
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of versatile ligand precursors based on phosphonium ylides for palladalactam formation and catalytic investigation†
Cheng-Po
Kao
,
Jhen-Yi
Lee
,
Min-Cheng
Tang
and
Hon Man
Lee
 *
*
Department of Chemistry, National Changhua University of Education, Changhua, 500, Taiwan. E-mail: leehm@cc.ncue.edu.tw
First published on 30th May 2024
Abstract
A new series of ligand precursors designed for the synthesis of palladalactams has been developed. These precursors are easily accessible through a one-step reaction involving 2-chloro-N-phenylacetamide and a wide choice of various monophosphines, offering tunable electronic and steric properties within the ligand framework. The stability of both ligand precursors and resulting palladalactams in ambient air enhances their practical applicability. A newly synthesized palladalactam, featuring an electron-donating triethylphosphine moiety on the anionic phosphonium ylide ligand scaffold exhibited promising catalytic activities in the Mizoroki–Heck coupling reaction between aryl chlorides and alkenes. Theoretical calculations further affirmed that the ligand system in the complex is the most electron-donating, forming the strongest Pd–C bond compared to other complexes with alternative phosphine moieties.
Introduction
Transition metal complexes involving phosphonium ylides have been attracting interest over several decades.1–16 Recently, Gessner et al. introduced a pioneering class of palladium complexes featuring ylide-functionalized phosphine ligands known as YPhos (Fig. 1).17–22 These ligands exhibit exceptional electron-donating properties, attributed to the additional contribution from the ylide group to the phosphorus center. They resemble N-heterocyclic carbene (NHC) ligands,23–29 being neutral in their free states and serving as strong σ-donors and excellent Lewis bases for metals. Indeed, their donor capacity exceeds classical phosphines and even common NHC ligands.17 Moreover, the extent of electron donation can be conveniently modulated by altering the substitution pattern at the ylidic carbon atom. Consequently, palladium complexes with YPhos have demonstrated outstanding catalytic activities in Pd-catalyzed reactions, particularly with unreactive aryl chloride substrates, surpassing those of palladium complexes with electron-donating phosphines such as XPhos and common NHC ligands (IMes, and IPr, etc.).18–20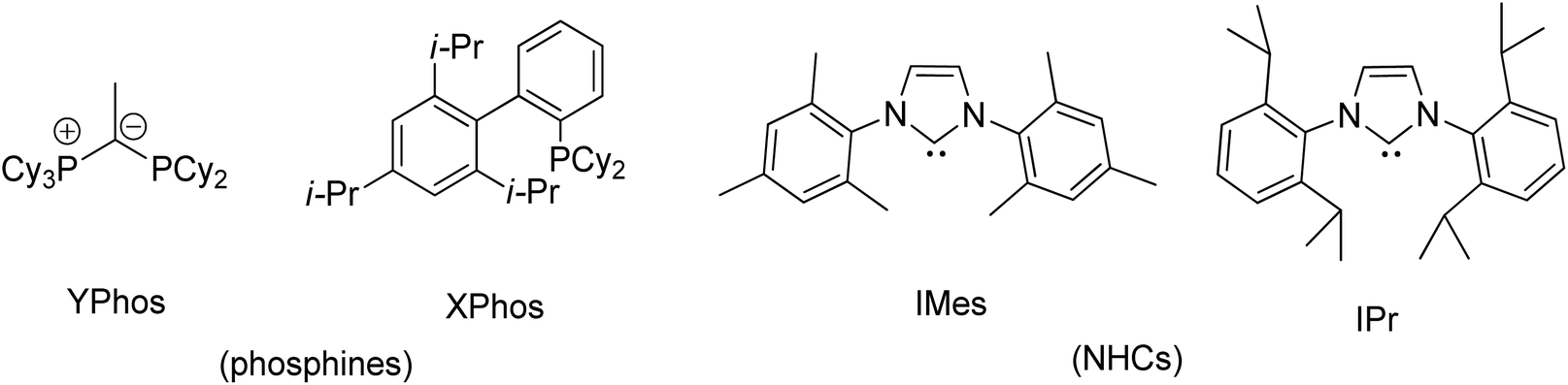 | ||
| Fig. 1 Commercially available YPhos, XPhos, IPr, and IMes ligands. | ||
Meanwhile, palladalactams, known for their distinctive four-membered metalacyclic ring system, have long been documented but remain exceptionally rare in the literature (Fig. 2).30–34 In exploring palladium complexes with mesoionic NHC ligands, we have concurrently uncovered an intriguing class of palladalactams.33,34 The precursors of the ligand systems can be easily synthesized through a one-step reaction involving various imidazole derivatives and organic amides. Drawing inspiration from Gessner's works,22 we propose a novel approach where the imidazole derivatives in our ligand system are substituted with diverse phosphines, leading to an unprecedented series of phosphonium chlorides (Scheme 1). Upon deprotonation with bases, these compounds will generate anionic phosphonium ylide ligands. Stabilizing these new ylides involves charge delocalization, encompassing the ylidic carbon, the amidate nitrogen, the carbonyl group, and the N-aryl ring. Like YPhos, the degree of electron donation in our new ligand can be modulated by the selection of different phosphine groups. We anticipate these ylides will serve as effective ligands for synthesizing innovative palladalactams (see Fig. 2).
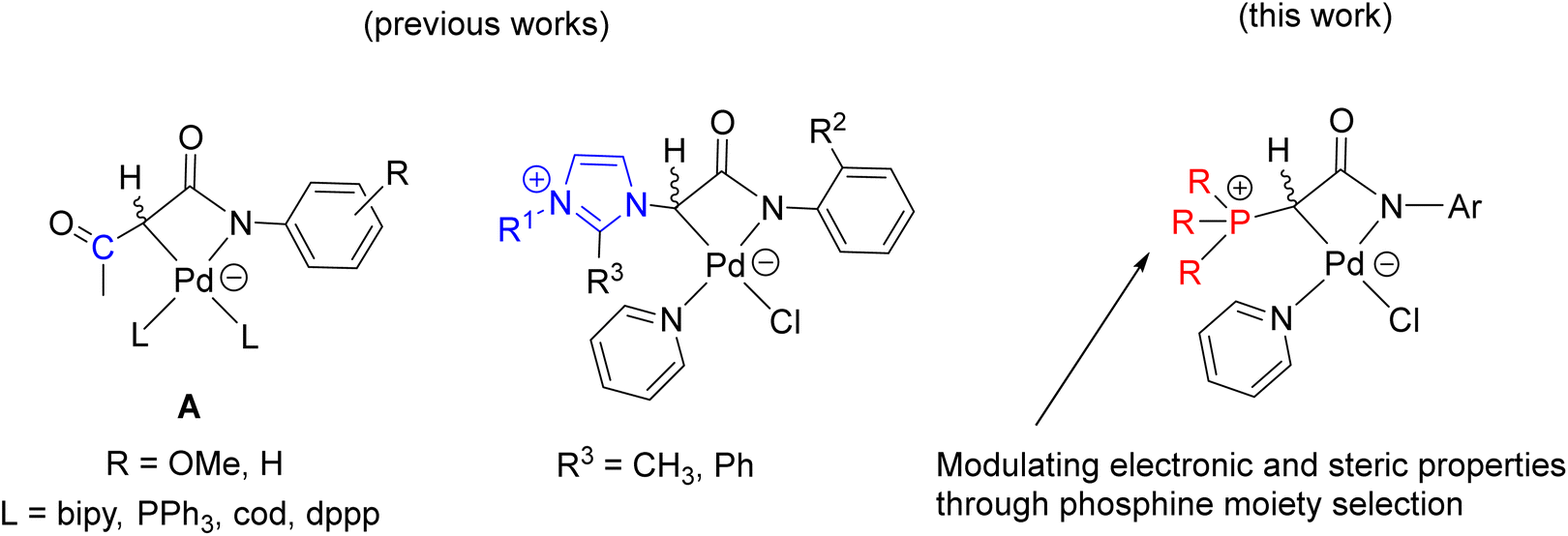 | ||
| Fig. 2 Palladalactams derived from functionalized acetanilide. | ||
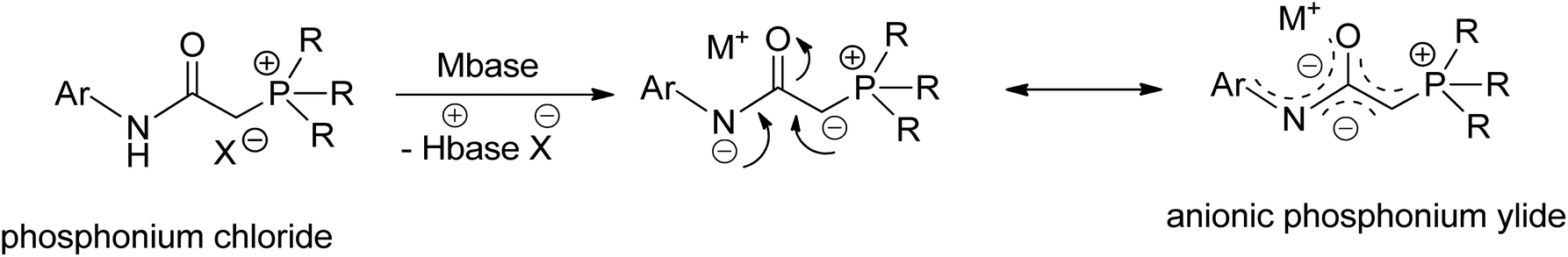 | ||
| Scheme 1 The formation of an anionic stabilized phosphonium ylide in the presence of a base. | ||
This work details synthesizing a new series of phosphonium chlorides with diverse phosphine groups, showcasing their excellence as ligand precursors for preparing novel palladalactams. We further assess the catalytic efficiency of these new palladium complexes in the Mizoroki–Heck coupling reaction. In contrast to other coupling reactions, the Mizoroki–Heck coupling reaction typically necessitates higher temperatures (>100 °C), emphasizing the importance of a robust bonding between the palladium center and the ligand to prevent ligand dissociation.35 While Pd complexes with NHC ligands have been widely employed,36,37 exploring other robust palladium complexes with non-NHC ligands is a worthwhile endeavor. In many instances of Mizoroki–Heck coupling reactions catalyzed by Pd NHC complexes, a high Pd loading, and more reactive and often costlier aryl bromides or iodides are typically needed as substrates.36–45 Among our newly prepared complexes, the one containing an electron-donating trialkylphosphine moiety in the ligand system exhibited the most promising catalytic activity in the coupling of electron-deficient aryl chlorides with alkenes. Theoretical calculations revealed that the ligand system in this complex is the most electron-donating, forming the strongest Pd–C and Pd–N bonds compared to other complexes featuring alternative phosphine moieties.
Results and discussion
Syntheses of ligand precursors and palladalactam complexes
The synthesis of ligand precursors is illustrated in Scheme 2. In a standard reaction, the ligand precursor was formed by reacting 2-chloro-N-phenylacetamide with phosphine in acetonitrile at an elevated temperature of 80 °C. Various phosphine ligands, including trialkylphosphines and triarylphosphines, were successfully employed, resulting in good yields ranging from 76% to 84%. A salt metathesis reaction between compound 1e and sodium tetrafluoroborate yielded the ligand precursor 1e′ with a BF4 counter-anion, achieving a yield of 81%. All the ionic salt products are only sparingly soluble in common organic solvents. But they dissolve readily in highly polar solvents such as DMSO and DMF. Notably, compounds 1a and 1d were derived from triethylphosphine and tricyclohexylphosphine, respectively. Despite the air sensitivity of these trialkylphosphines, both compounds 1a and 1d exhibit stability in the presence of air after formation. The unprotected OH groups in compounds 1e and 1e′ are well-tolerated during both synthesis and subsequent complexation reactions, likely facilitated by the formation of intramolecular OH⋯H hydrogen bonds, as revealed in the X-ray structures of their palladium complexes (vide infra).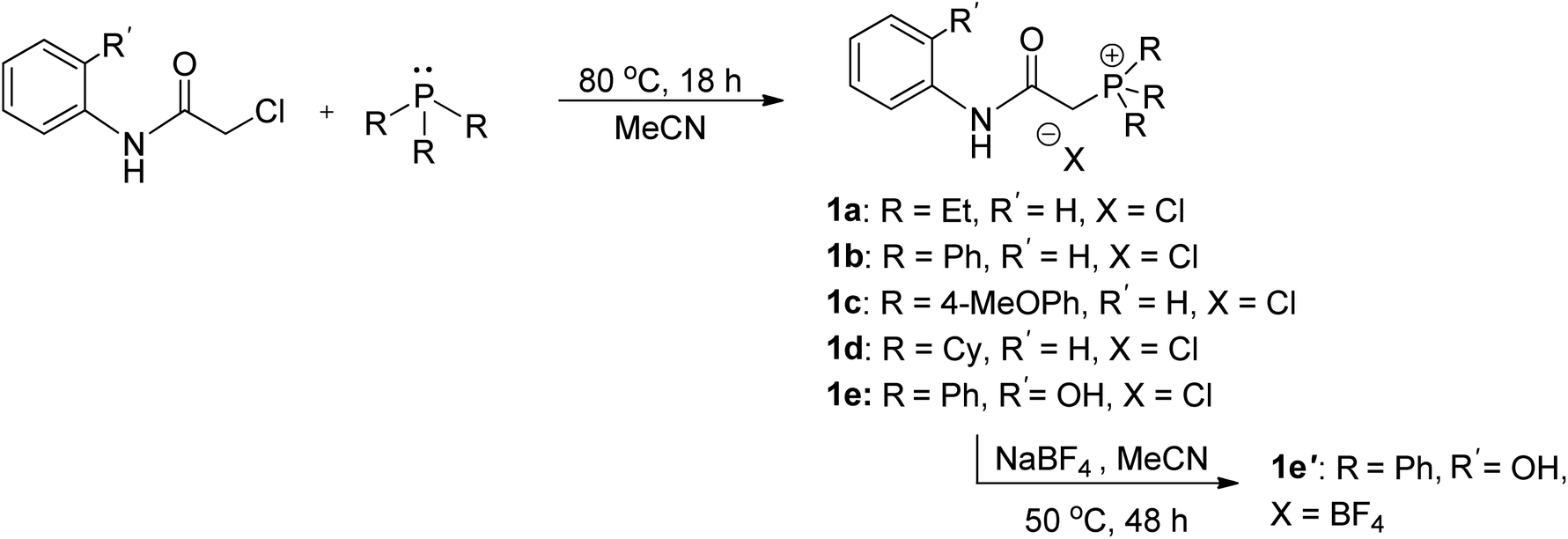 | ||
| Scheme 2 Synthesis of ligand precursors. | ||
The methylene signals of all products appear as doublets, attributed to the two-bond coupling with adjacent 31P nuclei, with a constant coupling of 15 Hz in most cases except for 1d (12 Hz). The electron-donating nature of the phosphine moiety significantly influences the chemical shift of methylene signals. In cases involving the electron-donating trialkylphosphine moiety, a pronounced shielding effect occurs, resulting in signals resonating at an upfield chemical shift, precisely at 3.98 and 4.03 ppm in 1a and 1d, respectively. Conversely, compounds with triphenylphosphine moieties exhibit downfield chemical shifts above 5 ppm (1b, 5.43; 1c, 5.12; 1e, 5.32; 1e′, 5.26 ppm). Moreover, compounds 1a and 1d showcase a distinct upfield shift of the α-carbon signals (1a, 27.2; 1d, 23.8 ppm) compared to the others (1b, 32.6; 1c, 33.3; 1e, 32.4; 1e′, 32.5), mirroring the trends observed in the methylene signals. These signals, appearing as doublets due to one-bond coupling with 31P NMR nuclei, exhibit smaller coupling constants for electron-donating phosphine moieties (1a, 49.8 Hz; 1d, 45.2 Hz) compared to those with triphenylphosphine moieties within the range of 52.8–60.3 Hz. On the contrary, the formation of phosphonium salts, resulting in formal positive charges on the phosphorus atoms, induces deshielding of the 31P nuclei, leading to a downfield shift in phosphorous signals towards positive chemical shifts. Ligand precursors 1a and 1d, bearing electron-donating triethylphosphine and tricyclohexylphosphine, manifest more pronounced downfield shifts at 38.3 and 33.4 ppm, respectively, in contrast to those with triphenylphosphine at 21.8 ppm (1b, 1e, and 1e′). Conversely, the electron-donating 4-methoxy group on the phenyl ring causes a slightly more upfield shift of the 31P NMR signal in compound 1c compared to 1b with a triphenylphosphine moiety (19.6 vs. 21.8 ppm). Fig. 3 shows the correlation between the 31P and methylene 1H NMR signal chemical shifts in the new ligand precursors.
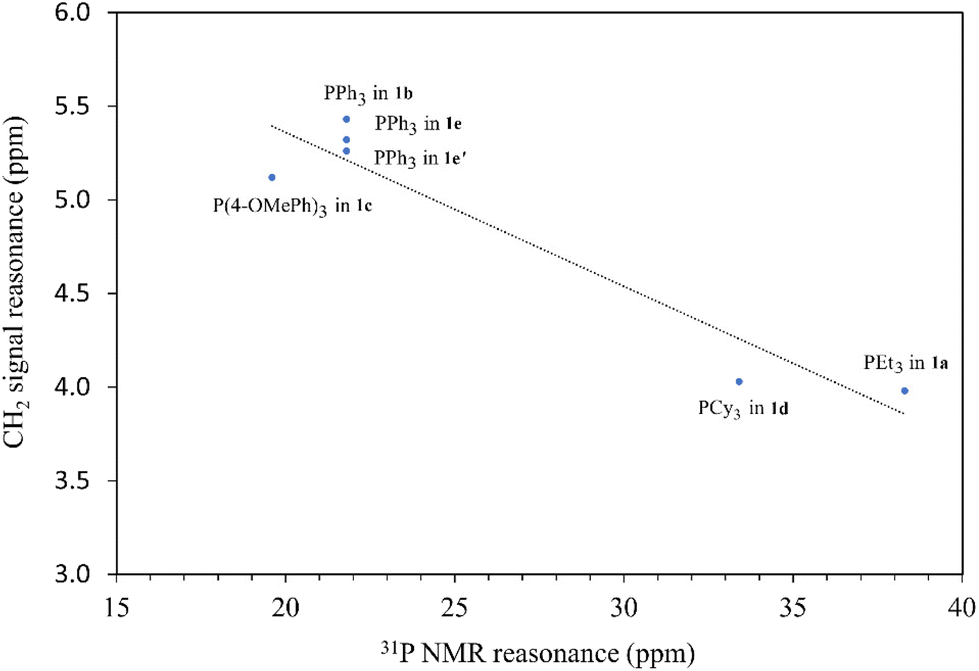 | ||
| Fig. 3 The correlation between the 31P and methylene 1H NMR signal chemical shifts in the new ligand precursors. | ||
The complexation reaction to prepare metallalactams A necessitated the addition of Ag2O to proceed,30,32 presumably via a transmetalation step. In contrast, in our case, direct complexation between the ligand and palladium precursors can be carried out without the need for Ag2O addition. Scheme 3 illustrates the synthesis of new palladalactams using the ligand precursors. In a typical reaction, a solution of chloride salts and PdCl2 in pyridine, with K2CO3 as a base at room temperature, leads to the formation of compounds 2a–d. Compound 2e, featuring a hydroxy group on the phenyl ring of the triphenylphosphine moieties, was synthesized using Pd(OAc)2 and NaOAc as the base. The complexes were obtained in good yields, ranging from 54% to 74%. Confirmation of the coordination of a pyridine ligand around the palladium center in these complexes is attained through the presence of pyridine signals in their 1H NMR spectra. Upon coordination, the 31P NMR signals of the complexes shift downfield. The difference in chemical shifts between the complex and the ligand is more minor for those containing trialkylphosphine (Δ = 1.2 ppm for 2a and Δ = 2.1 ppm for 2d), whereas the shift is much more substantial (ca. 6.3 ppm) for those containing PPh3 or P(4-OMePh)3 moieties. Interestingly, the doublets corresponding to the coordinated CH ylide carbons in 2a and 2d were detected at −2.9 and −8.1 ppm, respectively, in stark contrast to all other complexes where contrasting downfield signals with positive ppm values ranging from 7.0 to 9.2 were observed. These distinctive negative ppm values in 2a and 2d are likely attributed to the electron-donating property of the PEt3 and PCy3 moieties, resulting in a higher electron density on the carbon atom compared to ligands with PPh3 moieties, thus shielding the 13C nuclei. It is worth mentioning that the 13C resonances of the CH ylide carbon atoms in all of our new complexes are significantly upfield compared to the 19.5 ppm observed in the reported palladium complex based on an anionic C,C,C-phosphonium ylide core pincer ligand.11
 | ||
Scheme 3 Synthesis of palladalactams. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) For the synthesis of 2e, Pd(OAc)2 and NaOAc were used. For the synthesis of 2e, Pd(OAc)2 and NaOAc were used. | ||
Compounds 2a–e incorporate pyridine solvent as ligands due to its coordinative nature. Attempting the reaction between ligand precursor 1b and PdCl2 in less coordinative DMF solvent resulted in the formation of dimeric complex 3b. Interestingly, the ylide carbon doublet was observed at 16.4 ppm, significantly downfield compared to all the other complexes. Similarly, the chloride anion demonstrates a coordinative effect. Using ligand precursor 1e′ with the non-coordinative BF4 anion resulted in the formation of ionic complex 4e, with two pyridine ligands coordinated around the palladium center. The ylide carbon doublet, similar to those of complexes 2, was observed at 7.2 ppm. Yields for the two compounds are 53 and 32%, respectively. The exact structures of all the new complexes were confirmed by single-crystal X-ray diffraction studies. Notably, these complexes are air-stable and slightly soluble in common organic solvents such as CHCl3 and CH2Cl2.
Structural descriptions
The structures of 2a–c, 2e, 3b, and 4e were successfully established by single-crystal X-ray diffraction studies (Fig. 4–6 and Fig. S1–S4 in the ESI†). In the case of 2d, the complex converted to the dimeric complex 3d, apparently due to the slow de-coordination of pyridine ligand during the crystal growth process. Generally, the palladium centers in complex 2 adopt a distorted square planar coordination geometry. In these complexes, the pyridine ligand is positioned trans to the amidate N atom, while the chloride ligand is trans to the coordinated C atom. The four bond angles around the palladium centers are highly distorted from the ideal 90°. This distortion is attributable to the small bite angle of the bidentate ligand. For example, in 2a, the N1–Pd1–C8 is just 67.53(12)°. However, the summation of bond angles around the Pd center (360.2°) is very close to the ideal 360°.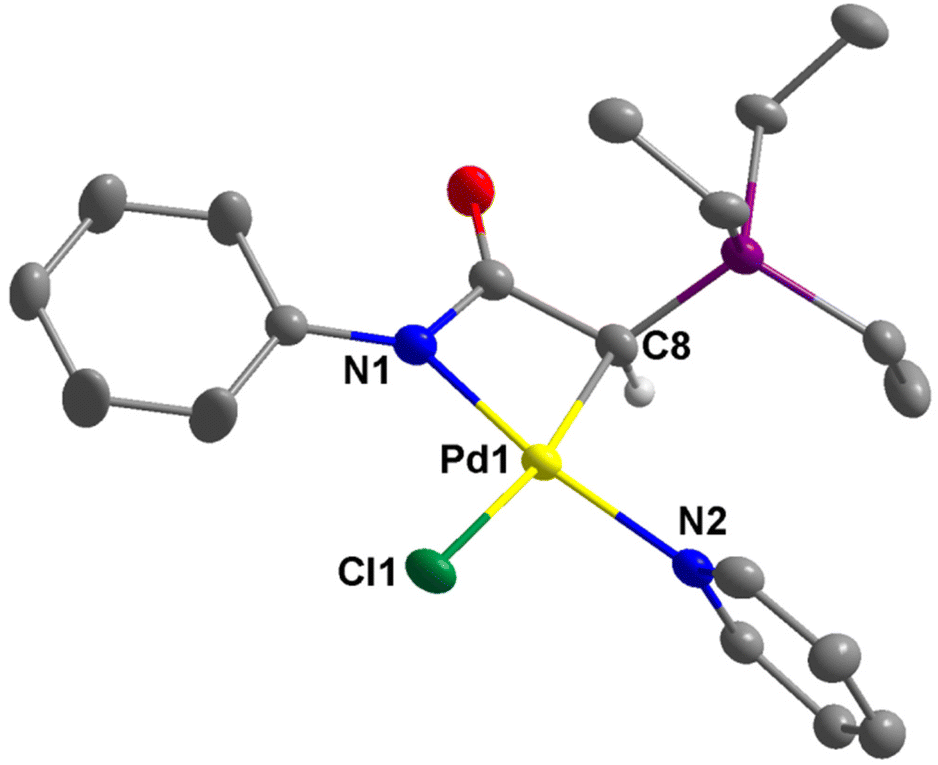 | ||
| Fig. 4 Thermal ellipsoid plot of 2a (50% probability). Hydrogen atoms except that on C8 are omitted for clarity. Selected bond distances (Å) and angles (°): Pd1–C8, 2.053(3); Pd1–Cl1, 2.3963(8); Pd1–N1, 2.023(3); Pd1–N2, 2.047(3); C8–Pd1–N1, 67.53(12); N1–Pd1–Cl1, 104.12(9); Cl1–Pd1–N2, 88.38(8); N2–Pd1–C8, 100.17(12); N1–Pd1–N2, 167.35(12); C8–Pd1–Cl1, 170.48(9). | ||
 | ||
| Fig. 5 Thermal ellipsoid plot of 3d (50% probability). Hydrogen atoms except that on C8 are omitted for clarity. Selected bond distances (Å) and angles (°): Pd1–C8, 2.026(4); Pd1–N1, 2.034(4); Pd1–Cl1, 2.3491(11); Pd1–Cl1#1, 2.4167(11); Cl1#1–Pd1–N1, 108.66(11); N1–Pd1–C8, 68.07(16); C8–Pd1–Cl1, 100.38(13); Cl1–Pd1–Cl#1, 83.94(4); N1–Pd1–Cl1, 166.95(11); Cl1#1–Pd1–C8, 176.67(13). Symmetry code #1: 1 − x, 1 − y, 1 − z. | ||
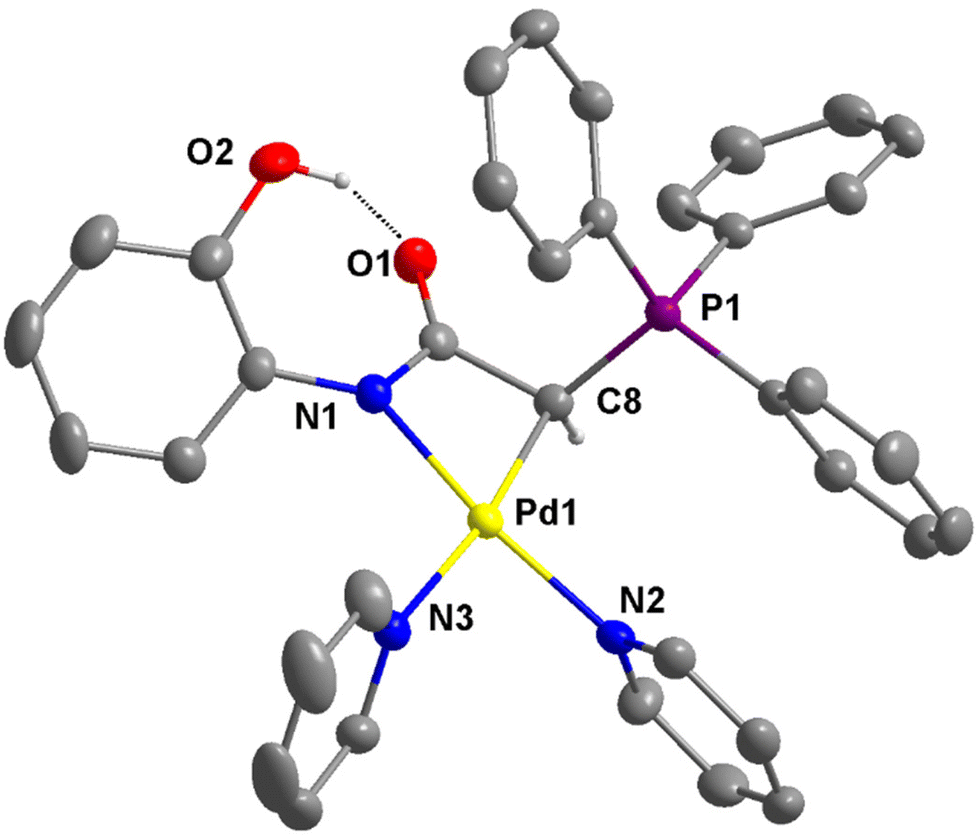 | ||
| Fig. 6 Thermal ellipsoid plot of the cationic portion of 4e (50% probability). Hydrogen atoms except that on C8 and O2 are omitted for clarity. Selected bond distances (Å) and angles (°): Pd1–C8, 2.048(3); Pd1–N1, 2.029(2); Pd1–N2, 2.043(2); Pd1–N3, 2.111(3); C8–Pd1–N1, 67.28(11); N1–Pd1–N3, 104.41(10); N2–Pd1–N3, 87.28(10); N2–Pd1–C8, 100.91(11); C8–Pd1–N3, 171.57(11); N1–Pd1–N2, 167.53(10). The intramolecular hydrogen bond is indicated by a broken line. | ||
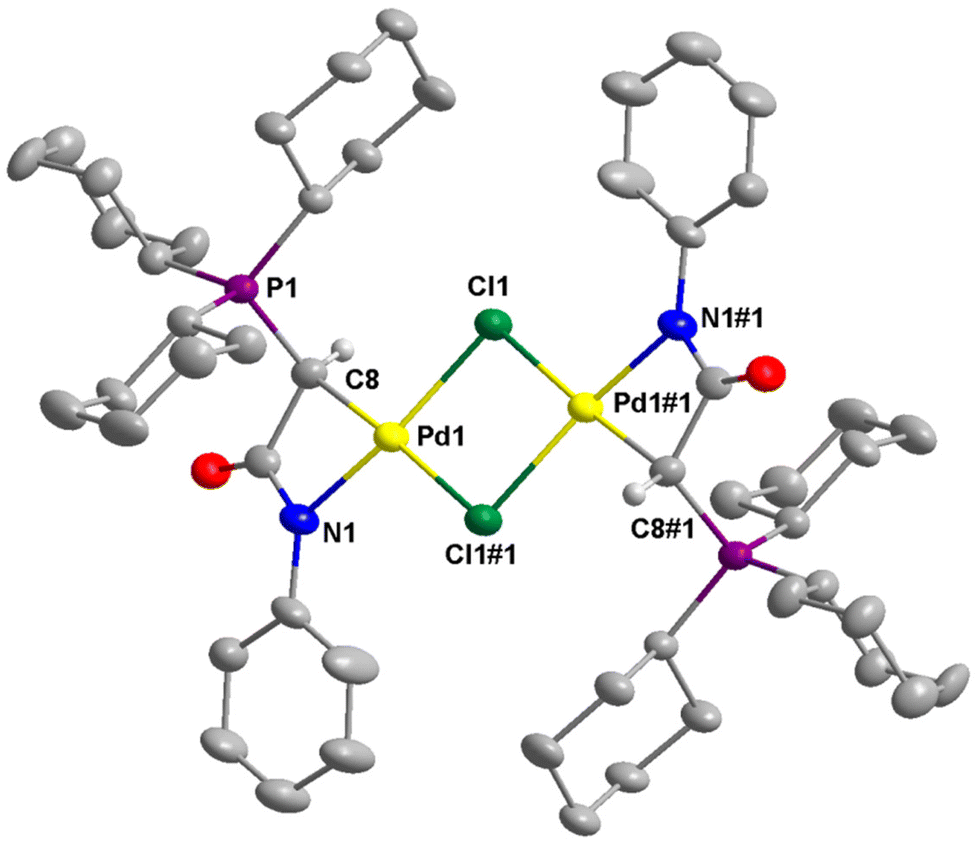
The molecular structures of dimeric palladium complexes 3d and 3b are shown in Fig. 5 and Fig. S4,† respectively. Each dimeric palladium complex is positioned on an inversion center in these structures. The square planar coordination geometry is highly distorted, as observed in the monomeric complexes. In both structures, the Pd–Cl bond trans to the C atom is longer compared to that trans to the N atom (e.g., 2.416(11) vs. 2.349(11) Å in 3d), indicating a higher trans influence from the coordinated carbon atom. The nonbonding Pd⋯Pd distances in 3d and 3b are 3.571 and 3.467 Å, respectively.
The molecular structure of the cationic portion of 4e is shown in Fig. 6. The two unequal Pd–N bond distances are the most notable structural feature. The bond distance trans to the coordinated carbon atom is longer at 2.111(3) Å, while that trans to the nitrogen atom is much shorter at 2.043(2) Å. The greater trans influence exerted by the carbon atom compared to the nitrogen atom in the structure of 4e is consistent with that observed in 3d and 3b, as well as the reported structure of [Pd{NPhC(O)CHC(O)Ph}(bipy)].32 Similar to the structure of 2e (see Fig. S3†), an intramolecular OH⋯O hydrogen bond is also present in 4e. The O⋯O distances of 2.564 and 2.607 Å, respectively, are observed in these structures.
Complex 3d, featuring the bulky PCy3 moiety, exhibits the shortest Pd–C bond distance of 2.026(4) Å compared to the others, which range from 2.048 to 2.082 Å. Notably, to the best of our knowledge, the Pd–C bond in complex 3d is the shortest among compounds containing a Pd–CHPY moiety, where Y represents a non-hydrogen atom.3,6,11,30,32,46,47 For instance, the Pd–C bond distances are 2.070(7) and 2.0546 Å in the two reported structures of palladalactams derived from acetoacetanilides.30,32 The sizeable trans influence of the coordinated carbon in 3d results in the longest trans Pd–Cl bond of 2.4167(11) Å among the complexes, varying from 2.3624 to 2.3963 Å. Importantly, the installation of bulky PCy3 moiety in 3d resulted in the ligand showing the largest C–Pd–N bite angle of 68.07(16)°, while all others have smaller angles in the range of 66.99 to 67.53°. This indicates that not only the electronic effect but also the steric property of the new ligand framework can be easily modulated by selecting different phosphines. Additionally, the C–Pd–N bite angles of these phosphine-based palladalactams are larger than those of 65.4 and 66.6° in the two reported imidazole-based palladalactams (see Fig. 1).33
Catalytic activities
The catalytic application of the new palladium complexes in the Mizoroki–Heck coupling reaction of aryl halides with alkene was investigated. Table 1 presents the screening of the catalytic precursors. The benchmark reaction involves the coupling of 4-chloroacetophenone and styrene under our previously reported reaction conditions.48,49 Entries 1, 2, 4, and 7 highlight that complexes 2a, 2b, 2d, and the ionic complex 4e are highly promising catalysts, delivering excellent yields of the product (>95%) under a mild 0.2 mol% Pd loading. Surprisingly, entries 3 and 6 reveal that complexes 2c and 3b are inactive. The Pd loading was then reduced to 0.1 mol% to differential the catalytic activities of the four complexes. Entries 8 and 10 show that complexes 2a and 2d, featuring electron-donating trialkylphosphine moieties, outperform complexes 2b and 4e, with complex 2a slightly more active than 2d. To further confirm the efficiency of 2a, the Pd loading was further decreased to 0.05 mol%. Under such a low Pd loading, complex 2a, bearing the electron-donating PEt3 moiety, still delivers a decent 49% yield of the product, while complex 2d shows no activity at all (entries 12 vs. 13).|
|
|||
|---|---|---|---|
| Entry | Cat. | Pd (mol%) | Yield (%) |
| a Reaction conditions: 1.4 mmol of styrene, 1.0 mmol of aryl halide, 1.1 mmol of NaOAc, 2 g of TBAB, Pd cat., 140 °C. Yields are determined by using 1,3,5-trimethoxybenzene as the internal standard. Ratios of anti/gem isomers in parenthesis. b For dimeric complex 3b, 0.1 mol% of the complex was used. | |||
| 1 | 2a | 0.2 | 97 (94/6) |
| 2 | 2b | 0.2 | 95 (95/5) |
| 3 | 2c | 0.2 | 0 |
| 4 | 2d | 0.2 | 97 (94/6) |
| 5 | 2e | 0.2 | 88 (94/6) |
| 6 | 3b | 0.2b | 0 |
| 7 | 4e | 0.2 | 97 (94/6) |
| 8 | 2a | 0.1 | 95 (93/7) |
| 9 | 2b | 0.1 | 89 (94/6) |
| 10 | 2d | 0.1 | 91 (93/7) |
| 11 | 4e | 0.1 | 32 (93/7) |
| 12 | 2a | 0.05 | 49 (94/6) |
| 13 | 2d | 0.05 | 0 |
Time–yield curves of the reaction catalyzed by precatalysts 2a, 2b, and 2d, as shown in Fig. S5,† clearly indicate that complex 2a is the most efficient complex. Interestingly, there is an indication period of approximately 30 minutes in each instance, implying that the complexes are not active species. To verify this, catalytic solutions containing the complexes but without substrates in DMSO-d6 were heated at 140 °C for 30 minutes. Subsequently, the solutions were analyzed using 31P{1H} NMR spectroscopy (Fig. S6 and S7†). In the cases of 2a and 2d containing trialkylphosphines, an additional small signal, approximately 1–2 ppm upfield to those of catalytic precursors, was observed. These extra peaks were tentatively identified as the active palladium(0) species [LPd0]− resulting from the reduction of complexes 2a and 2d. It is worth noting that after heating the solutions, palladium black was observed for all three solutions. We hypothesize that DMSO is a less effective solvent for stabilizing the palladium(0) species. In contrast, neat TBAB at high temperatures is much better at stabilizing the species. Conversely, in the case of 2b, no such extra species was observed by the 31P NMR spectroscopy, likely due to its poorer stability in DMSO solvent than the other two complexes. Noteworthy, [LPd0]− from 2a possesses the strongest Pd–C and Pd–N bonds and is the most electron-donating complex according to our theoretical calculation (vide infra).
With the optimal catalyst precursor 2a in hand, we embarked on refining the reaction conditions for further enhancement. Various combinations of bases and solvents were tested at different temperatures (Table 2). The initial attempts (entries 1–4) revealed that complex 2a was inactive in an aqueous solvent containing the ionic salt tetrabutylammonium bromide (TBAB) with K3PO4 as the base (entry 1). Similarly, the complex showed inactivity in pure organic solvents DMF and NMP using NEt3 and K2CO3 as bases at temperatures below 130 °C (entries 2–4). Entry 5 indicates the complex's activity in DMA as the solvent with 10 mol% TBAB and K2CO3 as the base, yielding a decent 56% product yield. Utilizing the molten salt TBAB at 140 °C as the solvent, an optimum 97% yield of coupled product was achieved using NaOAc as the base in 2 h (entry 6). However, substituting TBAB with DMA led to a significant reduction in yield (entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Temp. (°C) | Time (h) | Yield (%) |
| a Reaction conditions: 1.0 mmol of 4-chloroacetonphenone, 1.4 mmol of styrene, 0.2 mol% 2a. Yields determined by NMR using 1,3,5-trimethoxybenzene as the internal standard. Ratios of anti/gem isomers in parenthesis. b K3PO4 (1.5 mmol), TBAB (0.5 mmol), H2O (2.0 mL). c NEt3 (1.5 mmol), DMF (2.0 mL). d K2CO3 (2.0 mmol), DMF (6.0 mL). e K2CO3 (1.1 mmol), NMP (3.0 mL). f K2CO3 (1.0 mmol), TBAB (10 mol%), DMA (4.0 mL). g NaOAc (1.1 mmol), TBAB (2.0 g). h NaOAc (2.0 mmol), DMA (3.0 mL). | |||||
| 1b | K3PO4 | TBAB/H2O | 110 | 12 | 0 |
| 2c | NEt3 | DMF | 120 | 2 | 0 |
| 3d | K2CO3 | DMF | 80 | 2 | 0 |
| 4e | K2CO3 | NMP | 130 | 2 | 0 |
| 5f | K2CO3 | TBAB/DMA | 120 | 6 | 56 (98/2) |
| 6g | NaOAc | TBAB | 140 | 2 | 97 (94/6) |
| 7h | NaOAc | DMA | 140 | 2 | 19 (95/5) |
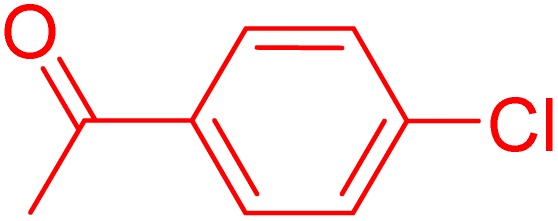
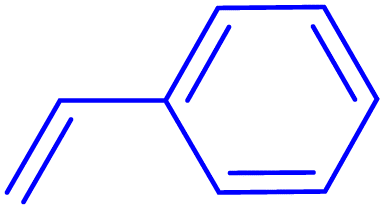
Following the optimization of reaction conditions, a thorough exploration of the substrate scope was undertaken (Table 3). Notably, employing a reduced Pd loading of 0.1 mol% yielded comparable results to the higher 0.2 mol% loadings, promoting the preference for the former, as indicated in entries 1 vs. 8 of Table 1. Entries 1–13 showcase the couplings of a wide range of aryl chlorides with varying electronic properties with styrene. Generally, good to excellent yields of coupled products can be achieved with electron-deficient aryl chlorides in 2 h (entries 1–6). Compared to the para-substituted 4-chloroacetophenone, an extended reaction time of 12 h was required for the sterically hindered meta-substituted isomer (entries 3 vs. 7). However, the catalyst system is less efficient in utilizing electron-neutral or electron-rich aryl chlorides. Entries 8 shows that a low 36% yield of trans-stilbene was obtained from chlorobenzene in 12 h. Even with an increased Pd loading of 0.2 mol%, a mere 12% yield was obtained using the highly unreactive 4-chloroanisole as the substrate. For substrates featuring methoxy substituents on the phenyl rings, aryl bromides, known for their enhanced reactivity, were investigated. A significant improvement was observed, with an 84% yield of 4-methoxystilbene achieved using the bromo analog (entries 9 vs. 10). Additionally, a decent 61% yield was attained when 3,4,5-trimethoxyphenyl bromide was employed as the substrate. For sterically hindered meta- and ortho-methoxyphenyl bromides, reduced product yields of 54% and 28%, respectively, were obtained (entries 12 and 13). Expanding beyond styrene, the catalyst system proved highly efficient with n-butyl acrylate as the alkene partner (entries 14–18). For examples, quantitative yields of coupled products were achieved in 6 h when 4-chlorobenzonitrile and 4-chloroacetophenone were used (entries 14 and 15). Notably, a very good 87% yield of the coupled product was achieved with the electron-rich 4-bromoanisole as the substrate, employing an increased 0.2 mol% Pd loading (entry 19).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar–X | Alkene | Product | Time (h) | Yield (%) |
| a Reaction conditions (unless otherwise specified): 1.0 mmol aryl halide, 1.4 mmol alkene, 1.1 mmol NaOAc, 2 g of TBAB, 0.1 mol% 2a, 140 °C, 2–12 h. Isolated yield. b 0.2 mol% 2a. | |||||
| 1 |
|
|
|
2 | 99 |
| 2 |
|
|
|
2 | 98 |
| 3 |
|
|
|
2 | 94 |
| 4 |
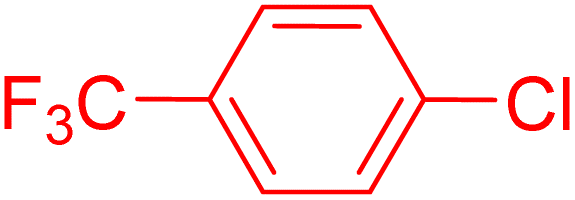
|
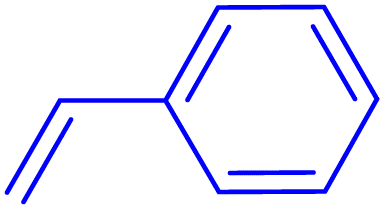
|
|
2 | 90 |
| 5 |
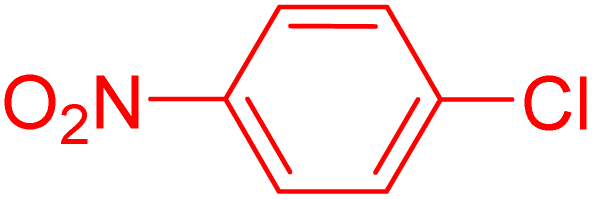
|
|
|
2 | 78 |
| 6 |
|
|
|
2 | 75 |
| 7 |
|
|
|
12 | 43 |
| 8 |
|
|
|
12 | 36 |
| 9 |
|
|
|
12 | 12b |
| 10 |
|
|
|
12 | 84 |
| 11 |
|
|
|
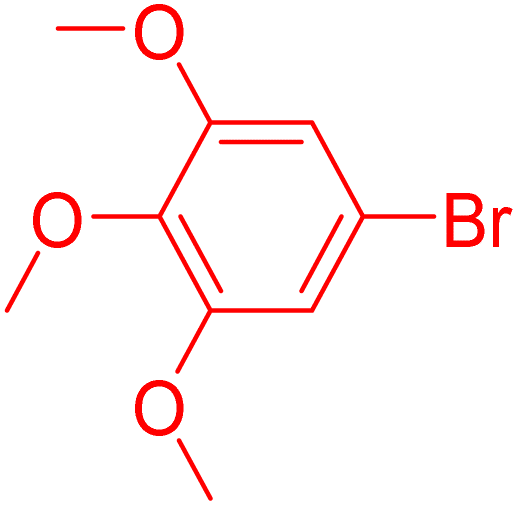
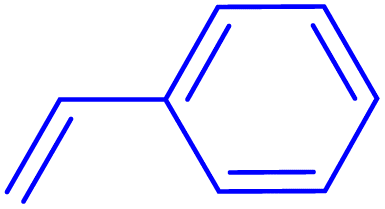
12 | 61 |
| 12 |
|
|
|
12 | 54 |
| 13 |
|
|
|
12 | 28b |
| 14 |
|
|
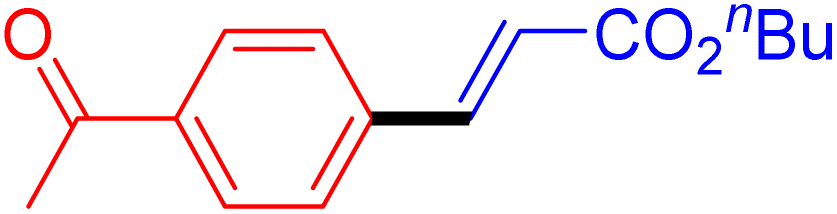
|
6 | 99b |
| 15 |
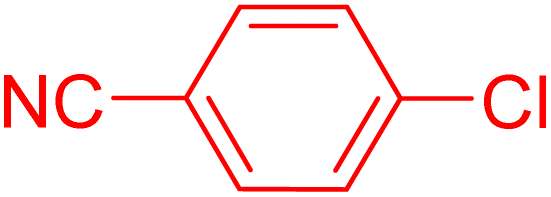
|
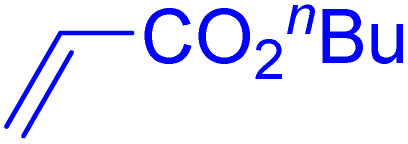
|
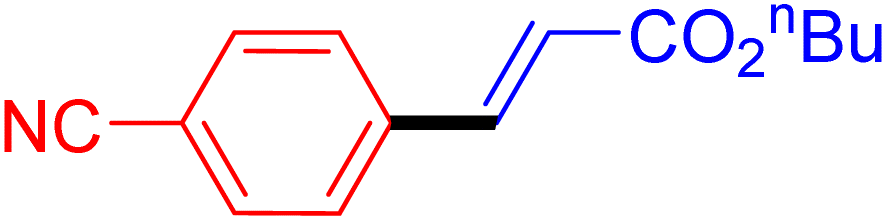
|
6 | 99b |
| 16 |
|
|
|
6 | 90b |
| 17 |
|
|
|
6 | 77b |
| 18 |
|
|
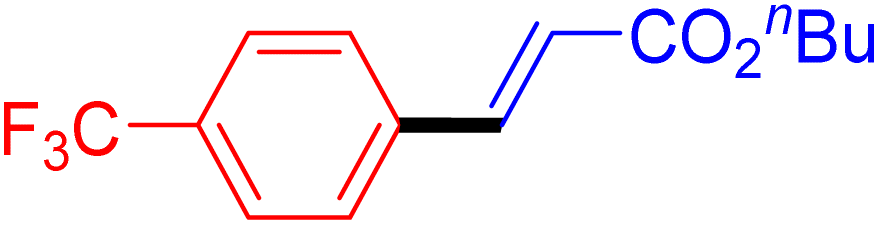
|
6 | 72b |
| 19 |
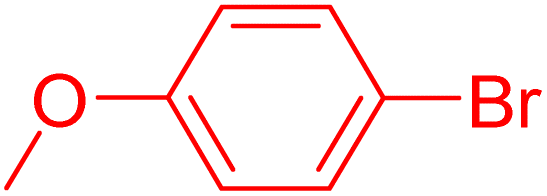
|
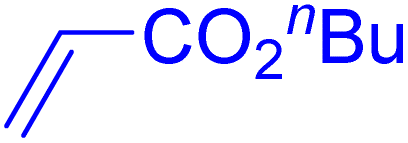
|
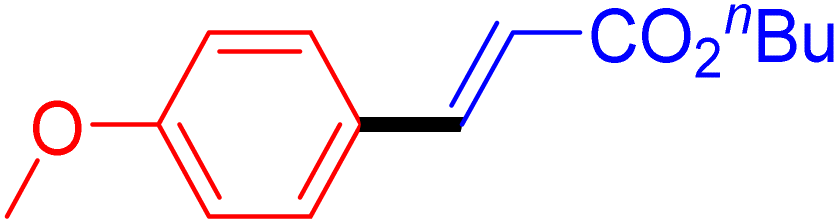
|
12 | 87b |
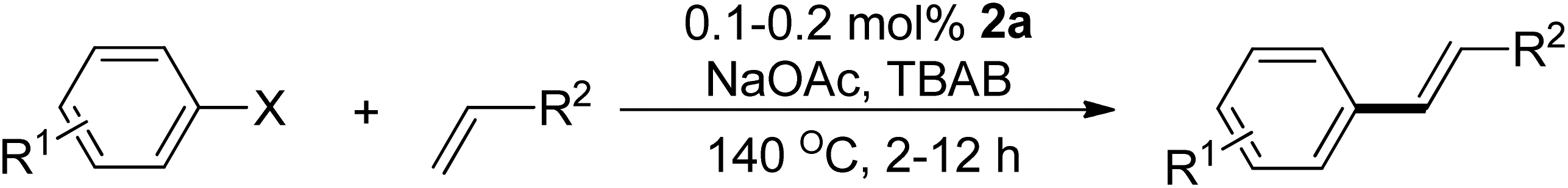
The catalytic performance of complex 2a was compared with other reported palladium complexes in the literature (Table 4). The selected catalyst systems were tested for catalyzing the coupling reaction between 4-chloroacetophenone and styrene. Turnover frequencies (TOFs) were estimated from reported data to compare the catalytic efficiency among different complexes, using the mole percentage of Pd loading, the reaction yield, and the reaction time. Complex 2a displayed the highest TOF of 475 h−1 among all the selected catalyst systems (entry 1). The ligand-free palladium acetate displayed the next highest TOF (395 h−1) (entry 2).50 The reported tetranuclear Pd aNHC complex was highly effective in utilizing electron-rich aryl chloride substrates.48 However, it shows a lower TOF value of 248 h−1 compared to complex 2a in the reaction with the electron-deficient substrate (entries 1 vs. 3). The reported C,C-type palladacycles also displayed a good TOF value of 235 h−1 (entry 4). All other catalyst systems, including Pd-NHC complexes such as PEPPSI complex PdCl2(IPr)(3-py),49 PdBr2(bis-NHC),51 and Pd-pyrimidine-based NHC,52etc., displayed much inferior TOF values, all below 100 h−1 (entries 5–12).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Complex type | Mol% Pd | Time (h) | Yield (%) | TOF (h−1) | Ref. |
| a NMR yield. b GC yield. c Isolated yield. | ||||||
| 1 | Palladalactam 2a | 0.1 | 2 | 95a | 475 | This work |
| 2 | Ligand-free Pd(OAc)2 | 0.1 | 2 | 79a | 395 | 50 |
| 3 | Tetranuclear Pd aNHC | 0.2 | 2 | 99a | 248 | 48 |
| 4 | C,C-type palladacycles | 0.2 | 2 | 94a | 235 | 50 |
| 5 | Oxime-based palladacycle | 0.5 | 2 | 100b | 100 | 53 |
| 6 | PdCl2/amino-chroman-4-one | 0.3 | 3.5 | 87c | 83 | 54 |
| 7 | PdCl2(IPr)(3-py) | 0.2 | 2 | 26a | 65 | 49 |
| 8 | PdBr2(bis-NHC) | 0.01 | 30 | — | 38 | 51 |
| 9 | Benzothiazole-oxime PdCl2 | 0.5 | 10 | 87b | 17 | 55 |
| 10 | PEPPSI-type Pd-NHC | 1 | 15 | >99b | 6.6 | 56 |
| 11 | Pd-pyrimidine-based NHC | 1 | 20 | 88b | 4.4 | 52 |
| 12 | [Pd2(Xantphos)2(4,4′-SC12H8S)]2(OTf)4 | 2 | 24 | 15a | 0.3 | 57 |
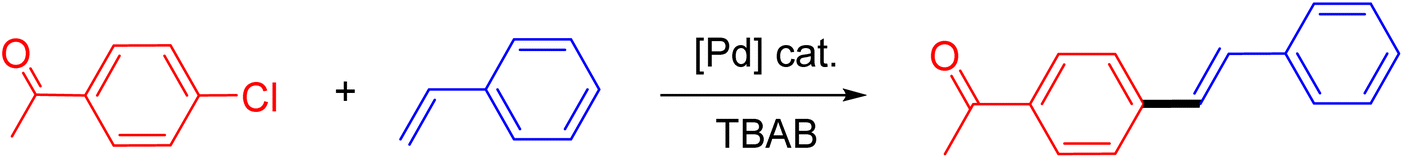
Computational studies
Finally, the Wiberg bond indices (WBIs) were calculated using natural bond orbital (NBO) analysis58 to determine the bond orders of the Pd–C and Pd–N bonds between the coordinated bidentate ligands and metal centers in compounds 2a–d (Table 5). Due to the strong electron-donating property of the PEt3 moiety, the Pd–C and Pd–N bonds in complex 2a are the strongest among the four complexes. The Pd–C bond in complex 2c is the second strongest. Furthermore, to understand the different donating abilities of the bidentate ligands, a charge decomposition analysis (CDA) was performed on [LPd0]− (L = bidentate ligand) (see also Table 5).59 [LPd0]− is believed to be the active species resulted from the in situ reduction of the Pd(II) pre-catalyst in the catalytic cycle. The CDA results confirmed that, again, the ligand containing PEt3 moiety in 2a is the most electron-donating, consistent with the NMR data described above.| Complexes | WBI (Pd–C) | WBI (Pd–N) | [LPd0]− | d | b | r | (d + b) | b/(d + b) |
|---|---|---|---|---|---|---|---|---|
| a At the M06-L/def2TZVP level. b Number of electrons donated from the ligand to the Pd atom. c Number of electrons donated from the Pd atom to the ligand. d Closed-shell interaction. | ||||||||
| 2a | 0.6269 | 0.4375 | [LaPd0]− | 0.192374 | 0.121512 | −0.138286 | 0.313886 | 38.7% |
| 2b | 0.6169 | 0.4330 | [LbPd0]− | 0.181107 | 0.142346 | −0.176867 | 0.323453 | 44.0% |
| 2c | 0.6214 | 0.4319 | [LcPd0]− | 0.180921 | 0.140253 | −0.176829 | 0.321174 | 43.7% |
| 2d | 0.6168 | 0.4271 | [LdPd0]− | 0.178887 | 0.131640 | −0.151839 | 0.310527 | 42.4% |
Conclusion
A new series of ligand precursors has been successfully developed to prepare palladalactams. These precursors, synthesized via a simple one-step reaction between 2-chloro-N-phenylacetamide and various monophosphines, offer a versatile platform for tailoring the electronic and steric properties of ligand frameworks. Additionally, their exceptional stability in ambient air enhances their practical applicability. The successful synthesis of a new palladalactam, incorporating an electron-donating triethylphosphine moiety, underscores the catalytic potential of these ligand systems, particularly in the Mizoroki–Heck coupling reaction. Theoretical calculations further support the superior electron-donating ability of this ligand, highlighting its role in forming robust Pd–C bonds. Our ongoing efforts in fine-tuning the ligand framework aim to optimize catalytic performance across a range of Pd-catalyzed reactions. This research sets the stage for the continued exploration and application of tailored ligand systems based on phosphonium ylides in homogeneous catalysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the National Science and Technology Council of Taiwan for the financial support of this work (112-2113-M-018-008).References
- H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1983, 22, 907–927 CrossRef.
- J. Vicente, M.-T. Chicote, I. Saura-Llamas, M.-J. López-Muñoz and P. G. Jones, J. Chem. Soc., Dalton Trans., 1990, 3683–3689, 10.1039/DT9900003683.
- L. R. Falvello, S. Fernández, R. Navarro, A. Rueda and E. P. Urriolabeitia, Inorg. Chem., 1998, 37, 6007–6013 CrossRef CAS PubMed.
- L. R. Falvello, S. Fernández, R. Navarro, A. Rueda and E. P. Urriolabeitia, Organometallics, 1998, 17, 5887–5900 CrossRef CAS.
- A. Spannenberg, W. Baumann and U. Rosenthal, Organometallics, 2000, 19, 3991–3993 CrossRef CAS.
- R. Zurawinski, B. Donnadieu, M. Mikolajczyk and R. Chauvin, J. Organomet. Chem., 2004, 689, 380–386 CrossRef CAS.
- L. R. Falvello, J. C. Ginés, J. J. Carbó, A. Lledós, R. Navarro, T. Soler and E. P. Urriolabeitia, Inorg. Chem., 2006, 45, 6803–6815 CrossRef CAS PubMed.
- S. J. Sabounchei, M. Hosseinzadeh, S. Salehzadeh, F. Maleki and R. W. Gable, Inorg. Chem. Front., 2017, 4, 2107–2118 RSC.
- E. Serrano, R. Navarro, T. Soler, J. J. Carbó, A. Lledós and E. P. Urriolabeitia, Inorg. Chem., 2009, 48, 6823–6834 CrossRef CAS PubMed.
- R. Taakili, C. Lepetit, C. Duhayon, D. A. Valyaev, N. Lugan and Y. Canac, Dalton Trans., 2019, 48, 1709–1721 RSC.
- R. Taakili, C. Barthes, C. Lepetit, C. Duhayon, D. A. Valyaev and Y. Canac, Inorg. Chem., 2021, 60, 12116–12128 CrossRef CAS PubMed.
- D. A. Valyaev and Y. Canac, Dalton Trans., 2021, 50, 16434–16442 RSC.
- D. A. Petrone, K. M. Szkop, L. Miao, P. St. Onge, Z.-W. Qu, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2021, 60, 18547–18551 CrossRef CAS PubMed.
- Y. Shi, B.-W. Pan, J.-S. Yu, Y. Zhou and J. Zhou, ChemCatChem, 2021, 13, 129–139 CrossRef CAS.
- M. El Kadiri, A. Chihab, R. Taakili, C. Duhayon, D. A. Valyaev and Y. Canac, Organometallics, 2022, 41, 456–466 CrossRef CAS.
- M. Ameskal, R. Taakili, E. S. Gulyaeva, C. Duhayon, J. Willot, N. Lugan, C. Lepetit, D. A. Valyaev and Y. Canac, Inorg. Chem., 2023, 62, 20129–20141 CrossRef CAS PubMed.
- T. Scherpf, C. Schwarz, L. T. Scharf, J.-A. Zur, A. Helbig and V. H. Gessner, Angew. Chem., Int. Ed., 2018, 57, 12859–12864 CrossRef CAS PubMed.
- P. Weber, T. Scherpf, I. Rodstein, D. Lichte, L. T. Scharf, L. J. Gooßen and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 3203–3207 CrossRef CAS PubMed.
- X.-Q. Hu, D. Lichte, I. Rodstein, P. Weber, A.-K. Seitz, T. Scherpf, V. H. Gessner and L. J. Gooßen, Org. Lett., 2019, 21, 7558–7562 CrossRef CAS PubMed.
- J. Tappen, I. Rodstein, K. McGuire, A. Großjohann, J. Löffler, T. Scherpf and V. H. Gessner, Chem. – Eur. J., 2020, 26, 4281–4288 CrossRef CAS PubMed.
- A. Sarbajna, V. S. V. S. N. Swamy and V. H. Gessner, Chem. Sci., 2021, 12, 2016–2024 RSC.
- S. Lapointe, A. Sarbajna and V. H. Gessner, Acc. Chem. Res., 2022, 55, 770–782 CrossRef CAS PubMed.
- W. A. Herrmann, K. Öfele, D. von Preysing and S. K. Schneider, J. Organomet. Chem., 2003, 687, 229–248 CrossRef CAS.
- E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- W. Henderson, J. Fawcett, R. D. W. Kemmitt, C. Proctor and D. R. Russell, J. Chem. Soc., Dalton Trans., 1994, 3085–3090, 10.1039/DT9940003085.
- W. Henderson, B. K. Nicholson and A. G. Oliver, Polyhedron, 1994, 13, 3099–3104 CrossRef CAS.
- W. Henderson, A. G. Oliver, C. E. F. Rickard and L. J. Baker, Inorg. Chim. Acta, 1999, 292, 260–265 CrossRef CAS.
- J.-Y. Lee, Y.-H. Huang, S.-Y. Liu, S.-C. Cheng, Y.-M. Jhou, J.-H. Lii and H. M. Lee, Chem. Commun., 2012, 48, 5632–5634 RSC.
- S.-J. Chen, Y.-D. Lin, Y.-H. Chiang and H. M. Lee, Eur. J. Inorg. Chem., 2014, 1492–1501 CrossRef CAS.
- M. Oestreich, The Mizoroki–Heck Reaction, John Wiley & Sons, Ltd, 2009 Search PubMed.
- K. R. Balinge and P. R. Bhagat, C. R. Chim., 2017, 20, 773–804 CrossRef CAS.
- P. J. Anju, M. Neetha and G. Anilkumar, ChemistrySelect, 2022, 7, e202103564 CrossRef CAS.
- T. Mino, Y. Shirae, Y. Sasai, M. Sakamoto and T. Fujita, J. Org. Chem., 2006, 71, 6834–6839 CrossRef CAS PubMed.
- G.-R. Peh, E. A. B. Kantchev, C. Zhang and J. Y. Ying, Org. Biomol. Chem., 2009, 7, 2110–2119 RSC.
- S. Inomata, H. Hiroki, T. Terashima, K. Ogata and S.-i. Fukuzawa, Tetrahedron, 2011, 67, 7263–7267 CrossRef CAS.
- A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2017, 36, 1981–1992 CrossRef CAS.
- D. Borah, B. Saha, B. Sarma and P. Das, J. Chem. Sci., 2020, 132, 51 CrossRef CAS.
- S. Chakrabortty, M. Kaur, M. Adhikari, K. K. Manar and S. Singh, Inorg. Chem., 2021, 60, 6209–6217 CrossRef CAS PubMed.
- O. Bysewski, A. Winter and U. S. Schubert, Inorganics, 2023, 11, 164 CrossRef CAS.
- R. O. Pankov, D. O. Prima, A. Y. Kostyukovich, M. E. Minyaev and V. P. Ananikov, Dalton Trans., 2023, 52, 4122–4135 RSC.
- S. Stallinger, C. Reitsamer, W. Schuh, H. Kopacka, K. Wurst and P. Peringer, Chem. Commun., 2007, 510–512, 10.1039/B609723E.
- B. S. Aweke, C.-H. Yu, M. Zhi, W.-C. Chen, G. P. A. Yap, L. Zhao and T.-G. Ong, Angew. Chem., Int. Ed., 2022, 61, e202201884 CrossRef CAS PubMed.
- J.-Y. Lee, Y.-S. Su, Y.-S. Wang and H. M. Lee, Adv. Synth. Catal., 2019, 361, 4714–4726 CrossRef CAS.
- C.-H. Hung, W.-Y. Zheng and H. M. Lee, Organometallics, 2021, 40, 702–713 CrossRef CAS.
- C. H. Lo and H. M. Lee, Organometallics, 2018, 37, 1150–1159 CrossRef CAS.
- M. A. Taige, A. Zeller, S. Ahrens, S. Goutal, E. Herdtweck and T. Strassner, J. Organomet. Chem., 2007, 692, 1519–1529 CrossRef CAS.
- J. Ye, W. Chen and D. Wang, Dalton Trans., 2008, 4015–4022, 10.1039/B801264D.
- D. A. Alonso, C. Nájera and M. C. Pacheco, Adv. Synth. Catal., 2002, 344, 172–183 CrossRef CAS.
- D. Khan and I. Parveen, Eur. J. Org. Chem., 2021, 4946–4957 CrossRef CAS.
- K. M. Dawood, Tetrahedron, 2007, 63, 9642–9651 CrossRef CAS.
- Y.-C. Lin, H.-H. Hsueh, S. Kanne, L.-K. Chang, F.-C. Liu, I. J. B. Lin, G.-H. Lee and S.-M. Peng, Organometallics, 2013, 32, 3859–3869 CrossRef CAS.
- P. A. Mane, S. Neogy and S. Dey, Appl. Organomet. Chem., 2020, 34, e5405 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, 2001 Search PubMed.
- S. Dapprich and G. Frenking, J. Phys. Chem., 1995, 99, 9352–9362 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2340510–2340517. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00862f |
| This journal is © The Royal Society of Chemistry 2024 |