refers to a change from a bulk 4-fold to 3-fold coordination of Co2+ ions on the clean surface. Upon adsorption of water, the cosine distribution shows three peaks. A peak at −0.33 (109.5°) that can be assigned to tetrahedrally coordinated Co2+ ions (4-fold coordination) and two peaks at −0.97 (166°) and 0.07 (86°) originate from 5-fold coordinated Co2+. While these two peaks could both be assigned to the bond angles between surface oxygens, Co2+ and water oxygens (Os–Co2+–Ow), the one at 0.07 is assigned exclusively to the bond angles between the Co2+ and water molecules only (Ow–Co2+–Ow).
3.2. B-termination
State of dissociation of water and active sites occupation. Snapshots of the final equilibrium trajectory of the structure of water on the B-termination after a simulation of 20 ps are shown in Fig. 6(A). This termination shows the Co3+ and Co2+ ions on the outermost surface layer. Water binds mostly to the Co3+ ions and only a small amount of water molecules bind to the Co2+ ions. Two different types of oxygen atoms are present on the surface, the 4-fold coordinated oxygens, Os1, which bridge the Co3+ and Co2+ ions in layer L0, and the 3-fold coordinated oxygens, Os2, which are only connected to the Co3+ ions in layer L2. These can be distinguished in Fig. 1 (bottom right) where the OS1 atoms are located on the edges of the hexagons, while the Os2 atoms are located in the center of the hexagons. Of the 32 oxygen atoms in layer L1, 24 can be assigned to type Os1 and 8 to type Os2. A proton is transferred to all 3-fold coordinated Os2 atoms upon dissociation of a water molecule. No hydrogen bonds to the surface oxygen atoms are observed.
 | ||
| Fig. 6 (A) The B-terminated Co3O4 (111) topmost surface layers in contact with a water film of 24 molecules. Top, side and front views of the final equilibrium trajectory. (B) Density profiles of the topmost surface cobalt and oxygens, as well as the oxygens and hydrogens of the water molecule. The center of mass of layer L1 is chosen as the reference. | ||
The density profiles for the oxygens and hydrogens of the water and hydroxide molecules are shown in Fig. 6(B). The oxygens show two peaks centered at 2.7 Å and 3.7 Å. These can be assigned to the water and hydroxide molecules chemisorbed to the cobalt atoms (2.7 Å) and to the physisorbed (3.7 Å) molecules, respectively. Four different types of hydrogen atoms are present at the interface. Hydrogen atoms represented by the peak at 0.85 Å originate from the protons transferred to the surface Os2. Water and hydroxide molecules adsorbed on the cobalt atoms form hydrogen bonds among each other; the hydrogens involved in this bonding constitute the peak located at 2.7 Å. The peak at 3.4 Å represents hydrogens which either form hydrogen bonds to the physisorbed water molecules or those of hydroxide molecules. Hydrogens of the physisorbed water molecules are furthest away from the surface (4.7 Å).
Fig. 7 shows the radial distribution functions for the Co2+ and Co3+ atoms with respect to the oxygens of the water and hydroxide molecules. It can be seen that water and hydroxide molecules adsorb on the Co2+ and Co3+ ions. This is supported by the peaks extending from 1.8 Å to 2.5 Å. Hydroxide molecules are located at 1.95 Å and are about 0.2 Å closer to the cobalt atoms than the water molecules. The integrals reported in Table 4 show that a total of 17.5 molecules are adsorbed on the Co3+ ions, of which 7.8 are hydroxide molecules and 9.7 are intact water molecules. Only one water molecule adsorbs on the Co2+ ions.
 | ||
| Fig. 7 Radial density distribution of the Co2+ and Co3+ ions in layers L0 and L1 to the oxygen atoms of the water molecule Ow for the B-terminated Co3O4(111) surface. | ||
| Atom type | Integration range (Å) | Integral | Assignment |
|---|---|---|---|
| Ow | 0.0–3.2 | 20.1 | H2O/OH − Co2+/Co3+ |
| Ow | 3.2–6.0 | 3.9 | H2O physisorbed |
| H | 0.0–1.2 | 8.0 | H + Os |
| H | 1.2–3.1 | 19.4 | H–Ow–H − Ow |
| H | 3.1–4.2 | 19.2 | H–Ow–H − Ow/Ow–H |
| H | 4.2–6.0 | 1.4 | H2O physisorbed |
The hydrogen bond network. The radial density distribution of the different oxygens to the hydrogen atoms and the orientation of the different OH vectors are shown in Fig. 8. Like on the A-termination, the segment of the radial distribution function extending from 0.8 Å to 1.3 Å can be assigned to the hydrogens forming intramolecular bonds, while those in the range between 1.3 Å and 2.2 Å can be assigned to those involved in hydrogen bonding. Exactly 8 hydrogen atoms were transferred to the 8 Os2. No further protons were transferred to the surface (see Fig. S2 in the ESI†). The water molecules form only hydrogen bonds with each other and not with the surface. This can be explained by the fact that after transferring protons to the surface Os2, all oxygens on the surface are 4-fold coordinated (see Table 5). This is also supported by the orientation of the OH vectors. Only a few OH vectors point in the direction of the surface. Most OH vectors of the water molecules are perpendicular to the surface normal and form hydrogen bonds within the water film. The OH vectors of the hydroxide molecules and the transferred protons point out of the surface plane.
 | ||
| Fig. 8 (A) Radial density distribution of the different oxygen atoms Ow, Os1 and Os2 to the hydrogen atoms. (B) Orientation of the different OH vectors H2Ow, OwH and OsH for the B-terminated Co3O4 (111) surface. | ||
| Atom type | Integration range (0.0–1.3 Å) | Integration range (1.3–2.2 Å) |
|---|---|---|
| Ow | 40.10 | 30.34 |
| Os1 | 0.00 | 0.00 |
| Os2 | 8.00 | 0.00 |
| O | 48.10 | 30.34 |
 | ||
| Fig. 9 Radial density distribution of the Co2+ (A) and Co3+ (B) atoms of position L0 to the oxygen atoms Os for the B-terminated Co3O4 (111) surface. (C and D) Cosine of the bond angles between the topmost cations and their nearby oxygen atoms with an illustration of the distorted octahedral coordinative environment. In the presence of water, the water oxygens are also considered. The prominent binding motifs of water are also displayed (E, F and G). | ||
| Coverage | Co2+–Os | Co3+–Os | Co2+–Ow | Co3+–Ow | Co2+–O | Co3+–O |
|---|---|---|---|---|---|---|
| 0 | 3.0 | 3.0 | — | — | 3.0 | 3.0 |
| 24 | 3.0 | 3.0 | 0.1 | 2.2 | 3.1 | 5.2 |
From Fig. 9(C) that shows the O–Co–O bond angle distribution, it is seen that the O–Co2+–O bond angles shift to flatter values on the clean surface, similar to what was observed in the case of the A-terminated surface. While, the cosine of the O–Co3+–O bond angles for the clean surface shows only one peak located at 0.1 (84°), as illustrated in Fig. 9(D), three peaks can be observed in the bulk and are assigned to the opposing oxygen atoms (180°), the oxygen atoms of the neighboring tetrahedral voids (<90°) and those of the opposite tetrahedral voids (>90°). In the case of the (111) surface, the opposite tetrahedral voids are not occupied, which explains why only one peak can be observed (see Fig. 9 (D)).
It can be seen that the angle distribution for the O–Co2+–O bonds barely changes with water adsorption. This can be explained by the fact that a water molecule is bound to only one of the Co2+ atoms, which can be seen in the range around 0 (90°). In contrast, the bond angles of the Co3+ atoms change significantly. Upon adsorption of water, the cosines of the bond angles converge to their bulk counterparts, with three peaks slightly above and below 0 (>90° and <90°), and one peak near −1 (180°). Due to the anion parameter of cobalt oxide of 0.263 (vs. 0.25 in the ideal case), the octahedral coordination of the Co3+ ion is distorted, which is reflected in the bond angles of the coordinating oxygen that are found above and below 90°.
The (111) plane separates the oxygen atoms, which are closer together due to the widened tetrahedral voids and therefore have a bond angle below 90°. The distortion of the octahedral void is illustrated in Fig. 9(D). In the upper picture, the origin of the angles α (<90°) and β (>90°) is shown on the left-hand side of the angle distributions. From its perspective view, only 4 oxygen and Co2+ atoms of the octahedral void are visible, because the additional 2 oxygens in the bottom left and top right are perfectly aligned. This can be seen in the second picture below, which shows the structure from a slightly tilted perspective. On the clean (111) surface, only the narrower bond angles can be observed on the topmost layer. The following bonds can therefore be assigned to the three peaks on the surface with 24 water molecules: the peak at 0.2 (78°) corresponds to the Os–Co3+–Os bond angles, while those at −0.1 (84°) and −1 (180°) correspond the Os–Co3+–Ow bond angles. In addition, a minimum can be observed at 0 in the bulk. This peak is significantly higher on the surface occupied by water.
4. Conclusions
Ab initio molecular dynamics simulations were used to investigate the monolayer regime of water on the A and B terminations of the Co3O4 (111) surface. The structure of interfacial water and the magnitude as well as the driving force of water induced relaxations of the underlying substrates were investigated. We also looked at the proton transfer to the surface, the occupation of active sites and the hydrogen bond network at the interface. The B-termination is found to be more reactive with a larger dissociation degree and higher occupation of active sites.On the clean A-terminated surface, Co2+–Os bonds are shortened compared to bulk values and the Co2+ ions are coordinated with oxygens in a trigonal-planar geometry. These effects are reduced by the adsorption of water. 1.2 water molecules bind per Co2+. On the 8 surface adsorption sites, only 9.9 water or hydroxide molecules out of 24 are chemisorbed. A dissociation degree of 13% is observed in the contact layer with water. During dissociation, the water molecules transfer their proton to the 3-fold coordinated Os1 and 3-fold coordinated Os2. However the surface OH formed upon proton transfer to Os2 is not stable and the H+ recombines to an OH– in the water film shortly after. The binding mode of water on this surface combines Co2+–Ow covalent bonds and hydrogen bonds to the surface. 12.7% of the H-bonding is formed with surface Os1 and 19.8% with Os2. The hydrogen bond network is dominated by the interaction among the water molecules in the aqueous film (67.4%). Of the 9.9 chemisorbed water molecules, 1.25 are on an average dissociated. This corresponds to a degree of dissociation of 13% in the contact layer.
On the B-terminated surface, a shortening of the Co2+–Os with respect to bulk bonds and a nearly tetragonal-planar coordination of the Co2+ atoms are also observed. Similarly, like on the A-termination, these features are barely lifted upon adsorption of water. The Co3+–Os bonds are also shortened on the clean surface, but water adsorption elongates these bonds beyond the bulk value. This is explained by the fact that water molecules barely bind to the Co2+ ions, whereas on the Co3+ ions, the occupation number reads 2–3. This results in the formation of octahedrally coordinated Co3+. Such a complex was observed experimentally in ref. 21. 17.5 water adsorbates out of 24 are present in the partially dissociated contact layer, with a degree of dissociation of 46% per surface cell. The corresponding 8 water molecules that dissociate transfer their proton exclusively to the Os2 atoms, leaving them saturated. No hydrogen bonds to the surface are observed and the adsorbate–substrate interaction is almost exclusively made of Co3+–Ow bonds. Also, the hydrogen bond network at the interface involves only molecules in the water film. This is a capital information that could be of importance for studies on water electrolysis as the O–H binding and spectroscopic signatures are known to depend on the surface composition and the electrochemical environment at the interface.32
Data availability
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.Author contributions
Both authors have contributed equally to the work: planning, discussions and writing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 388390466 – TRR 247 within the work of Project A6. The authors gratefully acknowledge computing time granted by the Center for Computational Sciences and Simulation (CCSS) of the Universität of Duisburg-Essen and provided on the supercomputer magnitUDE (DFG grants INST 20876/209-1 FUGG, INST 20876/243-1 FUGG) at the Zentrum für Informations- und Mediendienste (ZIM).References
- S. Anke, G. Bendt, I. Sinev, H. Hajiyani, H. Antoni and I. Zegkinoglou, et al., Selective 2-propanol oxidation over unsupported Co3O4 spinel nanoparticles: mechanistic insights into aerobic oxidation of alcohols, ACS Catal., 2019, 9(7), 5974–5985 CrossRef CAS.
- Y. Cai, J. Xu, Y. Guo and J. Liu, Ultrathin, polycrystalline, two-dimensional Co3O4 for low-temperature Co oxidation, ACS Catal., 2019, 9(3), 2558–2567 CrossRef CAS.
- X. Deng and H. Tüysüz, Cobalt-oxide-based materials as water oxidation catalyst: recent progress and challenges, ACS Catal., 2014, 4(10), 3701–3714 CrossRef CAS.
- J. P. Llorca, J.-A. Dalmon and N. Homs, Transformation of Co3O4 during ethanol steam-re-forming. activation process for hydrogen production, Chem. Mater., 2004, 16(18), 3573–3578 CrossRef CAS.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng and G. Q. Lu, et al., Mesoporous Co3O4 and au/Co3O4 catalysts for low-temperature oxidation of trace ethylene, J. Am. Chem. Soc., 2010, 132(8), 2608–2613 CrossRef CAS PubMed.
- V. R. Shinde, S. B. Mahadik, T. P. Gujar and C. D. Lokhande, Supercapacitive cobalt oxide (Co3O4) thin films by spray pyrolysis, Appl. Surf. Sci., 2006, 252(20), 7487–7492 CrossRef CAS.
- F. Zasada, W. Piskorz, P. Stelmachowski, A. Kotarba, J.-F. Paul and T. Płociński, et al., Periodic DFT and HR-STEM studies of surface structure and morphology of cobalt spinel nanocrystals. retrieving 3D shapes from 2D images, J. Phys. Chem. C, 2011, 115(14), 6423–6432 CrossRef CAS.
- A. H. Omranpoor and S. Kenmoe, 2-Propanol Activation on the Low Index Co3O4 Surfaces: A Comparative Study Using Molecular Dynamics Simulations, Catalysts, 2024, 14, 25 CrossRef CAS.
- H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Co3O4 nanocrystals with predominantly exposed facets: synthesis, environmental and energy applications, J. Mater. Chem., 2013, 1, 14427–14442 RSC.
- X. Wang, L. Ding, Z. Zhao, W. Xu, B. Meng and J. Qiu, Novel hydrodesulfurization nano-catalysts derived from Co3O4 nanocrystals with different shapes, Catal. Today, 2011, 175(1), 509–514 CrossRef CAS.
- A. H. Omranpoor, A. Bera, D. Bullert, M. Linke, S. Salamon, S. Webers, H. Wende, E. Hasselbrink, E. Spohr and S. Kenmoe, 2-Propanol interacting with Co3O4 (001): A combined vSFS and AIMD study, J. Chem. Phys., 2023, 158, 164703 CrossRef CAS PubMed.
- X. L. Xu and J. Q. Li, DFT studies on H2O adsorption and its effect on CO oxidation over spinel Co3O4 (110) surface, Surf. Sci., 2011, 605(23), 1962–1967 CrossRef CAS.
- G. Fickenscher, C. Hohner, T. Xu and J. Libuda, Adsorption of D2O and CO on Co3O4(111) :Water Stabilizes Coadsorbed CO, J. Phys. Chem. C, 2021, 125(48), 26785–26792 CrossRef CAS.
- A. Omranpoor, T. Kox, E. Spohr and S. Kenmoe, Influence of temperature, surface composition and electrochemical environment on 2-propanol decomposition at the Co3O4 (001)/H2O interface, Appl. Surf. Sci. Adv., 2022, 12, 100319 CrossRef.
- D. H. Douma, K. N. Nono, A. H. Omranpoor, A. Lamperti, A. Debernardi and S. Kenmoe, Probing the local environment of active sites during 2-propanol oxidation to acetone on the Co3O4 (001) surface: Insights from first principles O K-edge XANES spectroscopy, J. Phys. Chem. C, 2023, 127, 5351–5357 CrossRef CAS.
- J. Chen and A. Selloni, Water adsorption and oxidation at the Co3O4 (110) surface, J. Phys. Chem. Lett., 2012, 3(19), 2808–2814 CrossRef CAS.
- F. Creazzo, D.-R. Galimberti, S. Pezzotti and M.-P. Gaigeot, DFT-MD of the (110)- Co3O4 cobalt oxide semiconductor in contact with liquid water, preliminary chemical and physical insights into the electrochemical environment, J. Chem. Phys., 2019, 150(4), 041721 CrossRef PubMed.
- T. Kox, E. Spohr and S. Kenmoe, Impact of solvation on the structure and reactivity of the Co3O4 (001)/H2O interface: insights from molecular dynamics simulations, Front. Energy Res., 2020, 8, 312 Search PubMed.
- E. Budiyanto, S. Zerebecki, C. Weidenthaler, T. Kox, S. Kenmoe, E. Spohr, S. DeBeer, O. Rdiger, S. Reichenberger, S. Barcikowski and H. Tüysüz, Impact of single-pulse, low-intensity laser post-processing on structure and activity of mesostructured cobalt oxide for the oxygen evolution reaction, ACS Appl. Mater. Interfaces, 2021, 13(44), 51962–51973 CrossRef CAS PubMed.
- M. Schwarz, S. Mohr, C. Hohner, K. Werner, T. Xu and J. Libuda, Water on Atomically-Defined Cobalt Oxide Surfaces Studied by Temperature-Programmed IR Reflection Absorption Spectroscopy and Steady State Isotopic Exchange, J. Phys. Chem. C, 2019, 123(13), 7673–7681 CrossRef CAS.
- G. Yan, T. Wähler, R. Schuster, M. Schwarz, C. Hohner, K. Werner, J. Libuda and P. Sautet, Water on Oxide Surfaces: A Triaqua Surface Coordination Complex on Co3O4 (111), J. Am. Chem. Soc., 2019, 141(14), 5623–5627 CrossRef CAS PubMed.
- G. Yan and P. Sautet, Surface Structure of Co3O4 (111) under Reactive Gas-Phase Environments, ACS Catal., 2019, 9(7), 6380–6392 CrossRef CAS.
- F. Zasada, W. Piskorz, S. Cristol, J.-F. Paul, A. Kotarba and Z. Sojka, Periodic density functional theory and atomistic thermodynamic studies of cobalt spinel nanocrystals in wet environment: molecular interpretation of water adsorption equilibria, J. Phys. Chem. C, 2010, 114(50), 22245–22253 CrossRef CAS.
- S. Kenmoe and P. U. Biedermann, Water adsorbate phases on ZnO and impact of vapor pressure on the equilibrium shape of nanoparticles, J. Chem. Phys., 2018, 148(5), 054701 CrossRef PubMed.
- S. Kenmoe, D. H. Douma, A. T. Raji, B. Mpassi-Mabiala, T. Goetsch, F. Girgsdies, A. Knop-Gericke, R. Schloegl and E. Spohr, X-ray absorption near-edge structure (XANES) at the O K-edge of bulk Co3O4 : experimental and theoretical studies, Nanomaterials, 2022, 12(6), 921 CrossRef CAS PubMed.
- J. Chen, X. Wu and A. Selloni, Electronic structure and bonding properties of cobalt oxide in the spinel structure, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245204 CrossRef.
- A. Montoya and B. S. Haynes, Periodic density functional study of Co3O4 surfaces, Chem. Phys. Lett., 2011, 502(1), 63–68 CrossRef CAS.
- T. D. Kuhne, et al., CP2K: an electronic structure and molecular dynamics software package – Quickstep: efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152(19), 194103 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- J. Hubbard and B. H. Flowers, Electron correlations in narrow energy bands, Proc. R. Soc. London, Ser. A, 1963, 276(1365), 238–257 Search PubMed.
- T. Bookholt, X. Qin, B. Lilli, D. Enke, M. Huck, D. Balkenhohl, K. Rüwe, J. Brune, J. P. Klare, K. Küpper, A. Schuster, J. Bergjan, M. Steinhart, H. Gröger, D. Daum and H. Schäfer, Increased Readiness for Water Splitting: NiO-Induced Weakening of Bonds in Water Molecules as Possible Cause of Ultra-Low Oxygen Evolution Potential, Small, 2024, 2310665 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01335b |
| This journal is © The Royal Society of Chemistry 2024 |