
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular engineering of 3-arylated tetrazo[1,2-b]indazoles: divergent synthesis and structure–property relationships†
Asmae
Bousfiha‡
a,
Oumaima
Abidi‡
a,
Louis
Lemetayer
 b,
Navya
Sood
a,
Iogann
Tolbatov
a,
Fabien
Barrois
a,
Ahmad
Daher
a,
Hélène
Cattey
a,
Marie
Cordier
b,
Muriel
Hissler
b,
Jean-Cyrille
Hierso
a,
Paul
Fleurat-Lessard
*a,
Pierre-Antoine
Bouit
*b and
Julien
Roger
*a
b,
Navya
Sood
a,
Iogann
Tolbatov
a,
Fabien
Barrois
a,
Ahmad
Daher
a,
Hélène
Cattey
a,
Marie
Cordier
b,
Muriel
Hissler
b,
Jean-Cyrille
Hierso
a,
Paul
Fleurat-Lessard
*a,
Pierre-Antoine
Bouit
*b and
Julien
Roger
*a
aInstitut de Chimie Moléculaire de l'Université de Bourgogne, UMR 6302 – Université Bourgogne (UB), 9, Avenue Alain Savary, 21078 Dijon, France. E-mail: julien.roger@u-bourgogne.fr; Paul.Fleurat-Lessard@u-bourgogne.fr
bCNRS, ISCR – UMR 6226, Univ. Rennes, 35000 Rennes, France. E-mail: pierre-antoine.bouit@univ-rennes1.fr
First published on 31st May 2024
Abstract
The synthetic scope of 3-arylated tetrazo[1,2-b]indazoles is reported based on a Pd-catalyzed Liebeskind–Srogl cross-coupling reaction followed by an N-cyclisation process. The reactivity of the nitrogen atoms was used to further diversify these N-rich polyaromatic tetrazo[1,2-b]indazoles in a panel of reactions (protonation, selective oxidation, metallations). Selective ortho-C–H activation/functionalization on the heterocycle was also demonstrated with three transition metals (TM = Pd, Ir and Rh). The effects of all these molecular engineering strategies, particularly the N-modifications, on the optical and redox properties of the 3-arylated tetrazoindazoles were studied experimentally and theoretically. This study highlights the diversity of molecular structures and electronic properties offered by the tetrazo[1,2-b]indazole platform.
Introduction
Heterocycles containing nitrogen atoms are key building blocks for various applications ranging from biomedicine and agrochemistry to functional materials (explosives, optoelectronics). In the case of the latter application, aza-aromatics have shown great potential. Indeed, the presence of nitrogen atoms makes those compounds more electron-deficient than their all carbon-based counterparts and gives them excellent electron affinity. In addition, the specific reactivity of the nitrogen atom can be utilised to fine-tune their optical/redox properties at the molecular level, as well as those of the corresponding molecular materials.1 Nitrogen-containing heteroaromatics, including among many others pyrazino[2,3-b:5,6-b′]diindolizines (A, Scheme 1) and azapyridazines (B, Scheme 1) have been developed for applications in optoelectronics or bioimaging.2 However, in this field, the development of efficient synthetic approaches for innovative building blocks remains highly desirable, notably for safety and more atom-economy reasons. In this context, we have recently developed an unprecedented synthesis of tetrazo[1,2-b]indazoles (TzIn, C and D, Scheme 1) through the N–N cyclization of azido-functionalized s-tetrazines (Scheme 1, bottom).3,4 These compounds exhibit a wide range of physico-chemical properties, depending on their molecular structure: low reduction potential, UV-vis absorption up to the near-infrared and intense fluorescence. However, this procedure gave limited access to structurally diverse tetrazo[1,2-b]indazoles D. This was due to the use of Pinner type synthesis to obtain the key fluoro-functionalized tetrazines (Tz) E (Scheme 1). Indeed, Pinner-type synthesis gives limited yields of ortho-functionalized products because of steric hindrance and side-reactivities especially when fluorine atoms are needed in ortho-position.5 Moreover, the synthesis of unsymmetrical Tz in the presence of two different benzonitrile derivatives also furnished their corresponding symmetrical s-tetrazines. To overcome this issue, we envisaged a two steps strategy from 3-thiomethyltetrazines F (Scheme 1) as the key intermediate. This strategy allows for the divergent synthesis of arylated tetrazo[1,2-b]indazoles by employing a Pd-catalyzed Liebeskind–Srogl cross-coupling followed by a well-controlled N-cyclisation process. | ||
| Scheme 1 N-rich heteroaromatics (top) and strategic routes to dissymmetric 3-aryltetrazo[1,2-b]indazoles (bottom, this work). | ||
We report the synthesis and characterization of a library of 3-arylated tetrazoindazoles. We explore the N-reactivity in various reactions (protonation, oxidation, coordination with transition metals). Furthermore, we demonstrate the selective ortho-C–H activation/functionalization on the TzIn scaffold using these TMs (Pd, Ir and Rh). Finally, we investigated the impact of these molecular engineering strategies on the optical and redox properties of these TzIn both experimentally and theoretically using density functional theory (DFT).
Results and discussion
Synthetic route to 3-aryltetrazo[1,2-b]indazoles
The recently disclosed heteroaromatic N-rich tetrazo[1,2-b]indazole is accessible from ortho-fluorinated aryl-s-tetrazine by a cascade azidation and intramolecular cyclisation process (Scheme 1, bottom).3 Herein, we developed the access to diverse 3-arylated TzIn by a new synthetic route taking advantage of the divergent method reported by Fox et al. for the synthesis of unsymmetrical Tz.6 In this approach, the key step is the Pd-catalyzed Liebeskind–Srogl cross-coupling on the 3-(thiomethyl)-6-(2-fluorophenyl)-tetrazine F (Scheme 1). In this key synthon F, the fluorine function, used for nucleophilic azidation substitution is introduced in the early steps of the synthesis. The pallado-catalyzed Liebeskind–Srogl cross-coupling reaction allowed the introduction of different aryls (Scheme 2).7 The reaction afforded moderate to high isolated yields of 61–95% for 1–4 with para substituents (H, OMe, tBu and NPh2), while the meta-OMe 5 gave a 55% isolated yield. The symmetrical bis-3,6-(2-fluorophenyl)tetrazine 6 was obtained in the low isolated yield of 17% compared to our previous synthetic route, confirming that such a strategy is only versatile for preparing unsymmetrical building blocks.5 As previously observed by Fox and coworkers,6,7e the bulky polyaromatic hydrocarbons and heteroarenes were found more challenging with lower conversions (Scheme 2). The 1-naphthyl and 9-phenanthryl were coupled in 32% and 53% yields for 7 and 8, respectively. Finally, the 2-pyridenyl, 3-thienyl and 2-thienyl provided 9–11 in 22%, 50% and 23% isolated yields, respectively. | ||
| Scheme 2 Palladium-catalyzed Liebeskind–Srogl cross-coupling reaction for the synthesis of 3-aryl-6-(2-fluorophenyl)tetrazines. | ||
The 3-arylated tetrazo[1,2-b]indazoles 12–22 were synthesized in the presence of 3 equivalents of NaN3 in DMF using microwave irradiation (μW) for one hour at 130 °C (Scheme 3), following our previously reported procedure.3 Under these conditions, the 3-phenyl-tetrazo[1,2-b]indazole 12 was isolated in 44% yield. The presence of donating substituents in the para (OMe, tBu and NPh2) or meta positions (OMe) improved the cyclization process slightly with 52%, 64%, 55% and 47% isolated yields for 13–16, respectively. To favor the formation of the monocyclic 3-(2-fluorophenyl)-tetrazo[1,2-b]indazole 17 and limit the formation of the bis-tetrazo[1,2-b]indazoles,5 the amount of sodium azide was reduced to only 1 equivalent to allow the formation of 17 in 47% isolated yield. The polyaromatic-substituted tetrazo[1,2-b]indazoles 18 and 19 were obtained in 41% and 64% isolated yields, respectively, after modifying the cyclization protocol for the 9-phenanthryl derivative (1.1 equivalents of NaN3 at 110 °C). The 3-heteroarylated tetrazo[1,2-b]indazoles 20–22 were isolated in 59%, 24% and 36% isolated yields, respectively, principally due to a lack of reactivity and solubility.
 | ||
Scheme 3 Synthesis of the corresponding 3-aryl-tetrazo[1,2-b]indazoles. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 equiv. NaN3; b1.1 equiv. NaN3 at 110 °C. 1 equiv. NaN3; b1.1 equiv. NaN3 at 110 °C. | ||
This synthetic methodology, which combines a Pd-catalyzed Liebeskind–Srogl cross-coupling with an N3-induced cyclization process, has significantly expanded the library of tetrazo[1,2-b]indazoles that was limited to poorly functionalized systems.3
Reactivity of the nitrogen core
The 3-(2-fluorophenyl)-tetrazo[1,2-b]indazole (17) was selected as a model to study the reactivity of these substrates (Schemes 4–7). First the reaction with acid was tested. Protonation of 17 using an excess of trifluoroacetic acid in CDCl3 was evidenced by 1H and 19F NMR (see the ESI, Fig. S-3 and S-4†), illustrating that 17 is indeed prone to protonation. According to our DFT computation (see the ESI, Table S-Th1†), 17 should be protonated only on nitrogen N1 (see Scheme 4 for numbering). Then N-oxide formation was tested, as such reactivity is characteristic of aromatic amino-heterocycles.8 In the presence of an excess of trifluoroacetic anhydride and H2O2, the oxidation of 17 led to the formation of two products in a 50/50 ratio. X-ray diffraction allowed those products to be assigned to the derivatives where the N-oxidation had occurred selectively at N1 (17-O1) and N2 (17-O2) with 35% and 32% isolated yields (Scheme 4).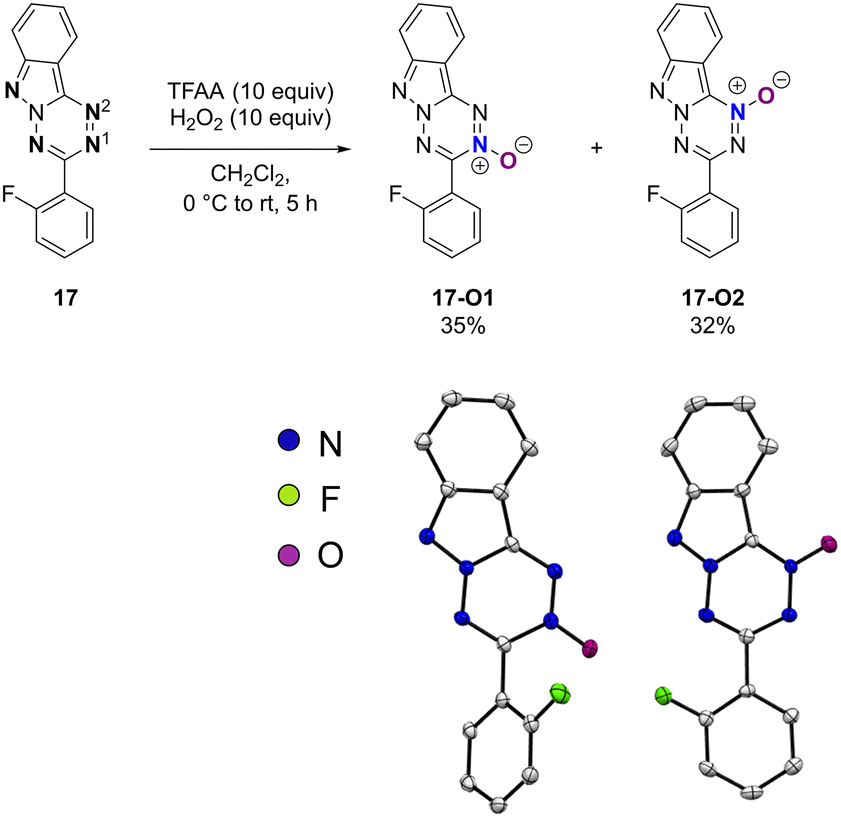 | ||
| Scheme 4 N-oxide formation from 17. Molecular structures of 17-O1 (left) and 17-O2 (right) (thermal ellipsoids at 30% probability, H atoms and CH2Cl2 are omitted for clarity). | ||
 | ||
| Scheme 5 Palladacycle synthesis with the 3-(2-fluorophenyl)-tetrazo[1,2-b]indazole (17). Only one isomer for Pd is represented for clarity. | ||
 | ||
| Scheme 6 Iridacycle and rhodacycle synthesis with the 3-(2-fluorophenyl)-tetrazo[1,2-b]indazole (17). Molecular structures of Ir (left) and Rh (right) (thermal ellipsoids at 30% probability, H atoms and CH2Cl2 are omitted for clarity). | ||
 | ||
| Scheme 7 N-directed palladium-catalyzed C–H bond functionalization of 17. Conversion based on the consumption of 17 determined by 1H and 19F NMR (CDCl3, 500 MHz and 470 MHz respectively), isolated yields in brackets. | ||
Selective synthesis of metallacycles (M = Pd, Ir and Rh) from 17
We previously demonstrated that the use of the N-tetrazoindazole core as a directing group allows for N-directed palladium-catalyzed halogenation/acetoxylation reactions.3 Here we unambiguously show that the functionalization occurs with full selectivity on the ortho-position despite the presence of four different nitrogen atoms (Scheme 5, in color) which could potentially direct the C–H bond activation at three different ortho-positions (Scheme 5, H1, H2 or H5). To clarify the origin of this selectivity trend, the formation of the metallacycle was studied computationally and experimentally.9,10The dimeric palladacycle Pd2 was isolated in 75% yield by precipitation after reaction of [Pd(OAc)2] in HOAc (Scheme 5). Our attempts to crystallize the corresponding complex were unsuccessful, so we envisaged synthesizing the monomeric palladacycle by a ligand exchange (with chloride) followed by the addition of pyridine. Based on 19F and 1H NMR spectroscopies in solution and the X-ray analysis in the solid state, we identified the product as a mixture of two cis/trans isomeric complexes with an 85/15% ratio (Pd) in favor of the coordination of the palladium at the N1 and at the C1 of the fluorinated aryl with the pyridine ligand in the cis position with respect to the tetrazine core (Scheme 5 and Fig. 1).
 | ||
| Fig. 1 Molecular structure of Pd (thermal ellipsoids at 30% probability, H atoms and one CH2Cl2 are omitted for clarity). | ||
The iridacycle Ir and rhodacycle Rh were synthesized from the corresponding [MCl2Cp*]2 (M = Ir or Rh) precursor, using KOAc as the base in MeOH with 33% and 31% isolated yields, respectively (Scheme 6, top).11 The X-ray diffraction study of the corresponding metallacycles unambiguously confirmed the coordination mode at N1 and C1 (Scheme 6, bottom).
A computational study was then conducted to understand the high selectivity of the metallation. The free energies of palladacycles formed at various nitrogens were computed at the B3LYP/6-311++G(2df,2pd) level. The relative energies for the formed palladacycles Pd(TzInd)(Cl)(pyridine) were 0.0 kcal mol−1 for N1cis and 0.6 kcal mol−1 for N1trans, 9.3 for N3cis and 12.5 for N3trans, and more than 24.5 kcal mol−1 for the other nitrogen atoms (Fig. 2, see Fig. S-Th1 in the ESI† for all isomers). The metallacycles formed at N2 are so unstable that the Pd-based fragment prefers to shift to the carbon atom adjacent to N2, thus lowering its energy by ∼1 kcal mol−1 (see the ESI, Fig. S-Th1†).
 | ||
| Fig. 2 Metallacycles Pd formed at various nitrogens of the tetrazo[1,2-b]indazole with the indication of their relative Gibbs free energies (in kcal mol−1). Color scheme: Pd (sky blue), Cl (green), F (light green), N (blue), C (grey). | ||
Applications to transition metal N-directed C–H activation/functionalization
Due to the selective activation of the ortho-C–H bond on the N-containing scaffold by transition metals, we evaluated its potential for post-functionalization (Schemes 7 and 8). New C–I and C–OAc were successfully inserted in the ortho-position of the aryl of the tetrazo[1,2-b]indazole by Pd catalysis in the presence of the appropriate electrophilic source (Scheme 7).12 | ||
| Scheme 8 N-directed oxidative cross-coupling (hetero)arylation of 17. Conversion based on the consumption of 17 determined by 1H and 19F NMR (CDCl3, 500 MHz and 470 MHz respectively), isolated yields in brackets. | ||
The iodination occurred in the presence of [Pd(OAc)2] and N-iodosuccinimide (NIS) in acetic acid with 66% isolated yield for 23 after 30 minutes under microwave irradiation. Under similar conditions, the more challenging acetoxylation takes place with phenyl iodane diacetate (PIDA) as the electrophilic reagent affording 24 in 51% isolated yield.8,13
We then extended our C–H activation toolbox by exploring the use of other transition metals. Based on the abilities of rhodium and iridium in the N-directed oxidative cross-coupling functionalization,14,15 the direct (hetero)arylation of the 3-(2-fluorophenyl)-tetrazo[1,2-b]indazole 17 was demonstrated in two oxidative cross-coupling reactions (Scheme 8). These reactions took place with [Cu(OAc)2] as the oxidant in dichloroethane (DCE) at 140 °C.16 To couple the 4-t-butylphenyl boronic acid, the rhodium catalyst [RhCl2Cp*]2 was employed with success to provide 25 in 58% isolated yield. The most challenging dehydrogenative coupling reaction with the 2-methylthiophene was also demonstrated with [IrCl2Cp*]2 to form 26 in 49% isolated yield.17
Spectroscopic and redox properties of 3-aryltetrazo-[1,2-b]indazoles
The spectroscopic properties of 12–22 were investigated in diluted CH2Cl2 solutions (c = 1 × 10−5 mol L−1, Fig. 3 and Table S-1 in the ESI†). The electrochemical properties of 12–22 were investigated by cyclic voltammetry in dichloromethane solutions (see the ESI, Fig. S-3 and S-4†). | ||
| Fig. 3 UV-vis absorption in CH2Cl2 of 12 (blue), 13 (grey), 15 (orange) (left); 17 (green), 17-O1 (red), 17-O2 (yellow) and Ir (purple) (right). | ||
First, the effect of the (hetero)aryl in the 3-position was investigated (Fig. 3, and Fig. S-3 and S-4 in the ESI†). The tetrazo[1,2-b]indazoles 12–22 all display a large absorption with moderate extinction coefficient in the visible range (λmax ∼ 435 nm) assigned to a π–π* transition, regardless of the lateral substituent. Only the presence of the strongly electron donating diphenylamine (DPA) in 15 allows a red-shift of the absorption to 459 nm through the introduction of sufficient charge transfer from the NPh2 to the electron-deficient TzIn core (see the ESI, Fig. S-Th2†). This is also supported by cyclic voltammetry as 15 is the only compound to display an oxidation wave under these conditions (Eox(15) = +1.18 V vs. SCE). Such low potential oxidation is characteristic of DPA based derivatives. Please note that all 3-aryl-substituted TzIn display quasi-reversible reduction at low potential in the −0.8 to 0.9 V vs. SCE range, as previously observed with such derivatives.4
As the effect of the 3-substituent is rather limited, we then investigated the effect of N-substitution. While protonation (Fig. S-4†) and oxidation of N2 has a limited impact on the absorption (λmax(17-O2) = 428 nm), the oxidation of N1 leads to a strong red-shift (λmax(17-O1) = 517 nm). This difference is well reproduced by TD-DFT (see the ESI, Fig. S-Th4†). The red-shift is accompanied by an increase of the reduction potential (Ered(17-O1) = −0.81 V vs. SCE, Ered(17-O2) = −0.89 vs. SCE) (see Fig. S-6†).
Finally, the insertion of transition metals via the presence of metallacycles also induces a stronger redshift (λmax(Ir) = 675 nm, Fig. 3, and Fig. S-1 and S-2 in the ESI†). These low energy absorptions are characteristic of MLCT transitions in cyclometalated complexes (see also NTO in Fig. S-Th3†).18
Note that none of these TzIn show luminescence either in diluted solution or in the solid state.
As a conclusion, the specific chemistry of the N-atom of these novel heteroaromatics is a powerful way to tune their redox properties.
Conclusions
In conclusion, we have reported a two steps protocol for the divergent synthesis of 3-arylated tetrazo[1,2-b]indazoles 12–22, based on a Pd-catalyzed Liebeskind–Srogl cross-coupling reaction followed by our previously reported N-cyclisation process. The N atoms of the TzIn scaffold remain reactive as demonstrated by protonation, selective oxidation (17-O1 and 17-O2) and metalations with Pd, Ir and Rh. Additionally, using these transition metals, selective ortho-C–H activation/functionalization on the heterocycle was also demonstrated (23–26). The effects of all these molecular engineering strategies on the properties of the 3-arylated tetrazoindazoles were studied experimentally and theoretically, revealing that both optical and redox properties can be finely tuned. This study highlights the diversity of the molecular structures and the electronic properties offered by the tetrazo[1,2-b]indazole platform and opens up interesting perspectives for its integration into optoelectronic devices.Experimental section
Procedure for the synthesis of 3-(2-fluorophenyl)-6-aryl-[1,2,4,5]-tetrazine
An oven-dried Schlenk tube equipped with a magnetic stirring bar was sequentially charged with of 3-(methylthio)-6-(2-fluorophenyl)-[1,2,4,5]-tetrazine (1.0 equiv.), [PdCl2(dppf)] (15 mol%), arylboronic acid (2.0 equiv.) and Ag2O (2.0 equiv.). After 3 cycles of vacuum purging with argon, DMF [0.1 M] solvent was added by syringe. After heating under argon at 60 °C for 20 h, the DMF was removed by rotary evaporation under vacuum. The crude was purified by column chromatography.Procedure for the synthesis of 3-(aryl)-[1,2,4,5]-tetrazo[1,2-b]indazole
As a typical experiment, the 3-aryl-6-(2-fluorophenyl)-[1,2,4,5]-tetrazine (1.0 equiv.) and sodium azide (3.0 equiv.) were introduced in a microwave reaction vessel equipped with a magnetic stirring bar. The DMF [0.125 M] was added, and the reaction mixture was heated at 130 °C under air for 1 h. After cooling down to room temperature, the solvent was removed under vacuum. The crude product was purified by silica gel column chromatography to afford the corresponding product.Procedure for the direct halogenation/acetoxylation of 3-(2-fluorophenyl)-[1,2,4,5]-tetrazo[1,2-b]indazole
As a typical experiment, an oven-dried Schlenk tube equipped with a magnetic stirring bar was charged with 3-(2-fluorophenyl)-[1,2,4,5]-tetrazo-[1,2-b]-indazole (1.0 equiv.), NIS or PIDA (2.0 equiv.), [Pd(OAc)2] (10 mol%), and HOAc [0.5 M] under air. The mixture was stirred at 110 °C for 30 min under microwave irradiation (200 Watts). The solvent was removed under vacuum and the crude product was purified by column chromatography on silica using an appropriate ratio of the eluent to afford the desired product.Procedure for the direct arylation of 3-(2-fluorophenyl)-[1,2,4,5]-tetrazo[1,2-b]indazole
As a typical experiment, an oven-dried Schlenk tube equipped with a magnetic stirring bar was charged with 3-(2-fluorophenyl)-[1,2,4,5]-tetrazo-[1,2-b]-indazole (1.0 equiv.), 4-tertiobutylphenyl boronic acid or 2-methylthiophene (3.0 equiv.), [MCl2Cp*]2 (5 mol%, M = Rh or Ir), AgSbF6 (20 mol%), Cu(OAc)2 (3.0 equiv.) and dichloroethane [0.5 M] under air. The mixture was stirred at 140 °C for 5 h or 24 h. The solvent was removed under vacuum and the crude product was purified by column chromatography on silica using an appropriate ratio of eluent to afford the desired product.Procedure for the metallacycle formation by o-C–H activation of 3-(2-fluorophenyl)-[1,2,4,5]-tetrazo-[1,2-b]-indazole
N-oxides (17-O1 and 17-O2)
In a dry Schlenk tube under argon was added H2O2 (1.55 ml, 15.1 mmol, 10 equiv.) to trifluoromethanesulfonic anhydride (2.1 ml, 15.1 mmol, 10 equiv.) in dry CH2Cl2 (10 ml), and the mixture was stirred for 5 min at 0 °C. Then 3-(2-fluorophenyl)-[1,2,4,5]-tetrazo-[1,2-b]-indazole 17 (400 mg, 1.5 mmol, 1 equiv.) was added to the solution at 0 °C and stirred for 5 h. The solvent was removed under vacuum. The product was purified through silica gel column chromatography (CH2Cl2/petroleum ether: 40/60) to afford 17-O2 (135 mg, 32%, yellow powder) as the first eluted compound and 17-O1 (148 mg, 35%, red powder) as the second.Author contributions
Asmae Bousfiha: investigation, methodology, supervision, validation. Oumaima Abidi: investigation, methodology, validation. Louis Lemetayer: investigation, validation. Navya Sood: investigation. Iogann Tolbatov: formal analysis (DFT). Fabien Barrois: formal analysis (DFT). Ahmad Daher: investigation. Hélène Cattey: formal analysis (crystal analysis). Marie Cordier: formal analysis (crystal analysis). Muriel Hissler: funding acquisition, conceptualization, writing – review & editing. Jean-Cyrille Hierso: funding acquisition, conceptualization, writing – review & editing. Paul Fleurat-Lessard: funding acquisition, investigation, conceptualization, supervision, writing – original draft. Pierre-Antoine Bouit: funding acquisition, investigation, conceptualization, supervision, writing – original draft. Julien Roger: funding acquisition, investigation, conceptualization, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS, Conseil Régional de Bourgogne (PARI and FEDER programs), by the COMUE UBFC (ISITE UB180013.MUB.IS_SmarTZ; R. J., I. T. and PhD grant for A. D.), by the ANR JCJC program 2018 FITFUN (ANR-18-CE07-0015; R. J. and PhD grant for O. A.) and ANR Pi-Aza (ANR-21-CE07-0024-01, P.-A. B., A. B. and PhD grant for L. L.). Calculations were performed using HPC resources from DSI-CCUB at the Université de Bourgogne. The authors thank the PACSMUB platform for analyses (SATT SAYENS) especially M.-J. Penouilh and Q. Bonnin.Notes and references
- (a) U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed; (b) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2016, 117, 3479–3716 CrossRef PubMed; (c) G. Clavier and P. Audebert, Chem. Rev., 2010, 110, 3299–3314 CrossRef CAS PubMed; (d) P. Audebert, E. Kroke, C. Posern and S. H. Lee, Chem. Rev., 2021, 121, 2515–2544 CrossRef CAS PubMed.
- (a) C. L. Anderson, T. Zhang, M. Qi, Z. Chen, C. Yang, S. J. Teat, N. S. Settineri, E. A. Dailing, A. Garzón-Ruiz, A. Navarro, Y. Lv and Y. Liu, J. Am. Chem. Soc., 2023, 145, 5474–5485 CrossRef CAS PubMed; (b) G. K. Rastogi, M. L. Deb and P. K. Baruah, Chem. Commun., 2023, 59, 9642–9645 RSC; (c) D. Sirbu, J. Diharce, I. Martinic, N. Chopin, S. V. Eliseeva, G. Guillaumet, S. Petoud, P. Bonnet and F. Suzenet, Chem. Commun., 2019, 55, 7776–7779 RSC; (d) S. Maier, N. Hippchen, F. Jester, M. Dodds, M. Weber, L. Skarjan, F. Rominger, J. Freudenberg and U. H. F. Bunz, Angew. Chem., Int. Ed., 2023, 62, e20221403 CrossRef PubMed.
- A. Daher, A. Bousfiha, I. Tolbatov, C. D. Mboyi, H. Cattey, T. Roisnel, P. Fleurat-Lessard, M. Hissler, J.-C. Hierso, P.-A. Bouit and J. Roger, Angew. Chem., Int. Ed., 2023, 62, e202300571 CrossRef CAS PubMed.
- For other examples of N–N bond formation through azide cyclization, see: (a) R. A. Carboni and J. E. Castle, J. Am. Chem. Soc., 1962, 84, 2453–2454 CrossRef CAS; (b) O. Yu. Smirnov, A. M. Churakov, Yu. A. Strelenko and V. A. Tartakovsky, Russ. Chem. Bull., 2008, 57, 2180–2184 CrossRef CAS; (c) A. A. Konnov, M. S. Klenov, A. M. Churakov, Y. A. Strelenko, A. O. Dmitrienko, L. N. Puntus, K. A. Lyssenko and V. A. Tartakovsky, Asian J. Org. Chem., 2018, 7, 2534–2543 CrossRef CAS; (d) S. Gutierrez, A. Arnault, V. Ferreira, A. Artigas, D. Hagebaum-Reignier, Y. Carissan, Y. Coquerel, M.-A. Hiebel and F. Suzenet, J. Org. Chem., 2022, 87, 13653–13662 CrossRef PubMed.
- C. Testa, E. Gigot, S. Genc, R. Decreau, J. Roger and J.-C. Hierso, Angew. Chem., Int. Ed., 2016, 55, 5555–5559 CrossRef CAS PubMed.
- Y. Xie, Y. Fang, Z. Huang, A. M. Tallon, C. W. am Ende and J. M. Fox, Angew. Chem., Int. Ed., 2020, 59, 16967–16973 CrossRef CAS PubMed.
- For other examples of cross-coupling reactions on s-tetrazine, see: (a) Z. Novak and A. Kotschy, Org. Lett., 2003, 5, 3495–3497 CrossRef CAS PubMed; (b) N. Leconte, A. Keromnes-Wuillaume, F. Suzenet and G. Guillaume, Synlett, 2007, 204–210 CAS; (c) L. Pellegatti, E. Vedrenne, J.-M. Leger, C. Jarry and S. Routier, Tetrahedron, 2010, 66, 4683–4389 CrossRef; (d) C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, G. Clavier, L. Vignau and P. Audebert, New J. Chem., 2015, 39, 9700–9713 RSC; (e) W. D. Lambert, Y. Fang, S. Mahapatra, Z. Huang, C. W. am Ende and J. M. Fox, J. Am. Chem. Soc., 2019, 141, 17068–17074 CrossRef CAS PubMed; (f) E. Ros, A. Prades, D. Forson, J. Smyth, X. Verdaguer, L. Ribas de Pouplana and A. Riera, Chem. Commun., 2020, 56, 11086–11089 RSC; (g) Y. Qu, P. Pander, O. Vybornyi, M. Vasylieva, R. Guillot, F. Miomandre, F. B. Dias, P. Skabara, P. Data, G. Clavier and P. Audebert, J. Org. Chem., 2020, 85, 3407–3416 CrossRef CAS PubMed; (h) L. V. Hoff, S. D. Schnell, A. Tomio, A. Linden and K. Gademann, Org. Lett., 2021, 23, 5689–5692 CrossRef CAS PubMed.
- A. Bousfiha, S. Fournier, H. Cattey, P. Fleurat-Lessard, C. H. Devillers, D. Lucas, J.-C. Hierso and J. Roger, Organometallics, 2024, 43, 807–816 CrossRef CAS.
- (a) D. Li, P. Wu, N. Sun, Y.-J. Lu, W.-L. Wong, Z. Fang and K. Zhang, Curr. Org. Chem., 2019, 23, 616–627 CrossRef CAS; (b) H. Wie, H. Gao and J. Shreeve, Chem. – Eur. J., 2014, 20, 16943–16952 CrossRef PubMed.
- C. D. Mboyi, C. Testa, S. Reeb, S. Genc, H. Cattey, P. Fleurat-Lessard, J. Roger and J.-C. Hierso, ACS Catal., 2017, 7, 8493–8501 CrossRef CAS.
- Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC.
- (a) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (b) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS PubMed.
- D. Kalyani and M. S. Sanford, Org. Lett., 2005, 7, 4149–4152 CrossRef CAS PubMed.
- S. Basak, J. P. Biswas and D. Maiti, Synthesis, 2021, 53, 3151–3179 CrossRef CAS.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- (a) T. Vogler and A. Studer, Org. Lett., 2008, 10, 129–131 CrossRef CAS PubMed; (b) X.-L. Lyu, S.-S. Huang, Y.-Q. Huang, Y.-Q. Li, H.-J. Song, Y.-X. Liu and Q.-M. Wang, J. Org. Chem., 2020, 85, 10271–10282 CrossRef CAS PubMed.
- G. Tan, Q. You and J. You, ACS Catal., 2018, 8, 8709–8714 CrossRef CAS.
- Y. Chi and P. T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and analytical data, NMR data and X-ray crystallographic data. CCDC 2307023 (17-O1), 2307024 (17-O2), 2332725 (Rh), 2332726 (Pd) and 2332727 (Ir). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01122h |
| ‡ A. B. and O. A. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |