
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear NHC–gold(I)–thiolato and –alkynyl complexes: synthesis, anticancer activity, and catalytic activity in lactonization reactions†
Xinyuan
Ma‡
ae,
Yuan
Zhao‡
 a,
Isabella
Caligiuri
b,
Flavio
Rizzolio
bc,
Nestor
Bracho Pozsoni
a,
Kristof
Van Hecke
a,
Thomas
Scattolin
*d and
Steven P.
Nolan
*a
a,
Isabella
Caligiuri
b,
Flavio
Rizzolio
bc,
Nestor
Bracho Pozsoni
a,
Kristof
Van Hecke
a,
Thomas
Scattolin
*d and
Steven P.
Nolan
*a
aDepartment of Chemistry, Centre for Sustainable Chemistry, Ghent University, Krijgslaan 281, S3, 9000 Ghent, Belgium. E-mail: steven.nolan@ugent.be
bPathology Unit, Centro di Riferimento Oncologico di Aviano (C.R.O.) IRCCS, via Franco Gallini 2, 33081, Aviano, Italy
cDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari, Campus Scientifico Via Torino 155, 30174 Venezia-Mestre, Italy
dDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: thomas.scattolin@unipd.it
eChemical Science and Technology Research Institute, Sinochem Group, 20 Xueyuan Road, Haidian District, 100083 Beijing, China
First published on 17th April 2024
Abstract
A series of novel dinuclear NHC–gold–thiolato and –alkynyl complexes bearing aromatic linkers were successfully synthesized by an efficient and simple synthetic route. The catalytic activity of these complexes was tested in a lactonization reaction. The reaction proceeds in high efficiency, in short reaction time and under mild conditions, and is complementary to existing methods. Furthermore, the digold(I)–thiolato derivatives exhibit remarkable cytotoxicity towards several cancer cell lines.
Introduction
In the last decades, due to various side-effects exhibited by platinum-based anticancer drugs,1–3 much research has focused on the exploration of the potential anticancer properties of other metal complexes bearing a wide range of organic ligands.4,5 Encouragingly, some gold,6–11 ruthenium12,13 and palladium14–17 complexes have shown excellent in vitro, ex vivo and in vivo antitumor activity. More importantly, these not only proceed via a completely different mechanism of action from conventional platinum drugs, but they are often effective against cisplatin-resistant cancer cells,18 a fact that has attracted a great deal of attention.Mononuclear gold(I) complexes are of importance in medicinal chemistry as they can be used for the treatment of rheumatic diseases,19 but they have also great potential as antibiotics and chemotherapeutic agents.7,8 Intensive studies in recent decades have demonstrated that anticancer gold complexes have a wide range of cellular targets, such as mitochondria and specific proteins that exert relevant functional roles (e.g. thioredoxin reductase (TrxR), poly(ADP-ribose) polymerase (PARP-1), chelating proteins, proteasome, and protein tyrosine phosphatases).6
In recent years, numerous gold-phosphine complexes have been reported to exhibit excellent cytotoxicity as well as good stability under physiological conditions.20–22 In addition, such research has shown that the biological activity of these complexes can be further enhanced by employing thiolato and alkynyl ligands.23,24 Among the gold(I)–thiolato complexes, (1-thio-beta-D-glucopyranos-2,3,4,6-tetraacetato-S-triethylphosphine)gold(I), known as auranofin, is one of the most famous anti-arthritic agents.25 Furthermore, it has also recently been approved by the FDA for use in clinical phase II for cancer treatment.26 Equally intriguing in this research area are mononuclear gold(I)–alkynyl complexes, although they have not yet been approved for clinical use. This class of compounds can include both phenylacetylene-derived as well as hormone-derived alkynyl ligands. Encouraging results from preclinical studies have been obtained in the case of gold(I)–alkynyl complexes bearing phosphines and N-heterocyclic carbenes (NHCs) as ancillary ligands.24,27 The latter are usually considered as an attractive alternative to tertiary phosphines, owing to their ability to form strong bonds with transition metals.28
In particular, the high stability of gold–NHC complexes even in physiological environments makes NHCs promising ligands in metallodrug design.6–11,29 In this context, the Barnard group reported for the first time the anticancer properties of cationic mononuclear [Au(NHC)2]+ and dinuclear [Au2(bisNHC)2]2+ complexes more than 20 years ago.30 More recently, we have proposed a simple synthetic entryway into mononuclear gold(I)–thiolato and gold(I)–alkynyl complexes with good antitumor activity on different cancer cell lines.27,31
Upon examining the existing literature, the development of straightforward synthetic procedures to dinuclear gold(I)–thiolato and –alkynyl complexes bearing NHC ligands, and the exploration of their structural and functional properties, remain largely unexplored compared to their mononuclear counterparts.
For this reason, inspired by the mentioned research, we have devised a simple synthetic approach for producing these two categories of digold complexes and investigated their anticancer activity as well as their catalytic activity in lactonization reactions.
It should be remembered that lactones are basic molecular structures commonly found in natural product frameworks.32 These cyclic molecules serve diverse biological functions, ranging from bacterial quorum sensing signalling33 to enzyme inhibition by the lactone core's ring opening.34 Consequently, lactones hold substantial importance not only as industrially relevant compounds but also as potential candidates for antibacterial and antitumor medications.
Although the synthesis of δ- and γ-lactones is well-documented,35 there is a notable scarcity of reports detailing efficient strategies for their formation using digold(I) catalysts. Probably, the most important example comes from our laboratories, in which [{Au(IPr)}2(μ-OH)][BF4] was employed as an efficient precatalyst.36 However, this compound differs significantly from the complexes investigated in this study as, due to its positive charge, it is known to act as a dual catalyst (Au–NHC+ as an acidic moiety and [Au(NHC)OH] as a basic function).
Therefore, this work aims primarily to explore the catalytic and antitumor properties of novel neutral digold(I) complexes bearing diNHC and thiolato/alkynyl ligands, utilizing a synthetic protocol employing an extremely weak and inexpensive base such as sodium acetate under extremely mild and aerobic conditions. The objective is to enrich our understanding of the synthesis of digold(I) species, especially the activation of C–H and S–H bonds under mild conditions, and to expand the collection of derivatives possessing interesting applications, thus partially fill the gap with classical mononuclear gold–NHC complexes.
Results and discussion
Based on our previous reports,37,38 we initiated our investigation with the model substrate of [(IPr)o-xylene(AuBr)2] 1a with 2.2 equiv. of thiophenol and 6 equiv. of NaOAc in EtOH.It should be remembered that sodium acetate was also successfully used by our group instead of potassium carbonate for the synthesis of the [Au(NHC)Cl] complexes, as well as for their post-functionalization.39 To our delight, the desired complex [(IPr)o-xylene(AuSPh)2] 2a complex was obtained in 85% yield after 6 hours of mixing at room temperature (Table 1, entry 1). Encouraged by this result, an optimization of the reaction conditions was undertaken. Reducing the amount of NaOAc to 4 equiv. resulted in an 86% yield of 2a (Table 1, entry 2). When utilizing 2 equiv. of thiophenol instead of 2.2 equiv., a lower 74% yield was isolated after 7 hours (Table 1, entry 3). Various solvents were also screened. Acetone and EtOAc as solvents led to a significant reduction in the yield of 2a, affording 47% and trace yield, respectively (Table 1, entries 4 and 5). Next, we turned our attention to optimizing the base. When employing K2CO3 or NEt3 as the base, only 45 to 67% of 2a were obtained, even with prolonged reaction times (Table 1, entries 6 and 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Thiophenol (equiv.) | NaOAc (equiv.) | Solvent | Time (h) | Yieldb (%) |
| a Unless otherwise noted, standard reaction conditions: 1a (0.05 mmol, 1 equiv., 56 mg), thiophenol, base, in 1 mL of solvent at RT. b Isolated yields. c K2CO3 instead of NaOAc. d NEt3 instead of NaOAc. | |||||
| 1 | 2.2 | 6 | EtOH | 6 | 85 |
| 2 | 2.2 | 4 | EtOH | 7 | 86 |
| 3 | 2 | 4 | EtOH | 7 | 74 |
| 4 | 2.2 | 4 | Acetone | 7 | 47 |
| 5 | 2.2 | 4 | EtOAc | 7 | Trace |
| 6 | 2.2 | 4c | EtOH | 6 | 67 |
| 7 | 2.2 | 4d | EtOH | 6 | 45 |

Having optimized reaction conditions, a series of dinuclear NHC–gold(I)–thiolato complexes bearing different aromatic linkers (2a–2e) was synthesized (Scheme 1). Most of these can be isolated in good to excellent yields (83–91%). The structure of compound 2a was confirmed through X-ray diffraction studies on single crystals grown by slow vapor diffusion of the antisolvent (pentane) into saturated solutions of dichloromethane containing 2a at 4 °C (Scheme 1). A notable exception to this general high yield procedure was found for the NHC with para-aryl linker (2e), where isolated yields were significant lower compared to the ones bearing ortho- or meta-aryl linkers. This observation was also made in the previously observed reactivity/isolation of dinuclear NHC–gold(I) carbazolyl complexes.38
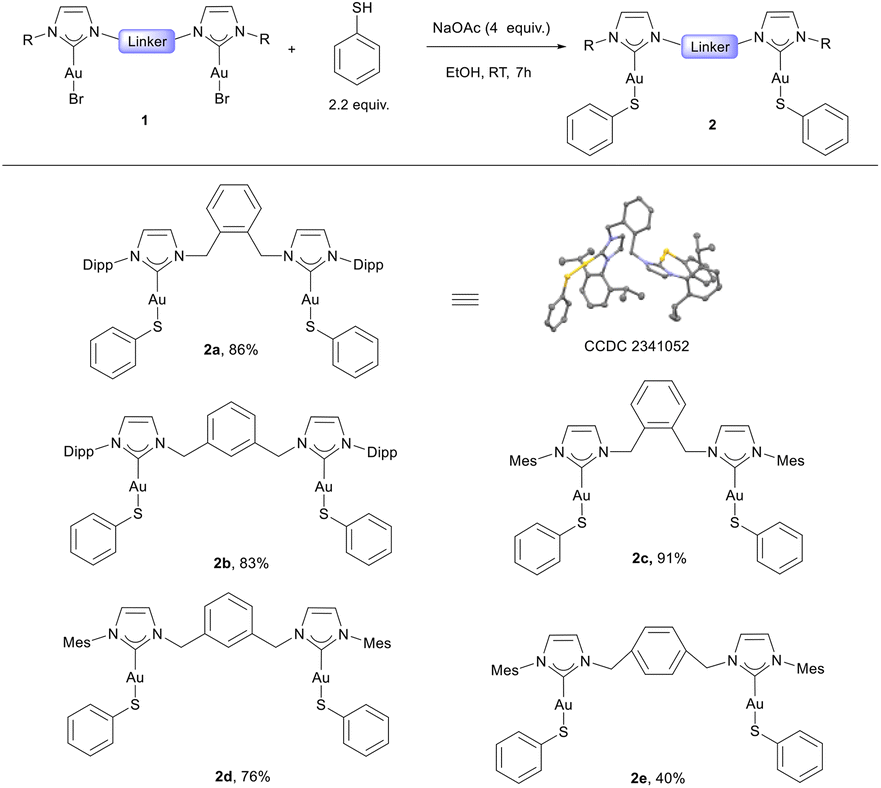 | ||
| Scheme 1 Synthetic reaction scope for dinuclear bridging NHC–Au–thiolate complexes. Unless otherwise noted, standard reaction conditions: 1 (0.05 mmol), thiophenol (2.2 equiv.), NaOAc (4 equiv.) in EtOH (1 mL) at RT for 7 h; yields of isolated products. | ||
We next turned our attention to the synthesis of dinuclear NHC–gold(I)–alkynyl complexes using the weak base route. In the presence of 6 equiv. of NaOAc and 4 equiv. of alkyne, the [(NHC){Au(alkynyl)}2] complexes 3a–b were synthesized in excellent yields (Scheme 2). It is worth noting that a dinuclear [IPro-xylene{Au(bisalkynyl)}2] complex 3b was obtained in 82% yield using more forceful conditions (at 80 °C after 12 h), but also highlights the thermal stability of such complexes under synthetic conditions.
 | ||
Scheme 2 Synthetic reaction scope for dinuclear bridging NHC–Au–alkynyl complexes. Unless otherwise noted, standard reaction conditions: 1a (0.10 mmol), alkyne (4 equiv.), NaOAc (6 equiv.) in EtOH (0.8 mL) at 40 °C for 7 h; yields of isolated products. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 80 °C for 12 h. 80 °C for 12 h. | ||
After successfully synthesizing these dinuclear gold complexes, their catalytic activity was tested in an intramolecular cyclization reaction (Table 2), which represents an example of an efficient and atom-economical method to obtain lactones.36 To our delight, all tested dinuclear NHC–gold(I)–thiolato complexes appear to display excellent catalytic activity with hexo-5-acetic acid, as the model substrate. Complexes 2b and 2d with meta-xylene linkers performed very well by using 0.5 mol% catalyst loading after only 30 min of reaction time, affording the corresponding lactone in excellent yield (Table 2, entries 5 and 8). Among the catalysts tested, an 87% isolated yield was obtained by employing 2b. Moreover, 0.5 mol% of 2a or 2c afforded the lactone in excellent yield after 2.5 hours and 1 hour, respectively (Table 2, entries 4 and 6). When 2e was used as catalyst, an 89% yield of the product was observed after only 1 hour (Table 2, entry 9).
|
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Time (h) | NMR yieldb (%) |
| a Unless otherwise noted, standard reaction conditions: alkynoic acid (0.5 mmol), catalyst (0.5 mol%) at 80 °C. b NMR yield determined with 1,3,5-trimethoxybenzene as internal standard (yield of isolated product in bracket). c Catalyst (1 mol%) at 60 °C. d Catalysts (1 mol%) at 80 °C. | |||
| 1c | 2a | 12 | Trace |
| 2d | 2a | 2 | 100 |
| 3 | 2a | 2 | 75 |
| 4 | 2a | 2.5 | 100 |
| 5 | 2b | 0.5 | 100 (87) |
| 6 | 2c | 1 | 100 |
| 7 | 2c | 0.5 | 84 |
| 8 | 2d | 0.5 | 100 |
| 9 | 2e | 1 | 89 |
| 10d | [Au(PPh3)Cl] | 0.5 | Trace |
| 11d | [Au(IPr)(SPh)] | 0.5 | 100 |
| 12 | 1b | 0.5 | Trace |
| 13 | 3a | 0.5 | 80 |
| 14 | 3b | 0.5 | 75 |

Interestingly, the digold–alkynyl complexes 3a–b have also shown promising catalytic activity, albeit lower to the digold–thiolato complex 2b, achieving yields ranging from 75 to 80% of the desired product. For completeness, we also tested the catalytic activity of two mononuclear gold(I) complexes such as [Au(IPr)(SPh)] and [Au(PPh3)Cl], as well as the digold precursor 1b. [Au(IPr)(SPh)] exhibited excellent catalytic activity, while both 1b and [PPh3AuCl] were practically inactive. Therefore, these experiments highlight how the presence of digold diNHC complexes functionalized with alkynyl or thiolato moieties represents an efficient combination for catalysing this type of reaction. The high catalytic activity of NHC–gold complexes (mononuclear or dinuclear) bearing thiolato or alkynyl ligands can be explained by their greater tendency to form cationic gold species in solution compared to their halide counterparts.
With the aim of exploring the potential anticancer properties of our digold–thiolato and digold–alkynyl complexes, we subjected a range of human tumour cell lines (ovarian cancer A2780 and its cisplatin-resistant variant A2780cis, colon cancer HCT116, lung cancer A549, triple-negative breast cancer MDA-MB231) to a 96-hour treatment with both the synthesized compounds and cisplatin (positive control).
Initially, we assessed the stability of the digold derivatives in DMSO-d6 using NMR spectroscopy. After 24 hours, no significant changes were observed in the spectra of these complexes, confirming their structural integrity after such treatment.
The antiproliferative activity data of the compounds tested are illustrated in Table 3, presenting the half inhibitory concentrations (IC50) values. It is important to point out that, in the case of digold–alkynyl complexes 3a–b, a negligible cytotoxicity (>100 μM) was observed in the preliminary tests and therefore these were not investigated further.
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| A2780 | A2780cis | HCT116 | A549 | MDA-MB231 | |
| a Data after 96 h incubation time. Stock solutions in DMSO for all complexes; stock solutions in H2O for cisplatin. A2780 (cisplatin-sensitive ovarian cancer cells), A2780cis (cisplatin-resistant ovarian cancer cells), HCT116 (colon cancer cells), A549 (lung cancer cells), MDA-MB231 (triple-negative breast cancer). b Data obtained from ref. 40. | |||||
| Cisplatin | 1.1 ± 0.1 | 11 ± 3 | 21 ± 7 | 9 ± 1 | 15 ± 1 |
| [Au(IMes)Cl]b | 5 ± 2 | 5 ± 2 | — | — | — |
| 2a | 3.7 ± 0.1 | 1.5 ± 0.4 | 2.8 ± 0.2 | 5.2 ± 0.3 | 4.0 ± 0.5 |
| 2b | 0.4 ± 0.1 | 2.4 ± 0.2 | 4.0 ± 0.1 | 7.7 ± 0.1 | 4.5 ± 0.3 |
| 2c | 0.3 ± 0.1 | 1.0 ± 0.3 | 4.4 ± 0.2 | 4.4 ± 0.2 | 4.4 ± 0.2 |
| 2d | 0.12 ± 0.03 | 0.11 ± 0.01 | 1.0 ± 0.1 | 5.8 ± 0.2 | 1.2 ± 0.7 |
| 2e | 2.6 ± 0.4 | 2.3 ± 0.1 | 5.0 ± 0.8 | >100 | 5 ± 1 |
All compounds generally exhibited excellent cytotoxicity in all tumour lines examined, with IC50 values in the micro- or sub-micromolar range (0.1–8 μM). Notably, the tested compounds present an activity comparable, or even up to an order of magnitude higher, than those of cisplatin and [Au(IMes)Cl]. Based on the data available in the literature, the latter has been chosen as the reference mononuclear gold(I) complex.40 Our results seem to suggest that the presence of two gold(I) centers provides superior antitumor activity compared to their mononuclear congeners. As a further confirmation, in the case of mononuclear NHC–Au–SR complexes with classical thiophenolates (e.g., [Au(IMes)(S-4-COOH-Ph)]), no activity was observed on in vitro breast and colon cancer models.41
Returning to the compounds investigated in this study, particularly interesting are the data obtained on cisplatin-resistant cancer (A2780cis) and triple-negative breast cancer (MDA-MB231) cells. It should be remembered that cisplatin-resistant ovarian cancer refers to a condition where cancer cells develop resistance to cisplatin. This resistance poses a significant challenge in the management of ovarian cancer, limiting the effectiveness of treatment options.42 Overcoming cisplatin resistance typically involves exploring alternative chemotherapy drugs, targeted therapies, or combination treatments tailored to the individual patient's cancer characteristics. Regarding the triple-negative breast cancer, it is a subtype of breast cancer characterized by the absence of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) expression.43 For these reasons, it is usually more aggressive and has a poorer prognosis compared to other types of breast cancer.
All tested digold complexes have been found to be more active than cisplatin towards these two types of tumors, especially complexes 2c and 2d, which exhibit superior cytotoxicity by up to an order of magnitude compared to the reference drug.
In terms of structure–activity relationships (SARs), it is clear, especially by comparing complexes 2c–e with each other, that the presence of a meta-xylene spacer is a generally favourable condition for the anticancer activity of the compounds. In contrast, the digold–thiolato derivative bearing a para-xylene spacer (2e) is the compound with the lowest cytotoxicity. This compound is almost inactive towards A549 colon cancer cells (IC50 > 100 μM). Further in vitro and in vivo studies using these complexes are ongoing in our laboratories.
Conclusions
We have easily synthesized a novel class of dinuclear NHC gold(I)–thiolato and –alkynyl complexes bearing aromatic linkers via an efficient and operationally simple synthetic route. These dinuclear gold complexes exhibit high efficiency in an intramolecular lactonization reaction only requiring low catalyst loading and short reaction times. Moreover, the digold(I)–thiolate derivatives 2a–e exhibited excellent cytotoxicity towards a wide range of cancer cell lines. The antiproliferative activity is significant in the case of complexes bearing a meta-xylene spacer. In most cases the IC50 values are lower, up to an order of magnitude, with respect to those of cisplatin and [Au(IMes)Cl], thus suggesting the beneficial role of two gold(I) centers in the anticancer activity of such complexes. In the case of the biological results, our aim is to delve deeper into understanding their mechanism of action and assessing their cytotoxicity in more complex biological systems, such as organoids and animal models. This work is ongoing and will be reported in due course.Experimental
General information
All manipulations were carried out under air in scintillation vials. Solvents and reagents were used as received without any further purification or distillation. 1H NMR and 13C NMR were recorded in CDCl3, DMSO-d6 or CD2Cl2 at room temperature on Bruker spectrometer (300 MHz or 400 MHz). Chemical shifts (ppm) are referenced to the residual solvent peak. Coupling constants (J) are given in hertz. Abbreviations used in the designation of the signals: s = singlet, br s = broad singlet, d = doublet, br d = broad doublet, dd = doublet of doublets, ddd = doublet of doublets of doublets, m = multiplet, td = triplet of doublets, tt = triplet of triplets, q = quadruplet, qt = quadruplet of triplets, hept = heptet. Elemental analyses were performed at Université de Namur, rue de Bruxelles, 55 B-5000 Namur, Belgium. The synthetic route to [NHC(AuBr)2] complexes has been previously reported.31General synthetic procedure leading to dinuclear NHC–Au(I)–thiolato complexes (2)
A 4 mL vial equipped with a screw cap and a stirring bar was charged, under air, with the corresponding [NHC(AuBr)2] (0.1 mmol, 1 equiv.), thiophenol (0.22 mmol, 2.2 equiv.), NaOAc (0.4 mmol, 4 equiv.) and EtOH (1 mL). The reaction mixture was stirred at room temperature for 7 hours. The solvent was then removed under vacuum, and purification of the product was carried out by filtration through a microfilter using pentane/dichloromethane. The solution was then concentrated to 1 mL, and 10 mL of pentane were added to precipitate the product as a colourless solid. After collection by filtration on a frit, the compound was dried under vacuum overnight.
1H NMR (300 MHz, CDCl3) δ 7.53 (t, J = 7.8 Hz, 2H, Dipp C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.46–7.39 (m, 2H, o-xylene C), 7.30 (d, J = 7.8 Hz, 4H), 7.07 (dd, J = 5.6, 3.5 Hz, 2H, o-xylene C), 7.02 (t, J = 2.4 Hz, 4H), 6.97 (dd, J = 5.9, 3.2 Hz, 2H), 6.89–6.72 (m, 6H, Ph C), 5.56 (s, 4H, C2), 2.55 (dt, J = 13.6, 6.8 Hz, 4H, C(CH3)2), 1.28 (d, J = 6.9 Hz, 12H, CH(C3)2), 1.17 (d, J = 6.9 Hz, 12H, CH(C3)2).
), 7.46–7.39 (m, 2H, o-xylene C), 7.30 (d, J = 7.8 Hz, 4H), 7.07 (dd, J = 5.6, 3.5 Hz, 2H, o-xylene C), 7.02 (t, J = 2.4 Hz, 4H), 6.97 (dd, J = 5.9, 3.2 Hz, 2H), 6.89–6.72 (m, 6H, Ph C), 5.56 (s, 4H, C2), 2.55 (dt, J = 13.6, 6.8 Hz, 4H, C(CH3)2), 1.28 (d, J = 6.9 Hz, 12H, CH(C3)2), 1.17 (d, J = 6.9 Hz, 12H, CH(C3)2).
13C NMR (101 MHz, CDCl3) δ 146.1, 134.3, 133.7, 132.0, 130.7, 129.4, 128.4, 127.6, 124.4, 124.3, 122.5, 52.3 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 28.8 (H(CH3)2), 24.6 (CH(H3)2), 24.4 (CH(H3)2).
H2), 28.8 (H(CH3)2), 24.6 (CH(H3)2), 24.4 (CH(H3)2).
Elemental analysis calcd for C50H56Au2N4S2 (%): C, 51.28; H, 4.82; N, 4.78. Found: C, 51.31; H, 4.69; N, 4.39.
1H NMR (300 MHz, CDCl3) δ 7.58–7.48 (m, 3H), 7.38 (dt, J = 9.6, 4.2 Hz, 3H), 7.30 (d, J = 7.9 Hz, 4H), 7.25 (d, J = 1.3 Hz, 2H), 7.11–7.02 (m, 4H), 6.95–6.78 (m, 8H), 5.45 (s, 4H, C2), 2.44 (dt, J = 13.7, 6.8 Hz, 4H, C(CH3)2), 1.27 (d, J = 6.8 Hz, 12H, CH(C3)2), 1.13 (d, J = 6.9 Hz, 12H, CH(C3)2).
13C NMR (101 MHz, CDCl3) δ 145.8, 136.5, 134.1, 131.8, 130.3, 129.8, 127.6, 127.3, 124.0, 123.6, 122.2, 54.2 (H2), 28.4 (H(CH3)2), 24.2 (CH(H3)2), 24.1 (CH(H3)2).
Elemental analysis calcd for C50H56Au2N4S2 (%): C, 51.28; H, 4.82; N, 4.78. Found: C, 51.74; H, 5.19; N, 4.82.
1H NMR (300 MHz, DMSO-d6) δ 7.73 (s, 2H), 7.61 (d, J = 1.8 Hz, 2H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.41 (dd, J = 5.7, 3.3 Hz, 2H, m-xylene C), 7.13 (s, 4H, Mes C), 7.06–6.99 (m, 2H), 6.82 (dd, J = 12.9, 5.7 Hz, 10H, Ph C), 5.72 (s, 4H, C2), 2.35 (s, 6H, C3), 2.05 (s, 12H, C3).
CH), 7.41 (dd, J = 5.7, 3.3 Hz, 2H, m-xylene C), 7.13 (s, 4H, Mes C), 7.06–6.99 (m, 2H), 6.82 (dd, J = 12.9, 5.7 Hz, 10H, Ph C), 5.72 (s, 4H, C2), 2.35 (s, 6H, C3), 2.05 (s, 12H, C3).
13C NMR (75 MHz, DMSO-d6) δ 138.9, 135.1, 134.7, 134.6, 131.0, 129.0, 128.5, 127.5, 127.1, 123.4, 50.5 (H2), 20.7 (H3), 17.5 (H3).
Elemental analysis calcd for C44H44Au2N4S2 (%): C, 48.62; H, 4.08; N, 5.15. Found: C, 48.99; H, 4.23; N, 5.14.
1H NMR (400 MHz, CDCl3) δ 7.43 (s, 1H, m-xylene C), 7.41–7.37 (m, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.18–7.15 (m, 6H), 7.00 (s, 4H, Mes C), 6.89–6.85 (m, 8H, Ph C), 5.43 (s, 4H, C2), 2.38 (s, 6H, C3), 2.05 (s, 12H, C3).
13C NMR (101 MHz, CDCl3) δ 183.8 (NN), 139.7 (Mes), 136.8 (m-xylene), 135.2 (Mes), 135.0, 132.2, 130.2, 129.5 (Mes H), 127.9, 127.7 (Ph H), 127.4 (m-xylene H), 122.7, 122.6, 120.9, 54.5 (H2), 21.4 (H3), 18.1 (H3).
Elemental analysis calcd for C44H44Au2N4S2 (%): C, 48.62; H, 4.08; N, 5.15. Found: C, 48.89; H, 4.57; N, 5.34.
1H NMR (300 MHz, CD2Cl2) δ 7.38 (s, 4H), 7.16 (d, J = 1.8 Hz, 2H), 7.06 (m, 8H), 6.98 (d, J = 1.9 Hz, 2H), 6.86 (m, J = 6.8 Hz, 6H), 5.50 (s, 4H), 2.39 (s, 6H), 2.09 (s, 12H).
13C NMR (75 MHz, CD2Cl2) δ 176.2 (NN), 140.3, 137.0, 135.3, 135.2, 130.4, 129.7, 128.1, 127.8, 123.3, 121.6, 54.9 (H2), 21.3 (H3), 18.1 (H3).
Elemental analysis calcd for C44H44Au2N4S2 (%): C, 48.62; H, 4.08; N, 5.35. Found: C, 48.48; H, 4.37; N, 5.75.
General synthetic procedure leading to dinuclear bridging NHC–Au(I)–alkynyl complexes (3)
A 4 mL vial equipped with a screw cap and a stirring bar was charged, under air, with the corresponding [NHC(AuBr)2] (0.1 mmol, 1 equiv.), alkyne (0.4 mmol, 4 equiv.), NaOAc (0.6 mmol, 6 equiv.) and EtOH (1 mL). The reaction mixture was stirred at 40 °C for corresponding time. Volatiles were then removed under vacuum and purification of the product was carried out by filtration through a microfilter with pentane/dichloromethane (2 mL and 20 mL). The solvent was then concentrated to near precipitation, warmed to rt, and pentane (10 mL) was added to precipitate the complex, affording a colourless microcrystalline solid. After collection on a fritted filter, the compound was dried under vacuum overnight.
1H NMR (300 MHz, CDCl3) δ 7.51–7.43 (m, 2H), 7.41–7.28 (m, 11H), 7.17–7.00 (m, 9H), 6.96 (d, J = 1.8 Hz, 2H), 5.71 (s, 4H, C2), 2.54 (dd, J = 13.6, 6.8 Hz, 4H, C(CH3)2), 1.35 (d, J = 6.9 Hz, 12H, CH(C3)2), 1.17 (d, J = 6.9 Hz, 12H, CH(C3)2).
13C NMR (101 MHz, CDCl3) δ 190.2 (NN), 146.2, 134.6, 134.1, 132.5, 130.9, 129.6, 129.4, 128.2, 126.6, 126.1, 125.1, 124.7, 121.6, 105.1, 52.4 (H2), 29.0 (H(CH3)2), 25.1 (CH(H3)2), 24.7 (CH(H3)2).
HRMS (ESI): calcd for C54H56Au2N4 [M]+: 1154.3836. Found: 1154.3815.
1H NMR (300 MHz, CDCl3) δ 7.51–7.42 (m, 2H, Dipp C), 7.43–7.35 (m, 2H, o-xylene C), 7.26 (dt, J = 3.4, 2.3 Hz, 10H), 7.07–7.00 (m, 4H, NC, o-xylene C), 6.73 (d, J = 8.9 Hz, 4H), 5.62 (s, 4H, C2), 3.75 (s, 6H, OC3), 2.49 (dq, J = 13.6, 6.8 Hz, 4H, C(CH3)2), 1.31 (d, J = 6.8 Hz, 12H, CH(C3)2), 1.17 (d, J = 6.8 Hz, 12H, CH(C3)2).
13C NMR (75 MHz, CDCl3) δ 188.7 (NN), 159.3 (OCH3), 145.7 (Dipp, iPr), 134.0, 133.9, 133.5, 130.6 (Dipp, H), 129.3 (o-xylene H), 128.6, 128.2 (o-xylene H), 124.8, 124.2 (Dipp, H), 121.2, 115.4, 113.8, 86.7, 74.7, 69.8, 55.3 (OH3), 52.1 (H2), 28.6(H(CH3)2), 24.7 (CH(H3)2), 24.3 (CH(H3)2).
Elemental analysis calcd for C60H60Au2N4O2 (%): C, 57.05; H, 4.79; N 4.44. Found: C, 56.51; H, 5.13; N, 4.04.
Typical procedure for catalytic lactonization of alkynoic acid
A 4 mL vial equipped with a screw cap and a stirring bar was charged under air with hexo-5-acetic acid and the catalyst. The reaction mixture was stirred at the indicated temperature for the indicated time. The yield of the product was determined using NMR with 1,3,5-trimethoxybenzene as the internal standard.Analytical data obtained agree with the literature.33
Cytotoxicity assays
Cancer cell lines (A2780, A2780cis, HCT116, A549, MDA-MB231), were cultured following the supplier's guidelines and maintained at 37 °C in a humidified atmosphere containing 5% CO2. In 96-well plates, 1 × 103 cancer cells were seeded and treated after 24 hours with six different concentrations of digold complexes 2a–e (0.001, 0.01, 0.1, 1, 10, 100 μM). It is important to note that stock solutions (10 mM) of all digold complexes were prepared using DMSO as a solvent. After 96 hours of treatment, cell viability was assessed using a CellTiter-Glo assay (Promega, Madison, WI, USA) with Tecan M1000 or Synergy H1 microplate readers. IC50 values were determined from logistical dose–response curves using GraphPad Prism software. Triplicate measurements were taken to calculate averages, and standard deviations are represented by error bars.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the Special Research Fund (BOF) of Ghent University (project grants to SPN) and from the FWO. YZ and XM thank the China Scholarship Council (CSC) (project 202206870016, project 201908530217) for PhD fellowships.Notes and references
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- K. A. Doucette, K. N. Hassell and D. C. Crans, J. Inorg. Biochem., 2016, 165, 56–70 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- I. Ott, in Advances in Inorganic Chemistry, ed. P. J. Sadler and R. van Eldik, Academic Press, 2020, vol. 75, pp. 121–148 Search PubMed.
- A. Bergamo, P. J. Dyson and G. Sava, Coord. Chem. Rev., 2018, 360, 17–33 CrossRef CAS.
- C. Schmidt, L. Albrecht, S. Balasupramaniam, R. Misgeld, B. Karge, M. Brönstrup, A. Prokop, K. Baumann, S. Reichl and I. Ott, Metallomics, 2019, 11, 533–545 CrossRef CAS PubMed.
- C. Schmidt, B. Karge, R. Misgeld, A. Prokop, R. Franke, M. Brönstrup and I. Ott, Chem. – Eur. J., 2017, 23, 1869–1880 CrossRef CAS PubMed.
- E. Bortolamiol, F. Visentin and T. Scattolin, Appl. Sci., 2023, 13, 5561 CrossRef CAS.
- S. M. Meier-Menches, B. Neuditschko, K. Zappe, M. Schaier, M. C. Gerner, K. G. Schmetterer, G. Del Favero, R. Bonsignore, M. Cichna-Markl, G. Koellensperger, A. Casini and C. Gerner, Chem. – Eur. J., 2020, 26, 15528–15537 CrossRef CAS PubMed.
- J.-J. Zhang, M. A. Abu el Maaty, H. Hoffmeister, C. Schmidt, J. K. Muenzner, R. Schobert, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2020, 59, 16795–16800 CrossRef CAS PubMed.
- T. A. C. A. Bayrakdar, T. Scattolin, X. Ma and S. P. Nolan, Chem. Soc. Rev., 2020, 49, 7044–7100 RSC.
- A. Gatti, A. Habtemariam, I. Romero-Canelón, J.-I. Song, B. Heer, G. J. Clarkson, D. Rogolino, P. J. Sadler and M. Carcelli, Organometallics, 2018, 37, 891–899 CrossRef CAS PubMed.
- K. Lin, Z.-Z. Zhao, H.-B. Bo, X.-J. Hao and J.-Q. Wang, Front. Pharmacol., 2018, 9, 1323–1332 CrossRef CAS PubMed.
- T. Scattolin, E. Bortolamiol, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio, L. Cavallo, B. Dereli, M. V. Mane, S. P. Nolan and F. Visentin, Chem. Commun., 2020, 56, 12238–12241 RSC.
- T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. – Eur. J., 2020, 26, 11868–11876 CrossRef CAS PubMed.
- T. Scattolin, I. Caligiuri, N. Mouawad, M. El Boustani, N. Demitri, F. Rizzolio and F. Visentin, Eur. J. Med. Chem., 2019, 179, 325–334 CrossRef CAS PubMed.
- A. R. Kapdi and I. J. S. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751–4777 RSC.
- E. Boros, P. J. Dyson and G. Gasser, Chem, 2020, 6, 41–60 CAS.
- L. Messori and G. Marcon, Met. Ions Biol. Syst., 2004, 41, 279–304 CAS.
- T. Zou, C. T. Lum, C.-N. Lok, W.-P. To, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 5810–5814 CrossRef CAS PubMed.
- J. J. Liu, P. Galettis, A. Farr, L. Maharaj, H. Samarasinha, A. C. McGechan, B. C. Baguley, R. J. Bowen, S. J. Berners-Price and M. J. McKeage, J. Inorg. Biochem., 2008, 102, 303–310 CrossRef CAS PubMed.
- C. K. Mirabelli, D. T. Hill, L. F. Faucette, F. L. McCabe, G. R. Girard, D. B. Bryan, B. M. Sutton, J. O. L. Barus, S. T. Crooke and R. K. Johnson, J. Med. Chem., 1987, 30, 2181–2190 CrossRef CAS PubMed.
- W. Henderson, B. K. Nicholson and E. R. T. Tiekink, Inorg. Chim. Acta, 2006, 359, 204–214 CrossRef CAS.
- J.-J. Zhang, M. A. Abu el Maaty, H. Hoffmeister, C. Schmidt, J. K. Muenzner, R. Schobert, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2020, 59, 16795–16800 CrossRef CAS PubMed.
- C. K. Mirabelli, D. T. Hill, L. F. Faucette, F. L. McCabe, G. R. Girard, D. B. Bryan, B. M. Sutton, J. O. Bartus, S. T. Crooke and R. K. Johnson, J. Med. Chem., 1987, 30, 2181–2190 CrossRef CAS PubMed.
- G.-X. Hou, P.-P. Liu, S. Zhang, M. Yang, J. Liao, J. Yang, Y. Hu, W.-Q. Jiang, S. Wen and P. Huang, Cell Death Dis., 2018, 9, 1–15 CrossRef CAS PubMed.
- T. Scattolin, P. Lippmann, M. Beliš, K. Van Hecke, I. Ott and S. P. Nolan, Appl. Organomet. Chem. DOI:10.1002/aoc.6624.
- T. Scattolin, A. A. Logvinov, N. V. Tzouras, C. S. J. Cazin and S. P. Nolan, Organometallics, 2023, 42, 2692–2730 CrossRef CAS.
- T. Scattolin, G. Tonon, E. Botter, S. G. Guillet, N. V. Tzouras and S. P. Nolan, Chem. – Eur. J., 2023, 29, e202301961 CrossRef CAS PubMed.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642–1647 CrossRef CAS PubMed.
- M. S. Filho, T. Scattolin, P. Dao, N. V. Tzouras, R. Benhida, M. Saab, K. V. Hecke, P. Lippmann, A. R. Martin, I. Ott and S. P. Nolan, New J. Chem., 2021, 45, 9995–10001 RSC.
- S. Schulz and S. Hotling, Nat. Prod. Rep., 2015, 32, 1042–1066 RSC.
- M. A. Churchill, H. Sibhatu and C. Uhlson, in Quorum Sensing, ed. K. P. Rumbaugh, Humana Press, 2011, vol. 692, pp. 159–171 Search PubMed.
- I. K. Monika and J. P. Balbina, Mini-Rev. Med. Chem., 2005, 5, 73–95 CrossRef PubMed.
- D. Campeau, D. F. L. Rayo, A. Mansour, K. Muratov and F. Gagosz, Chem. Rev., 2020, 121, 8756–8867 CrossRef PubMed.
- D. Gasperini, L. Maggi, S. Dupuy, R. M. P. Veenboer, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Adv. Synth. Catal., 2016, 358, 3857–3862 CrossRef CAS.
- I. I. Hashim, T. Scattolin, N. V. Tzouras, L. Bourda, K. V. Hecke, I. Ritacco, L. Caporaso, L. Cavallo, S. P. Nolan and C. S. J. Cazin, Dalton Trans., 2021, 51, 231–240 RSC.
- X. Ma, V. A. Voloshkin, E. A. Martynova, M. Beliš, M. Peng, M. Villa, N. V. Tzouras, W. Janssens, K. V. Hecke, P. Ceroni and S. P. Nolan, Catal. Sci. Technol., 2023, 13, 4168–4175 RSC.
- T. Scattolin, N. V. Tzouras, L. Falivene, L. Cavallo and S. P. Nolan, Dalton Trans., 2020, 49, 9694–9700 RSC.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- J. Fernández-Gallardo, B. T. Elie, T. Sadhukha, S. Prabha, M. Sanaú, S. A. Rotenberg, J. W. Ramos and M. Contel, Chem. Sci., 2015, 6, 5269–5283 RSC.
- M. Song, M. Cui and K. Liu, Eur. J. Med. Chem., 2022, 232, 114205 CrossRef CAS PubMed.
- P. Zagami and L. A. Carey, npj Breast Cancer, 2022, 8, 95 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2341052. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00890a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |