
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation-dependent Lewis acidity in chalcogen adducts of Sb/P frustrated Lewis pairs†
Jonas
Krieft
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Norbert W.
Mitzel
 *
*
Chair of Inorganic and Structural Chemistry, Center for Molecular Materials CM2, Faculty of Chemistry, Bielefeld University, Universitätsstraße 25, Bielefeld 33615, Germany. E-mail: mitzel@uni-bielefeld.de
First published on 24th June 2024
Abstract
The reactions of the frustrated Lewis pair (F5C2)2SbCH2P(tBu)2 with oxygen, sulphur, selenium and tellurium led to the mono-oxidation products (F5C2)2SbCH2P(E)(tBu)2 (E = O, S, Se, Te). Further oxidation of these chalcogen adducts with tetrachloro-ortho-benzoquinone (o-chloranil) gave (F5C2)2Sb(CH2)(μ-E)P(tBu)2·CatCl (CatCl = o-O2C6Cl4) with a central four-membered ring heterocycle for E = O, S, and Se. For E = Te the elimination of elemental tellurium led to an oxidation product with two equivalents of o-chloranil, (F5C2)2SbCH2P(tBu)2·2CatCl, which is also accessible by reaction of (F5C2)2SbCH2P(tBu)2 with o-chloranil. The synthesised compounds were characterised by NMR spectroscopy and X-ray structure analyses, and the structural properties were analysed in the light of the altered Lewis acidity due to the oxidation of the antimony atoms.
Introduction
The early contributions of Gutmann, Olah and Gillespie on antimony-containing Lewis acids demonstrated their reaction potential for synthesis and formation of donor–acceptor systems.1 It might seem obvious to integrate antimony-containing reagents as Lewis acidic functions into the chemistry of frustrated Lewis pairs (FLPs) and thus to extend the diversity of these intra- or intermolecular systems. The first known FLPs were mainly based on boron- or aluminium-containing functions as Lewis acidic parts and phosphorus- or nitrogen-containing functions as complementary Lewis basic parts. Owing to the suppressed acid/base adduct formation, FLPs can be used to activate C–H bonds, hydrogen and small molecules or for the reduction of CO2.2–4 However, the range of reactive Lewis acid–base combinations has increased over the last 20 years.4–8 The application of weaker B Lewis acids and stronger N Lewis bases, the so called “inverse” FLP by Krempner et al.,3,9 provides a further impetus to the still expanding FLP chemistry.Antimony, as a versatile Lewis acid function, exhibits a particularly high calculated fluoride ion affinity in both oxidation states V and III, depending on the substituents.10 Therefore it has a broader applicability as Lewis acid than other pnictogens.11 In addition to the theoretical considerations mentioned above, Gabbaï et al. performed impressive experimental studies on e.g. perfluorinated stibonium cations and stiboranes, which are air stable and able to activate strong element–fluorine bonds or complex formaldehyde; both in aqueous media.12,13 In other contributions, Gabbaï, Matile and Cozzolino highlighted the strong binding and chelation ability of stibanes, distibanes and -stiboranes towards anions like halides.14,15 Our group has recently presented rigid systems, based on anthracene, substituted with strong Lewis acidic Sb(C2F5)2 functions, which allow the chelate-binding of halide ions and various Lewis bases in the form of pnictogen bonds.16 In antimony-containing systems the Lewis acidity can be varied both by oxidation of the antimony atom from III to V and by the electron withdrawing substituents. The apparent influence of the oxidation state on the Lewis acidity of organoantimony compounds has been investigated in terms of the distinctness of the σ-holes.14,17 Gabbaï showed that both stiboranes and stibonium cations with sufficiently pronounced Lewis acidity can interact with oxygen atoms to form heterocycles (Scheme 1).18–20 Even with lower Lewis acidity of the antimony atom, the formation of such (four-membered) antimony- and chalcogen-containing heterocycles was demonstrated.21
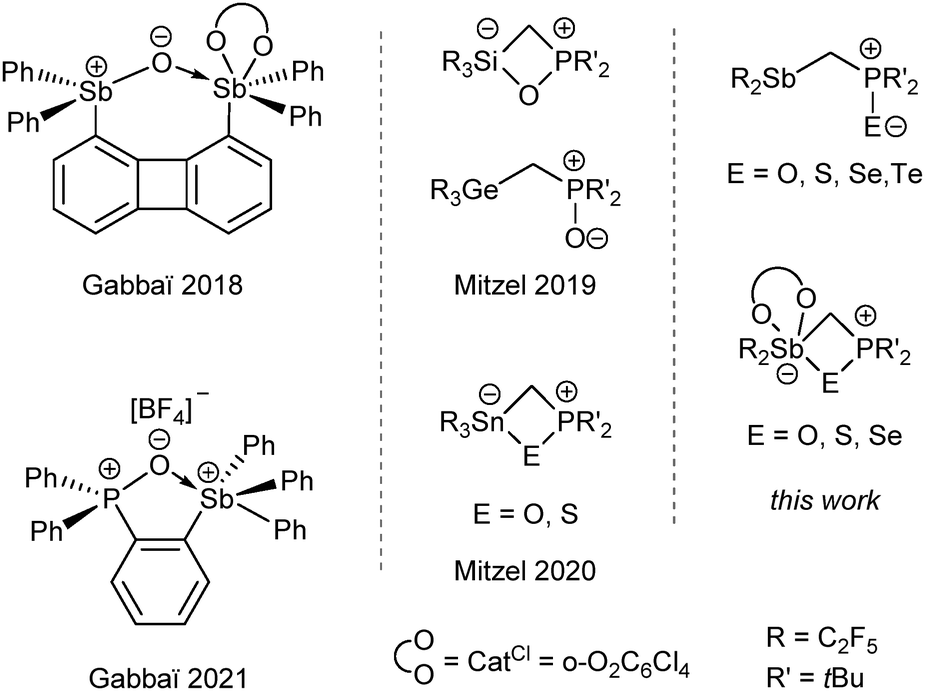 | ||
| Scheme 1 Different interaction motifs of a stiborane and a stibonium cation with oxygen atoms by Gabbaï19,20 and various structural motifs of FLP chalcogen adducts.8,22 | ||
In previous work in our group, we gained insight into the interaction of tetrel-based Lewis acids, (F5C2)3ECH2P(tBu)2 with E = Si, Ge, Sn, with oxygen. Depending on the Lewis acidity, a four-membered heterocycle is obtained while the backbone of the FLP is deformed (Scheme 1).8,22 In this work, we intended to investigate the intramolecular interaction of analogously structured, Sb-based Lewis acids in the oxidation states III and V with chalcogen atoms.
The C2F5 group as one of the most electron withdrawing organyl ligands, is particularly suitable to increase the Lewis acidity of stibanes and stiboranes.11 Naumann and Hoge, among others, introduced this substituent into pnictogen-based systems, resulting in highly reactive reagents.23 We have used the C2F5 substituent to generate sufficient Lewis acidity in the aforementioned tetrel-based FLP.6–8 Especially the tin-containing FLP shows a broad spectrum of reactivity towards small molecules such as hydrogen, carbon dioxide or unsaturated systems.7,22,24
Results and discussion
Recently we introduced the FLP (F5C2)2SbCH2P(tBu)2 (1).25 Its antimony(III)-based Lewis acid function turned out to be relatively soft, in accordance with the qualitative predictions of the HSAB concept.26This is one of the reasons why we studied the reaction of the FLP with the different chalcogen elements (or substitutes) to explore the behaviour of 1 under oxidative conditions.
Because of the decomposition or over-oxidation that occurs on contact with molecular oxygen, N2O was used instead. Under elimination of molecular nitrogen, the oxygen oxidation product (F5C2)2SbCH2P(O)(tBu)2 (2) is formed in which only the phosphorus atom is oxidised (Fig. 1 and Scheme 2).
 | ||
| Fig. 1 Molecular structure of compound 2 in the solid state. The asymmetric unit contains two molecules, so two different values are given. Ellipsoids are set at 50% probability; hydrogen atoms are omitted for clarity. Selected bond lengths and atom distances [Å] and angles [°]: Sb(1)–C(1) 2.260(3), Sb(1)–C(3) 2.236(4), Sb(1)–C(5) 2.174(3), P(1)–C(5) 1.812(3), P(1)–C(6) 1.844(4), P(1)–C(10) 1.858(3), P(1)–O(1) 1.509(3)/1.504(3), Sb(1)⋯O(1) 2.784(3)/2.888(3); C(1)–Sb(1)–C(3) 94.8(1), C(1)–Sb(1)–C(5) 91.6(2), C(1)–Sb(1)–O(1) 153.9(1), C(5)–Sb(1)–C(3) 91.2(2), C(5)–P(1)–C(6) 107.7(2), C(5)–P(1)–C(10) 107.3(2), C(6)–P(1)–C(10) 114.4(2), O(1)–P(1)–C(5) 106.0(2), O(1)–P(1)–C(6) 111.6(2), O(1)–P(1)–C(10) 109.3(2), P(1)–C(5)–Sb(1) 100.7(2)/102.2(2), Sb(1)–C(5)–P(1)–O(1) 19.5(1)/22.9(1). | ||
 | ||
| Scheme 2 Overview of the reactions of 1 with various chalcogens (or substitutes) and the subsequent oxidation of these products with o-chloranil, as well as the direct reaction of 1 with o-chloranil. | ||
It appears that the strong electron-withdrawing pentafluoroethyl substituents make the antimony atom so electron deficient that only the phosphorus atom is oxidised. However, the X-ray diffraction experiments indicate no formation of a four-membered ring by Sb–O interactions in the solid state; we can conclude that the Lewis acidity of the antimony function is too weak to deform the conformation of the Sb–C–P–O backbone. Instead, the oxidation has no effect on the angle of the bridging Sb–C–P unit; it is identical in the crystal structures of 1 and 2.
It is therefore not surprising that the distance (2.784(3)/2.888(3) Å) between the Sb and O atom is less than the sum of their van der Waals radii (∑rvdW(Sb–O) = 3.58 Å)27,28 but well above the sum of their covalence radii (∑rcov(Sb–O) = 2.03 Å).29 Another indicator for a possible Sb⋯O interaction is the P–O bond length, which should be longer if the Lewis acid interacts with the oxygen atom. For comparison we examined trimethylphosphane oxide and tris(tert-butyl)phosphane oxide (as a similar reference without the acid function). In trimethylphosphane oxide the P–O bond is shorter than in 2. Due to the greater steric demand of the tert-butyl groups, the P–O bond in the tert-butyl phosphane oxide is longer than in trimethylphosphane oxide; actually, it is identical to 2 (Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 with CatCl = o-O2C6Cl4. If two values are given, the asymmetric unit of the corresponding compound contains two molecules (or three for 7). (Please note, that tBu3P
22 with CatCl = o-O2C6Cl4. If two values are given, the asymmetric unit of the corresponding compound contains two molecules (or three for 7). (Please note, that tBu3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is an adduct with H2O2 with a hydrogen bond between the P atom and one H atom of H2O2)
O is an adduct with H2O2 with a hydrogen bond between the P atom and one H atom of H2O2)
| E | Me3PE |
tBu3PE |
(C2F5)2SbCH2P(E)tBu2 (2–5) | (C2F5)2Sb(μCH2)(μ-E)PtBu2·CatCl (6–8) | (C2F5)3Sn(μ-CH2)(μ-E)PtBu2 | ||
|---|---|---|---|---|---|---|---|
|
d(PE) [Å] |
d(PE) [Å] |
d(PE) [Å] |
d(Sb⋯E) [Å] | d(P–E) [Å] | d(Sb–E) [Å] | d(P–E) [Å] | |
| O | 1.489(6)30 | 1.510(1)32 | 1.509(3)/1.504(3) | 2.784(3)/2.888(3) | 1.567(1) | 2.158(1) | 1.532(2)21 |
| S | 1.969(1)31 | 1.962(3)33 | 1.982(2)/1.976(2) | 3.145(2)/3.108(1) | 2.036(2)/2.031(2) | 2.633(2)/2.619(2) | 2.010(2)21 |
| Se | 2.122(1)31 | 2.133(1)34 | 2.131(1) | 3.171(1) | 2.194(2)/2.187(2) | 2.748(1)/2.703(1) | — |
| Te | — | 2.368(4)35 | 2.382(1) | 3.362(1) | — | — | — |
This underlines the independence of the Lewis acidic function and therefore excludes any strong Sb⋯O interaction. The C(1)–Sb(1)–O(1) angle of 153.9(1)/150.4(1)°, which should be closer to 180° for a σ-hole interaction, also supports this. In other molecules, such as (F5C2)2SbCH2P(tBu)2·CS2, the CS2 adduct of 1,25 or anthracene-based poly Lewis acids with terminal Sb(C2F5)2 functions,16 we have presented examples of this σ-hole interaction between a Lewis acidic antimony(III) function and Lewis bases.
The torsion angle of the Sb–C–P–E motif of 2 is 19.5(1)/22.9(2)°, so the atoms involved are not in the same plane. In contrast to the previous synthesis of 2, the reactions of 1 with the elemental forms of sulphur, selenium and tellurium could be used to generate the corresponding oxidation products 3–5; (F5C2)2SbCH2P(E)(tBu)2 with E = S, Se, Te (Scheme 2 and Fig. 2). In all cases, only the phosphorus atom was oxidised. There was no formation of a four-membered ring, nor any significant deformation of the Sb–C–P backbone compared to 1, although the Sb–C–P angle in 2–5 increases slightly with increasing size of the chalcogen atom (Table 1).
 | ||
| Fig. 2 Molecular structures of compounds 3–5 in the solid state. If two values are given, the asymmetric unit contains two molecules. Ellipsoids are set at 50% probability; hydrogen atoms are omitted for clarity. Selected bond lengths and atom distances [Å] and angles [°]; negative value for torsion angle means rotation in opposite direction: 3: P–S 1.982(2)/1.976(2), Sb⋯S 3.145(2)/3.108(1), P–C–Sb 106.8(2)/106.0(2), Sb–C–P–S 25.0(3); 4: P–Se 2.131(1), Sb⋯Se 3.171(1), P–C–Sb 107.6(1), Sb–C–P–Se −22.2(1); 5: P–Te 2.382(1), Sb⋯Te 3.362(1), P–C–Sb 109.1(1), Sb–C–P–Te 27.7(1). | ||
The Sb⋯E distances are all well below the sum of their corresponding van der Waals radii (∑rvdW(Sb–S) = 3.86 Å, ∑rvdW(Sb–Se) = 3.96 Å, ∑rvdW(Sb–Te) = 4.12 Å),27,28 but also clearly above the sum of their respective covalence radii (∑rcov(Sb–S) = 2.43 Å, ∑rcov(Sb–Se) = 2.56 Å, ∑rcov(Sb–Te) = 2.76 Å).29 As mentioned above for the P–O bond length, the other P–E bond lengths in 3 and 4 are also slightly longer than in the trimethylphosphane chalcogenides.
Even shorter is the P–S bond length in tris(tert-butyl)phosphane sulphide. However, for 4 and 5 the P–E bond lengths are more similar to their corresponding tert-butyl analogues, respectively. In addition to the natural elongation of the P–E bond with increasing atomic number of the chalcogen, the distance between the antimony and the chalcogen atom also increases (Table 1).
The oxygen oxidation products of the analogous tetrel-based FLPs, (F5C2)3ECH2P(O)(tBu)2, form a four-membered heterocycle for E = Si and Sn. For E = Ge the same open-chain structural motif as described above for the Sb(III) compound 2 is obtained (Scheme 1).8 In the latter two cases the Lewis acidity is apparently insufficient to show a significantly strong interaction between the Lewis acid function and the chalcogen atom. The molecular structures in the solid-state are supposedly more informative than investigations in the liquid phase, because they are usually more close to the ground state structure of a molecule. As with the Si and Sn analogues, there is no additional splitting of the resonances of the C2F5 or tert-butyl substituents due to formation of the four-membered heterocycle.8,22
Since the oxidation of Sb(III) to Sb(V) generally increases the Lewis acidity, this should also increase the interaction strength between the Lewis acid function and the chalcogen atom in compounds 2–5. Therefore, we exposed the mono-oxidation products 2–5 to a different and stronger oxidant. Tetrachloro-ortho-benzoquinone (o-chloranil) is a well-established reagent for the oxidation of stibanes36 and proved to be suitable for this purpose. The change in colour of the oxidant solution, which was deep red before it was added to the mono-oxidations products 2–5, allowed the reaction to be followed macroscopically. As expected, the antimony atom was oxidised. The significant increase in acidity of the antimony site led to the formation of a four-membered SbCPE ring with strong deformation of the Sb–C–P backbone.
Fig. 3 shows the molecular structure of compound 6 ((Cl4C6O2)(F5C2)2SbCH2P(μ-O)(tBu)2) in the solid state; analogous structural motifs were obtained for compounds 7 (S) and 8 (Se).
 | ||
| Fig. 3 Molecular structure of compound 6 in the solid state. Ellipsoids are set at 50% probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] of 6: Sb(1)–C(1) 2.240(2), Sb(1)–C(3) 2.279(2), Sb(1)–C(5) 2.194(2), Sb(1)–O(1) 2.158(1), Sb(1)–O(2) 2.009(1), Sb(1)–O(3) 2.044(1), P(1)–C(5) 1.809(2), P(1)–C(6) 1.853(2), P(1)–C(10) 1.844(2), P(1)–O(1) 1.567(1); C(1)–Sb(1)–C(3) 93.2(1), C(1)–Sb(1)–C(5) 154.8(1), C(5)–Sb(1)–C(3) 96.3(1), O(1)–Sb(1)–C(1) 83.8(1), O(1)–Sb(1)–C(3) 103.5(1), O(1)–Sb(1)–C(5) 71.2(1), O(1)–Sb(1)–O(2) 173.3(1), O(1)–Sb(1)–O(3) 93.1(1), O(2)–Sb(1)–C(1) 98.6(1), O(2)–Sb(1)–C(3) 82.6(1), O(2)–Sb(1)–C(5) 105.7(1), O(2)–Sb(1)–O(3) 80.9(1), O(3)–Sb(1)–C(1) 85.3(1), O(3)–Sb(1)–C(3) 163.0(1), O(3)–Sb(1)–C(5) 92.1(1), C(5)–P(1)–C(6) 113.6(1), C(5)–P(1)–C(10) 111.3(1), C(6)–P(1)–C(10) 114.2(1), O(1)–P(1)–C(5) 97.0(1), O(1)–P(1)–C(6) 109.5(1), O(1)–P(1)–C(10) 109.9(1), P(1)–C(5)–Sb(1) 91.5(1), P(1)–O(1)–Sb(1) 100.0(1). | ||
Due to the ring closure, the Sb–C–P angles in the molecules 6–8 are significantly more acute than in their respective open-chain precursors 2–5. Along with this deformation and the formation of the four-membered ring, the P–E bond lengths and the Sb–E distances have also undergone drastic changes (Fig. 4). The former are all longer than in the corresponding precursors 2–4 and also longer than in the tris(tert-butyl)phosphane chalcogenides. For 2 and 3, a comparison with the oxygen and sulphur adducts of the aforementioned Sn/P-FLP, which contain the same central structural motif, is possible: The P–E bond lengths are in a similar range in both cases (Table 1). The distances between the Lewis acidic Sb(V) atom and the chalcogen atom in compounds 6–8 are significantly shorter than in the corresponding precursors 2–4 and only slightly above the sum of the covalence radii of the atoms involved (the differences are: ∑rvdW(Sb,E) − d(Sb–E) = 0.13 Å for 6, 0.20 Å for 7, 0.19 Å for 8).
 | ||
| Fig. 4 Molecular structures of compounds 7 and 8 in the solid state. The asymmetric units contain more than one molecule, respectively. Ellipsoids are set at 50% probability; hydrogen atoms are omitted for clarity. Selected bond lengths and atom distances [Å] and angles [°]: 7: P–S 2.036(2)/2.031(2), Sb–S 2.633(2)/2.619(2), P–C–Sb 101.2(3), P–S–Sb 81.9(1); 8: P–Se 2.194(2)/2.187(2), Sb–Se 2.748(1)/2.703(1), P–C–Sb 102.5(3), P–Se–Sb 78.2(1)/79.4(1). | ||
An additional test, whether the ring formation could be precluded by donor solvents e.g. pyridine, demonstrated, that even in the presence of an excess of donor solvent, the four-membered ring is formed by oxidation with o-chloranil.
In contrast, the Te oxidation product 5 behaves differently. Unlike reactants 2–4, the oxidation of 5 with o-chloranil does not lead to a four-membered ring, but to the elimination of tellurium. The result is compound 9, which contains a second o-chloranil unit that occupies the binding position between the Sb and P atoms of the FLP, resulting in a seven-membered ring (Fig. 5). The antimony atom is at the center of an octahedral coordination environment. The Sb–C–P angle, now part of the seven-membered ring, is smaller than in both reactant 5 and FLP 1; it has an angle of 118.4(2)°. As an alternative synthetic route to the unexpectedly formed compound 9, we found the reaction of 1 with two equivalents of o-chloranil. Interestingly, compound 9 is selectively formed at room temperature with both excess and deficient oxidant. To understand how the reaction proceeds, a solution of 1 was mixed with a dilute solution of o-chloranil under kinetic control at −60 °C. Theoretically, three different products are conceivable when one molecule of 1 reacts with one molecule of the oxidant. Surprisingly, neither the stiborane (product of the oxidation of the Sb atom) nor the phosphorane (product of the oxidation of the P atom) are formed, but rather compound 10, in which a molecule of the oxidant is found in the binding pocket of 1, forming the seven-membered heterocycle mentioned above (Fig. 6).
 | ||
| Fig. 5 Molecular structure of compound 9 in the solid state. Ellipsoids are set at 50% probability; hydrogen atoms are omitted for clarity. Only the non-disordered of the two molecules per asymmetric unit is depicted. Selected bond lengths [Å] and angles [°]: Sb(1)–C(1) 2.233(4), Sb(1)–C(3) 2.247(4), Sb(1)–C(5) 2.181(3), Sb(1)–O(1) 2.023(2), Sb(1)–O(2) 2.027(3), Sb(1)–O(3) 2.044(2), P(1)–C(5) 1.794(4), P(1)–C(6) 1.846(4), P(1)–C(10) 1.833(3), P(1)–O(4) 1.587(2); C(1)–Sb(1)–C(3) 93.7(2), C(1)–Sb(1)–C(5) 98.7(2), C(5)–Sb(1)–C(3) 89.7(2), O(1)–Sb(1)–C(1) 88.0(1), O(1)–Sb(1)–C(3) 94.5(2), O(1)–Sb(1)–C(5) 171.9(2), O(1)–Sb(1)–O(2) 81.2(1), O(1)–Sb(1)–O(3) 89.4(1), O(2)–Sb(1)–C(1) 169.1(1), O(2)–Sb(1)–C(3) 88.4(2), O(2)–Sb(1)–C(5) 92.0(2), O(2)–Sb(1)–O(3) 89.6(1), O(3)–Sb(1)–C(1) 89.1(1), O(3)–Sb(1)–C(3) 175.2(1), O(3)–Sb(1)–C(5) 86.1(2), C(5)–P(1)–C(6) 107.5(2), C(5)–P(1)–C(10) 110.6(2), C(6)–P(1)–C(10) 116.5(2), O(4)–P(1)–C(5) 113.8(2), O(4)–P(1)–C(6) 99.2(2), O(4)–P(1)–C(10) 109.0(2), P(1)–C(5)–Sb(1) 118.4(2). | ||
 | ||
| Fig. 6 Molecular structure of compound 10 in the solid state. Ellipsoids are set at 50% probability; hydrogen atoms are omitted for clarity. Only one of the two molecules per asymmetric unit is depicted. Selected bond lengths [Å] and angles [°]: Sb(1)–C(1) 2.242(2), Sb(1)–C(3) 2.316(2), Sb(1)–C(5) 2.186(2), Sb(1)–O(1) 2.316(1), P(1)–C(5) 1.782(2), P(1)–C(6) 1.847(2), P(1)–C(10) 1.848(2), P(1)–O(2) 1.593(1); C(1)–Sb(1)–C(3) 90.2(1), C(1)–Sb(1)–C(5) 92.4(1), C(5)–Sb(1)–C(3) 83.5(1), O(1)–Sb(1)–C(1) 78.4(1), O(1)–Sb(1)–C(3) 156.8(1), O(1)–Sb(1)–C(5) 77.0(1), C(5)–P(1)–C(6) 108.2(1), C(5)–P(1)–C(10) 111.0(1), C(6)–P(1)–C(10) 116.3(1), O(2)–P(1)–C(5) 111.4(1), O(2)–P(1)–C(6) 99.2(1), O(2)–P(1)–C(10) 110.2(1), P(1)–C(5)–Sb(1) 113.6(1). | ||
This is different from the behaviour described by Gabbaï et al. for the oxidation of a related Sb/P system, o-C6H4(PPh2)(SbAr2) with Ar = C6H5 and C6F5, in which only the antimony atom is oxidized and bears a chelating o-C6Cl4O2 ligand after the reaction.13
The obvious reason for this difference is the extreme electronegativity of the C2F5 ligands (even if compared to C6F5), which makes the antimony(III) function in 1 so electron deficient, that the phosphorus atom becomes the site more easily oxidised. 10 is an adduct of FLP 1 with an o-chloranil molecule and has thus lost its reactive potential to bind and activate other molecules. Like 9, 10 contains a seven-membered ring as a structural motif. The Sb–C–P angle of 113.6(1)/115.1(1)° is between that of free FLP 1 (110.6(1)°) and that of compound 9 (118.4(2)°). The same applies to the Sb⋯P distance (1: 3.306(1) Å, 9: 3.420(1) Å, 10: 3.327(1)/3.352(1) Å). In contrast to the tetrahedrally surrounded phosphorus atom, the antimony atom is bisphenoidally surrounded due to the lone pair on the antimony atom in oxidation state III, as is also typical for other adducts of FLP 1.24
When o-chloranil is added to 10, compound 9 is selectively formed. This is the second step in the reaction sequence of FLP 1 to 9 at room temperature with two equivalents of oxidant.
As with the bond lengths and angles of the molecular structures in the solid state of compounds 1–10, some trends can be seen in the NMR spectroscopic chemical shifts of the substances in solution. Both the methylene protons and the resonance of the phosphorus nucleus are good indicators of the influence of different oxidants in these systems. The resonance of the methylene protons becomes increasingly low-field shifted as the atomic number of the chalcogen increases (Table 2). By oxidising the antimony atom, these proton resonances undergo a significant shift of about 2 ppm towards low field. An opposite tendency, i.e. a shift towards the high field, is observed in the resonances of the phosphorus nuclei of 3–5.
| Compound | 1H δ(CH2) [ppm] | 31P δ [ppm] | Compound | 1H δ(CH2) [ppm] | 31P δ [ppm] |
|---|---|---|---|---|---|
| 1 | 2.17 | 15.9 | 6 | 4.38 | 88.4 |
| 2 | 2.66 | 63.7 | 7 | 4.68 | 78.9 |
| 3 | 2.79 | 76.6 | 8 | 4.92 | 73.2 |
| 4 | 2.93 | 73.0 | 9 | 3.09/2.89 | 110.0 |
| 5 | 3.18 | 45.4 | 10 | 2.90 | 111.4 |
Compound 2 behaves contrary to this trend. The change in molecular symmetry caused by the oxidation with o-chloranil leads to the splitting of the pentafluoroethyl groups into two signal sets in 19F NMR for 7–9 and for 8 and 9 also the splitting into two signal sets for the tert-butyl groups in 1H NMR.
Conclusions
We have found that the phosphorus atom in the FLP (F5C2)2SbCH2P(tBu)2 (1) is selectively oxidised by reaction with N2O or the elemental chalcogens sulphur, selenium and tellurium. The solid state molecular structure data of the products (F5C2)2SbCH2P(E)(tBu)2 (E = O, S, Se, Te; 2–5) do not indicate any interaction between the antimony(III) Lewis acid and the Lewis basic chalcogen atom; the different size and electrostatics of the bound chalcogen atoms do not seem to affect the nature of product formed. Increasing the Lewis acidity by oxidation of Sb(III) to Sb(V) strengthens the interaction between the Lewis acid function and the Lewis basic chalcogen atom, resulting in the formation of four-membered heterocycles and, in some cases, strong deformation of the Sb–C–P backbone. Selective oxidation of the antimony atom with o-chloranil is not possible due to the electron deficiency caused by the C2F5 groups; it is rather the phosphane function of 1 which is oxidised first. Only in a second oxidation step is the Sb(III) function oxidised.Author contributions
J. Krieft: investigation, methodology, validation, visualisation, writing (original draft), B. Neumann: investigation (SCXRD), H.-G. Stammler: investigation (SCXRD), N. W. Mitzel: funding acquisition, project administration, supervision, reviewing and editing.Data availability
The data published in this contribution are available as ESI,† submitted with the manuscript. Crystallographic data have been deposited with the Cambridge Crystal Structure Database (CCDC).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jan-Niklas Bollnow for lab assistance, Marco Wißbrock for recording NMR spectra and Barbara Teichner for performing elemental analyses. This work was founded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, grant MI 477/44-1, project number 461833739).References
- (a) M. Baaz, V. Gutmann and O. Kunze, Monatsh. Chem., 1962, 93, 1142–1161 CrossRef; (b) G. A. Olah and R. H. Schlosberg, J. Am. Chem. Soc., 1968, 90, 2726–2727 CrossRef CAS; (c) G. A. Olah, J. Org. Chem., 2005, 70, 2413–2429 CrossRef CAS PubMed; (d) J. Bacon and R. J. Gillespie, J. Am. Chem. Soc., 1971, 93, 6914–6919 CrossRef CAS; (e) J. Y. Calves and R. J. Gillespie, J. Am. Chem. Soc., 1977, 99, 1788–1792 CrossRef CAS.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed; (b) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed; (c) D. W. Stephan, Dalton Trans., 2009, 17, 3129–3136 RSC; (d) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed; (e) O. J. Metters, S. J. K. Forrest, H. A. Sparkes, I. Manners and D. F. Wass, J. Am. Chem. Soc., 2016, 138, 1994–2003 CrossRef CAS PubMed.
- C. Manankandayalage, D. K. Unruh and C. Krempner, Chem. - Eur. J., 2021, 27, 6263–6273 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- (a) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (b) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed; (c) C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925–3928 CrossRef CAS PubMed; (d) C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2013, 52, 4256–4259 CrossRef CAS PubMed; (e) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (f) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (g) W. Uhl, J. Possart, A. Hepp and M. Layh, Z. Anorg. Allg. Chem., 2017, 643, 1016–1029 CrossRef CAS; (h) L. Wickemeyer, N. Aders, A. Mix, B. Neumann, H.-G. Stammler, J. J. Cabrera-Trujillo, I. Fernández and N. W. Mitzel, Chem. Sci., 2022, 13, 8088–8094 RSC.
- B. Waerder, M. Pieper, L. A. Körte, T. A. Kinder, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2015, 54, 13416–13419 CrossRef CAS PubMed.
- P. Holtkamp, F. Friedrich, E. Stratmann, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 5114–5118 CrossRef CAS PubMed.
- T. A. Kinder, R. Pior, S. Blomeyer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2019, 25, 5899–5903 CrossRef CAS PubMed.
- (a) H. Li, A. J. A. Aquino, D. B. Cordes, F. Hung-Low, W. L. Hase and C. Krempner, J. Am. Chem. Soc., 2013, 135, 16066–16069 CrossRef CAS PubMed; (b) S. Mummadi, D. K. Unruh, J. Zhao, S. Li and C. Krempner, J. Am. Chem. Soc., 2016, 138, 3286–3289 CrossRef CAS PubMed; (c) S. Mummadi, A. Brar, G. Wang, D. Kenefake, R. Diaz, D. K. Unruh, S. Li and C. Krempner, Chem. – Eur. J., 2018, 24, 16526–16531 CrossRef CAS PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- (a) M. Schorpp, R. Yadav, D. Roth and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202207963 CrossRef CAS PubMed; (b) M. Á. García-Monforte, M. Baya, D. Joven-Sancho, I. Ara, A. Martín and B. Menjón, J. Organomet. Chem., 2019, 897, 185–191 CrossRef; (c) D. Sharma, S. Balasubramaniam, S. Kumar, E. D. Jemmis and A. Venugopal, Chem. Commun., 2021, 57, 8889–8892 RSC; (d) B. L. Murphy and F. P. Gabbaï, J. Am. Chem. Soc., 2023, 145, 19458–19477 CrossRef CAS PubMed.
- B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564–9567 CrossRef CAS PubMed.
- D. Tofan and F. P. Gabbaï, Chem. Sci., 2016, 7, 6768–6778 RSC.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- (a) L. M. Lee, M. Tsemperouli, A. I. Poblador-Bahamonde, S. Benz, N. Sakai, K. Sugihara and S. Matile, J. Am. Chem. Soc., 2019, 141, 810–814 CrossRef CAS PubMed; (b) J. Qiu, B. Song, X. Li and A. F. Cozzolino, Phys. Chem. Chem. Phys., 2017, 20, 46–50 RSC; (c) Di You, B. Zhou, M. Hirai and F. P. Gabbaï, Org. Biomol. Chem., 2021, 19, 4949–4957 RSC.
- (a) J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2023, 62, e202310439 CrossRef CAS PubMed; (b) J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. Sci., 2023, 14, 13551–13559 RSC.
- (a) M. Yang, D. Tofan, C.-H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 13868–13872 CrossRef CAS PubMed; (b) A. M. Christianson and F. P. Gabbaï, Organometallics, 2017, 36, 3013–3015 CrossRef CAS.
- J. E. Smith, H. Yang and F. P. Gabbaï, Organometallics, 2021, 40, 3886–3892 CrossRef CAS.
- V. M. Gonzalez, G. Park, M. Yang and F. P. Gabbaï, Dalton Trans., 2021, 50, 17897–17900 RSC.
- C.-H. Chen and F. P. Gabbaï, Dalton Trans., 2018, 47, 12075–12078 RSC.
- (a) B. Eberwein and J. Weidlein, Z. Anorg. Allg. Chem., 1976, 420, 229–239 CrossRef CAS; (b) M. J. Begley, D. B. Sowerby, D. M. Wesolek, C. Silvestru and I. Haiduc, J. Organomet. Chem., 1986, 316, 281–289 CrossRef CAS; (c) J. M. Mercero, X. Lopez, J. E. Fowler and J. Ugalde, J. Phys. Chem. A, 1997, 101, 5574–5579 CrossRef CAS; (d) I. Ghesner, L. Opris, G. Balazs, H. J. Breunig, J. E. Drake, A. Silvestru and C. Silvestru, J. Organomet. Chem., 2002, 642, 113–119 CrossRef CAS; (e) O. Kysliak and J. Beck, Z. Anorg. Allg. Chem., 2013, 639, 2860–2867 CrossRef CAS.
- P. Holtkamp, T. Glodde, D. Poier, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2020, 59, 17388–17392 CrossRef CAS PubMed.
- (a) D. Naumann, G. Nowicki and K.-J. Sassen, Z. Anorg. Allg. Chem., 1997, 623, 1183–1189 CrossRef CAS; (b) M. B. Murphy-Jolly, L. C. Lewis and A. J. M. Caffyn, Chem. Commun., 2005, 4479–4480 RSC; (c) S. Solyntjes, J. Bader, B. Neumann, H.-G. Stammler, N. Ignat'ev and B. Hoge, Chem. – Eur. J., 2017, 23, 1557–1567 CrossRef CAS PubMed.
- (a) P. Holtkamp, J. Schwabedissen, B. Neumann, H.-G. Stammler, I. V. Koptyug, V. V. Zhivonitko and N. W. Mitzel, Chem. – Eur. J., 2020, 26, 17381–17385 CrossRef CAS PubMed; (b) P. Holtkamp, D. Poier, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2021, 27, 3793–3798 CrossRef CAS PubMed.
- J. Krieft, P. C. Trapp, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler, J.-H. Lamm and N. W. Mitzel, Chem. Sci., 2024, in revision Search PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- M. Engelhardt, C. L. Raston, C. R. Whitaker and A. H. White, Aust. J. Chem., 1986, 39, 2151–2154 CrossRef.
- J. Ramler and C. Lichtenberg, Chem. – Eur. J., 2020, 26, 10250–10258 CrossRef CAS PubMed.
- S. H. Ahn, K. J. Cluff, N. Bhuvanesh and J. Blümel, Angew. Chem., Int. Ed., 2015, 54, 13341–13345 CrossRef CAS PubMed.
- H. U. Steinberger, B. Ziemer and M. Meisel, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 835–837 CrossRef CAS PubMed.
- H. U. Steinberger, B. Ziemer and M. Meisel, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 323–324 CrossRef CAS PubMed.
- N. Kuhn, H. Schumann and G. Wolmershäuser, Z. Naturforsch., B: J. Chem. Sci., 1987, 42, 674–678 CrossRef CAS.
- (a) M. M. Sidky, M. R. Mahran and W. M. Abdou, Phosphorus Sulfur Relat. Elem., 1983, 15, 129–135 CrossRef CAS; (b) R. R. Holmes, R. O. Day, V. Chandrasekhar and J. M. Holmes, Inorg. Chem., 1987, 26, 157–163 CrossRef CAS; I.-S. Ke and F. P. Gabbaï, Inorg. Chem., 2013, 52, 7145–7151 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2350426–2350434. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01591f |
| This journal is © The Royal Society of Chemistry 2024 |