
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriptycene as a scaffold in metallocene catalyzed olefin polymerization†
Pavel S.
Kulyabin
 a,
Mikhail I.
Sharikov
a,
Vyatcheslav V.
Izmer‡
a,
Dmitry S.
Kononovich
a,
Georgy P.
Goryunov
a,
Nikita V.
Alexeev
a,
Dmitry V.
Uborsky
a,
Antonio
Vittoria
b,
Giuseppe
Antinucci
b,
Christian
Ehm
b,
Peter H. M.
Budzelaar
*b,
Roberta
Cipullo
b,
Vincenzo
Busico
*b and
Alexander Z.
Voskoboynikov
*a
a,
Mikhail I.
Sharikov
a,
Vyatcheslav V.
Izmer‡
a,
Dmitry S.
Kononovich
a,
Georgy P.
Goryunov
a,
Nikita V.
Alexeev
a,
Dmitry V.
Uborsky
a,
Antonio
Vittoria
b,
Giuseppe
Antinucci
b,
Christian
Ehm
b,
Peter H. M.
Budzelaar
*b,
Roberta
Cipullo
b,
Vincenzo
Busico
*b and
Alexander Z.
Voskoboynikov
*a
aDepartment of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia. E-mail: voskoboy@med.chem.msu.ru
bDipartimento di Scienze Chimiche, Università di Napoli Federico II, 80126 Napoli, Italy. E-mail: p.budzelaar@unina.it; busico@unina.it
First published on 13th May 2024
Abstract
A set of metallocene olefin polymerization catalysts bearing triptycene moieties in either position 4–5 (complexes Ty1–Ty5) or in position 5–6 (complexes Ty6–Ty8) of the basic dimethylsilyl-bridged bis(indenyl) system has been tested in propene polymerization and in ethene/1-hexene copolymerization. Comparison of the results with QSPR (quantitative structure–property relationship) predictions not parametrized for these exotic ligand variations demonstrates that trends can still be identified by extrapolation. Interestingly, Ty7, upon suitable activation, provides a highly isotactic polypropylene with an exceptional amount of 2,1 regio-errors (8%). The previously developed QSPR type models successfully predicted the low regioselectivity of this catalyst, despite the fact that the catalyst structure differs significantly from the benchmark set.
Introduction
Catalysis of olefin polymerization is all about control: control of stereochemistry,1–7 of regiochemistry (1,2 vs. 2,1 insertion),8–11 of molecular weight (insertion vs. β-H transfer and hydrogenolysis),2,12–15 of chemoselectivity (ethene vs. propene vs. α-olefins),16–20 of catalyst stability (propagation vs. deactivation),21–24 and catalyst activity.25–28 For typical catalyst classes the main factor is sterics.29,30 Stereoselectivity is completely determined by steric interactions between ligand, growing chain and incoming monomer.29,30 Regioselectivity and chemoselectivity have some contributions from electronic factors but are still dominated by sterics.16,17,31,32 Electronic factors are more relevant in MW (molecular weight) control.33 Thus, it stands to reason that catalyst optimization (within a given class) is a matter of precision tuning of the catalyst 3D structure. Enzymes achieve this kind of finetuning through their tertiary structure, which is mostly held together by hydrogen bridges.34,35 Human design of the 3D structure of polymerization catalysts cannot use this principle, if only because hydrogen bridges are acidic and would not survive in the alkylating medium of a polymerization reaction.27 One could imagine ligand structure being precisely architected using non-covalent interactions,36 but the art of ligand design has not yet progressed to the point where this is practical. Moreover, the approach seems ill suited for the high operating temperatures employed in industrial olefin polymerization (Tp > 100 °C). Thus, we must instead rely on covalent linking of building blocks, and this imposes severe limitations on the design of 3D structures. The importance of catalyst rigidity in olefin polymerization is well-documented,13,32,37–39 but achieving such rigid structures precisely covering the right distribution of steric bulk over space is nontrivial. Enforced coplanarity of catalyst fragments (e.g., cyclopentadienyl (Cp) → indenyl) is relatively easy to establish. Orthogonal connections (e.g., aryl – 2,6-Me2C6H3) are also easy to enforce, be it that usually some flexibility remains. Unrestricted connections (bonds having free rotation) are straightforward. For anything more complex than these, sophisticated and creative choices of building blocks are required. This is nicely illustrated by our recently reported two metallocene catalysts bearing triptycyl fragments in their skeleton, with the 9,10-dihydroanthracenyl moieties bridging the indenyl positions 4 and 5 (Ty1 and Ty2 in Fig. 1).13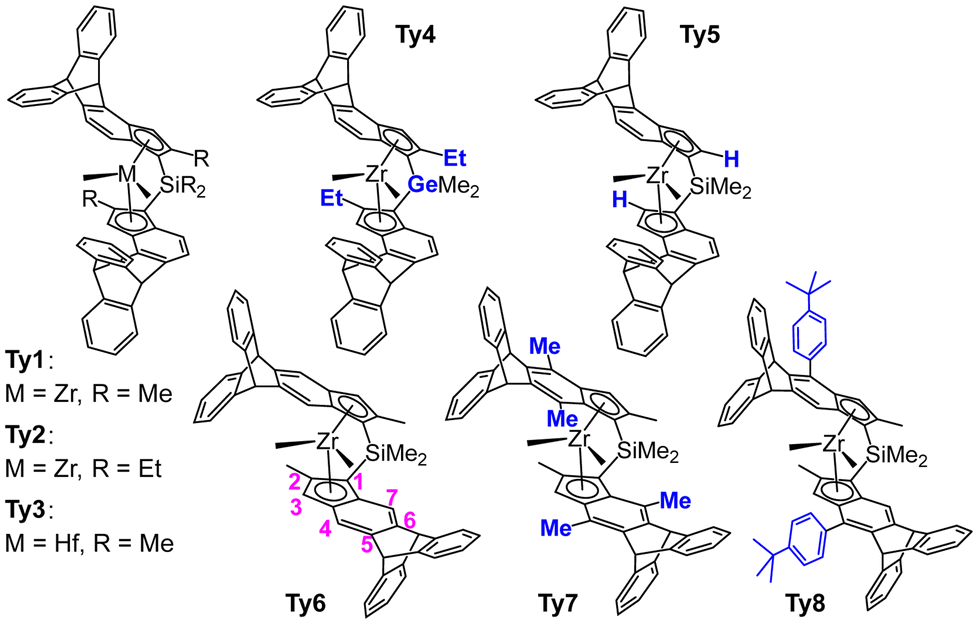 | ||
| Fig. 1 Triptycene based metallocenes tested in olefin polymerization catalysis. Ty1–Ty5: 4,5-bridged, Ty6–Ty8: 5,6-bridged. Modifications relative to the parent compounds Ty1 and Ty6 indicated in blue. Numbering of indenyl positions in magenta. Ty1/Ty2,13Ty3,40Ty4–Ty8 this work. | ||
In these complexes the ligands precisely cover space around the metal center to virtually suppress stereo- and regioerror insertion as well chain end epimerization and β-H transfer to monomer, thus providing highly regioregular isotactic polypropylene with a high molecular mass capability. This precision architecture was made possible by the unusual paddlewheel shape of the triptycyl unit,41 which allows fixing of ligand fragments at 120° angles. The relative rigidity of the resulting ligand backbone presumably is also an important factor.13
We consider the prediction13 of superior performance of catalysts Ty1 and Ty2 a success of our QSPR (quantitative structure–property relationship) type modelling.12,29,31,32 In order to broaden the applicability of our model in the future, covering aspects of catalyst structures not taken into account in the training set used to predict Ty1 and Ty2 performances, we embarked on the synthesis, although demanding, of several new triptycyl complexes (Fig. 1):
(a) Ty3, the Hf analog of Ty1, features a different central metal (Hf vs. Zr),37
(b) Ty4 features a variation of the metallocene bridge atom (Ge vs. Si) with respect to Ty2.
(c) Ty5 lacks the crucial 2-R-substituent, which is present in Ty1 and Ty2.
(d) Ty6–8 incorporate a 5,6 instead of 4,5 bridging triptycyl unit. Note that Ty7 shows in addition severe distortions of the ligand framework introduced by 7-Me substituents.
In this manuscript we report on the synthesis of the metallocenes of Fig. 1 and on their performance in propene polymerization and ethene/1-hexene copolymerization. Predictions by QSPR models are also discussed.
Results and discussion
Synthesis of metallocenes
We have developed a strategy for the synthesis of triptycene derived metallocenes that is quite general and requires in most cases easily available starting materials: variously halogenated toluenes or indanones. No evidence for a specific “triptycyl effect” has been observed. Triptycene moieties have been incorporated in sophisticated ligand frameworks in different ways, such as: (a) connected at C9 as a very bulky C3-symmetric t-alkyl substituent for Tpz type ligands,42 (b) acting as a spacer for wide-angle bidentate diphosphine ligands in hydroformylation,43,44 (c) attached at C1 as a very bulky and asymmetric aryl substituent at the N atoms of NHC ligands,45 (d) as an aromatic scaffold for mono-and dinucleating diphosphinamide ligands for ethene oligomerization.46Syntheses of metallocenes Ty1–3 was described in our preceding papers.13,40 Proligands for new 4,5-bridged triptycene precatalysts Ty4 and Ty5 were synthesized using the same malonate pathway, only differing in the reactants used: Me2GeCl2 for Ty4 and diethyl malonate for Ty5 (Scheme 1).
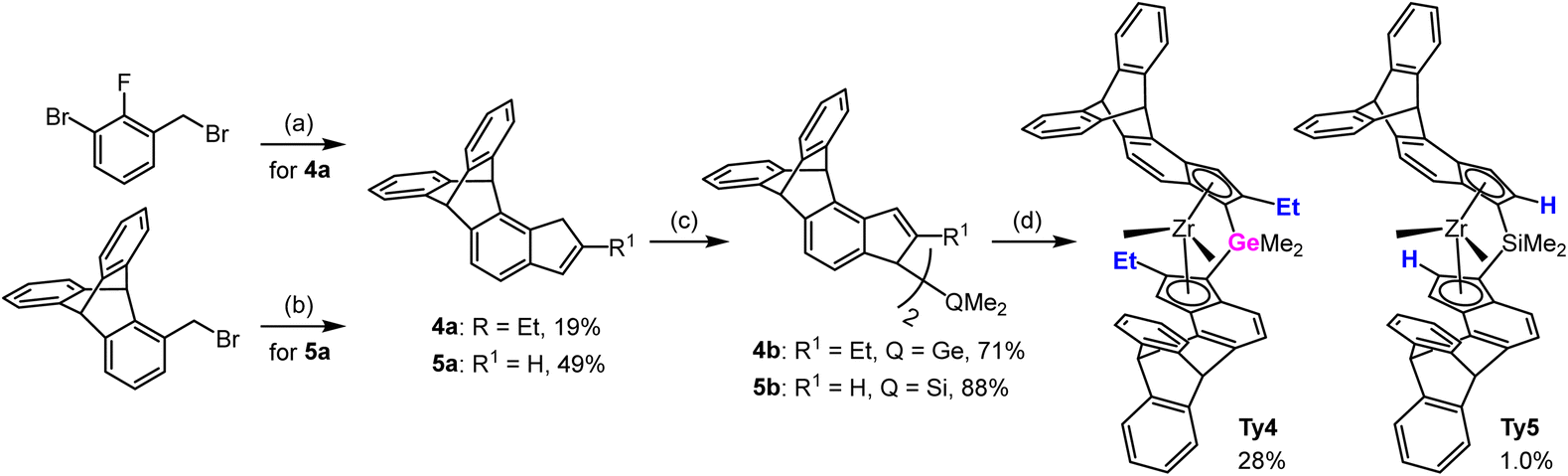 | ||
| Scheme 1 Syntheses of 4,5-bridged triptycene precatalysts. (a) see ref. 13 (b) 1. NaH, CH2(COOEt)2, THF; 2. KOH, water, reflux; 3. HClaq; 4. 160 °C; 5. H3PO4-P4O10, 130 °C; 6. NaBH4, MeOH-THF; 7. TsOH, PhMe, reflux; (c) for 4b: i. nBuLi, Et2O, ii. Me2SiCl2, N-methylimidazole; for 5b: nBuLi, Et2O, ii. CuCN, Me2SiCl2. (d) for Ty4: 1. i. nBuLi, Et2O, ii. ZrCl4; 2. MeMgBr, PhMe-Et2O; 3. LiCl, THF; for Ty5: 1. i. nBuLi, Et2O, ii. ZrCl4; 2. rac/meso separation, 3. MeMgBr, Et2O. | ||
The key step in the synthesis of indenes with 5,6-bridged triptycene moieties was the Diels–Alder reaction of the benzyne formed from the respective 5,6-dihalogen-substituted methoxyindanes Scheme 2). The ortho-dihalogen-substituted aryl moiety was either in the starting compound (6a) or introduced by bromination (7a to 7b) or diazotation (8a to 8b). After elimination of methanol from the intermediate methoxyindanes, the indenes thus obtained were converted to the Me2Si-bridged proligands via reaction of their lithium salts with Me2SiCl2 in the presence of CuCN or N-methylimidazole.47
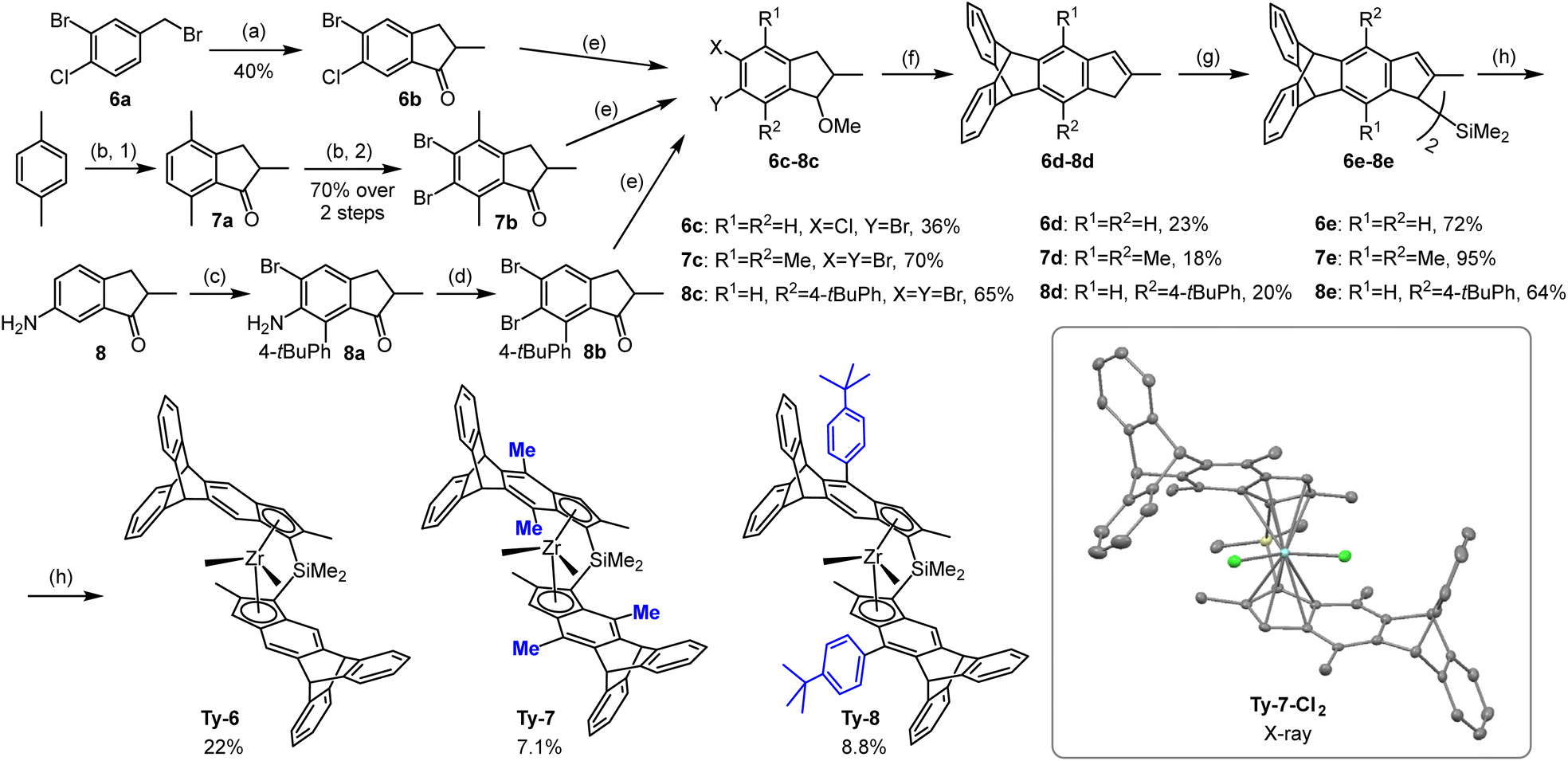 | ||
| Scheme 2 Syntheses of 5,6-bridged triptycene precatalysts: (a) 1. MeCH(COOEt)2, NaOEt, EtOH 2. KOH, water, reflux; 3. 180 °C; 4. SOCl2, CH2Cl2; 5. AlCl3, CH2Cl2. (b) 1. AlCl3, CH2Cl2; 2. one-pot: Br2, CH2Cl2. (c) 1. NBS, DMF; 2. 4-tBuPhB(OH)2, Cs2CO3, Pd(PPh3)4, dioxane/water, reflux; 3. NBS, DMF. (d) amyl nitrite, CuBr2, MeCN. (e) 1. NaBH4, MeOH-THF; 2. for 6c, 7c: KOH, MeI, DMSO; for 8c: NaH, MeI, THF. (f) 1. nBuLi, THF, anthracene, 2. for 6d: HCl (aq.), MeOH, reflux; for 7d, 8d: TsOH, PhMe, reflux. (g) For 6e, 7e: i. nBuLi, Et2O, ii. THF, CuCN, Me2SiCl2; for 8e: i. nBuLi, Et2O, ii. Me2SiCl2, N-methylimidazole (h) i. nBuLi, ZrCl4, Et2O; ii. rac/meso separation; 3. MeMgBr, Et2O. Insert: crystal structure of Ty7-Cl2: image generated by Mercury software with thermal ellipsoids at 50% probability level; hydrogen atoms and co-crystallized toluene molecules omitted for clarity. | ||
Most of the racemic dimethyl complexes (Ty5–Ty8) were obtained by reaction of the racemic dichloride precursors (separated from the isomeric meso side-products by crystallization in the previous step) with MeMgBr. For preparation of Me2Ge-bridged Ty4, we used LiCl-catalyzed isomerization of a mixture of dimethyl complexes rac/meso-Ty4 to the rac-Ty4,47 which was then isolated by crystallization.
The crystal structure of Ty7-Cl2, the dichloride precursor of Ty7, was elucidated by X-ray diffraction analysis. The asymmetric unit cell contains two molecules of co-crystallized toluene, located on left side with respect to the plane bisecting the left and right halves of the structure (inset in Scheme 2 and Fig. S1†) thus distorting the ligand surrounding in the respective area of the metallocene and the overall C2-symmetry of the complex.
Ty7-Cl2 has methyl groups at indenyl position 7, a feature for which there are relatively few examples in the literature. Repulsion between position 7 substituents and neighboring Si–Me groups affects the entire structure in a number of common ways:37,48,49 (1) the 7-methyls are pushed from the mean planes of the benzene rings they are connected to by 0.450/0.355 Å; (2) the Si–Me groups are pushed to the opposite direction resulting in rotation of the Me–Si–Me plane with respect to the Cl–Zr–Cl plane (torsion angles Me–Si–Zr–Cl are 10.85/10.30° vs. 1.36/1.64° in Ty113); (3) the ligand bite angle is reduced (57.58° vs. 59.54° in Ty1); (4) the Cp moieties are rotated with respect to each other (angle C2–Cpcent–Cpcent′–C2′ is 8.64° vs. 1.98° in Ty1); (5) the Zr–CCp distances change: e.g. Zr–C1/C1′ are elongated (2.618/2.599 vs. 2.583/2.576 Å in Ty1), whereas Zr–C3/C3′ are shorter (2.488/2.476 vs. 2.534/2.532 Å in Ty1).
Testing in iPP catalysis
The new metallocenes Ty4–Ty8 were tested in our high-throughput experimentation setup (described in detail in ref. 50 and 51) in propene polymerization at Tp = 60 °C and ppropene = 6.6 bar, as well as at Tp = 100 °C and ppropene = 7.9 bar, where possible. Conditions were held rigorously identical to those used previously for Ty1/Ty2,13 and Ty3,40 so numbers cited allow direct comparison. Results for the new catalysts Ty4–Ty8 are summarized in Table 1, together with data for previously studied systems (Ty1–Ty3) included here for comparison. It should be noted that the employed protocol emphasizes kinetic control over catalyst activity utilizing an activation delay to minimize undesired effects on polymer properties.52 Activities (see ESI†) are therefore not representative of the maximum activities attainable for these catalysts.| Catalyst | 60 °C | 100 °C | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 − σ, % | 2,1, % | 3,1, % | M n, kDa | PDI | 1 − σ, % | 2,1, % | 3,1, % | M n, kDa | PDI | |
| Other experimental conditions: solvent, toluene; scavenger/activator, tri-iso-butylaluminum/HNMe2Ph+[B(C6F5)4]− (TIBA/AB). Propene pressure at 60 °C: 6.6 bar, at 100 °C: 7.9 bar. All data are averages of at least duplicate experiments. n.d. not detected. For further details see ESI.† | ||||||||||
| Ty1 | ≤0.02 | 0.26 | n.d. | 1100 | 2.5 | 0.03 | 0.33 | 0.02 | 200 | 2.1 |
| Ty2 | 0.03 | 0.18 | n.d. | 1900 | 2.1 | 0.04 | 0.23 | 0.03 | 140 | 2.3 |
| Ty3 | 0.03 | 0.11 | n.d. | 1400 | 2.3 | 0.10 | 0.12 | 0.06 | 90 | 2.0 |
| Ty4 | ≤0.02 | 0.19 | n.d. | 880 | 2.6 | ≤0.02 | 0.24 | 0.03 | 150 | 2.5 |
| Ty5 | 0.05 | 1.0 | n.d. | 60 | 2.1 | not tested, molar mass capability too low | ||||
| Ty6 | 1.2 | 1.3 | 0.05 | 40 | 2.0 | |||||
| Ty7 | 0.43 | 7.9 | 0.1 | 25 | 2.0 | |||||
| Ty8 | 0.03 | 1.3 | 0.02 | 430 | 2.1 | 0.10 | 1.4 | 0.48 | 67 | 2.1 |
Both Ty1 and Ty2 feature remarkable stereoselectivity (even at elevated temperature) as well as good regioselectivity, high MW and good activity, as reported earlier. Ty3, which differs from Ty1 in the central metal (Hf vs. Zr), shows similar stereoerrors, higher molecular weight and higher regioselectivity. At elevated temperatures (Tp = 100 °C), both stereoselectivity and molar mass capability of Ty3 are lower than with Ty1 (see ESI†).
Ty4, which was synthesized to test the influence of a germanium-based bridge, shows similar performance to Ty2 in terms of regio- and stereoselectivity at Tp = 60 °C; the molar mass capability is similar. However, at elevated temperatures, the catalyst appears to be unstable, which might explain why despite initial promising reports by Kaminsky,53 Ge-based bridges have not attracted attention in the field. Ty5 lacks a substituent at indenyl position 2. This substitution is generally considered to be a requirement for obtaining decent MW.54 Our results support this, as illustrated by the observed MW for Ty1 that is 20 times higher than for Ty5.
The performance of the new 5,6-bridged systems Ty5–Ty8 differs dramatically from the 4,5-bridged systems studied before. This should not be too surprising: while use of the 4,5-bridged skeleton apparently puts steric bulk exactly where it is most effective, the use of the 5,6-linker creates bulk in a different and presumably less effective region of space. This is precisely why we want to include such sub-optimal species in our future model building.
In fact, the introduction of the 5,6-triptycene unit in Ty6 even lowers the molar mass capability compared to the parent 2-Me-indenyl ansa-metallocene,12 even though substituents at the positions 5 and 6 are known to increase molar mass capability.31,32,55 In Ty8, stereoselectivity is restored to the level of Ty1 by the addition of a bulky aryl group at the indenyl position 4 while molar mass capability is mostly but not fully restored.
Evidently, only the simultaneous introduction of substituents at the positions 4 and 5–6 constrains the active pocket enough to affect chain transfer transition states, and the 5,6-triptycene unit appears to be roughly as effective as a 6-Ph substituent in increasing molar mass capability.31 In contrast, regioselectivity of Ty8 is not restored to the Ty1 level.
Ty7 is, from a scientific point of view, the most interesting new catalyst. Fig. 2 reports the 13C NMR spectrum of a polypropylene obtained a 60 °C with Ty7. In addition to the three main resonances typical of a regioregular polypropylene,2 the set of resonances ascribed to the head-to-head/tail-to-tail enchainments are well visible.2 Interestingly the resonances of 3,1 units are hardly visible (<0.1%), suggesting that isomerization of a last 2,1 propene insertion is still blocked for Ty7 like it is for Ty1/Ty2.
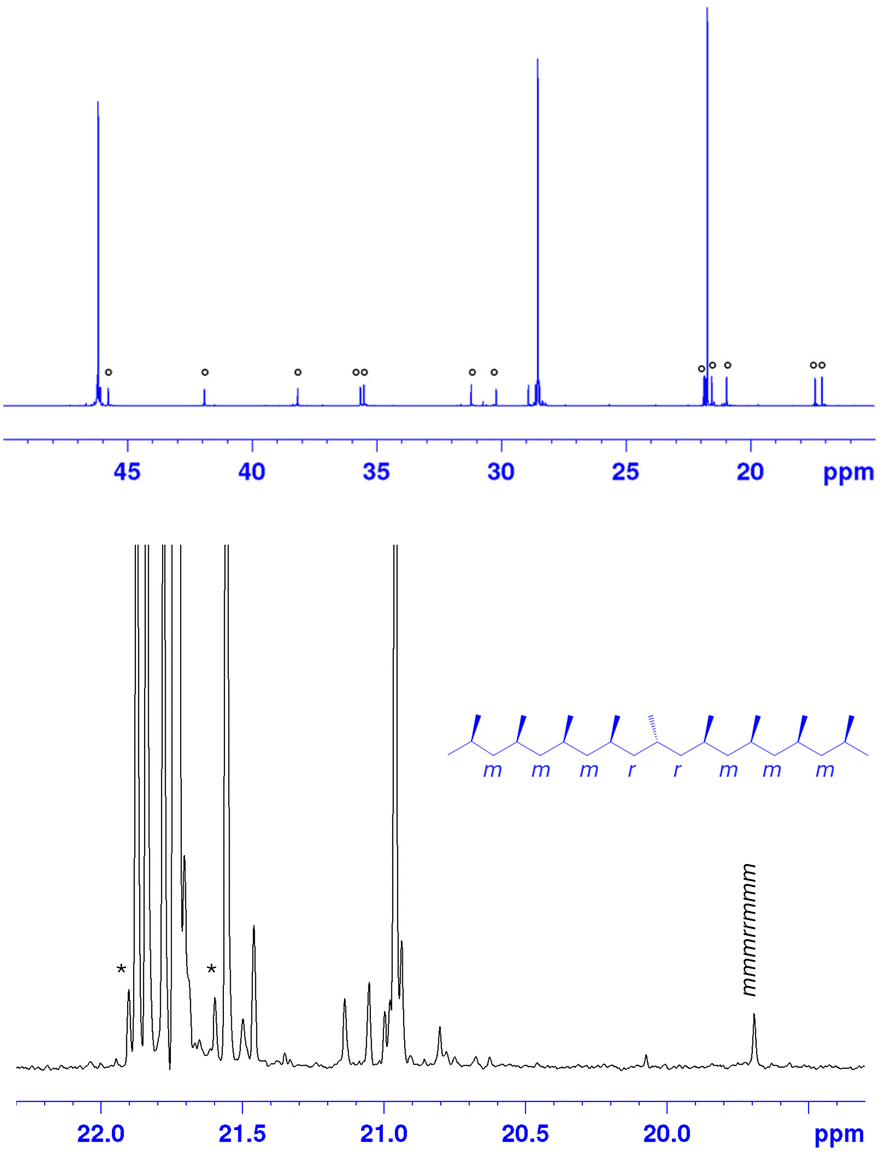 | ||
| Fig. 2 Full 13C NMR spectrum (top) and expansion of the methyl region (bottom) of highly regioirregular iPP made using catalyst Ty7. Peaks marked with * and o are due 13C–13C satellite band and to regioerrors, respectively. | ||
The expansion of the methyl region of the 13C NMR spectrum (Fig. 2) clearly shows that the sample is highly stereoregular, since the intensity of the peak at 19.7 ppm due to the isolated stereoerror is lower than that of the 13C–13C satellite band of the main resonance (marked with an asterisk). Overall, Ty7 produces, to our knowledge, an unprecedented polypropylene sample with an exceptionally high degree of regiodefects in the form of isolated 2,1 units: 7% (with only ∼0.4% stereoerrors). Typical values for e.g. Spaleck-type selectivity would be <1% stereo-errors and 1–2% 2,1e regiodefects.56 Resconi56–58 has reported up to 3.4% 2,1 regioerrors in polymers produced with rac-Me2Si(2-Me-4-Ph-5-OMe-6-tBu-Ind)2ZrCl2.55 The regioselectivity and 3,1-isomerization tendency of this catalyst is however tied to activation/immobilization conditions.31,57 This opens the door to study the effect of the presence of substantial amounts of 2,1 units in stereoregular polypropylene samples on material properties. On this we will report in due time.
Testing in ethene/1-hexene copolymerization catalysis
All catalysts were also tested in ethene/1-hexene copolymerization under standard conditions previously used for database building.17 Comonomer feeding ratio was adjusted to obtain copolymers with high 1-hexene incorporations to evaluate from a single 13C NMR spectrum the reactivity ratios values (see ESI† and Experimental section). Table 2 summarizes the rE and rH values, averaged on at least duplicate experiments.| Catalyst | r E | r H |
|---|---|---|
| Ty1 | 4.0 | 0.23 |
| Ty2 | 3.5 | 0.23 |
| Ty3 | 3.3 | 0.25 |
| Ty4 | 4.4 | 0.21 |
| Ty5 | 3.4 | 0.27 |
| Ty6 | 8.8 | 0.014 |
| Ty7 | 19 | 0.12 |
| Ty8 | 3.3 | 0.29 |
For the catalysts with considerable steric bulk around the active pocket, Ty1–Ty5 and Ty8, the comonomer affinity after ethene insertion is on par with the best performing C2-symmetric ansa-metallocene catalysts identified recently in a large screening (rE 3.2–4.6).59 Similarly, the comonomer affinity after 1-hexene insertion (rH = 0.21–0.29) is comparable to other ansa-metallocenes.
For the 4,5-triptycenes (Ty1–4), the reactivity ratios values are insensitive to changes in substituent pattern, and metal changes (Ty1vs.Ty3). This has been rationalized previously as indicative of a change in rate limiting step from insertion to olefin capture,17,20 which is typical for well performing catalysts of this class. The effects of the 5,6-triptycene pattern, notable in propene polymerization, are minimal with respect to the comonomer affinity. Only Ty6 and Ty7, which are also the worst performing catalysts in propene polymerization, show poorer comonomer affinity (higher rE and lower rH value). However, their performance is similar to that of their parent metallocene system17 (2-Me-indenyl)2SiMe2ZrCl2, and (2-4-di-Me-indenyl)2SiMe2ZrCl2, respectively.
How effective are QSPR predictions?
Catalysts Ty1 and Ty2 were “designed” based on a QSPR model built using many standard metallocene catalysts, none of which had any “exotic” substituents like the triptycyls discussed here.13,31 It was clear from the start that this reported model would not be capable of producing quantitatively accurate predictions for these new catalysts (nor for the remaining species Ty3–Ty8). However, it was good enough to guide us in the synthesis of the metallocenes Ty1 and Ty2 which showed unprecedented high-temperature performance.To understand if it was just a lucky coincidence or the model, although based on incomplete data set structures, can nevertheless help prioritize the search for new catalyst types, it is instructive to confront the experimental data for these triptycyl-bearing catalysts with the relevant predictions (Table 3).
| Catalyst | 1 − σ, % | regio tot % | M n, kDa | |||
|---|---|---|---|---|---|---|
| Obs. | Pred. | Obs. | Pred. | Obs. | Pred. | |
| Ty1 | ≤0.02 | ≤0.02 | 0.26 | 0.11 | 1100 | 2700 |
| Ty2 | 0.03 | ≤0.02 | 0.18 | 0.08 | 1900 | 2500 |
| Ty3 | 0.03 | ≤0.02 | 0.11 | 0.19 | 1400 | 2200 |
| Ty4 | ≤0.02 | ≤0.02 | 0.19 | 0.06 | 880 | 4400 |
| Ty5 | 0.05 | ≤0.02 | 1.0 | 0.1 | 60 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Ty6 | 1.2 | 1.75 | 1.35 | 1.82 | 40 | 580 |
| Ty7 | 0.43 | 0.92 | 7.9 | 22.8 | 25 | 350 |
| Ty8 | 0.03 | 0.04 | 1.32 | 0.77 | 430 | 2100 |
Predictions for catalysts Ty1–Ty2 and Ty8, bearing some resemblance to the catalysts used to build the QSPR models, are in good agreement with the observed experimental performance, especially for stereoselectivity. Regioselectivity (measured as regiotot, i.e. the total sum of all regioerrors) and molar mass capability, in terms of number average molecular weight, Mn, deviate up to a factor of 2–4 from the predictions, which isn't much in terms of absolute energy differences (RTln2 = 0.5 kcal mol−1 at 333 K). Moreover, for these catalysts the reactions are very challenging to control at such a low polymerization temperature, and the uncertainty in the experimental data for molar mass capability is therefore substantial.40
Conversely Ty3–Ty7 incorporate more drastic changes, for which no training was done whatsoever, e.g., changes in central metal or bridging element, and lack of 2-R or 4-R substituent. However, stereoselectivity predictions are in relatively good agreement with experiment, with deviations not exceeding a factor of ≈2.
Qualitatively, predictions for Ty3 (Hf) and Ty4 (Ge) regioselectivity and molar mass capability are good; however, the predicted direction of the Hf-effect (decreasing regioselectivity) is wrong,40 as is the Ge-effect (no effect observed but regioselectivity increase predicted). This isn't surprising: the models were build relying exclusively on silyl bridged zirconocenes. In the grand scheme, this calls into question the applicability of the electronic descriptor e−NPA,MCl2 (NPA charge on the MCl2 fragment) for across-the-board models.
The substituent pattern changes in Ty5 (missing 2-R substituent) and Ty6 (missing 4-R substituent) challenge especially the molar mass capability model. Molar mass capability is overestimated by a factor of 220 for Ty5 and a factor of 15 for Ty6. The linear QSPR models we have developed treat substituent effects as additive, essentially reflecting the gradual evolution in catalyst development. For example, since the discovery of the 2-Me effect,54 there was no reason to go back to 2-H systems. The disagreements could indicate that only the simultaneous steric pressure from two sides induces effects, for that without one of the “anchors”, the space demanding β-hydrogen transfer to monomer TS can simply escape in one direction. From a model building perspective, this indicates that “if/then” situations might exist and should be incorporated into the models. Alternatively, electronic factors could dominate for these catalysts which are unimportant in the training set and therefore not considered in the molar mass QSPR model. Indeed, Ty5 is much less electron-poor than the other Ty catalysts and the catalysts in the training set of the QSPR model (e−NPA,MCl2 0.343 vs. 0.38–0.42 for other Ty catalysts and metallocenes in the training set).
The 7-Me substituents in Ty7, not present in the original ansa-zirconocene training set, significantly distort the catalyst backbone, something not present in the original ansa-zirconocene training set. The molar mass capability model fails for Ty7 but the regioselectivity model reproduces the very low regioselectivity fairly well (22.8 vs. 8.0 mol% regiotot), considering that the performance is significantly outside of the model training set (max. 1.4 mol%). Importantly, based on the model we can trace the origin of this peculiar regioselectivity to an increased steric bulk in the open quadrant originating from the distortion of the relative positions of the indenyl fragments (Fig. 3) due to the 7-Me substituents.
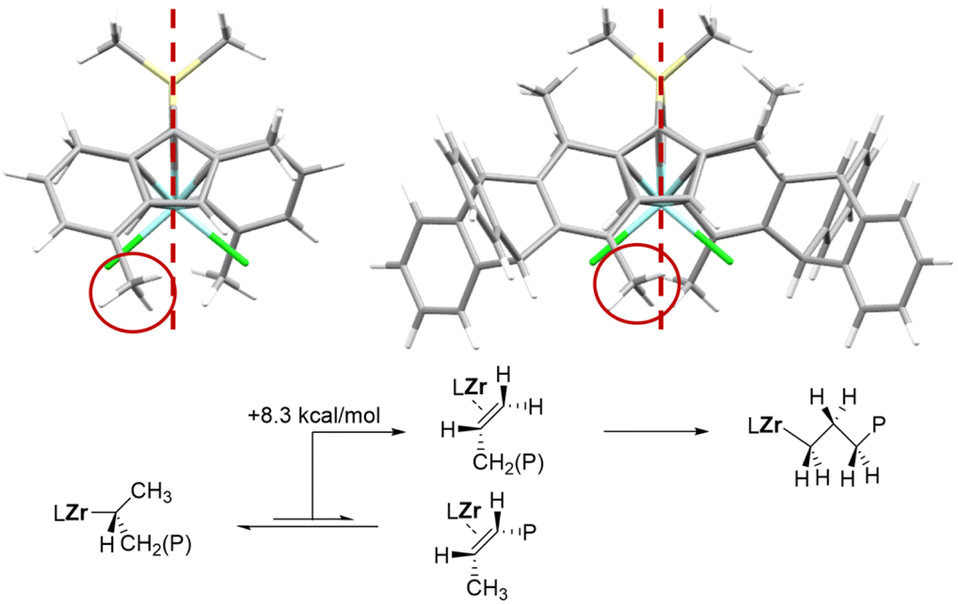 | ||
| Fig. 3 Top: change in relative orientation of the indenyl fragments repositions the 4-Me substituents. Comparison of rac-Me2Si(2-Me-4-Me-indenyl)2ZrCl2 and Ty7. The resulting interference with 1,2 transition states is responsible for the low regioselectivity of Ty7. Red dashed lines indicate the middle plane of the active pocket, separating the two active sites. Bottom: the two possible β-hydrogen eliminations after 2,1 insertion in Ty7. | ||
Due to the backbone distortion, the 4-Me substituent moves partially into the open quadrant interfering with the 1,2-insertion transition state, while 2,1 insertion transition state is almost unaffected. Salan-type catalysts with very bulky ortho-substituents show decreased regioselectivity for similar reasons.
As a side note, the isomerization of 2,1 to 3,1 units requires β-hydrogen elimination from the CH3 unit, rotation, and reinsertion.2 DFT calculations (for details see ESI†) indicate that unproductive β-hydrogen elimination from the CH2 unit is preferred over elimination from the CH3 unit leading to 3,1-isomerization by ΔΔG‡ = 8.1 kcal mol−1, explaining why for Ty7 “all” regioerrors are present the form of head-to-head/tail-to-tail enchainments.
Interestingly, the QSPR predictions for the copolymerization reactivity ratios rE and rH are much more effective (see ESI†). However, as some of us have previously shown, likely, insertion is not rate limiting in most of these cases and changes in the active pocket therefore have much smaller, if any, effects on these parameters.17,20
Conclusions
In conclusion, we report on the synthesis of several exotic triptycyl complexes and on their performance in propene polymerization and ethene/1-hexene copolymerization. Installing a triptycene motif in position 5,6 deteriorates the catalyst performance in propene polymerization with respect to the 4,5 analogues, but not in ethene/1-hexene copolymerization. The comparison between experimental results and predictions based on QSPR models built using standard metallocene catalysts shows that, as expected, the main performance indicators are not always quantitatively reproduced. However, this shouldn't be surprising; several of the tested triptycene catalyst features are far outside the original training set such as metal changes, missing R-substituents, or significant distortions. However, the models can qualitatively predict the performance for modestly “extrapolating” candidate catalysts. This is the case, for example for Ty7, producing an isotactic polypropylene with an unprecedent high level of 2,1-insertions.The continuing interest in this topic ensures we will keep on improving our understanding of the intimate details of olefin polymerization. QSPR type model building can be a useful tool in this effort, in favourable cases pointing us to interesting effects we might not have found solely through inspired substituent choices. To broaden the applicability of QSPR type models, experimental databases need to be grown that also cover unusual substituent patterns. Importantly, metal changes are ill reproduced by current models, indicating that electronic descriptors need to be identified that describe electronic effects of the ligand framework and differences in central metal equally well. The development of improved models applicable to more than one catalyst class/central metal will critically depend on the identification of such descriptors.
Experimental
Inorganic synthesis
The synthesis of zirconocenes Ty4–Ty8 is detailed in the ESI.† The synthesis of Ty1,13Ty2,13 and Ty340 has been reported before. Starting compounds 1-(bromomethyl)triptycene,13 2-ethylcyclopenta[a]triptycene (2),13 6-amino-2-methylindanone (8),60 were prepared according to literature procedures. Ethereal solvents were distilled from sodium/benzophenone. Toluene and hexane were sparged with argon and dried over molecular sieves 4 A. Other reagents were purchased from commercial sources and used as received. All manipulations with compounds, which are sensitive to moisture and air, were performed either in an atmosphere of argon using a standard Schlenk technique or in an inert atmosphere (Ar) of glove box (MBraun). GC/MS analysis was performed on Agilent Technologies 7890A paired with Agilent Technologies 5975C MSD GC/MS system and on Agilent Technologies 8890 GC/5977C MSD system. High-resolution mass spectra (HRMS) were recorded on an Agilent Technologies 6530 Q-TOF LC/MS system paired with Agilent 1260 HPLC and using Agilent JetStream ion source. NMR spectra were recorded on Bruker AVANCE (400 MHz) or Agilent Technologies 400-MR (400 MHz) NMR spectrometers and can be found in the ESI.† C, H, N microanalyses were done using PerkinElmer 2400 Series II CHNS/O elemental analyzer.X-ray crystallography
X-ray experiments were carried out using Bruker D8 Quest with Photon III detector diffractometer (λ(Mo-Kα) = 0.71073 Å, graphite monochromator, ω-scans) at 100 K. The structure was solved by direct methods and refined by the full-matrix least-squares procedure in anisotropic approximation for non-hydrogen atoms. All hydrogen atoms were placed in geometrically calculated positions and included in the refinement using riding model. The details of data collection and crystal structure refinement for which we used SAINT Plus,61 SADABS62 and SHELXL-2018/363 program packages, are summarized in Table S1.† The crystallographic data for Ty7-Cl2 has been deposited with the Cambridge Crystallographic Data Center, CCDC 2308227† (https://www.ccdc.cam.ac.uk). For further details, see the ESI.†Polymerization experiments and polymer characterization
Prior to the execution of a polymerization library, the PPR modules undergo ‘bake-and-purge’ cycles overnight (8 h at 90–140 °C with intermittent dry N2 flow), to remove any contaminants and left-overs from previous experiments. After cooling to glovebox temperature, the module stir tops are taken off, and the 48 cells are fitted with disposable 10 mL glass inserts (pre-weighed in a Mettler-Toledo Bohdan Balance Automator) and stir paddles (see Table S1 for details†). The stir tops are then set back in place, and N2 in the reactors is replaced with propene or ethene (ambient pressure). The cells are then loaded with the appropriate amounts of solvent containing TIBA as a scavenger (see Table S1 for details). For ethene/1-hexene copolymerization experiments, the desired amount of 1-hexene (Table S2.2†) is injected into the cells.
The system is then thermostated at the desired polymerization temperature and pressurized with monomer (ethene or propene, see Table S1†). At this point, the catalyst injection sequence is started; aliquots of (a) a solvent ‘chaser’, (b) a toluene solution of catalyst (variable amount, see Table S2.1 and S2.2†), (c) a toluene solution of the activator trityl borate or anilinium borate, variable ratio, see Table S2.1 and S2.2,† and (d) a solvent ‘buffer’, all separated by nitrogen gaps, are uploaded into the needle and subsequently injected into the destination cell in reverse order, thus starting the reaction. The pre-catalysts were injected into the PPR cells without pre-activation. The polymerization is left to proceed under stirring (800 rpm) at constant temperature and pressure with feed of gaseous monomer on demand until the desired monomer consumption has been reached (for reaction time, see Table S2.1 and S2.2†), and quenched by over-pressurizing the cell with 50 psi (3.4 bar) of dry air (preferred over other possible catalyst quenchers because in case of cell or quench line leakage oxygen is promptly detected by the dedicated glove-box sensor).
Computational and modelling details
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Lomonosov Moscow State University Program of Development for opportunity to use NMR equipment, and Dr K. A. Lyssenko for his assistance with the X-ray structure determination analysis.References
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1346 CrossRef CAS PubMed.
- V. Busico and R. Cipullo, Prog. Polym. Sci., 2001, 26, 443–533 CrossRef CAS.
- V. Busico and R. Cipullo, J. Am. Chem. Soc., 1994, 116, 9329–9330 CrossRef CAS.
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS.
- M. K. Leclerc and H. H. Brintzinger, J. Am. Chem. Soc., 1995, 117, 1651–1652 CrossRef CAS.
- J. A. Ewen, J. Am. Chem. Soc., 1984, 106, 6355–6364 CrossRef CAS.
- J. A. Ewen, J. Mol. Catal. A: Chem., 1998, 128, 103–109 CrossRef CAS.
- V. Busico and R. Cipullo, J. Organomet. Chem., 1995, 497, 113–118 CrossRef CAS.
- M. Toto, L. Cavallo, P. Corradini, G. Moscardi, L. Resconi and G. Guerra, Macromolecules, 1998, 31, 3431–3438 CrossRef CAS.
- L. Resconi, F. Piemontesi, I. Camurati, O. Sudmeijer, I. E. Nifant'ev, P. V. Ivchenko and L. G. Kuz'mina, J. Am. Chem. Soc., 1998, 120, 2308–2321 CrossRef CAS.
- G. Talarico, V. Busico and L. Cavallo, J. Am. Chem. Soc., 2003, 125, 7172–7173 CrossRef CAS PubMed.
- C. Ehm, A. Vittoria, G. P. Goryunov, P. S. Kulyabin, P. H. M. Budzelaar, A. Z. Voskoboynikov, V. Busico, D. V. Uborsky and R. Cipullo, Macromolecules, 2018, 51, 8073–8083 CrossRef CAS.
- P. S. Kulyabin, G. P. Goryunov, M. I. Sharikov, V. V. Izmer, A. Vittoria, P. H. M. Budzelaar, V. Busico, A. Z. Voskoboynikov, C. Ehm, R. Cipullo and D. V. Uborsky, J. Am. Chem. Soc., 2021, 143, 7641–7647 CrossRef CAS PubMed.
- V. Busico, R. Cipullo, F. Cutillo, N. Friederichs, S. Ronca and B. Wang, J. Am. Chem. Soc., 2003, 125, 12402–12403 CrossRef CAS PubMed.
- G. Urciuoli, F. Zaccaria, C. Zuccaccia, R. Cipullo, P. H. M. Budzelaar, A. Vittoria, C. Ehm, A. Macchioni and V. Busico, Polymers, 2023, 15, 1378 CrossRef CAS PubMed.
- D. V. Uborsky, D. Y. Mladentsev, B. A. Guzeev, I. S. Borisov, A. Vittoria, C. Ehm, R. Cipullo, C. Hendriksen, N. Friederichs, V. Busico and A. Z. Voskoboynikov, Dalton Trans., 2020, 49, 3015–3025 RSC.
- C. Ehm, A. Vittoria, G. P. Goryunov, V. V. Izmer, D. S. Kononovich, O. V. Samsonov, P. H. M. Budzelaar, A. Z. Voskoboynikov, V. Busico, D. V. Uborsky and R. Cipullo, Dalton Trans., 2020, 49, 10162–10172 RSC.
- N. Friederichs, B. Wang, P. H. M. Budzelaar and B. B. Coussens, J. Mol. Catal. A: Chem., 2005, 242, 91–104 CrossRef CAS.
- F. Zaccaria, C. Ehm, P. H. M. Budzelaar and V. Busico, ACS Catal., 2017, 7, 1512–1519 CrossRef CAS.
- F. Zaccaria, R. Cipullo, P. H. M. Budzelaar, V. Busico and C. Ehm, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2807–2814 CrossRef CAS.
- C. Ehm, P. H. M. Budzelaar and V. Busico, J. Catal., 2017, 351, 146–152 CrossRef CAS.
- C. Ehm, R. Cipullo, M. Passaro, F. Zaccaria, P. H. M. Budzelaar and V. Busico, ACS Catal., 2016, 6, 7989–7993 CrossRef CAS.
- F. Zaccaria, C. Ehm, P. H. M. Budzelaar, V. Busico and R. Cipullo, Organometallics, 2018, 37, 2872–2879 CrossRef CAS.
- F. Zaccaria, C. Zuccaccia, R. Cipullo, P. H. M. Budzelaar, A. Macchioni, V. Busico and C. Ehm, Organometallics, 2018, 37, 4189–4194 CrossRef CAS.
- R. Cipullo, P. Melone, Y. Yu, D. Iannone and V. Busico, Dalton Trans., 2015, 44, 12304–12311 RSC.
- P. A. Wilson, M. H. Hannant, J. A. Wright, R. D. Cannon and M. Bochmann, Macromol. Symp., 2006, 236, 100–110 CrossRef CAS.
- M. Bochmann, Organometallics, 2010, 29, 4711–4740 CrossRef CAS.
- D. V. Uborsky, M. I. Sharikov, G. P. Goryunov, K. M. Li, A. Dall'Anese, C. Zuccaccia, A. Vittoria, T. Iovine, G. Galasso, C. Ehm, A. Macchioni, V. Busico, A. Z. Voskoboynikov and R. Cipullo, Inorg. Chem. Front., 2023, 10, 6401–6406 RSC.
- G. Antinucci, B. Dereli, A. Vittoria, P. H. M. Budzelaar, R. Cipullo, G. P. Goryunov, P. S. Kulyabin, D. V. Uborsky, L. Cavallo, C. Ehm, A. Z. Voskoboynikov and V. Busico, ACS Catal., 2022, 12, 6934–6945 CrossRef CAS.
- A. Poater and L. Cavallo, Dalton Trans., 2009, 8885–8890 RSC.
- C. Ehm, A. Vittoria, P. G. Goryunov, V. V. Izmer, S. D. Kononovich, V. O. Samsonov, R. Di Girolamo, H. M. P. Budzelaar, Z. A. Voskoboynikov, V. Busico, V. D. Uborsky and R. Cipullo, Polymers, 2020, 12, 1005–1005 CrossRef CAS PubMed.
- C. Ehm, A. Vittoria, G. P. Goryunov, V. V. Izmer, D. S. Kononovich, P. S. Kulyabin, R. Di Girolamo, P. H. M. Budzelaar, A. Z. Voskoboynikov, V. Busico, D. V. Uborsky and R. Cipullo, Macromolecules, 2020, 53, 9325–9336 CrossRef CAS.
- C. Ehm, P. H. M. Budzelaar and V. Busico, Eur. J. Inorg. Chem., 2017, 2017, 3343–3349 CrossRef CAS.
- C. I. Branden and J. Tooze, Introduction to Protein Structure, Garland Publishing, New York, 1990–1991 Search PubMed.
- J. Kyte, Structure in Protein Chemistry, Garland Publishing, New York, 1995 Search PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- A. Schöbel, E. Herdtweck, M. Parkinson and B. Rieger, Chem. – Eur. J., 2012, 18, 4174–4178 CrossRef PubMed.
- M. R. Machat, D. Lanzinger, A. Pöthig and B. Rieger, Organometallics, 2017, 36, 399–408 CrossRef CAS.
- G. P. Goryunov, M. I. Sharikov, A. N. Iashin, J. A. M. Canich, S. J. Mattler, J. R. Hagadorn, D. V. Uborsky and A. Z. Voskoboynikov, ACS Catal., 2021, 11, 8079–8086 CrossRef CAS.
- A. Vittoria, G. P. Goryunov, V. V. Izmer, D. S. Kononovich, O. V. Samsonov, F. Zaccaria, G. Urciuoli, P. H. M. Budzelaar, V. Busico, A. Z. Voskoboynikov, D. V. Uborsky, C. Ehm and R. Cipullo, Polymers, 2021, 13, 2621 CrossRef CAS PubMed.
- C. Cobzaru, S. Hild, A. Boger, C. Troll and B. Rieger, Coord. Chem. Rev., 2006, 250, 189–211 CrossRef CAS.
- T. Fillebeen, T. Hascall and G. Parkin, Inorg. Chem., 1997, 36, 3787–3790 CrossRef CAS PubMed.
- C. Azerraf, O. Grossman and D. Gelman, J. Organomet. Chem., 2007, 692, 761–767 CrossRef CAS.
- O. Grossman, C. Azerraf and D. Gelman, Organometallics, 2006, 25, 375–381 CrossRef CAS.
- A. Kaps and H. Plenio, Eur. J. Inorg. Chem., 2023, 26, e202300178 CrossRef CAS.
- S. Barman, E. A. Jaseer, N. Garcia, M. Elanany, M. Khawaji, W. Xu, S. Lin, H. Alasiri, M. N. Akhtar and R. Theravalappil, Chem. Commun., 2022, 58, 10044–10047 RSC.
- P. S. Kulyabin, V. V. Izmer, G. P. Goryunov, M. I. Sharikov, D. S. Kononovich, D. V. Uborsky, J. A. M. Canich and A. Z. Voskoboynikov, Dalton Trans., 2021, 50, 6170–6180 RSC.
- W. Kaminsky, O. Rabe, A. M. Schauwienold, G. U. Schupfner, J. Hanss and J. Kopf, J. Organomet. Chem., 1995, 497, 181–193 CrossRef CAS.
- J.-C. Buffet, T. A. Q. Arnold, Z. R. Turner, P. Angpanitcharoen and D. O'Hare, RSC Adv., 2015, 5, 87456–87464 RSC.
- V. Murphy, X. Bei, T. R. Boussie, O. Brümmer, G. M. Diamond, C. Goh, K. A. Hall, A. M. Lapointe, M. Leclerc, J. M. Longmire, J. A. W. Shoemaker, H. Turner and W. H. Weinberg, Chem. Rec., 2002, 2, 278–289 CrossRef CAS PubMed.
- T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe, M. Leclerc, C. Lund, V. Murphy, J. A. W. Shoemaker, U. Tracht, H. Turner, J. Zhang, T. Uno, R. K. Rosen and J. C. Stevens, J. Am. Chem. Soc., 2003, 125, 4306–4317 CrossRef CAS PubMed.
- C. Ehm, A. Mingione, A. Vittoria, F. Zaccaria, R. Cipullo and V. Busico, Ind. Eng. Chem. Res., 2020, 59, 13940–13947 CrossRef CAS.
- W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907–3945 CrossRef CAS.
- U. Stehling, J. Diebold, R. Kirsten, W. Roell, H. H. Brintzinger, S. Juengling, R. Muelhaupt and F. Langhauser, Organometallics, 1994, 13, 964–970 CrossRef CAS.
- I. E. Nifant'ev, P. V. Ivchenko, V. V. Bagrov, A. V. Churakov and P. Mercandelli, Organometallics, 2012, 31, 4962–4970 CrossRef.
- K. Reichelt, M. Parkinson and L. Resconi, Macromol. Chem. Phys., 2016, 217, 2415–2430 CrossRef CAS.
- M. S. Kuklin, V. Virkkunen, P. M. Castro, L. Resconi and M. Linnolahti, Eur. J. Inorg. Chem., 2015, 2015, 4420–4428 CrossRef CAS.
- D. Tranchida and L. Resconi, Polym. Cryst., 2018, 1, e10022 Search PubMed.
- A. Vittoria, A. Meppelder, N. Friederichs, V. Busico and R. Cipullo, ACS Catal., 2017, 7, 4509–4518 CrossRef CAS.
- E. Alcalde, N. Mesquida, J. Frigola, S. López-Pérez and R. Mercè, Org. Biomol. Chem., 2008, 6, 3795–3810 RSC.
- SAINT Plus. Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. Vittoria, V. Busico, F. D. Cannavacciuolo and R. Cipullo, ACS Catal., 2018, 8, 5051–5061 CrossRef CAS.
- A. Vittoria, A. Mingione, R. A. Abbate, R. Cipullo and V. Busico, Ind. Eng. Chem. Res., 2019, 58, 14729–14735 CrossRef CAS.
- A. Vittoria, A. Meppelder, N. Friederichs, V. Busico and R. Cipullo, ACS Catal., 2020, 10, 644–651 CrossRef CAS.
- C. Ehm, P. H. M. Budzelaar and V. Busico, J. Organomet. Chem., 2015, 775, 39–49 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed.
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2006, 125, 074110 CrossRef PubMed.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- P. Schwerdtfeger, ChemPhysChem, 2011, 12, 3143–3155 CrossRef CAS PubMed.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- C. Ehm, R. Cipullo, P. H. M. Budzelaar and V. Busico, Dalton Trans., 2016, 45, 6847–6855 RSC.
- F. D. Cannavacciuolo, R. Yadav, A. Esper, A. Vittoria, G. Antinucci, F. Zaccaria, R. Cipullo, P. H. M. Budzelaar, V. Busico, G. P. Goryunov, D. V. Uborsky, A. Z. Voskoboynikov, K. Searles, C. Ehm and A. S. Veige, Angew. Chem., Int. Ed., 2022, e202202258 CAS.
- J. L. Whitten, J. Chem. Phys., 1973, 58, 4496 CrossRef CAS.
- E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41–51 CrossRef CAS.
- M. Feyereisen, G. Fitzgerald and A. Komornicki, Chem. Phys. Lett., 1993, 208, 359–363 CrossRef CAS.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valence and Bonding - A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, UK, 2005 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev. Comput. Mol. Sci, 2012, 2, 1–42 Search PubMed.
- Theoretical Chemistry Institute, ed. E. D. Glendening, A. E. Reed, J. E. Carpenter, F. Weinhold and NBO 3.1, University of Wisconsin, Madison, WI, 2001 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of proligands, NMR spectra of synthetic intermediates and metal complexes, full polymerization results, details for the QSPR models and DFT calculations. CCDC 2308227. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01170h |
| ‡ Deceased |
| This journal is © The Royal Society of Chemistry 2024 |