
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphate selective binding and sensing by halogen bonding tripodal copper(II) metallo-receptors in aqueous media†
Hena
Bagha
a,
Robert
Hein
a,
Jason Y. C.
Lim
 a,
William K.
Myers
a,
Mark R.
Sambrook
b and
Paul D.
Beer
*a
a,
William K.
Myers
a,
Mark R.
Sambrook
b and
Paul D.
Beer
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk; Tel: +44(0)1865 285142
bCBR Division, Dstl Porton Down, Salisbury, SP4 0JQ, UK. E-mail: MSAMBROOK@mail.dstl.gov.uk; Tel: +44(0)1980 952077
First published on 3rd July 2024
Abstract
Combining the potency of non-covalent halogen bonding (XB) with metal ion coordination, the synthesis and characterisation of a series of hydrophilic XB tripodal Cu(II) metallo-receptors, strategically designed for tetrahedral anion guest binding and sensing in aqueous media is described. The reported metallo-hosts contain a tripodal C3-symmetric tris-iodotriazole XB donor anion recognition motif terminally functionalised with tri(ethylene glycol) and permethylated β-cyclodextrin functionalities to impart aqueous solubility. Optical UV-vis anion binding studies in combination with unprecedented quantitative EPR anion titration investigations reveal the XB Cu(II) metallo-receptors exhibit strong and selective phosphate recognition over a range of other monocharged anionic species in competitive aqueous solution containing 40% water, notably outperforming a hydrogen bonding (HB) Cu(II) metallo-receptor counterpart. Electrochemical studies demonstrate further the capability of the metallo-receptors to sense anions via significant cathodic perturbations of the respective Cu(II)/Cu(I) redox couple.
Introduction
The supramolecular recognition of anions in organic and aqueous media, lies key to the development of artificial receptors for their selective detection, sequestration and transport.1 Such hosts find important applications in a plethora of biological, medical and environmental fields, for example in the development of bioimmunoassays,2,3 in drug delivery systems4,5 and in the remediation of contaminated water.6,7 Overcoming competitive anion hydration is however one of the main challenges in receptor design, with this problem being especially acute for very hydrophilic anions positioned low in the Hofmeister series such as phosphate.8 Leading effective strategies so far have centred on the use of highly positively charged hosts, in the form of either organic, biologically inspired molecules, which establish multiple strong electrostatic and hydrogen bonding (HB) interactions to bind selectively the target tetrahedral guest via topological complementarity,8,9 or inorganic metallo-complexes, usually transition metal or lanthanide based, in which the ‘hard’ metal cation serves as a strong anchoring point for the similarly ‘hard’ target phosphate oxoanionic species.10,11 The synergetic combination of these two strategies has also been exploited, although to a lesser extent, in the design of mixed metallo-receptors,12–14 with Anslyn's phosphate-selective C3-symmetrical HB Cu(II) tripods being particularly elegant representative examples.15Defined as the attractive non-covalent interaction between an electron-deficient halogen atom and a Lewis base, halogen bonding (XB), has come to the fore in recent years as a remarkably powerful addition to the anion binding supramolecular tool-box.16 Its stringent directionality requirements compared to traditional HB interactions, coupled with greater hydrophobicity and lower solvent and pH dependency have led to XB host systems often outperforming their more prominent HB host analogues in terms of the strength and selectivity of anion binding.17,18 While these factors have also advantageously facilitated XB-mediated anion recognition in aqueous solvent media,19 impressively including in pure water,20–23 only a handful of XB receptors are known to be capable of selectively recognising phosphate, especially in aqueous containing environments.24–28 Motivated by this paucity, we recently showed a series of tris(iodotriazole)-containing XB Zn(II) tripodal metallo-receptors to be effective at preferentially binding inorganic phosphate over other more basic oxoanions and the halides in a competitive aqueous-acetonitrile mixture containing 10% water.29
With the aim of achieving selective phosphate recognition in an aqueous solution with increased water content, and anion sensing capability, herein we describe the synthesis of a series of tripodal C3-symmetrical tris-iodotriazole XB copper(II) metallo-receptor analogues containing hydrophilic polyether and permethylated β-cyclodextrin (pmβCD) functionalities (Fig. 1). Extensive optical UV-vis and unprecedented EPR anion titration investigations reveal the XB metallo-receptors to strongly and selectively bind phosphate over a range of anions in competitive 40% aqueous-containing solvent media. In addition, preliminary electrochemical anion sensing studies demonstrate significant cathodic perturbations of the respective metallo-receptors’ Cu(II)/(I) redox couple upon anion binding.
 | ||
| Fig. 1 General design of the ‘C3-symmetric’ XB anion recognition motif29 integrated into hydrophilic functionalised tripodal Cu(II)-receptor frameworks 1I·2OTf and 13I·2OTf with the proposed anion binding mode. A− = anion. (For the full chemical structure of the blue pmβCD ‘cone’ see Scheme 2.) | ||
Results and discussion
Synthesis of XB tripodal Cu(II) metallo-receptors
The target XB Cu(II) tripod 1I·2OTf with hydrophilic solubilising tri(ethylene glycol) (TEG) groups was initially prepared in conjunction with its tris(prototriazole)-containing HB Cu(II) counterpart 1H·2OTf. The modification of the tripodal termini of 1I·2OTf with alternative, permethylated β-cyclodextrin (pmβCD) functionalities enabled further systematic investigation into whether changing the solubilising functionalities played any role on the anion recognition capability of receptor 13I·2OTf.The first target tris(TEG)-appended XB pyridyl trimer ligand 6I was synthesised following the multistep synthetic route shown in Scheme 1. TEG-based terminal alkyne 3 was prepared by treating commercially-available TEG monomethyl ether with propargyl bromide under basic conditions using modified literature procedures30 and subsequently reacted with 6-azidomethyl pyridyl methanol 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 employing Zhu's one-pot ‘iodo-click’ copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) reaction protocol32 to afford the XB synthon 4I in good 64% yield after chromatographic purification. Chlorination with thionyl chloride generated 5I which was reacted under basic ammonia conditions using an excess of 20 molar equivalents of ammonium acetate, together with an equimolar amount of sodium carbonate to produce the desired XB trimer 6I in 60% yield. Upon conversion of 5I to 6I a pronounced upfield chemical shift change in the 1H NMR spectrum of the crude reaction mixture in CDCl3 was observed for the Ha′ methylene protons from 4.66 ppm to 3.84 ppm, characteristic of the tertiary N-amine region (Fig. S37 ESI†); a single peak at m/z = 1440.2 [M + H]+ in the electrospray ionisation (ESI) mass spectrum of the compound provided further confirmation of tripodal ligand formation (see Fig. S22†).
31 employing Zhu's one-pot ‘iodo-click’ copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) reaction protocol32 to afford the XB synthon 4I in good 64% yield after chromatographic purification. Chlorination with thionyl chloride generated 5I which was reacted under basic ammonia conditions using an excess of 20 molar equivalents of ammonium acetate, together with an equimolar amount of sodium carbonate to produce the desired XB trimer 6I in 60% yield. Upon conversion of 5I to 6I a pronounced upfield chemical shift change in the 1H NMR spectrum of the crude reaction mixture in CDCl3 was observed for the Ha′ methylene protons from 4.66 ppm to 3.84 ppm, characteristic of the tertiary N-amine region (Fig. S37 ESI†); a single peak at m/z = 1440.2 [M + H]+ in the electrospray ionisation (ESI) mass spectrum of the compound provided further confirmation of tripodal ligand formation (see Fig. S22†).
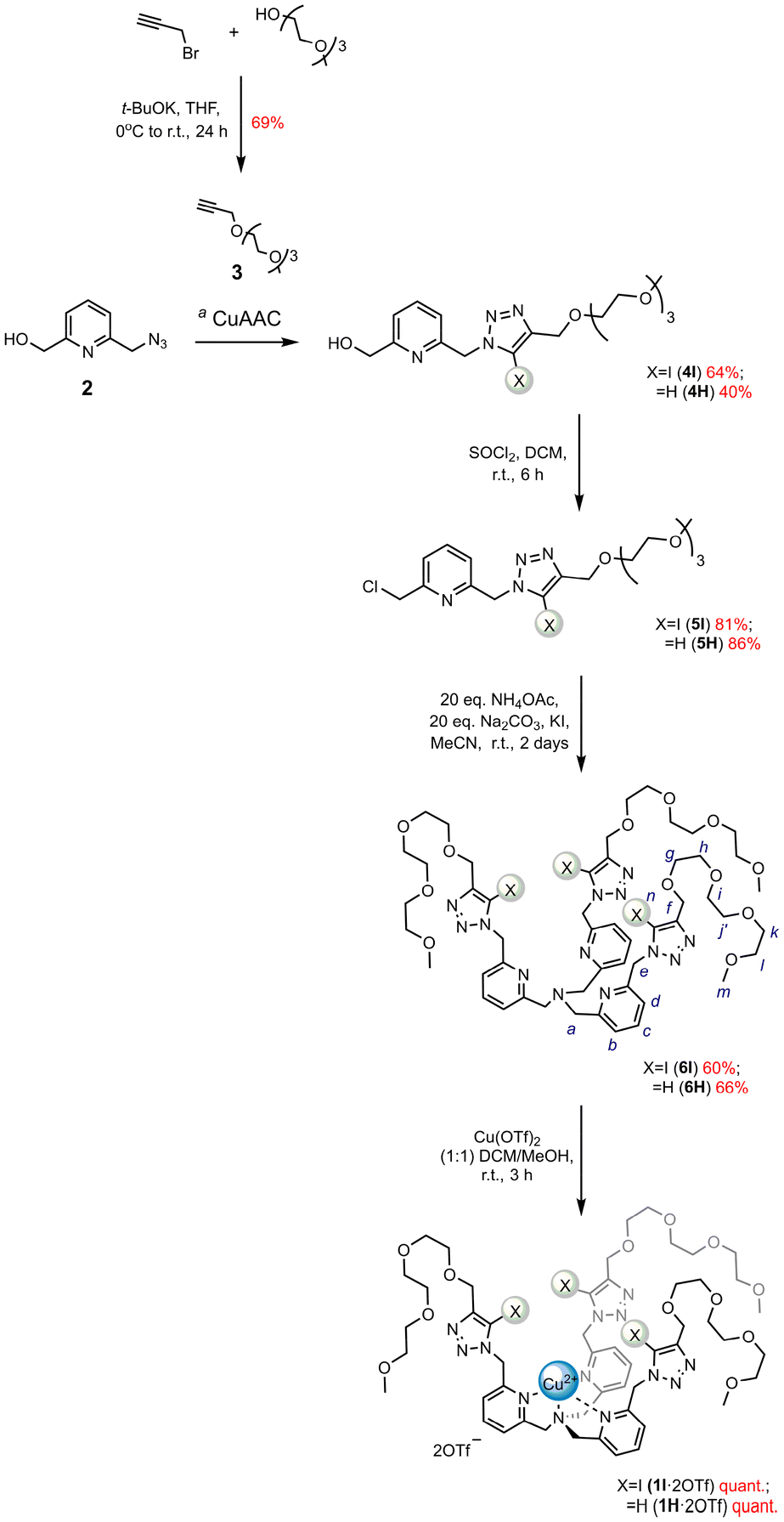 | ||
| Scheme 1 Synthesis of XB and HB receptors 1I·2OTf and 1H·2OTf. aFor X = I: Cu(ClO4)2·6H2O, TBTA (cat.), DBU, NaI, MeCN/THF (1:1), r.t., 16 h; for X = H: [Cu(MeCN)4]PF6, DIPEA, DCM, r.t., 48 h. | ||
Complexation of 6I with a stoichiometric quantity of copper(II) triflate afforded the XB Cu(II)-host 1I·2OTf. A UV-vis spectroscopy titration experiment revealed the addition of aliquots of the copper salt to an acetonitrile solution of 6I led to the appearance of a broad peak λabs,max (ε/M−1 cm−1) at 640 nm (161) and shoulder peak at 766 nm (98) as well as a visible colour change of the sample solution from colourless to light blue attributed to arise from chelated metal complexation (Fig. 2A).33 Monitoring the change in absorbance at 640 nm upon addition of increasing equivalents of copper(II) triflate verified the expected 1:1 stoichiometry for ligand to metal binding (Fig. 2B). Qualitative analysis of the binding isotherm indicated very strong Cu2+ binding affinity, which was too high for quantitative association constant determination in the organic acetonitrile solvent medium. Thus, an Isothermal titration calorimetry (ITC) titration was conducted in unbuffered water, with aliquots of the copper(II) triflate metal salt added to an aqueous solution of 6I determining an association constant of Ka = 1.55 × 106 M−1, as well as the relevant thermodynamic parameters revealing metal complexation to be both enthalpically (ΔH = −29.8 kJ mol−1) and entropically (−TΔS = −5.56 kJ mol−1) driven (see the ESI for full ITC details and Fig. S45†).
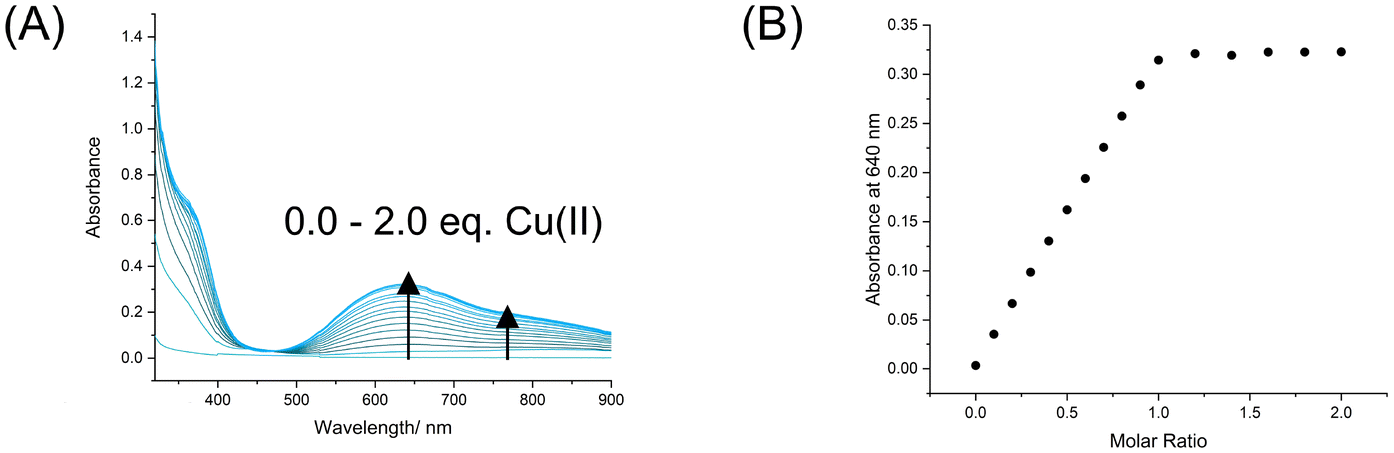 | ||
| Fig. 2 (A) UV-vis spectra of 6I upon addition of increments of Cu(OTf)2 up to 2.0 molar equiv. ([host] = 2 mM, CH3CN, T = 293 K), and (B) the corresponding mole ratio plot. | ||
The counterpart tris(prototriazole)-containing HB Cu(II)-tripod 1H·2OTf was also prepared via analogous procedures (Scheme 1) and both isolated paramagnetic metallo-receptors characterised via UV-vis, EPR, HR-MS as well as by electrochemical methods (vide infra).
The second XB Cu(II) tripod 13I·2OTf was prepared via adaptation of the synthetic methodology used to construct 1I·2OTf (Scheme 2). The pmβCD-derived terminal alkyne was first prepared via reaction of the activated ester succinimidyl-3-propiolate 734 with amine 819 and a catalytic amount of 4-dimethylaminopyridine (DMAP) overnight to afford 9 in 70% yield after chromatographic purification. An iodo-mediated one-pot CuAAC reaction between 2 and 9 afforded compound 10I, which was followed by chlorination with thionyl chloride to generate 11I. Reaction with a large excess of 30 molar equivalents of ammonium acetate and sodium carbonate produced 12I in 53% yield after purification via preparative TLC.‡ A diagnostic, significant upfield shift of the singlet signal (4.64 ppm) of the key Ha′ methylene protons of monomer 11I to the main pmβCD region was evidenced in the comparative 1H NMR of trimer 12I in CDCl3 (see Fig. 3 and S38 ESI†) which along with ESI-MS analysis (m/z = 2627.9 [M + Na + H]2+) affirmed the successful transformation to the XB tripodal assembly (Fig. S36†). Complexation of 12I with copper(II) triflate quantitatively afforded XB Cu(II)-host 13I·2OTf. Using an analogous ITC titration protocol to 6I, the Cu(II)-binding properties of 12I were studied in unbuffered water, determining an association constant of Ka = 1.26 × 105 M−1 where metal complexation is predominantly enthalpically favoured (ΔH = −28.9 kJ mol−1; −TΔS = −0.27 kJ mol−1). 13I·2OTf was also characterised by UV-vis, EPR and HR-MS methods. The electronic spectrum of 13I·2OTf in CH3CN/H2O 6:4 v/v (red dashed line, Fig. 5A) displays a pronounced maximum absorption peak λmax (ε/M−1 cm−1) at 645 nm (182) with a much broader shoulder peak at ca. 770 nm (119) attributed to arise again from the relevant d–d transitions of the XB ligand chelated to the Cu(II) center.33
 | ||
| Scheme 2 Synthesis of XB receptor 13I·2OTf. aCuAAC: Cu(ClO4)2·6H2O, TBTA (cat.), DBU, NaI, MeCN/THF (1:1), r.t., 16 h. | ||
 | ||
| Fig. 3 Stacked 1H NMR spectra of (A) 12I and (B) synthon 11I in CDCl3 (500 MHz, T = 298 K). See Scheme 2 for proton assignment. (The methylene He protons of both compounds are diastereotopic and hence appear as an AB quartet signal.) | ||
UV-vis anion binding studies
Quantitative UV-vis titration experiments were performed by adding increasing quantities of anions as their tetrabutylammonium (TBA) salts to separate solutions of 1I·2OTf and 1H·2OTf in the CH3CN/H2O 6:4 v/v solvent mixture (buffered with 10 mM HEPES at pH 6.3). Both receptors exhibited very similar molar extinction coefficients and peak absorption wavelengths λabs,max (ε/M−1 cm−1) at 647 nm (117) and 775 nm (91) (shoulder) for 1I·2OTf and 639 nm (125) and 760 nm (98) (shoulder) for 1H·2OTf, suggesting that the more electron-deficient, iodotriazole encompassing tripodal ligand framework of 1I·2OTf did not significantly affect the electronic energy levels of the respective Cu(II) core in mixed aqueous–organic media (Fig. S39 and S41†).
The interactions of Br−, Cl−, OAc− and H2PO4− anions with the Cu(II) center of 1I·2OTf and 1H·2OTf led to naked-eye visible colour changes of the host solution from light blue to green, with the presence of an isosbestic point in each of their corresponding UV-vis titration spectra. In each case, a decrease in intensity of the absorption band at ca. 640 nm concomitant with an increase in intensity of the broad shoulder peak at ca. 770 nm was observed, presumably due to ligand field effects and further implying a similar mode of anion binding within the XB/HB tripodal host interior (Fig. S47†). With the halides, the spectral changes of the latter lower energy band were greater than the former, higher energy band (Fig. 4A) whereas upon addition of oxoanions, both absorption bands exhibited comparable spectral perturbations (Fig. 4B). In contrast, negligible spectral changes were seen during the titrations of NO3− indicating that the anion was not interacting appreciably with either receptor.
 | ||
| Fig. 4 UV-vis anion titration spectra of 1I·2OTf in the presence of up to 100 equivalents of (A) TBA Br, (B) TBA H2PO4 and (C) TBA PMP ([host] = 2 mM, CH3CN/H2O 6:4 buffered with 10 mM HEPES at pH 6.3, T = 293 K). | ||
Global spectral fitting of the UV-vis titration data using the BindFit35 software monitoring the change in absorbance as a function of anion concentration determined anion association constants summarised in Table 1 (for the corresponding isotherms see Fig. S48 and S49†). All anions were found to bind predominantly via a 1:2 host:guest stoichiometry with the second binding constant (K12) being of much lower magnitude than the first (K11) suggesting weak peripheral association of the second anion, likely driven by the electrostatic attraction of the host's divalent Cu(II)-metallocentre. Notably, 1I·2OTf displays strong and selective recognition for H2PO4−,§ with an affinity over six times greater than weakest bound Cl−. Given that the hydration energy of phosphate is the largest of all the anions tested (Table 1), the observed selectivity thus attests to the favourable size and shape match of the TEG-appended XB metallo-tripodal receptor's binding pocket with the target tetrahedral guest (Fig. 4B). In addition 1I·2OTf exhibited a distinct Hofmeister preference for Br− > Cl−. Importantly, the HB receptor 1H·2OTf although also being phosphate selective, displayed attenuated anion binding in all cases, with very modest discrimination between Br− and Cl−. The large difference between the magnitudes of the phosphate association constants of the two receptors in particular, with the value calculated for 1I·2OTf over three-times greater than 1H·2OTf, provides a very rare demonstration of the superior ability of XB to enhance hard oxoanion recognition in highly competitive 40% aqueous–organic solvent mixtures. Compared to the current cadre of reported XB phosphate receptors,24–291I·2OTf therefore constitutes a very potent XB host capable of discriminating phosphate in aqueous media.
| Anion | ΔGhydb (kJ mol−−1) |
1I·2OTf | 1H·2OTf |
|---|---|---|---|
|
a A 1:2 host–guest binding model, unless otherwise stated, was used to determine Ka values from global fitting of the UV-vis titration spectra, monitoring the change in absorption from 800–850 nm, using BindFit;35 anions added as their TBA salts; ([host] = 2 mM, CH3CN/H2O 6:4, T = 293 K); errors (±) in parenthesis (<11%).
b Values reported by Marcus.36
c Titration not performed as I− is easily oxidised in the presence of Cu(II).
d NB: no binding. Spectral changes too little to accurately determine association constants.
e Multiple complex binding equilibria. Unable to fit data to 1:1, 1:2 or 2:1 host–guest models.
|
|||
| Cl− | −340 | K 11 = 379 (15) | 121 (3) |
| K 12 = 16 | |||
| Br− | −315 | K 11 = 834 (38) | K 11 = 108 (2) |
| K 12 = 23 | K 12 = 7 | ||
| I− | −275 | —c | —c |
| NO3− | −300 | NBd | NBd |
| OAc− | −365 | K 11 = 435 (20) | —e |
| K 12 = 21 | |||
| H2PO4− | −465 | K 11 = 2816 (309) | K 11 = 745 (25) |
| K 12 = 3 | K 12 = 1 | ||
| PMP− | n/a | K 11 = 1094 (68) | K 11 = 772 (63) |
| K 12 = 73 | K 12 = 11 | ||
The demonstration of selective inorganic monophosphate binding by both XB 1I·2OTf and HB 1H·2OTf receptors prompted us to also investigate their ability to bind a simple phosphate-ester and a higher-order polyphosphate species, specifically pinacolyl methyl phosphonate (PMP), the innocuous hydrolysis product of the ‘G-series’ chemical warfare nerve agent soman (GD)38 and inorganic pyrophosphate (PPi), respectively. Upon the addition of TBA PMP, pronounced optical perturbations analogous to those elicited with H2PO4− were evidenced in the individual absorption spectra of 1I·2OTf (Fig. 4C) and 1H·2OTf (Fig. S47†) and the organophosphonate found to associate with the host similarly with a 1:2 host–guest stoichiometry (Table 1). Yet again, the XB receptor proved to be the stronger complexant, binding PMP− with a larger affinity than the protic counterpart thus highlighting the applicability of XB as a potent supramolecular interaction to further exploit for the enhanced recognition of its parent GD compound as well as other, more toxic ‘G-series’ substances.39 The lower association constant value of PMP− with 1I·2OTf compared to H2PO4− is unsurprising given the steric demands of the much larger aliphatic group functionalised phosphonate, resulting in a less complementary fit within the XB host's tetrahedral cavity: possibly, the alkyl substituent protons of the organophosphonate's phosphate ‘head’ might impose a steric clash with one of the voluminous iodine atoms of a convergent iodotriazole binding unit (red lines, Fig. 4C).
In contrast, the addition of aliquots of TBA3 PPi to 1I·2OTf elicited explicitly larger spectral changes with a dramatic intensity decrease in both of the absorption bands of the XB Cu(II)-host (Fig. S47†). Compared to the other anions tested, only unidirectional changes were observed with PPi3− suggesting that it was being bound via a different coordination stoichiometry perhaps simultaneously ‘bridging’ the metal centers of two individual XB hosts, in a similar manner as when coordinating to the structurally related bimetallic Zn(II)–dipicolylamine compounds and/or di-nuclear Cu(II) tripodal amine complexes.40–42 Attempts to fit the titration data to various binding models of 2:1 as well as 1:1 and 1:2 host–guest stoichiometries unfortunately failed to determine quantitative association constant data.
Similar spectral perturbations of the host's d–d bands were observed upon the addition of aliquots of TBA H2PO4 to 13I·2OTf as 1I·2OTf, with the progressive decrease in intensity of the initial higher energy absorption band (at 645 nm) and comparable increase in intensity of the initial lower energy shoulder peak (at ca. 770 nm) suggestive of a similar mode of anion binding within the tripodal XB cavity (Fig. 5A). Global spectral fitting of the UV-vis titration data using the BindFit35 software notably revealed an enhancement in the binding of H2PO4− with 13I·2OTf (Fig. 5B), with the determined 1:2 stoichiometric association constants of K11 = 9442 ± 944 M−1 and K12 = 40 ± 2 M−1 being ca. thrice as large in magnitude compared to 1I·2OTf (cf.Table 1). The observed increased phosphate anion affinity may arise from the donor ability of the XB iodotriazoles being augmented via intramolecular N–H⋯I HB interactions from the neighbouring amide functionalities of the host's pmβCD-derived ‘arms’ which promote the synergistic binding of the target tetrahedral guest via multiple ‘hydrogen bonding-enhanced halogen bonding’ (HBeXB) interactions (Fig. 6).43,44
 | ||
| Fig. 5 (A) UV-vis titration spectra of 13I·2OTf (red dashed line) with up to 100 equivalents of TBA H2PO4 ([host] = 2 mM, CH3CN/H2O 6:4 buffered with 10 mM HEPES at pH 6.3, T = 293 K), (B) the corresponding isotherm monitoring the change in absorbance from 800–850 nm vs. equivalents of anion. | ||
 | ||
| Fig. 6 The envisioned binding mode of XB receptor 13I·2OTf with H2PO4− (intramolecular N–H⋯I HB indicated by orange dashed lines; N–H HBeXB interactions indicated by green dashed lines). | ||
EPR anion binding studies with 1I/H·2OTf
To complement the UV-vis anion titration experiments, the host–guest properties of 1I/H·2OTf towards Br− and H2PO4− were also investigated via EPR spectroscopy. The X-band continuous wave (CW) EPR spectra of both ‘free’ receptors 1I·2OTf and 1H·2OTf were first recorded in the frozen buffered CH3CN/H2O 6:4 v/v solvent mixture at 85 K (see Fig. 7A and B, bold red lines and Fig. S39 and S41, ESI†). The XB host displayed an axial signature characterised by the principal g values of g∥ = 2.225 and g⊥ = 2.056 (A∥ = 183.0 × 10−4 cm−1) with the classic four-line splitting pattern in the parallel region arising from the hyperfine interactions between the unpaired Cu(II) electron and the two principle 63Cu and 65Cu paramagnetic nuclei.¶ Notably, these g∥ > g⊥ values are quite unusual for tris-(2-pyridylmethyl)amine (TPMA) based Cu(II)-complexes which typically present g⊥ > g∥ values that denote a 5-coordinate Cu(II) environment, where the metal cation is ligated by four nitrogen atoms of the tetradentate TPMA subunit and also one solvent molecule (either CH3CN or H2O), predominantly in a trigonal bipyramidal (TBP) geometry.45–47 The obtained A∥ value is also much larger than the ones classically encountered for TBP Cu(II)-centers (A∥ < 80 × 10−4 cm−1).48 With these observations in mind and having previously evidenced the additional N2-coordination of two of the iodotriazoles to the Zn(II) center in the X-ray crystallographic solid state structure of the tris(ferrocene) receptor analogue,29 a similar 6-coordinate Cu(II) centre of 1I·2OTf in frozen solution thus seems likely.|| Due to the lack of spectroscopic characterisation of such ‘4 + 2’ coordinated Cu(II)–TPMA complexes in the literature however, the unambiguous assignment of an octahedral coordination geometry is rather complicated. Receptor 1H·2OTf exhibited virtually identical EPR parameters of g∥ = 2.285 and g⊥ = 2.055 apart from a very small difference of the A∥ value (A∥ = 180.0 × 10−4 cm−1) suggesting, similarly to the UV-vis characterisation data, little influence of the iodotriazoles on the hyperfine coupling and hence degree of delocalisation of the unpaired d-electron from the respective paramagnetic Cu(II) center.
 | ||
| Fig. 7 CW EPR titration spectra of receptors (A) 1I·2OTf and (B) 1H·2OTf in the presence of increasing quantities of anions ([host] = 2 mM, CH3CN/H2O 6:4 buffered with 10 mM HEPES at pH 6.3, T = 85 K). EPR signature changes specifically in the perpendicular region of the titration spectra are indicated by the arrows. | ||
EPR anion titration investigations were performed by adding increasing aliquots of anions (up to 100 equivalents) as their TBA salts to separate solutions of the XB and HB Cu(II)-tripodal receptors and the CW EPR spectra recorded at 85 K (full experimental details of the EPR titration protocol are included in the ESI†). Characteristic changes in the EPR spectral signatures and intensities of 1I·2OTf and 1H·2OTf were afforded upon addition of the Br− and H2PO4− guests (Fig. 7). Firstly, in all cases, the original quadruple line-splitting pattern contained multiple overlapping contributions in both parallel and perpendicular regions (i.e. >four-line splitting) thus indicative of a ‘mixed’ equilibrium species (presumably the ‘free’ host and ‘bound’ host–guest adduct) present in frozen aqueous–organic solution. A pronounced overall reduction in intensity of the g⊥ contribution of both 1I·2OTf and 1H·2OTf with both anions was further qualitatively observed upon signal saturation – this is particularly evident in the perpendicular region of the XB receptor with Br− (Fig. 7A, red arrows). Indeed whilst these qualitative differences show that it is possible to detect anion binding through form of a simple ligand-exchange at the metal center using EPR spectroscopy, the complex signatures of the titration spectra require further extensive simulation work (beyond the scope of this study) to separate the overlapping contributions and hence extract the corresponding g values and hyperfine EPR parameters giving insight to the exact nature of the anion interaction and axial Cu(II) coordination site.
For this reason, a global spectral fitting strategy of the EPR titration data was adopted, monitoring the changes in EPR intensity (within the limits of the A∥ and g∥ extrema) as a function of anion concentration (Fig. 8) which allowed for the determination of association constants of 1:2 stoichiometry using the BindFit35 software as summarised in Table 2. Pleasingly, these EPR titration binding constants are in alignment with the results obtained from the corresponding UV-vis anion titrations: the XB receptor once more, not only exhibited preferential binding of H2PO4− over Br− but importantly also superior binding of anions than the HB counterpart in both cases (cf.Table 1). To the best of our knowledge, quantitative anion titrations with small artificial receptors such as these via EPR spectroscopy remain unprecedented in the literature – only calixarene-based azacryptand Cu(II) funnel complexes have been reported by Reinaud and co-workers for the quantitative EPR detection of neutral alcohol, amine and nitrile guests.45–47 This experimental data thus serves to highlight the applicability of EPR as a detection tool for the sensing of anionic guests as well as the determination of their association constants.
 | ||
| Fig. 8 EPR anion titration isotherms of receptor (A) 1I·2OTf and (B) 1H·2OTf, monitoring the change in signal intensity from 280–290 mT vs. equivalents of anion added ([host] = 2 mM, CH3CN/H2O 6:4 buffered with 10 mM HEPES at pH 6.3, T = 85 K). | ||
| Anion | 1I·2OTf | 1H·2OTf |
|---|---|---|
|
a A 1:2 host–guest binding model unless otherwise stated, was used to determine Ka values from global fitting of the EPR titration spectra, monitoring the change in signature intensity from 280–290 mT, using BindFit;35 anions added as their TBA salts; ([host] = 2 mM, CH3CN/H2O 6:4, T = 85 K); errors (±) in parenthesis (<11%).
|
||
| Br− | K 11 = 369 (22) | K 11 = 101 (7) |
| K 12 = 55 | K 12 = 16 | |
| H2PO4− | K 11 = 1641 (102) | K 11 = 743 (65) |
| K 12 = 36 | K 12 = 59 | |
Preliminary electrochemical anion sensing studies with 1I/H·2OTf
The success of 1I/H·2OTf as effective optical sensors and EPR probes for H2PO4− encouraged us to explore the possibility of exploiting the hosts’ cupric metal centre further for electrochemical phosphate sensing via the CuII/CuI redox couple. The redox properties of 1I·2OTf and 1H·2OTf were initially investigated via cyclic voltammetry (CV) in anhydrous CH3CN (0.1 M TBAClO4 supporting electrolyte solution) and pure water (0.05 M KPF6 supporting electrolyte solution) solvents as well as in various intermediate CH3CN/H2O mixtures, using a Ag/AgCl reference electrode (see ESI† for full reversibility details). Both receptors were found to display explicit solvent dependent redox behaviour.In anhydrous CH3CN, both 1I·2OTf and 1H·2OTf were electrochemically-active and displayed a well-defined, single quasi-reversible one-electron wave attributed to the CuII/CuI redox couple, with the more anodic E1/2 value of +74 mV of the XB host versus −179 mV of the HB counterpart (both vs. Fc/Fc+) consistent with the increased electron-withdrawing nature of the iodotriazoles disfavouring oxidation of the Cu center (Fig. S50 and S52†). The marked 253 mV difference between the two E1/2 values is particularly noteworthy and as previously suggested by the EPR characterisation data, indicates the N2-coordination of the XB/HB-triazoles to the metal core, which are thus able to directly exert their electronic influences on the redox properties of the Cu center in organic media. Contrastingly, whilst the XB and HB Cu(II)-receptors retained redox quasi-reversibility in various CH3CN/H2O solvent mixtures, a significantly distorted anodic peak was observed in each case particularly at higher scan rates. This was exacerbated in pure water (see Fig. S54 and S56†) and postulated to arise from the ‘static’ N2-ligation of the XB/HB-triazoles to the Cu center, as strongly suggested by the earlier investigative EPR data. Indeed, the more anodic E1/2 values of +174 mV of the XB host and +89 mV of the HB counterpart (both vs. Ag/AgCl) in pure water compared to those in CH3CN (where the triazole coordination is ‘dynamic’) lend credence to this hypothesis. Additionally, the much smaller 85 mV discrepancy between these E1/2 values likely results from the better stabilisation of the dicationic vs. monocationic Cu state due to the increased polarity of the all-aqueous solvent medium.
Owing to the complex voltammetric behaviour exhibited by 1I/H·2OTf in aqueous containing media, their redox anion sensing properties were probed only in anhydrous CH3CN (containing 0.1 M TBAClO4 as supporting electrolyte) using square-wave voltammetry (SWV). Due to precipitation problems with phosphate, preliminary electrochemical anion titrations were restricted to bromide. The addition of excess TBA Br (up to 100 equivalents) induced significant cathodic perturbations of the respective CuII/CuI redox couples of both XB and HB receptors arising from halide anion binding stabilising the Cu(II) oxidation state (Fig. 9). In both cases, a swift plateauing of the voltammetric response was observed further indicative of strong binding (Fig. 10). Notably however, the XB host exhibited a much larger magnitude of cathodic shift (−140 ± 5 mV) at saturation compared to the protic analogue (−80 ± 5 mV) clearly highlighting the dominant role played by the XB donor groups for redox anion sensing. The very large magnitudes of these cathodic shifts further enabled the quantitative analysis of the respective titration isotherms according to the established 1:1 host–guest stoichiometric Nernst model (eqn (1)):50
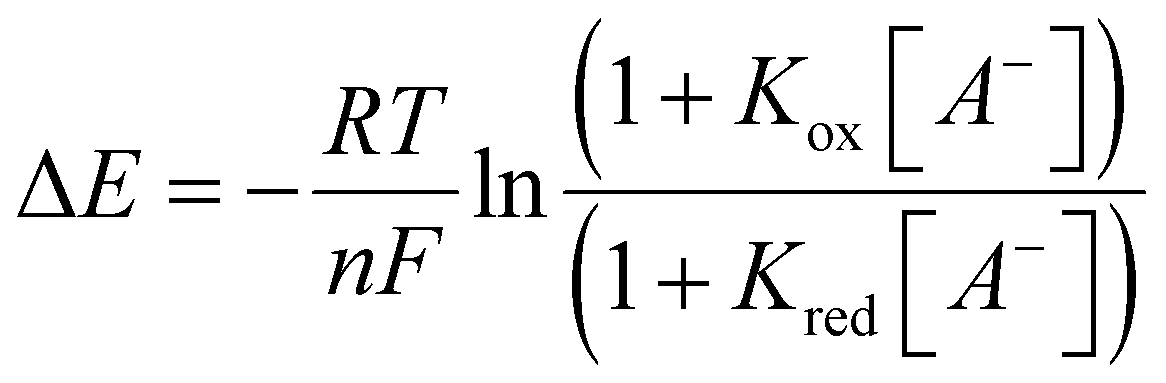 | (1) |
074/289 M−1) compared to the HB analogue (Kox/Kred = 15734/581 M−1). In particular, these amplified halide anion-induced voltammetric responses of 1I·2OTf and 1H·2OTf compared to those observed with their electrochemically active tris(ferrocene) Zn(II)-receptor counterparts29 convey not only the efficacy of the CuII/CuI redox couple as an electrochemical transduction entity but also the potency of utilising the host's metal epitope as a direct probe for the anion binding event (Fig. 4A) – which for self-evident reasons, exhibits a greater sensitivity to the recognition process as opposed to the through-bond communicative pathways between the anion binding cavity and redox reporter group.29
 | ||
| Fig. 9 Square wave voltammograms of receptors (A) 1I·2OTf and (B) 1H·2OTf with incremental quantities of TBA Br ([host] = 0.5 mM, electrolyte: 0.1 M TBAClO4 in anhydrous CH3CN, T = 293 K). All potentials taken in reference to the Ag/AgCl reference electrode (leak free). (The distorted red baseline in (A) arises from the need of having to adjust the potential range during the titration to prevent Br− oxidation.) | ||
 | ||
| Fig. 10 Electrochemical anion titration isotherms. Shift of the half-wave potential (ΔE1/2) of the CuII/CuI redox couple of 1I·2OTf (blue) and 1H·2OTf (red) in the presence of Br−. | ||
Conclusions
In conclusion, a series of strategically designed, hydrophilic XB tripodal Cu(II) metallo-receptors 1I·2OTf and 13I·2OTf were synthesised and demonstrated to be effective at selectively recognising and sensing inorganic monophosphate over a range of other monoanionic species in aqueous media containing 40% water. Extensive UV-vis spectrophotometric anion titration studies, notably accompanied by a naked-eye colour change response, revealed that 1I·2OTf bound H2PO4− strongly (Ka = 2816 M−1) with an affinity over six times greater than the weakest bound Cl−, which was attributed to the complementary size and shape match between the tetrahedral anion and the XB tripodal cavity. Importantly, the phosphate association constant value for 1I·2OTf was notably of a significantly larger magnitude than that obtained with the HB receptor counterpart 1H·2OTf further championing and advancing XB as a more effective interaction than HB for hard oxoanion binding in aqueous environments. Similarly augmented binding of the monophosphate-ester PMP− was evidenced with 1I·2OTf compared to 1H·2OTf thus also highlighting the applicability of XB as a worthwhile supramolecular interaction to potentially exploit for the enhanced recognition of toxic ‘G-series’ substances as well as their innocuous hydrolysis products. Furthermore, an impressive ca. three-fold enhancement in phosphate binding affinity was achieved with 13I·2OTf compared to 1I·2OTf, with the presence of the host's XB donor iodotriazole and conjugated HB donor NH amide functionalities bringing to light the synergistic interplay of these two, often competing, interactions in the binding process.Importantly both XB/HB Cu(II) receptors 1I/H·2OTf were also demonstrated to be highly sensitive EPR anion sensing probes, wherein the determination of quantitative anion binding constant data was achieved for the first time. The Cu(II) metallo-receptors also proved to be effective electrochemical anion sensors capable of sensing Br−via cathodic shift perturbations of their respective CuII/CuI redox couples. Further exploitation of the ‘C3-symmetric’ XB motif29 for XB-mediated oxoanion recognition and sensing applications in aqueous media is continuing in our laboratories.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. B. thanks Dstl for a DTA studentship.References
- L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Chem, 2022, 8, 46–118 CAS.
- S. Faulkner, S. J. A. Pope and B. P. Burton-Pye, Appl. Spectrosc. Rev., 2005, 40, 1–31 CrossRef CAS.
- S. E. Bodman and S. J. Butler, Chem. Sci., 2021, 12, 2716–2734 RSC.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- H. Cui and B. Xu, Chem. Soc. Rev., 2017, 46, 6430–6432 RSC.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- H. Wang, X. Ji, M. Ahmed, F. Huang and J. L. Sessler, J. Mater. Chem. A, 2019, 7, 1394–1403 RSC.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- S. Pal, T. K. Ghosh, R. Ghosh, S. Mondal and P. Ghosh, Coord. Chem. Rev., 2020, 405, 213128 CrossRef CAS.
- K. A. Jolliffe, Acc. Chem. Res., 2017, 50, 2254–2263 CrossRef CAS PubMed.
- M. V. R. Raju, S. M. Harris and V. C. Pierre, Chem. Soc. Rev., 2020, 49, 1090–1108 RSC.
- O. Francesconi, M. Gentili, F. Bartoli, A. Bencini, L. Conti, C. Giorgi and S. Roelens, Org. Biomol. Chem., 2015, 13, 1860–1868 RSC.
- S. J. Butler, Chem. – Eur. J., 2014, 20, 15768–15774 CrossRef CAS PubMed.
- R. Mailhot, T. Traviss-Pollard, R. Pal and S. J. Butler, Chem. – Eur. J., 2018, 24, 10745–10755 CrossRef CAS PubMed.
- S. L. Tobey, B. D. Jones and E. V. Anslyn, J. Am. Chem. Soc., 2003, 125, 4026–4027 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- R. Hein and P. D. Beer, Chem. Sci., 2022, 13, 7098–7125 RSC.
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, Angew. Chem., Int. Ed., 2019, 58, 13823–13827 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- S. P. Cornes, M. R. Sambrook and P. D. Beer, Chem. Commun., 2017, 53, 3866–3869 RSC.
- A. Borissov, I. Marques, J. Y. C. Lim, V. Félix, M. D. Smith and P. D. Beer, J. Am. Chem. Soc., 2019, 141, 4119–4129 CrossRef CAS PubMed.
- E. J. Mitchell, A. J. Beecroft, J. Martin, S. Thompson, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2021, 60, 24048–24053 CrossRef CAS PubMed.
- M. Cametti, K. Raatikainen, P. Metrangolo, T. Pilati, G. Terraneo and G. Resnati, Org. Biomol. Chem., 2012, 10, 1329–1333 RSC.
- B. Chowdhury, S. Sinha and P. Ghosh, Chem. – Eur. J., 2016, 22, 18051–18059 CrossRef CAS PubMed.
- F. Zapata, A. Caballero and P. Molina, Eur. J. Inorg. Chem., 2017, 2017, 237–241 CrossRef CAS.
- F. Zapata, L. González, A. Caballero, A. Bastida, D. Bautista and P. Molina, J. Am. Chem. Soc., 2018, 140, 2041–2045 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, New J. Chem., 2018, 42, 10472–10475 RSC.
- H. Bagha, R. Hein, J. Y. C. Lim, C. B. Durr, M. R. Sambrook and P. D. Beer, Chem. – Eur. J., 2024, 30, e202302775 CrossRef CAS PubMed.
- T. Shiraki, A. Dawn, Y. Tsuchiya and S. Shinkai, J. Am. Chem. Soc., 2010, 132, 13928–13935 CrossRef CAS PubMed.
- T. Zhang and E. V. Anslyn, Tetrahedron, 2004, 60, 11117–11124 CrossRef CAS.
- W. S. Brotherton, R. J. Clark and L. Zhu, J. Org. Chem., 2012, 77, 6443–6455 CrossRef CAS PubMed.
- P. J. Arnold, S. C. Davies, J. R. Dilworth, M. C. Durrant, D. V. Griffiths, D. L. Hughes, R. L. Richards and P. C. Sharpe, J. Chem. Soc., Dalton Trans., 2001, 736–746 RSC.
- J. Chao, W.-Y. Huang, J. Wang, S.-J. Xiao, Y.-C. Tang and J.-N. Liu, Biomacromolecules, 2009, 10, 877–883 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- M. R. Sambrook and S. Notman, Chem. Soc. Rev., 2013, 42, 9251–9267 RSC.
- D. H. Lee, S. Y. Kim and J.-I. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777–4780 CrossRef CAS PubMed.
- S. J. Butler and K. A. Jolliffe, Chem. – Asian J., 2012, 7, 2621–2628 CrossRef CAS PubMed.
- S. Watchasit, A. Kaowliew, C. Suksai, T. Tuntulani, W. Ngeontae and C. Pakawatchai, Tetrahedron Lett., 2010, 51, 3398–3402 CrossRef CAS.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A.-C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed.
- R. P. Orenha, S. S. P. Furtado, G. F. Caramori, M. J. Piotrowski, A. Muñoz-Castro and R. L. T. Parreira, New J. Chem., 2023, 47, 4439–4447 RSC.
- N. Le Poul, B. Douziech, J. Zeitouny, G. Thiabaud, H. Colas, F. Conan, N. Cosquer, I. Jabin, C. Lagrost, P. Hapiot, O. Reinaud and Y. Le Mest, J. Am. Chem. Soc., 2009, 131, 17800–17807 CrossRef CAS PubMed.
- G. Izzet, X. Zeng, H. Akdas, J. Marrot and O. Reinaud, Chem. Commun., 2007, 810–812 RSC.
- G. Thiabaud, A. Brugnara, M. Carboni, N. Le Poul, B. Colasson, Y. Le Mest and O. Reinaud, Org. Lett., 2012, 14, 2500–2503 CrossRef CAS PubMed.
- D. K. Chand and P. K. Bharadwaj, Inorg. Chem., 1996, 35, 3380–3387 CrossRef CAS PubMed.
- M. M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC.
- R. Oliveira, S. Groni, C. Fave, M. Branca, F. Mavré, D. Lorcy, M. Fourmigué and B. Schöllhorn, Phys. Chem. Chem. Phys., 2016, 18, 15867–15873 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01585a |
| ‡ The usual 20 equivalents of NH4OAc utilised in previous tripodal ligand formation procedures (see Scheme 1) was increased to 30 equivalents given the propensity of reagents to form inclusion complexes with the β-cyclodextrin ‘cone’. |
| § It should be noted that at the working pH 6.3 maintained in the UV-vis experiments, inorganic phosphate exists about 88% as H2PO4− and 12% as HPO42− (calculated using the HyperQuad37 software), hence here we have referred to the former dominant diprotic form as representative of the total monophosphate species. |
| ¶ (2NI + 1 = 4, where N is the number of nuclei felt by the unpaired electron and I is the nuclear spin quantum number, which for a Cu(II) nucleus is I = 3/2). |
| || The very fast relaxation times of the two dynamic T1 (spin–lattice) and T2 (spin–spin) relaxation processes often observed at room temperature (e.g. for transition-metal complexes) and crucial in the EPR experiment, inevitably lead to extensive broadening of the acquired spectrum such that effectively no EPR signal is observed (half linewidth = 1/T2 + 1/2T1, as also dictated by Heisenberg's uncertainty principle).49 Consequently, EPR measurements are routinely undertaken at cryogenic temperatures (i.e. in frozen solution) to increase the T1 and T2 times of such ‘fast relaxing systems’ thus leading to better-resolved EPR spectra. |
| This journal is © The Royal Society of Chemistry 2024 |