
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complex isomerism influencing the textural properties of organometallic [Cu(salen)] porous polymers: paramagnetic solid-state NMR characterization and heterogeneous catalysis†
David
Šorm
 *a,
Jan
Blahut
*b,
Bogdana
Bashta
a,
Ivana
Císařová
c,
Eva
Vrbková
d,
Eliška
Vyskočilová
d and
Jan
Sedláček
a
*a,
Jan
Blahut
*b,
Bogdana
Bashta
a,
Ivana
Císařová
c,
Eva
Vrbková
d,
Eliška
Vyskočilová
d and
Jan
Sedláček
a
aDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Hlavova 2030, Prague 2, 128 43, Czech Republic. E-mail: david.sorm@natur.cuni.cz
bInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 542/2, Prague 6, 160 00, Czech Republic. E-mail: blahutj89@gmail.com
cDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, Prague 2, 128 43, Czech Republic
dDepartment of Organic Technology, University of Chemistry and Technology Prague, Technická 5, Prague 6, 166 28, Czech Republic
First published on 25th June 2024
Abstract
Although organometallic porous polymer networks are recognized as promising heterogeneous catalysts, the relationship between ligand/monomer geometry and network parameters is usually not well understood due to the lack of atom-resolved characterization methods for the amorphous network matrix. In this work, a series of copper(II) salen-type metal complexes was synthesized, using trans- and cis-1,2-diaminocyclohexane segments, and thoroughly characterized by single-crystal X-ray diffraction and solution- and solid-state NMR spectroscopy. Terminal ethynyl groups of the complexes were then transformed into polyacetylene chains by coordination chain-growth homopolymerization, resulting in highly porous (458–655 m2 g−1) organometallic polymer networks with a copper(II) ion content of about 12 wt%. The presence of paramagnetic copper(II) moieties in these complexes and respective polymer networks required the application of tailored NMR techniques, which together with X-ray crystallography and DFT calculations of the paramagnetic NMR shifts made it possible to investigate the differences in the complex geometry in liquid, powder and crystalline form and compare it with the complex geometry in polymer networks. All prepared organometallic polymer networks were also tested as heterogeneous catalysts for styrene oxidation with uncommonly high substrate conversions and compared with their low-molecular-weight analogues. The high reusability of such heterogeneous polymer-based catalysts was also proven.
Introduction
Salen-type metal complexes, generally prepared by condensation of salicylaldehydes with various diamines followed by complexation of (usually) transition metal ions, are widely studied for their broad range of applications.1,2 For instance, metal salen complexes are used in various bioinorganic and medicinal applications,3 other metal salen complexes have been tested for their optical properties leading to the development of new LED systems4–7 and multimetallic salen particles are studied for their magnetic properties.8,9 However, by far the most widespread utilization of salen-type metal complexes is in the field of catalysis.1,10,11 Although diverse organometallic Schiff base complexes of various compositions have been reported,12–15 the Schiff base complexes with salen-type ligands strongly prevail in catalytic applications. A binuclear titanium(IV) salen complex was used for the cyanation of aldehydes,16 a cobalt(II) salen complex was employed for benzoyl fluoride promoted ring opening of epoxides,17 a chromium(III) salen complex was tested for nitroalkylation of aldehydes via the Henry reaction,18 an iron(III) salen complex was applied for sulfide oxidation,19 and a nickel(II) salen complex was utilized for Michael addition.20 Another widely studied reaction is the addition of CO2 to epoxides, resulting in the formation of cyclic carbonates. Various salen-type complexes with a wide spectrum of metal ions (Co, Cr, Zn, Cu, Ti, V, Sc, and Y) have been reported as effective catalysts of this addition.21–27Anchoring metal salen complexes to diverse, mostly porous supporting materials leads to the “heterogenization” of these catalysts resulting in their good separability from the reaction mixture, reusability, and sometimes enhanced selectivity or stability.28,29 In numerous instances, metal salen complexes were immobilized on porous supports like nanocomposites,30,31 polymer resins,32 heterostructured clays,33 graphene oxides34 or widely used mesoporous silica-based supports.35–39 Other complexes directly participated in the formation of porous solids while maintaining their catalytic activity. In these cases, metal salen complexes were integral components of porous materials such as covalent organic frameworks.40 The special category is represented by metal salen complexes incorporated as building blocks of porous organic polymers (POPs).41 POPs are a class of amorphous polymers characterized by a three-dimensional network structure possessing large specific surface areas given by permanent micro/mesoporosity. These characteristics arise from the rigidity of the network segments coupled with extensive cross-linking.42–46 An essential study in this field was published by Xie et al. in 2013: a metallosalen-containing POP was prepared by Sonogashira–Hagihara cross-coupling of a dibrominated cobalt(III) salen-based complex with a 1,3,5-triethynylbenzene cross-linker and subsequently used as a CO2 sorbent and at the same time as a heterogeneous catalyst for the transformation of CO2 into a cyclic carbonate.47 Subsequently, Xie et al. continued with a follow-up study using a POP based on a zinc(II) salen-type complex.48 Other authors inspired by these works prepared zinc(II) or cobalt(III) salen-based POPs with ionic imidazolium groups tested as heterogeneous catalysts for CO2 conversions.49,50 Another catalytic utilization of metallosalen-containing POPs was in halogenation,51 the Henry reaction52 or epoxide hydration.53
The characteristics and the catalytic activity of salen-type complexes can be influenced by the type of complexed metal ion, the substituents on the salicylidene segments of the ligand, and the character of the central diamine part of the ligand. The diamine part of the ligand can significantly affect the geometry of the complex. The most common diamines used for the synthesis of salen-type metal complexes are 1,2-ethylenediamine, 1,2-phenylenediamine, and 1,2-diaminocyclohexane.7,16–18,21,26,27 The last one can exist in two stereoisomers, cis and trans. Moreover, the trans stereoisomer is a chiral compound having two enantiomers with 1S,2S and 1R,2R configuration (Fig. 1). The enantiomerically pure trans-1,2-cyclohexanediamine can be used for the preparation of chiral salen-type metal complexes applied in enantioselective catalysis.16–18 Many authors referring to heterogenized salen-type metal complexes containing 1,2-diaminocyclohexane used mostly the enantiomerically pure trans variant or a racemic mixture.29 However, the effect of 1,2-diaminocyclohexane isomerism on the structure, texture, morphology, and catalytic properties of resulting materials was not studied. For these purposes, various NMR techniques represent powerful tools for the characterization of organometallic complexes in both solution and the solid state. Special NMR methods have proven to be effective even in the characterization of paramagnetic organometallic complexes. However, due to fast paramagnetically induced relaxations, a wide range of paramagnetically induced shifts, and their anisotropy, conventionally used pulse sequences typically fail. Thus, multiple experiments tailored to application on paramagnetic materials have been recently proposed.54 In particular, under fast magic-angle-spinning (MAS) conditions where 1H nuclei can be directly detected, the HSQC experiment with TEDOR recoupling provides excellent resolution and sensitivity.55 Additionally, a 1H–1H correlation experiment can be conducted on paramagnetic samples, under MAS conditions, correlating either single-quantum coherence with another single-quantum coherence (SQ–SQ) or with double-quantum coherence (SQ–DQ).56,57 The second approach is recommended for paramagnetic samples with broad resonances58 because of their diagonal-free properties. This means that the signal on the SQ–DQ diagonal appears only when multiple chemically equivalent atoms experience mutual dipolar couplings (e.g. the signal on the diagonal appears for a CH3 group but not for isolated CH). Back-to-back mixing using four phase-shifted 90° pulses in a rotor period (BaBa) is typically used for the SQ–DQ correlation in paramagnetic systems due to its extensive bandwidth.59
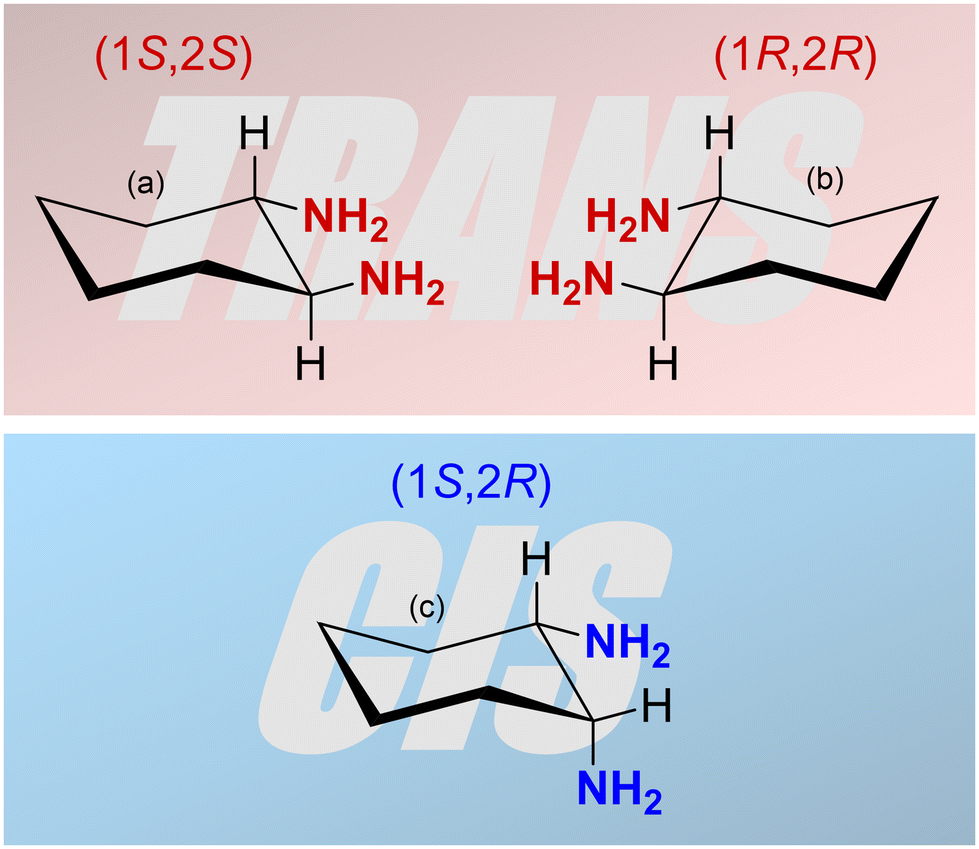 | ||
| Fig. 1 Three isomers of 1,2-diaminocyclohexane: (a) (1S,2S)-1,2-diaminocyclohexane, (b) (1R,2R)-1,2-diaminocyclohexane (both trans) and (c) (1S,2R)-1,2-diaminocyclohexane (cis). | ||
The vast majority of salen-based metal complexes containing POPs were prepared by step-growth copolymerization of the metal complexes with cross-linking comonomers.60 This approach introduces into the structure of the POP catalytically inactive chemical blocks (derived from cross-linking molecules) the only task of which is to form a network structure and help achieve a porous texture. Herein, we propose the concept of POP-type organometallic networks prepared by direct chain-growth insertion coordination homopolymerization of copper(II) salen-type complexes without using any supportive cross-linkers. Complexes used for this homopolymerization were prepared using isomerically well-defined 1,2-diaminocyclohexane building blocks, namely (i) enantiomerically pure trans-(1S,2S)-1,2-diaminocyclohexane, (ii) racemic trans-1,2-diaminocyclohexane and (iii) cis-1,2-diaminocyclohexane. The influence of isomerism of 1,2-diaminocyclohexane building blocks on the structure and properties of both paramagnetic copper(II) salen-type organometallic complexes and final POPs prepared from these complexes was investigated particularly by the abovementioned advanced NMR spectroscopy based on HSQC-TEDOR and BaBa SQ-DQ correlation combined with the quantum chemical prediction of paramagnetically induced shifts.55,61 In addition other characterization methods were used, namely, single-crystal X-ray diffraction, FTIR spectroscopy, circular dichroism, and N2 adsorption/desorption isotherms. The combination of a facile polymerization technique with a dexterous organometallic system led to the emergence of new, thoroughly characterized heterogeneous catalysts that are highly active in the oxidation of styrene.
Experimental
Materials
Acetylacetonate(norbornadiene)rhodium(I) [Rh(nbd)acac] (>98%), (1S,2S)-1,2-diaminocyclohexane (>98%), cis-1,2-diaminocyclohexane (>97%), and trans-1,2-diaminocyclohexane (>97%) (all from TCI Europe); 5-ethynyl-2-hydroxybenzaldehyde (95%, Advanced ChemBlocks Inc.); copper(II) acetate, Cu(OAc)2 (98%), tert-butylhydroperoxide solution (70 wt% in water), and styrene (≥99%, all from Merck); and dichloromethane (anhydrous, max. 0.001% of water, sealed under septum), ethanol (99.8%), methanol (≥99%), and dimethylformamide (99.8%) (all from VWR International) were used as obtained.Synthesis of N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane-type ligands
Triblock ligands consisting of two blocks of 5-ethynylsalicylaldehyde binding with one block of 1,2-diaminocyclohexane were synthesized as follows: 1 mmol of specific isomer of 1,2-diaminocyclohexane, either (i) (1S,2S)-1,2-diaminocyclohexane, (ii) trans-1,2-diaminocyclohexane or (iii) cis-1,2-diaminocyclohexane was dissolved in 1.5 ml of methanol and 2.4 mmol of 5-ethynyl-2-hydroxybenzaldehyde was dissolved in 9 ml of methanol. Both solutions were mixed together in a glass vial and the reaction mixture was stirred using a magnetic stirrer for 24 hours at room temperature. After that, the solvent was evaporated. The solid product was dissolved in 10 ml of pure ethanol and purified by recrystallization in a mixture of ethanol/water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The crystals were separated by filtration, washed with ethanol/water (2:1, v/v) mixture, and dried under vacuum. Three different ligands were prepared using this synthetic process: (i) (1S,2S)-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane (92% yield), L1, (ii) trans-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane (81% yield), L2 and (iii) cis-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane (73% yield), L3.
:1, v/v). The crystals were separated by filtration, washed with ethanol/water (2:1, v/v) mixture, and dried under vacuum. Three different ligands were prepared using this synthetic process: (i) (1S,2S)-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane (92% yield), L1, (ii) trans-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane (81% yield), L2 and (iii) cis-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane (73% yield), L3.
Synthesis of [Cu(N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane)] complexes
1 mmol of respective ligand, L1, L2, or L3 was dissolved in 24 ml of methanol and 1 mmol of copper(II) acetate was dissolved in 4 ml of distilled water. Both solutions were mixed together in a glass vial and stirred for 48 hours at room temperature. During this time a solid product was formed. The precipitate was filtered off and washed with methanol/water (6:1, v/v). The product was dried under vacuum. By following this procedure, three different complexes were synthesized: [Cu((1S,2S)-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane)], Cu-L1, where the complex was prepared in 93% yield; [Cu(trans-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane)], Cu-L2, where the complex was prepared in 98% yield; and [Cu(cis-N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane)], Cu-L3, where the complex was prepared in 96% yield. Crystals for single-crystal X-ray diffraction were grown by slow evaporation from a concentrated dimethylformamide solution (∼4 mg ml−1).
Transformation of [Cu(N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane)] complexes into organometallic polymer networks
Cu-L1, Cu-L2, and Cu-L3 complexes were transformed into organometallic polymer networks by homopolymerization. The overall polymerization concentrations were 0.05 mol dm−3 for the monomer and 0.007 mol dm−3 for the polymerization initiator. A typical polymerization procedure was as follows: 0.5 mmol of monomer (Cu-L1, Cu-L2, and Cu-L3) was dissolved in 9 ml of dry dichloromethane and 0.07 mmol of polymerization initiator [Rh(nbd)acac] was dissolved in 1 ml of dry dichloromethane. The solution of the monomer and solution of the polymerization initiator were mixed in a crimping vial, thoroughly stirred, blown by argon, and crimped under an argon atmosphere. The polymerization proceeded at 75 °C for 5 days, after which a gel-like solid block appeared. The solid product was separated by filtration, washed with dichloromethane, and dried under vacuum. Organometallic polymer networks P-Cu-L1, P-Cu-L2, and P-Cu-L3 were prepared in quantitative yields.Testing the catalytic activity of organometallic polymer networks
A glass vial was loaded with an organometallic polymer network applied as a catalyst (10 mg), styrene (1 mmol), tert-butylhydroperoxide solution (3 mmol), and dichloromethane (3 ml). The sealed glass vial was placed onto a magnetic stirrer equipped with a heating block and the reaction mixture was heated to 60 °C for 6 hours. In the case of the reuse experiment, the reaction mixture was centrifuged, the catalyst was removed, washed with dichloromethane three times, and used in the following experiment. Samples from the reaction mixture were filtered (0.45 μm filter) and analyzed by gas chromatography coupled with mass spectrometry.Techniques
The textural properties of the polymer networks were obtained by measuring N2 adsorption/desorption isotherms at 77 K. The measurement itself was preceded by degassing the sample (6 h temperature ramp to 383 K) using a Micromeritics SmartVacPrep instrument. The subsequent measurement of isotherms was carried out using Triflex V4.02 apparatus (Micromeritics). Adsorption and desorption isotherms were measured from p/p0 = 0 to p/p0 = 0.99 (p0 = 101325 Pa). The Brunauer, Emmett and Teller method was applied to obtain the value of the BET surface area, SBET, calculated from the amount of adsorbed nitrogen in the p/p0 range from 0.05 to 0.20, the volume of micropores, Vmi, was determined from the adsorbed amount of nitrogen at p/p0 = 0.1 and the total pore volume, Vtot, was determined from the adsorbed amount of nitrogen at p/p0 ∼ 0.99. Pore size distribution was determined using the N2-DFT model (calculation by density functional theory). The density of liquid nitrogen at 77 K considered for the calculation was ρ = 0.806 g cm−3. The morphology was studied by scanning electron microscopy using a Tescan Lyra3 microscope (accelerating voltage 10 kV). The content of copper(II) ions was determined by atomic absorption spectroscopy (the samples were mineralized in a Biotage Initiator microwave reactor using HNO3/HClO4 medium and then measured using a PerkinElmer model 3110 instrument). Diffuse reflectance infrared Fourier transform spectra were recorded using a Nicolet Magna IR 760 spectrometer with KBr background. A Bruker Q-TOF Compact instrument was used to obtain spectra from high-resolution mass spectrometry (HR-MS). UV/Vis and CD spectra were recorded on a J-810 spectrometer (Jasco, Japan) using a 1 mm cuvette. Specific optical rotation, [α], was measured using an Anton Paar MCP 5100 polarimeter at 293 K and wavelength 589 nm in CH2Cl2 solution. Single-crystal X-ray diffraction was measured on a Bruker D8 VENTURE Kappa Duo PHOTONIII with an IμS micro-focus sealed tube CuKα (λ = 1.54178) source at a temperature of 120(2) K. The structures were solved by direct methods62 and refined by full-matrix least squares based on F2 (ref. 63). The hydrogen atoms on carbon were fixed at idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2Ueq (pivot atom). Gas chromatography-mass spectrometry was performed using a GC-2010 Plus chromatograph (Shimadzu) equipped with a non-polar DB-5MS column (35 m, 0.2 μm i.d., 0.33 μm d.f.) coupled with a GCMS-QP 2010 Ultra (Shimadzu) mass spectrometer. The 1H and 13C solution-state NMR spectra of diamagnetic samples were acquired using a Bruker Avance III 400 MHz spectrometer. The 1H and 13C solution-state NMR spectra of paramagnetic samples were acquired on a Bruker Avance III 600 MHz spectrometer by conducting a direct-excitation spin-echo-detected experiment. The chemical shift was referenced to the residual signal of the solvent.64 The solid-state NMR spectra were acquired using a JEOL ECZ 600 MHz spectrometer with 3.2 mm and 1.0 mm HX MAS probes for slow and fast MAS experiments, respectively, both operating at room temperature (corresponding to 38–42 °C and 34–43 °C temperature gradients within the sample due to frictional heating under MAS conditions at 18 and 60 kHz in 1.0 and 3.2 mm rotors, respectively). A repetition time of 50 ms was used in all solid-state experiments and was demanded by hardware duty cycles rather than by the T1 value of the sample, which was typically an order of magnitude faster. The chemical shift of the adamantane CH2 peak (37.8 ppm) was used as an external secondary reference for calibration of the 13C spectra. The same correction of chemical shifts was used for 1H spectra as well. The 1D 1H and 13C NMR spectra were acquired by following a direct-excitation spin-echo-detected experiment with echo duration of a single rotor period. The nutation frequencies of the 1H/13C pulses of 350/290 kHz and 100/112 kHz were used with 1.0 mm and 3.2 mm probes, respectively.65 The 1H–13C 2D correlation experiments were conducted using the HSQC-TEDOR experiment55 with a dephasing and refocusing time of two rotor periods (32 μs) each. The 1H SQ-DQ experiment was performed using BaBa16 mixing59 with a dephasing and refocusing time of 128 μs each. The theoretical build-up curve of this experiment was calculated in SIMPSON.65 The DFT-based prediction of NMR shifts was calculated for geometries optimized at the deg-TZVP/pbe0 level in Turbomole 7.166 either in vacuo with the frozen position of non-hydrogen atoms on the position obtained from X-ray diffraction or using a COSMO/CHCl3 solvent model with the released coordinates of all atoms. The NMR shifts of nucleus K were calculated by the following equation (in ppm):where orbital (“diamagnetic”) shielding (σdia,
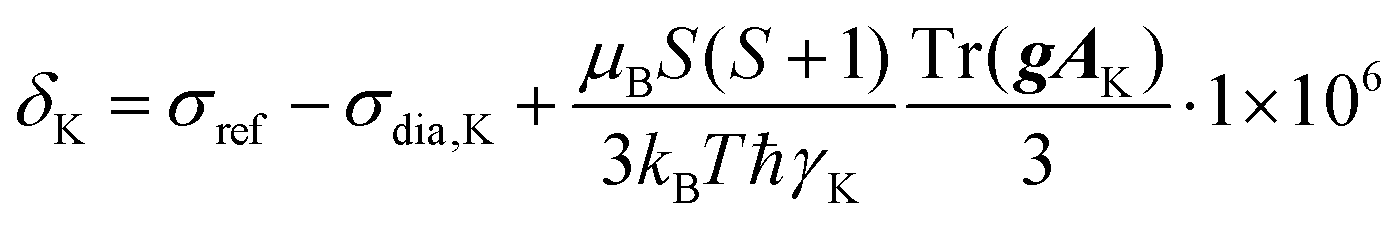 K), hyperfine coupling tensor (AK) and copper(II) g-tensor (g) were calculated (with ORCA 5.0.3)67 using the PBE functional with a variable admixture of Hartree–Fock exchange (HFX, 10–40%) with IGLO-III (H, N, C) and def2-TZVPD (Cu, O) base sets employing the auxiliary default base set RIJK def2/JK as well. TightSCF convergence criteria were used. The orbital shielding was calculated using the GIAO method. Hyperfine-coupling-tensor calculations called for both isotropic and dipolar contributions (aiso, adip flags) giving the origin of the Fermi contact and pseudo-contact shift, respectively. The first one strongly dominates in the studied system (Tables S1 and S2†). Because the electronic spin of copper is S = 1/2, no zero-field splitting is present in the system. Temperature (T) was set to 298.15 K unless stated otherwise. The shielding of the reference molecule (TMS) was calculated in the same way using 25% HFX.
K), hyperfine coupling tensor (AK) and copper(II) g-tensor (g) were calculated (with ORCA 5.0.3)67 using the PBE functional with a variable admixture of Hartree–Fock exchange (HFX, 10–40%) with IGLO-III (H, N, C) and def2-TZVPD (Cu, O) base sets employing the auxiliary default base set RIJK def2/JK as well. TightSCF convergence criteria were used. The orbital shielding was calculated using the GIAO method. Hyperfine-coupling-tensor calculations called for both isotropic and dipolar contributions (aiso, adip flags) giving the origin of the Fermi contact and pseudo-contact shift, respectively. The first one strongly dominates in the studied system (Tables S1 and S2†). Because the electronic spin of copper is S = 1/2, no zero-field splitting is present in the system. Temperature (T) was set to 298.15 K unless stated otherwise. The shielding of the reference molecule (TMS) was calculated in the same way using 25% HFX.
Results and discussion
This study deals with copper(II) organometallic complexes with various N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane (SALDAC) ligands. The SALDAC ligands were prepared by condensation of two molecules of 5-ethynyl-2-hydroxybenzaldehyde and one molecule of 1,2-diaminocyclohexane. Based on the mutual orientation of –NH2 groups, 1,2-diaminocyclohexane has trans and cis stereoisomers. The trans isomer is a chiral compound and can exist in the form of two enantiomers with configurations on their chiral centres 1S,2S and 1R,2R (Fig. 1). For the purposes of this study, we chose (i) the enantiomerically pure trans isomer, (1S,2S)-1,2-diaminocyclohexane, (ii) the racemic mixture of both trans enantiomers, trans-1,2-diaminocyclohexane and (iii) the cis isomer, cis-1,2-diaminocyclohexane. These were used to synthesize L1, L2, and L3 ligands (Scheme 1 and Fig. S1–S3†).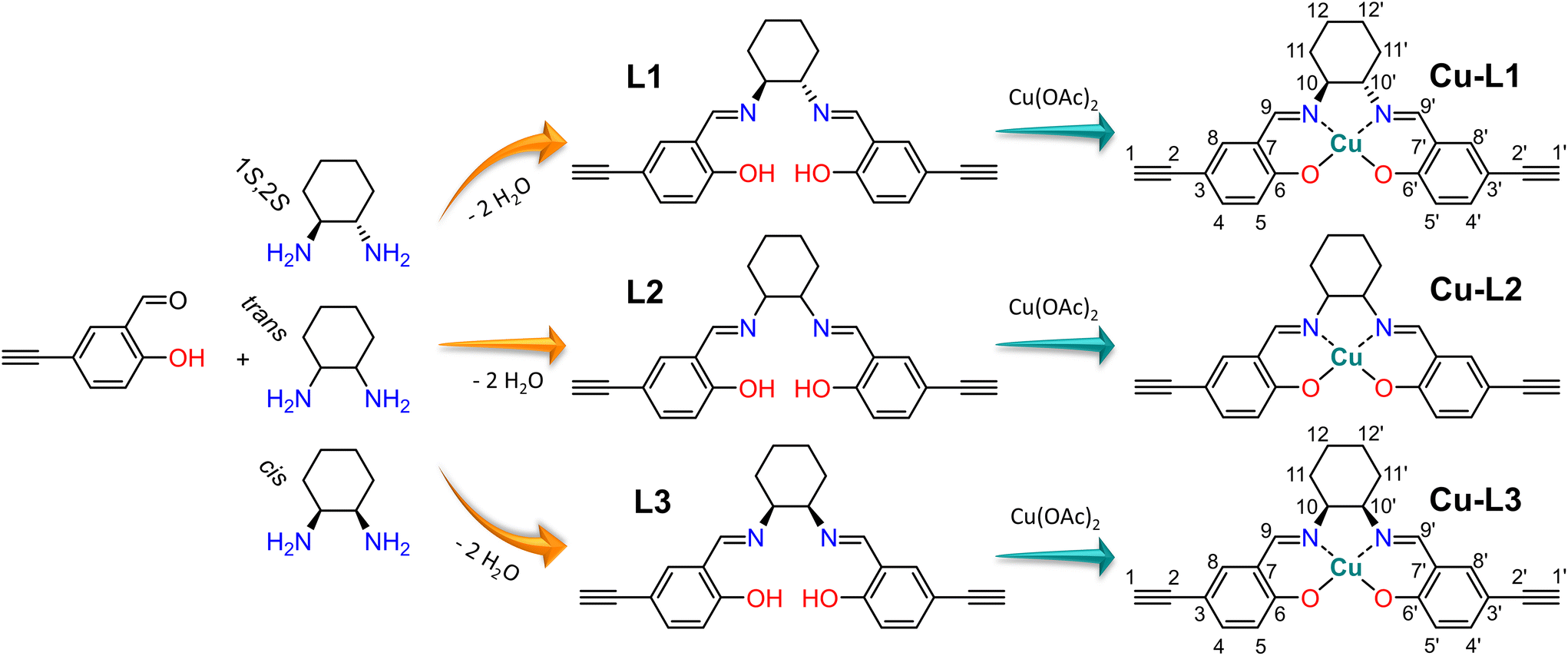 | ||
| Scheme 1 Synthesis of L1, L2 and L3 ligands by condensation of 5-ethynylsalicylaldehyde with the respective 1,2-diaminocyclohexane isomer, and their subsequent metalation with copper(II) ions upon the formation of Cu-L1, Cu-L2 and Cu-L3 complexes. | ||
The SALDAC ligands have a unique combination of functional groups suitable for the complexation of divalent metal ions (two imine groups and two hydroxy groups per molecule) and for polymerization into organometallic polymer networks (two terminal ethynyl groups per molecule). All three SALDAC ligands, L1, L2, and L3, were prepared in yields ranging from 73 to 92% and their proposed structures were confirmed by 1H and 13C{1H} NMR spectroscopy (Fig. S4–S9†), FTIR spectroscopy (Fig. 2) and HR-MS (see the ESI†).
 | ||
| Fig. 2 FTIR spectra of series (A) L1, Cu-L1 and P-Cu-L1, (B) L2, Cu-L2 and P-Cu-L2 and (C) L3, Cu-L3 and P-Cu-L3. | ||
Synthesis and characterization of copper(II) complexes
The prepared SALDAC ligands underwent complexation with copper(II) ions to form organometallic complexes. One molecule of L1, L2, and L3, respectively, reacted with one molecule of copper(II) acetate and formed Cu-L1, Cu-L2, and Cu-L3 complexes (Scheme 1). The reaction took place at room temperature in a mixture of methanol/water (6:1, v/v). All three complexes were prepared in yields above 90% (see the Experimental section).
FTIR spectroscopy confirmed the successful metalation of all three ligands: bands of –HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– stretching were shifted from ∼1633 cm−1 (in the FTIR spectra of ligands L1, L2, and L3) to ∼1628 cm−1 (in the FTIR spectra of complexes Cu-L1, Cu-L2 and Cu-L3) (Fig. 2). This shift is frequently discussed and reported upon the introduction of metal ions into Schiff base-type complexes.12,68 On the other hand, two bands proving the presence of ethynyl groups visible in the spectra of all three ligands (2100 cm−1 due to –C
N– stretching were shifted from ∼1633 cm−1 (in the FTIR spectra of ligands L1, L2, and L3) to ∼1628 cm−1 (in the FTIR spectra of complexes Cu-L1, Cu-L2 and Cu-L3) (Fig. 2). This shift is frequently discussed and reported upon the introduction of metal ions into Schiff base-type complexes.12,68 On the other hand, two bands proving the presence of ethynyl groups visible in the spectra of all three ligands (2100 cm−1 due to –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– stretching and 3300 cm−1 due to C–H stretching) also remained in the spectra of all three complexes. This confirmed that ethynyl groups of L1, L2, and L3 persist unaffected (after metalation) in Cu-L1, Cu-L2, and Cu-L3 complexes. Interestingly, the FTIR band at 3300 cm−1 was split in some spectra. In FTIR spectra of ligands L1 and L2, the bands of C–H stretching were split by 18 cm−1. This may reflect the conformational changes of L1 and L2 and the existence of two conformers of these trans-1,2-diaminocyclohexane-based ligands. The central cyclohexane segments of these conformers have both azomethine-linked substituents in either axial or equatorial positions. The conformational changes are, however, blocked once L1 and L2 are transformed into organometallic complexes Cu-L1 and Cu-L2. In the respective organometallic complexes, both substituents on the cyclohexane ring are locked in equatorial positions (confirmed by X-ray crystallography, Fig. S12†). This is consistent with the fact that the splitting of the C–H stretching band was not observed in FTIR spectra of Cu-L1 and Cu-L2 complexes. A different situation was observed in FTIR spectra of ligand L3 and complex Cu-L3. The cis isomer of the 1,2-disubstituted cyclohexane segment has one substituent in the equatorial and one in the axial position and its conformational change leads to an identical molecule. This is why in the spectrum of the L3 ligand only a single band of C–H stretching from ethynyl groups was visible. The splitting of the band of the C–H group in the spectrum of the Cu-L3 complex was most probably due to the fact that two ethynylsalicylidene segments present in the Cu-L3 complex were not identical as confirmed by the 1H-detected HSQC-TEDOR spectrum (vide infra, Fig. 4).
C– stretching and 3300 cm−1 due to C–H stretching) also remained in the spectra of all three complexes. This confirmed that ethynyl groups of L1, L2, and L3 persist unaffected (after metalation) in Cu-L1, Cu-L2, and Cu-L3 complexes. Interestingly, the FTIR band at 3300 cm−1 was split in some spectra. In FTIR spectra of ligands L1 and L2, the bands of C–H stretching were split by 18 cm−1. This may reflect the conformational changes of L1 and L2 and the existence of two conformers of these trans-1,2-diaminocyclohexane-based ligands. The central cyclohexane segments of these conformers have both azomethine-linked substituents in either axial or equatorial positions. The conformational changes are, however, blocked once L1 and L2 are transformed into organometallic complexes Cu-L1 and Cu-L2. In the respective organometallic complexes, both substituents on the cyclohexane ring are locked in equatorial positions (confirmed by X-ray crystallography, Fig. S12†). This is consistent with the fact that the splitting of the C–H stretching band was not observed in FTIR spectra of Cu-L1 and Cu-L2 complexes. A different situation was observed in FTIR spectra of ligand L3 and complex Cu-L3. The cis isomer of the 1,2-disubstituted cyclohexane segment has one substituent in the equatorial and one in the axial position and its conformational change leads to an identical molecule. This is why in the spectrum of the L3 ligand only a single band of C–H stretching from ethynyl groups was visible. The splitting of the band of the C–H group in the spectrum of the Cu-L3 complex was most probably due to the fact that two ethynylsalicylidene segments present in the Cu-L3 complex were not identical as confirmed by the 1H-detected HSQC-TEDOR spectrum (vide infra, Fig. 4).
The UV/Vis spectra of ligands L1, L2, and L3 showed the absorption band of the n → π*/π → π* transition belonging to the –HCN– group at λ = 333 nm. This band was red-shifted in UV/Vis spectra of complexes Cu-L1, Cu-L2, and Cu-L3 to λ = 377 nm (measured in dichloromethane, Fig. S10†). Similar spectroscopic behaviour was reported in our previous work.12 The circular dichroism (CD) spectra revealed the chiral character of the L1 ligand and the Cu-L1 complex, while no CD signals were observed for trans racemates L2 and Cu-L2 and cis isomers L3 and Cu-L3. The CD spectra in Fig. S11† showed signals at λ = 354 nm (spectrum of L1) and λ = 417 nm (spectrum of Cu-L1) relating to the chirality associated with the cyclohexane ring. Moreover, the CD spectrum of Cu-L1 displayed a positive Cotton effect (with a positive-to-negative CD couplet) in the range of λ = 430–680 nm, which can be ascribed to the presence of torsional helicity of the chiral organometallic complex.69 Along with that, a significant increase of specific rotation [α]D was observed when comparing L1 and Cu-L1 (from 19° to 632°, respectively, measured at 20 °C in dichloromethane, 1 mg ml−1).
Crystals for single-crystal X-ray diffraction were grown by slow evaporation from concentrated (∼4 mg ml−1) dimethylformamide solutions. Crystals of enantiomerically pure transCu-L1 (Fig. S12†) and cisCu-L3 (Fig. S13†) complexes (see more details in the ESI†) were obtained and measured. The racemic transCu-L2 complex did not crystallize. Measured single-crystal X-ray diffraction confirmed the proposed structures (Scheme 1) of organometallic complexes. For the Cu-L1 complex, two symmetrically independent molecules were observed (space group P21). This is a typical phenomenon for enantiomerically pure substances.70,71 Crystals of Cu-L3 were also formed by two molecules. Conformational changes of the cis-L3 ligand (from configuration 1S,2R to configuration 1R,2S and vice versa) were locked once the ligand was transformed into organometallic complex Cu-L3. This led to a racemic mixture of two centrosymmetric cis complexes (space group P21/n). The complexes differed in configuration on chiral centres (1S,2R vs. 1R,2S). Similar conformational locking of (non-metalated) cis-1,2-diaminocyclohexane-based compounds was described by van Beek and Samoshin.72 The structures of complexes Cu-L1 and Cu-L3 obtained from X-ray diffraction showed in both cases good steric accessibility of terminal ethynyl groups, promising for their efficient polymerization, together with good steric accessibility of coordinated copper(II) ions for substrate molecules, promising for catalytic application. The coplanarity of the cyclohexane ring within the Cu-L1 complex with the phenyl rings resulted in a reduced spatial occupancy of the entire complex in comparison with the Cu-L3 complex, in which the cyclohexane ring exhibited an out-of-plane orientation relative to the phenyl rings.
NMR spectroscopy provides a sensitive tool for comparison of organometallic complexes in the liquid state, in the solid form, as well as, within the polymer network. For the purpose of this advanced NMR analysis, only enantiomerically pure trans isomer Cu-L1 and cis isomer Cu-L3 were studied. The solution-state 1H spectrum of Cu-L1 (Fig. 3B) exhibited typical features of the paramagnetic compounds: we observed four distinct signals between 25 and −6.6 ppm with linewidths ranging from 22 Hz to 7.5 kHz and extremely short T1 relaxation times of ∼2 ms (global T1(1H) at 70 kHz MAS). Surprisingly, the 1H spectrum of solid Cu-L1 measured under fast MAS conditions (70 kHz, Fig. 3A) presents a similar resolution to the spectrum in solution with similar NMR shifts of all four observed signals. The structure of Cu-L1 therefore remained similar upon dissolution in aprotic solvents. Under the conditions of fast MAS, an irregular line-shape of resonance at 25 ppm was observed indicating the overlap of several lines (in contrast to the spectra at slower MAS; see Fig. S14†). Additionally, two bands between 10 and 0 ppm were composed of several overlapping signals. Similarly, the comparison between the solid-state 1H spectrum of Cu-L3 with its solution-state spectra indicates a similar structure in both phases (Fig. S15†). However, extensive signal overlap of multiple signals occurred in this case in the region between 15 and −10 ppm.
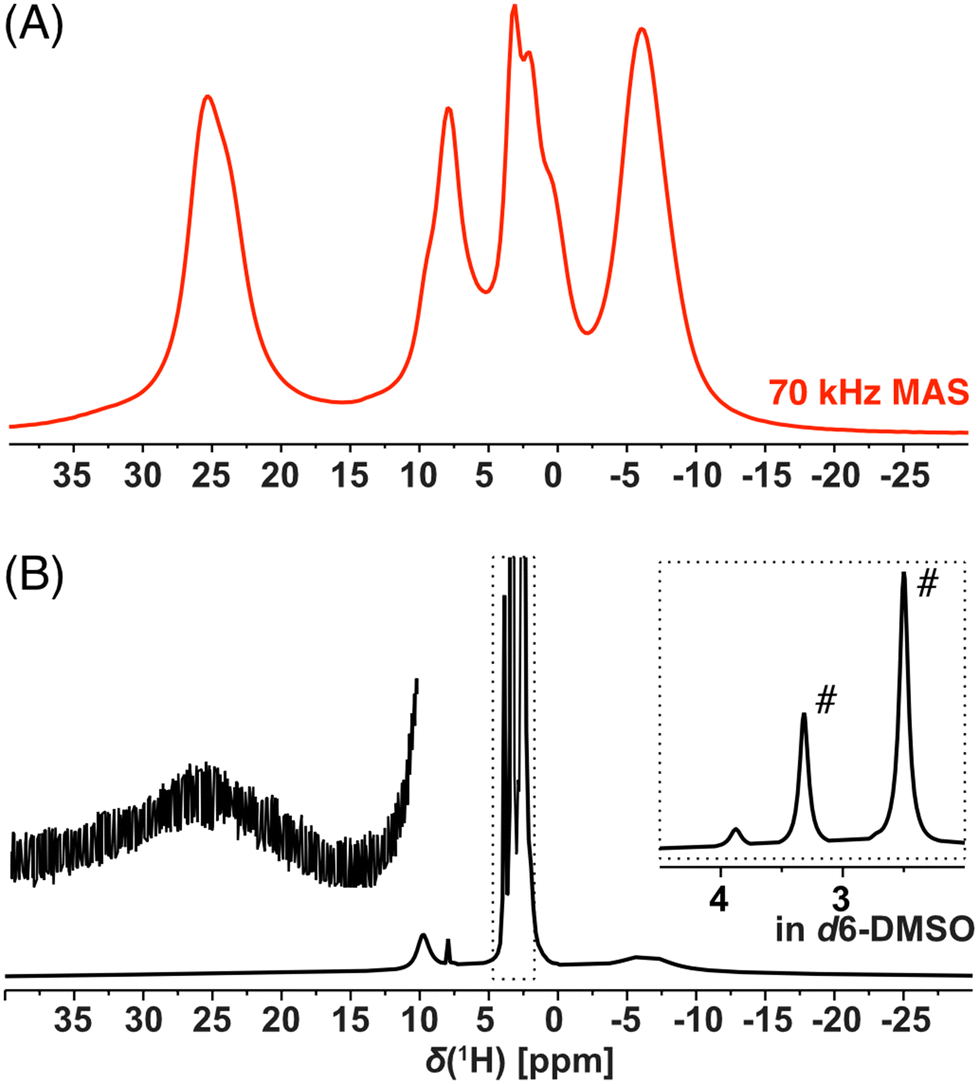 | ||
| Fig. 3 1H NMR spectra (14.09 T) of Cu-L1 acquired by the spin-echo experiment (A) on a solid-state powder sample under 70 kHz MAS conditions and (B) on the d6-DMSO solution (# indicate solvent and residual water signals). | ||
Fortunately, signal overlaps were efficiently resolved using 1H-detected 2D 1H–13C correlation experiments (HSQC-TEDOR).55 This 2D experiment can be readily measured (typically after approximately 4 h of experimental time) in the 13C natural abundance even for paramagnetic compounds (Fig. 4). This excellent sensitivity is mainly caused by a short T1 relaxation allowing short repetition time (here 55 ms due to limited hardware duty cycle) as well as strong 1H–13C dipolar interaction, which allows extremely short dephasing and rephasing times for the coherence transfer (2 × rotor period = 32 μs each). Extensive improvement of resolution is, for example, demonstrated in the signal at 25 ppm in the 1H spectrum of Cu-L1, which was clearly separated into the correlation of two CH pairs with very distinct 13C shifts (225 and 25 ppm).
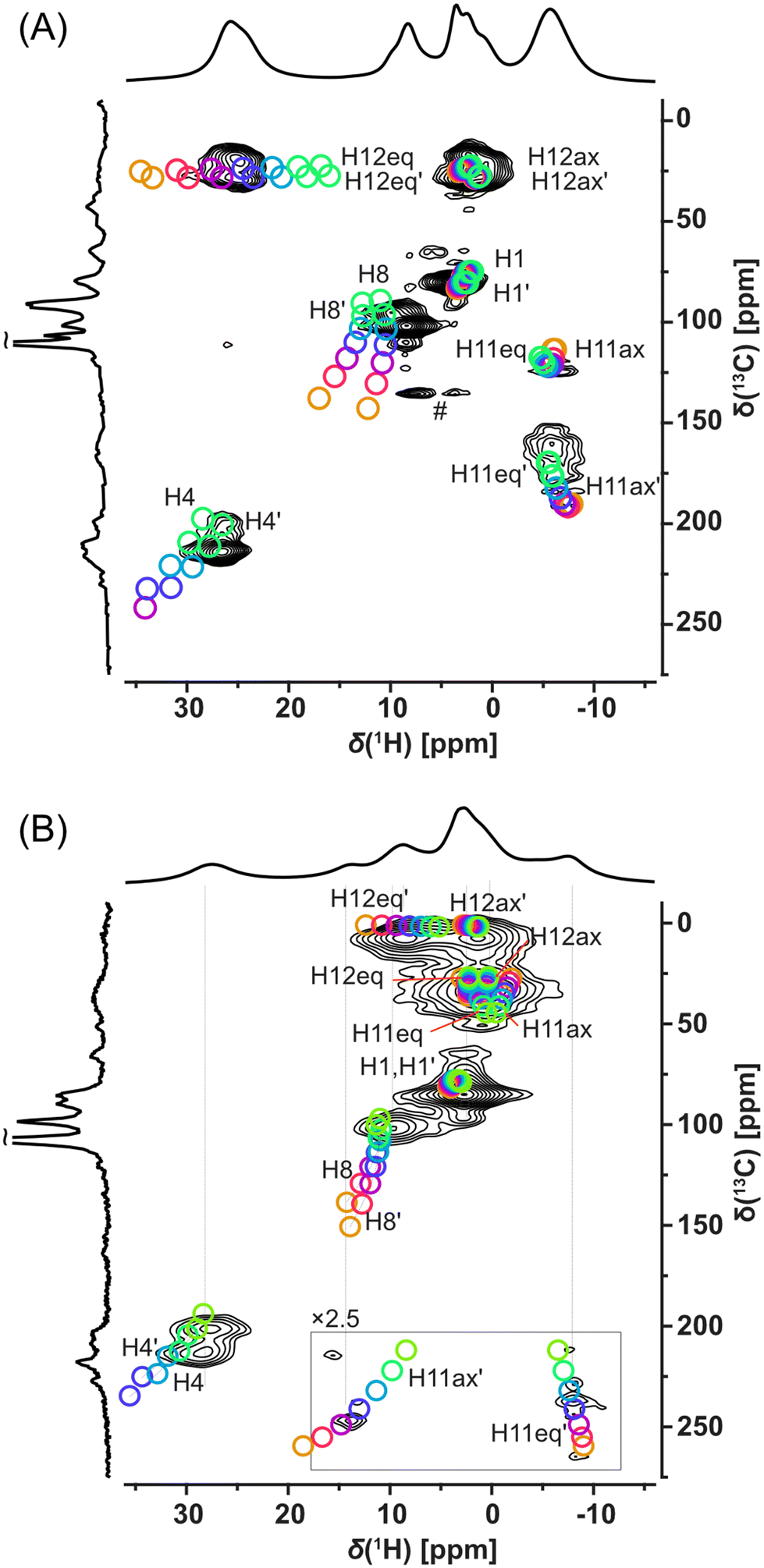 | ||
| Fig. 4 1H-detected HSQC-TEDOR spectra of (A) Cu-L1 and (B) Cu-L3 acquired at 62.5 kHz MAS and 14.09 T using a 1 mm rotor (black) compared with calculated 1H–13C signals using a variable Hartree–Fock exchange admixture for hyperfine coupling (from orange 10% to green 40% in 5% steps) for X-ray-based structure with reoptimized hydrogen position (# indicated an on-resonance T1 artefact). | ||
Additionally, these correlation experiments can be readily compared with DFT calculations providing unambiguous spectral assignment. Signals of all inequivalent CH pairs were detected for Cu-L1, apart from signals of the CH pair number 5, 9, and 10, which were too close to the copper(II) ion and were not detected due to fast T2 relaxation and extreme 1H and/or 13C NMR shifts predicted by calculation (Fig. S16 and S17;† see numbering of atoms in Scheme 1). Interestingly, the signals of equatorial and axial H12 appear at very distinct NMR shifts of 25 ppm and 3 ppm, respectively, which indicate the stronger contact shift for H12eq located in-plane with the paramagnetic copper(II) ion. The resolution was also extensively enhanced for the Cu-L3 sample using a 2D HSQC-TEDOR experiment (Fig. 4B).
Due to crystal packing, molecule Cu-L1 is not perfectly C2-symmetric in the solid state. This is most pronounced on different C11 and C11′ shifts but also other CH correlations are slightly split into pairs of overlapping signals. Compared to Cu-L1 with trans conformation, the cis conformation of the cyclohexane ring in Cu-L3 results in a much stronger deviation from the C2 symmetry of the complex, which was particularly emphasized by a distinct 13C NMR shift of C11 and C11′ (50 and 250 ppm). The difference in the 13C shift was also observable for C12 and C12′ as well as for C4 and C4′. In all cases, the calculated differences between NMR shifts of the primed and non-primed C–H pairs corresponded well to the experimental one.
Here, we recommend critical precautions during the analysis of the calculated NMR shift. The hyperfine coupling constant (which is the most critical parameter for paramagnetic-shift calculations in a system with a dominant Fermi-contact shift mechanism, as in this case, see Tables S1 and S2†) changes significantly with the variation of the exact Hartree–Fock exchange (HFX) admixture to the used density functional.55,73 On the one hand, for 10% HFX, the difference in the NMR shift of the C8 and C8′ was predicted to be almost 12 ppm while for 40% HFX the predicted difference was only about 3 ppm. The absence of splitting of the C8 signal in the experimental 2D spectra indicates that the admixture with higher HFX content provided a more realistic prediction for this nucleus. Also, the absolute value of the NMR shift was in better agreement with the admixture having the higher HFX content. On the other hand, the 1H shift of the equatorial H12 was best predicted with HFX about 25%. Therefore, several calculations in a range of HFX concentrations are recommended for appropriate sampling of this inaccuracy as indicated by our “rainbow plots” shown in Fig. 4.
The excellent resolution of the 1H spectra and completed assignment allowed us to fully understand the homonuclear 1H–1H SQ–DQ correlation spectra as well (Fig. 5 and Fig. S20†). The efficiency of double-quantum build-up and subsequent intensity of the observed signals of the SQ–DQ correlation depends on the strength of a dipolar coupling between correlated nuclei. The dipolar coupling decreases with the 3rd power of interatomic distance and therefore, for a given mixing time, the intensity of the SQ–DQ correlation signal steeply decreases with distance (inset of Fig. 5). Using a short mixing time (128 μs) the intensity of the SQ–DQ correlation signal for atoms separated by more than 5 Å decreases to below 10% of its maximal value making them effectively undetectable. However, experimental data show multiple correlations between 1H atoms separated by a distance longer than 5 Å within the Cu-L1 molecule – in an extreme case 17.9 Å for H1–H1′ correlation (intramolecular distances indicated as black values in Fig. 5). Observed correlations, therefore, indicate dense crystal packing of Cu-L1 and subsequent intermolecular contacts. This is in excellent agreement with the crystal structure detected by X-ray diffraction: close intermolecular proximities of involved 1H nuclei smaller than 5 Å were detected in the crystal structure for all observed SQ–DQ correlations (minimal distances indicated as red values in Fig. 5).
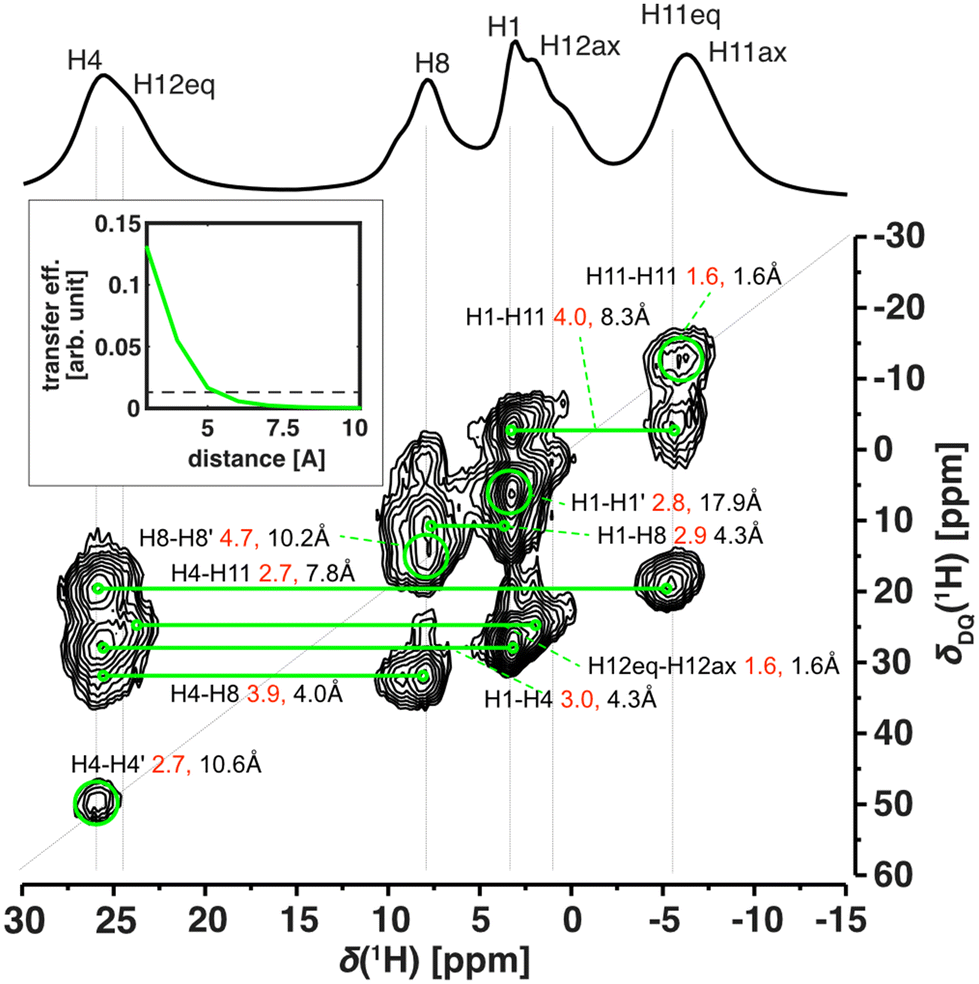 | ||
| Fig. 5 Homonuclear BaBa16 1H single–double quantum (SQ–DQ) correlation of Cu-L1 (62.5 kHz MAS, 14.09 T). Green lines and circles indicate assigned correlations. Shown distances correspond to the shortest distance found in the crystal structure (red) and the shortest intramolecular distance (black). The inset shows the simulated efficiency of the BaBa16 recoupling sequence with a recoupling time of 128 μs (similar to the experiment) as a function of 1H–1H distance with an indicated level of 10% efficiency (dashed line). | ||
The 1H–1H SQ–DQ correlation spectrum was also acquired for Cu-L3 (Fig. S20†) where, however, stronger signal overlaps hid most of the potential fine detail. The only two well-resolved correlations that indicated crystal packing via intermolecular proximities were between H4–H11ax′ and H4–H11eq′, which corresponded to distances of 2.4 and 3.5 Å in the single-crystal X-ray structure.
Altogether, we may conclude that 1H-detected solid-state NMR uniquely matches the structures in the single-crystal, in the powder sample, and in solution showing that the structure of these studied copper(II) complexes remained unchanged. This was also in line with calculation. The predicted NMR shifts of the structure fully optimized in the PCM solvent model match well with the predicted NMR shifts for the single-crystal structure (with reoptimized 1H positions), apart from subtle effects caused by the decrease of local symmetry due to crystal packing. This is true for both Cu-L1 and Cu-L3 (Fig. S16 and S18†).
Transformation of copper(II) complexes into organometallic polymer networks
Organometallic complexes Cu-L1, Cu-L2, and Cu-L3 were polymerized (in a chain-growth manner) by the transformation of their terminal ethynyl groups into polyacetylene main chains.45,46 The polymerizations were initiated by [Rh(nbd)acac] at 75 °C.74 As all three organometallic complexes had two polymerizable terminal ethynyl groups per molecule, their homopolymerization led to the formation of completely insoluble organometallic polymer networks P-Cu-L1, P-Cu-L2, and P-Cu-L3 (Scheme 2, all networks were prepared in quantitative yields).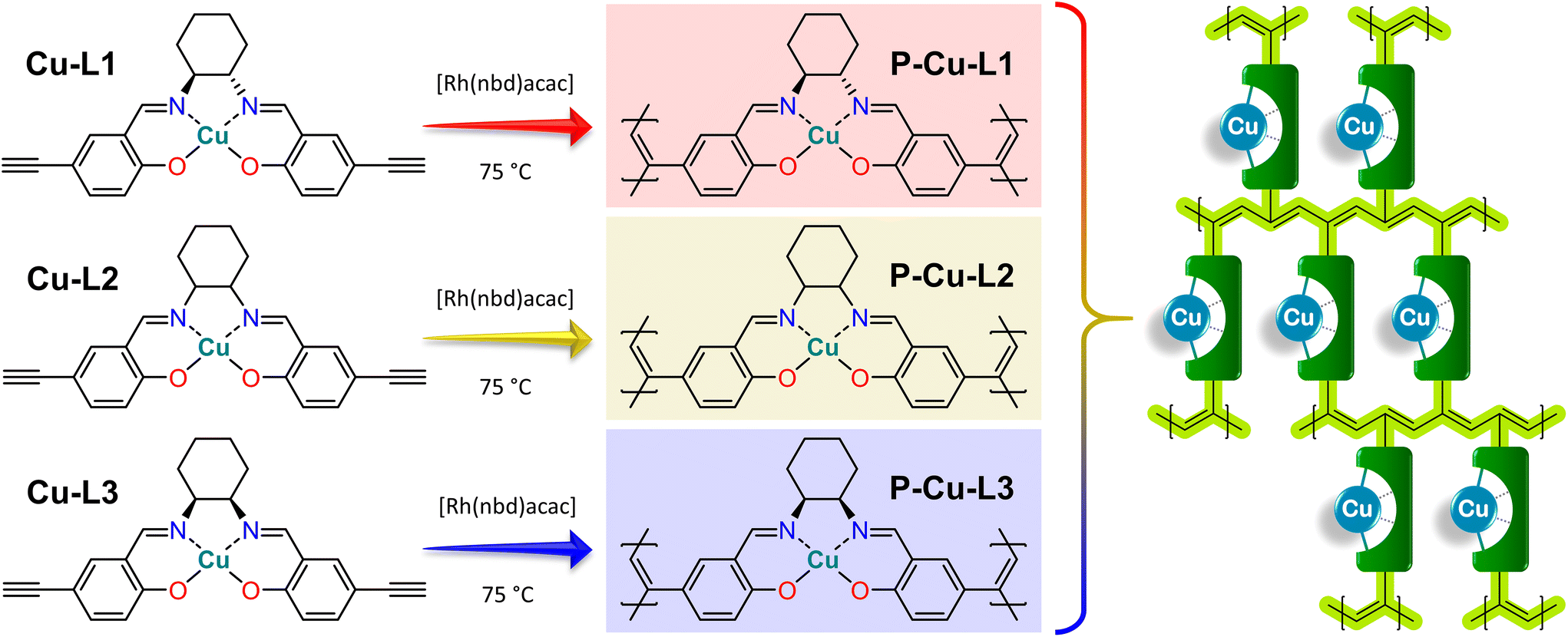 | ||
| Scheme 2 Homopolymerization of Cu-L1, Cu-L2 and Cu-L3 complexes leading to organometallic polymer networks P-Cu-L1, P-Cu-L2 and P-Cu-L3 and schematic representation of the incorporation of organometallic complexes as cross-linking-type building blocks into networks with conjugated polyene chains. | ||
All three networks contained almost the same amount of copper(II) ions, ranging from 12.0 to 12.2 wt% (Table 1). The FTIR spectra in Fig. 2 showed that roughly all terminal ethynyl groups of Cu-L1, Cu-L2, and Cu-L3 complexes were transformed upon polymerization into polymer networks P-Cu-L1, P-Cu-L2 and P-Cu-L3 (the bands at 3300 cm−1 and 2100 cm−1 were no more visible in their spectra). The band of –HCN– stretching at ∼1628 cm−1 from spectra of complexes remained at the same position in the spectra of all three polymer networks proving that the organometallic cores persist unaffected during polymerization. The polymer networks P-Cu-L1 and P-Cu-L3 were also studied by solid-state NMR. On comparing the 1H–13C correlation spectra of the polymer networks and related monomeric organometallic complexes (Fig. 6), the broadening of signals typical of the transformation of microcrystalline monomers into polymers can be seen. Additionally, the narrow signal of the acetylenic CH1 disappeared and was replaced by a broad resonance at 125 and 6.5 ppm in 13C and 1H spectra, respectively, which correspond to the vinylene groups in the polymer chains. The position of the most shifted CH4 resonance was the same in the spectra of complexes and final networks. This was another confirmation that the structure of the copper coordination environment remained unaltered upon polymerization.
 | ||
| Fig. 6 1H-detected HSQC-TEDOR spectra (62.5 kHz MAS, 14.09 T) of (A) Cu-L1 (black) vs.P-Cu-L1 (red) and (B) Cu-L3 (black) vs.P-Cu-L3 (blue). 1D horizontal and vertical traces show direct-excitation spin-echo detected 1H and 13C spectra acquired at 62.5 and 18 kHz, respectively. | ||
| Network | SALDAC isomer | Copper content [wt%] | S BET [m2 g−1] | V mi [cm3 g−1] | V tot [cm3 g−1] | V mi/Vtot |
|---|---|---|---|---|---|---|
| P-Cu-L1 | trans enantiomer | 12.2 | 522 | 0.22 | 0.39 | 0.56 |
| P-Cu-L2 | trans racemate | 12.1 | 458 | 0.19 | 0.34 | 0.57 |
| P-Cu-L3 | cis | 12.0 | 655 | 0.26 | 0.71 | 0.36 |
The results of FTIR and solid-state NMR spectroscopies agreed with the proposed structures (Scheme 2) of organometallic polymer networks P-Cu-L1, P-Cu-L2, and P-Cu-L3. All networks were composed exclusively of respective N,N′-bis(5-ethynylsalicylidene)-1,2-diaminocyclohexane ligands coordinating copper(II) ions (one ligand coordinated one metal ion) and polyacetylene chains.
Despite the fact that P-Cu-L1, P-Cu-L2, and P-Cu-L3 were cross-linked exclusively by metalated SALDAC segments (and no other cross-linker was used for the preparation), all these networks exhibited a micro/mesoporous texture and quite high BET areas (458–655 m2 g−1, Table 1). It is thus evident that organometallic segments not only introduced functionality into the networks but also served as rigid building blocks contributing to the formation of porous textures. The rigidity of the metalated copper(II) complex was essential to achieve porous textures with large BET areas. To prove this fact, we homopolymerized the non-metalated ligands, L1, L2, and L3, in the same way. Homopolymerization provided networks in quantitative yields, which were, however, non-porous.
Remarkably, the textural properties differed depending on the used SALDAC isomer. Networks P-Cu-L1 and P-Cu-L2 built from enantiomerically pure Cu-L1 and racemic Cu-L2 segments (both with trans configurations) showed BET areas of 522 and 458 m2 g−1, respectively. The N2 adsorption/desorption isotherms of both networks shown in Fig. 7 were similar with a pronounced N2 adsorption at low relative pressures (p/p0 < 0.1, filling of micropores), followed by only a small increase in the adsorbed amount of N2 as the relative pressure reached p/p0 ∼ 1. This shape of the N2 adsorption/desorption isotherms is characteristic for prevailingly microporous materials with a low content of mesopores. The characteristics of N2-DFT pore size distribution curves shown in Fig. 7 confirmed this finding, pointing to a high content of micropores (pore width < 2 nm) and a low contribution of mesopores (pore width > 2 nm) to the total porosity. In contrast, network P-Cu-L3 built from a segment with cis configuration showed a larger BET area (655 m2 g−1). The N2 adsorption/desorption isotherms on P-Cu-L3 (Fig. 7) again showed a pronounced N2 adsorption in the p/p0 < 0.1 region, which was followed by a continuous N2 trapping up to a relative pressure of p/p0 ∼ 1. Isotherms of this type are typical for micro/mesoporous materials with a significant contribution of mesopores. This was in good accordance with the N2-DFT pore size distribution shown in Fig. 7, indicating an increased contribution of mesopores to the total porosity. The differences in textural properties between networks with trans configuration, P-Cu-L1 and P-Cu-L2, and the network with cis configuration, P-Cu-L3, are noticeable also from total pore volumes (Fig. 7 and Table 1). Whilst the volume of micropores, Vmi, differed only slightly (0.19–0.26 cm3 g−1), the total pore volume, Vtot, showed more fundamental differences. Networks P-Cu-L1 and P-Cu-L2 had similar Vtot values (0.39 and 0.34 cm3 g−1, respectively). A much higher Vtot was observed for network P-Cu-L3 (0.71 cm3 g−1). The explanation for these disparities in textural properties can be based on single-crystal X-ray structures of organometallic complexes (Fig. S12 and S13†). We presume (based on FTIR and NMR results) that the geometries of organometallic segments remain unaffected by polymerization. The geometry of organometallic complex Cu-L1 detected by single-crystal X-ray diffraction is more planar and may result in a tighter packing of organometallic segments in the resulting polymer network, which does not mediate the formation of larger pores (mesopores) to such an extent. This organometallic segment and its racemic mixture are built into networks P-Cu-L1 and P-Cu-L2. In contrast, organometallic complex Cu-L3 showed a spatially more demanding geometry, which does not allow such a tight arrangement of organometallic segments, resulting in the formation of a wider range of pore sizes (high extent of micropores and mesopores) in network P-Cu-L3.
 | ||
| Fig. 7 (A) N2 adsorption (full symbols) and desorption (empty symbols) isotherms with pore volumes of micropores, Vmi, and total pore volumes, Vtot, for P-Cu-L1, P-Cu-L2 and P-Cu-L3. (B) N2-DFT calculated pore size distributions with relative cumulative pore volume, V/Vtot, for P-Cu-L1, P-Cu-L2 and P-Cu-L3. | ||
The type of SALDAC isomer used for the synthesis also affected the morphology of the resulting polymer networks. Scanning electron microscopy (SEM) confirmed the morphological similarity of networks P-Cu-L1 and P-Cu-L2 and dissimilarity with network P-Cu-L3. SEM images in Fig. 8 display rod-like particles of P-Cu-L1 and P-Cu-L2 networks, whereas partly aggregated spherically shaped particles were detected for network P-Cu-L3.
 | ||
| Fig. 8 SEM images of P-Cu-L1 (A), P-Cu-L2 (B) and P-Cu-L3 (C). | ||
Moreover, network P-Cu-L1 was characterized by powder X-ray diffraction (pXRD, Fig. S21†). The pXRD pattern showed no distinct signal, indicating the absence of long-range order and confirming the amorphous character of these networks. This finding was fully consistent with the formation of irregular cross-linking during the network formation via chain-growth polymerization.
Catalytic activity of organometallic polymer networks in styrene oxidation
The catalytic activity of the prepared organometallic polymer networks P-Cu-L1, P-Cu-L2, and P-Cu-L3 was evaluated in styrene oxidation, and compared with the catalytic activity of monomeric complexes Cu-L1, Cu-L2, and Cu-L3 (Table 2). In all cases, high styrene conversion was observed (80–91%, 6 h). Benzaldehyde was the most abundant product of all reactions. In the case of monomeric complexes used as catalysts, the selectivity to benzaldehyde was 21–32%. When polymer networks were used as catalysts the selectivity to benzaldehyde increased up to 50%. Another product that formed with remarkable selectivity was styrene oxide. In all cases, the selectivity to styrene oxide increased significantly when polymer networks were used as catalysts instead of monomeric complexes. We observed the increase from 5 to 41%, from 4 to 24% and from 2 to 28% using P-Cu-L1, P-Cu-L2, and P-Cu-L3, respectively. The increase in the selectivity after monomer polymerization was also observed in our previous study.12 In the case of polymer network P-Cu-L1, an interestingly high selectivity to benzaldehyde and styrene oxide (both 40 and 41%) was observed. This network was prepared from the enantiomerically pure complex with the chiral segment (1S,2S)-1,2-diaminocyclohexane. However, the enantioselectivity of P-Cu-L1 was not observed (styrene oxide was formed as a racemate). For verification, styrene oxidation was also performed (i) without a catalyst and (ii) with a non-metalated polymer network prepared by polymerization of L1. In both cases, no conversion of the substrate was observed. Similar types of materials have already been studied in styrene oxidation using H2O2 as an oxidizing agent. Maurya et al. studied the catalytic activity of similar copper(II) salen-type complexes encapsulated in zeolite-Y. In this case, a high selectivity to benzaldehyde was obtained (67%), however at low conversion of styrene (only 22%).75 Even higher selectivity to benzaldehyde (81%) was obtained using half-sandwich C-scorpionate copper(II) complexes on hydrochars, but again under lower styrene conversion (43%).76 Thus, our materials exhibited higher activity than those mentioned in previous studies and relatively high selectivity to benzaldehyde and, especially in the case of P-Cu-L1, also high selectivity to styrene oxide.| Catalyst | Styrene conversion [%] | Styrene oxide selectivity [%] | Benzaldehyde selectivity [%] | TON [mol molCu−1] |
|---|---|---|---|---|
|
|
||||
| Cu-L1 | 89 | 5 | 21 | 34 |
| P-Cu-L1 | 80 | 41 | 40 | 56 |
| Cu-L2 | 91 | 4 | 25 | 35 |
| P-Cu-L2 | 86 | 24 | 50 | 45 |
| Cu-L3 | 87 | 2 | 32 | 34 |
| P-Cu-L3 | 90 | 28 | 44 | 48 |
The advantage of heterogeneous catalysts based on organometallic polymer networks is their possible reuse in the reaction mixture. We tested the reusability of polymer network P-Cu-L1 in three consecutive cycles (Table S3†). The catalyst was simply removed from the reaction mixture, washed three times with dichloromethane, and used again as the catalyst for styrene oxidation. The possibility of catalyst reuse was confirmed and, moreover, an increase in styrene conversion in subsequent cycles was observed, accompanied by an increase in selectivity to benzaldehyde.
Conclusions
To summarize this work, three trios of a salen-type ligand, respective copper(II) complex, and organometallic polymer network were developed and prepared, namely (i) ligand L1, complex Cu-L1 and polymer network P-Cu-L1 were based on enantiomerically pure trans-(1S,2S)-1,2-diaminocyclohexane, (ii) L2, Cu-L2 and P-Cu-L2 were based on racemic trans-1,2-diaminocyclohexane and (iii) L3, Cu-L3 and P-Cu-L3 were based on the cis-1,2-diaminocyclohexane segment. The use of different isomers affected the final textural properties of the prepared polymer networks (microporous vs. micro/mesoporous texture, SBET 458–655 m2 g−1). The structures, geometries, and packings of organometallic complexes in solid states (crystal vs. powder) were investigated by single-crystal X-ray diffraction and advanced NMR techniques for paramagnetic compounds. The comparison with solid-state NMR spectra of polymer networks revealed the similar geometry of organometallic segments also in the networks and reflected the differences between individual isomers and their influence on the formation of the porous texture. Less spatially demanding trans isomers allowed tighter packing leading to lower SBET values, while the more voluminous cis isomer formed a polymer network with higher SBET. Finally, the catalytic activity of the prepared organometallic polymer networks was tested. Heterogeneously catalyzed styrene oxidation exhibited remarkably high styrene conversions (well above 80%) with high selectivity to the desired products (up to 41% to styrene oxide and up to 50% to benzaldehyde). All networks showed significantly increased selectivity compared to respective low-molecular organometallic complexes. Good reusability results in catalytic testing pointed to the promising potential of these materials.Author contributions
DŠ: investigation (development and synthesis of the materials), conceptualization, visualization, and writing – original draft; JB: formal analysis (NMR spectroscopy) and data curation; BB: formal analysis (nitrogen adsorption) and writing – review and editing; IC: formal analysis (X-ray crystallography) and data curation; EVr: investigation (catalysis); EVy: conceptualization (catalysis) and writing – review and editing; JS: conceptualization (development and synthesis of the materials), supervision, and writing – review and editing.Data availability
All results and experimental data are available in the manuscript and ESI.† The crystallographic data were deposited with the CCDC under numbers 2333975 and 2333976† for Cu-L1 and Cu-L3, respectively. Raw NMR data as well as I/O files from DFT calculations and SIMPSON simulations can be found in the Zenodo repository at https://doi.org/10.5281/zenodo.10973432.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (project no. 21-02183S, J. Sedláček; and project no. 24-10843S, J. Blahut) and the Science Foundation of Charles University (project no. 193223, D. Šorm). Computational resources were provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic. The authors kindly acknowledge the provided financial support.Notes and references
- Y.-C. Yuan, M. Mellah, E. Schulz and O. R. P. David, Chem. Rev., 2022, 122, 8841–8883 CrossRef CAS PubMed.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- A. Erxleben, Inorg. Chim. Acta, 2018, 472, 40–57 CrossRef CAS.
- A. M. G. Mutti, F. S. M. Canisares, W. B. S. Machini, A. M. Pires, M. F. S. Teixeira and S. A. M. Lima, Optik, 2021, 243, 167454 CrossRef CAS.
- G. Yu, Y. Liu, Y. Song, X. Wu and D. Zhu, Synth. Met., 2001, 117, 211–214 CrossRef CAS.
- O. Lavastre, I. Illitchev, G. Jegou and P. H. Dixneuf, J. Am. Chem. Soc., 2002, 124, 5278–5279 CrossRef CAS PubMed.
- A. C. W. Leung, J. H. Chong, B. O. Patrick and M. J. MacLachlan, Macromolecules, 2003, 36, 5051–5054 CrossRef CAS.
- T. Glaser, M. Heidemeier, R. Fröhlich, P. Hildebrandt, E. Bothe and E. Bill, Inorg. Chem., 2005, 44, 5467–5482 CrossRef CAS PubMed.
- T. Glaser, M. Heidemeier, S. Grimme and E. Bill, Inorg. Chem., 2004, 43, 5192–5194 CrossRef CAS PubMed.
- N. S. Venkataramanan, G. Kuppuraj and S. Rajagopal, Coord. Chem. Rev., 2005, 249, 1249–1268 CrossRef CAS.
- S. Shaw and J. D. White, Chem. Rev., 2019, 119, 9381–9426 CrossRef CAS PubMed.
- D. Šorm, B. Bashta, J. Blahut, I. Císařová, L. Dolejšová Sekerová, E. Vyskočilová and J. Sedláček, Eur. Polym. J., 2023, 184, 111772 CrossRef.
- W. Al Zoubi and Y. G. Ko, Appl. Organomet. Chem., 2017, 31, e3574 CrossRef.
- M. S. More, P. G. Joshi, Y. K. Mishra and P. K. Khanna, Mater. Today Chem., 2019, 14, 100195 CrossRef CAS PubMed.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- S. Lundgren, E. Wingstrand, M. Penhoat and C. Moberg, J. Am. Chem. Soc., 2005, 127, 11592–11593 CrossRef CAS PubMed.
- J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 3268–3269 CrossRef CAS PubMed.
- R. Kowalczyk, P. Kwiatkowski, J. Skarżewski and J. Jurczak, J. Org. Chem., 2009, 74, 753–756 CrossRef CAS PubMed.
- S. Liao and B. List, Adv. Synth. Catal., 2012, 354, 2363–2367 CrossRef CAS.
- E. F. DiMauro and M. C. Kozlowski, Org. Lett., 2001, 3, 1641–1644 CrossRef CAS PubMed.
- M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, ACS Catal., 2015, 5, 3398–3402 CrossRef CAS.
- V. Aomchad, S. Del Gobbo, P. Yingcharoen, A. Poater and V. D'Elia, Catal. Today, 2021, 375, 324–334 CrossRef CAS.
- Y.-M. Shen, W.-L. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559–1562 CrossRef CAS PubMed.
- A. Vidal-López, S. Posada-Pérez, M. Solà, V. D'Elia and A. Poater, Green Chem. Eng., 2022, 3, 180–187 CrossRef.
- F. Della Monica and C. Capacchione, Asian J. Org. Chem., 2022, 11, e202200300 CrossRef CAS.
- J. A. Castro-Osma, K. J. Lamb and M. North, ACS Catal., 2016, 6, 5012–5025 CrossRef CAS.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- C. Baleizão and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 CrossRef PubMed.
- M. Abd El Sater, N. Jaber and E. Schulz, ChemCatChem, 2019, 11, 3662–3687 CrossRef CAS.
- J. Rakhtshah, Coord. Chem. Rev., 2022, 467, 214614 CrossRef CAS.
- J. Min, W. Song, T. Hu, Y. Zhi, Z. Xia, T. Zhang, S. Shan and H. Su, Ceram. Int., 2021, 47, 35320–35332 CrossRef CAS.
- T. Maharana, N. Nath, H. C. Pradhan, S. Mantri, A. Routaray and A. K. Sutar, React. Funct. Polym., 2022, 171, 105142 CrossRef CAS.
- I. Kuźniarska-Biernacka, C. Pereira, A. P. Carvalho, J. Pires and C. Freire, Appl. Clay Sci., 2011, 53, 195–203 CrossRef.
- C. Xing, J. Deng, R. Tan, M. Gao, P. Hao, D. Yin and D. Yin, Catal. Sci. Technol., 2017, 7, 5944–5952 RSC.
- R. Ji, K. Yu, L.-L. Lou and S. Liu, J. Mol. Catal. A: Chem., 2013, 378, 7–16 CrossRef CAS.
- M. Halder, P. Bhanja, Md. M. Islam, A. Bhaumik and Sk. M. Islam, New J. Chem., 2018, 42, 11896–11904 RSC.
- S. Roy, P. Bhanja, Sk. Safikul Islam, A. Bhaumik and Sk. Manirul Islam, Chem. Commun., 2016, 52, 1871–1874 RSC.
- Md. M. Islam, P. Bhanja, M. Halder, S. K. Kundu, A. Bhaumik and Sk. M. Islam, RSC Adv., 2016, 6, 109315–109321 RSC.
- M. Shukla, K. C. Barick, H. G. Salunke and S. Chandra, Mol. Catal., 2021, 502, 111367 CrossRef CAS.
- W. Zhou, Q.-W. Deng, H.-J. He, L. Yang, T.-Y. Liu, X. Wang, D.-Y. Zheng, Z.-B. Dai, L. Sun, C. Liu, H. Wu, Z. Li and W.-Q. Deng, Angew. Chem., Int. Ed., 2023, 62, e202214143 CrossRef CAS PubMed.
- S. Kramer, N. R. Bennedsen and S. Kegnæs, ACS Catal., 2018, 8, 6961–6982 CrossRef CAS.
- S. Luo, Z. Zeng, H. Wang, W. Xiong, B. Song, C. Zhou, A. Duan, X. Tan, Q. He, G. Zeng, Z. Liu and R. Xiao, Prog. Polym. Sci., 2021, 115, 101374 CrossRef CAS.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- W. Song, Y. Zhang, C. H. Tran, H. K. Choi, D.-G. Yu and I. Kim, Prog. Polym. Sci., 2023, 142, 101691 CrossRef CAS.
- J. Sedláček and H. Balcar, Polym. Rev., 2017, 57, 31–51 CrossRef.
- L. Sekerová, M. Lhotka, E. Vyskočilová, T. Faukner, E. Slováková, J. Brus, L. Červený and J. Sedláček, Chem. – Eur. J., 2018, 24, 14742–14749 CrossRef PubMed.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- Y. Xie, T.-T. Wang, R.-X. Yang, N.-Y. Huang, K. Zou and W.-Q. Deng, ChemSusChem, 2014, 7, 2110–2114 CrossRef CAS PubMed.
- R. Luo, Y. Chen, Q. He, X. Lin, Q. Xu, X. He, W. Zhang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 1526–1533 CrossRef CAS PubMed.
- J. Li, Y. Han, H. Lin, N. Wu, Q. Li, J. Jiang and J. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 609–618 CrossRef CAS PubMed.
- D. Meng, J. Bi, Y. Dong, B. Hao, K. Qin, T. Li and D. Zhu, Chem. Commun., 2020, 56, 2889–2892 RSC.
- X. Sun, F. Meng, Q. Su, K. Luo, P. Ju, Z. Liu, X. Li, G. Li and Q. Wu, Dalton Trans., 2020, 49, 13582–13587 RSC.
- H. Li, J. Chen, J. Liu, C. Li, L. Liu and Q. Yang, ChemNanoMat, 2022, 8, e202100386 CrossRef CAS.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed.
- J. Blahut, L. Benda, A. L. Lejeune, K. J. Sanders, B. Burcher, E. Jeanneau, D. Proriol, L. Catita, P.-A. R. Breuil, A.-A. Quoineaud, A. J. Pell and G. Pintacuda, RSC Adv., 2021, 11, 29870–29876 RSC.
- Y. Nishiyama, G. Hou, V. Agarwal, Y. Su and A. Ramamoorthy, Chem. Rev., 2023, 123, 918–988 CrossRef CAS PubMed.
- Y. Nishiyama and N. T. Duong, J. Magn. Reson. Open, 2022, 10–11, 100062 CrossRef.
- V. D'Anna, S. Norsic, D. Gajan, K. Sanders, A. J. Pell, A. Lesage, V. Monteil, C. Copéret, G. Pintacuda and P. Sautet, J. Phys. Chem. C, 2016, 120, 18075–18087 CrossRef.
- K. Saalwächter, F. Lange, K. Matyjaszewski, C.-F. Huang and R. Graf, J. Magn. Reson., 2011, 212, 204–215 CrossRef PubMed.
- S. M. Elbert and M. Mastalerz, Org. Mater., 2020, 02, 182–203 CrossRef CAS.
- J. Vaara, S. A. Rouf and J. Mareš, J. Chem. Theory Comput., 2015, 11, 4840–4849 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- D. W. Juhl, Z. Tošner and T. Vosegaard, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 2020, vol. 100, pp. 1–59 Search PubMed.
- TURBOMOLE V7.1, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- W.-K. Dong, J.-G. Duan, Y.-H. Guan, J.-Y. Shi and C.-Y. Zhao, Inorg. Chim. Acta, 2009, 362, 1129–1134 CrossRef CAS.
- N. Kordestani, H. Amiri Rudbari, G. Bruno, S. Rosario, J. D. Braun, D. E. Herbert, O. Blacque, I. Correia, M. A. Zaman, M. M. Bindu, C. Janiak and M. Enamullah, Dalton Trans., 2020, 49, 8247–8264 RSC.
- R. K. Hylton, G. J. Tizzard, T. L. Threlfall, A. L. Ellis, S. J. Coles, C. C. Seaton, E. Schulze, H. Lorenz, A. Seidel-Morgenstern, M. Stein and S. L. Price, J. Am. Chem. Soc., 2015, 137, 11095–11104 CrossRef CAS PubMed.
- O. A. Lodochnikova, V. A. Startseva, L. E. Nikitina, A. V. Bodrov, A. E. Klimovitskii, E. N. Klimovitskii and I. A. Litvinov, CrystEngComm, 2014, 16, 4314–4321 RSC.
- C. van Beek and V. V. Samoshin, Tetrahedron Lett., 2022, 102, 153930 CrossRef CAS.
- A. Bertarello, L. Benda, K. J. Sanders, A. J. Pell, M. J. Knight, V. Pelmenschikov, L. Gonnelli, I. C. Felli, M. Kaupp, L. Emsley, R. Pierattelli and G. Pintacuda, J. Am. Chem. Soc., 2020, 142, 16757–16765 CrossRef CAS PubMed.
- O. Trhlíková, J. Zedník, H. Balcar, J. Brus and J. Sedláček, J. Mol. Catal. A: Chem., 2013, 378, 57–66 CrossRef.
- M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 270, 225–235 CrossRef CAS.
- T. A. G. Duarte, A. P. Carvalho and L. M. D. R. S. Martins, Catal. Today, 2020, 357, 56–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR characterization, X-ray structures, UV/Vis and CD spectra, and reuse of the catalyst. CCDC 2333975 (Cu-L1) and 2333976 (Cu-L3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01305k |
| This journal is © The Royal Society of Chemistry 2024 |