
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA detailed density functional theory exploration of the photodissociation mechanism of ruthenium complexes for photoactivated chemotherapy†
Daniele
Belletto
,
Fortuna
Ponte
*,
Gloria
Mazzone
 and
Emilia
Sicilia
*
and
Emilia
Sicilia
*
Department of Chemistry and Chemical Technologies, Università della Calabria, Ponte P. Bucci, 87036 Arcavacata di Rende, CS, Italy. E-mail: emilia.sicilia@unical.it; fortuna.ponte@unical.it
First published on 17th April 2024
Abstract
Polypyridyl Ru(II) complexes have attracted much attention due to their potential as light-activatable anticancer agents in photoactivated chemotherapy (PACT). The action of ruthenium-based PACT compounds relies on the breaking of a coordination bond between the metal center and an organic ligand via a photosubstitution reaction. Here, a detailed computational investigation of the photophysical properties of a novel trisheteroleptic ruthenium complex, [Ru(dpp)(bpy)(mtmp)]2+ (dpp = 4,7-diphenyl-1,10-phenanthroline, bpy = 2,2′-bipyridine and mtmp = 2-methylthiomethylpyridine), has been carried out by means of DFT and its time-dependent extension. All the aspects of the mechanism by which, upon light irradiation, the mtmp protecting group is released and the corresponding aquated complex, able to bind to DNA inducing cell death, is formed have been explored in detail. All the involved singlet and triplet states have been fully described, providing the calculation of the corresponding energy barriers. The involvement of solvent molecules in photosubstitution and the role played by pyridyl–thioether chelates as caging groups have been elucidated.
Introduction
Precise spatial and temporal control of the activity of externally applied chemical agents in their interaction with complex biological systems for therapeutic purposes is of special significance aiming at achieving a greater action specificity. Even though a large number of drugs able to be activated by specific stimuli, such as redox, pH, enzyme and temperature, have been proposed,1 differences in the cellular environment are very narrow and extremely sensitive compounds are required. Intense focused light sources accessible today, instead, make photoactivation a very attractive alternative and therapies relying on the use of chemotherapeutic compounds that respond to light of appropriate wavelength are currently the object of a great deal of attention. The term “photoactivated chemotherapy” (PACT) was proposed by Sadler et al. in 20092,3 and has been used since then to identify therapeutic strategies in oncology based on the use of biologically active metal complexes acting as prodrugs able to exert their cytotoxic action when irradiated. This kind of approach should overcome drawbacks such as dose escalation, drug resistance, and severe side effects of the drugs that are currently in use, which lack specificity and have low bioavailability.4,5 Three main modes of activation for inorganic compounds used in PACT are usually taken into consideration: (i) photoreduction in the cellular reducing environment to produce the corresponding reduced active complexes together with the released ligands; (ii) photosubstitution leading to a ligand replaced by a solvent molecule, typically water; (iii) photorelease of small biological effector molecules inactivated by coordination to a metal centre.PACT is often compared to photodynamic therapy (PDT) since both therapies rely on the use of light for activating drugs that are relatively non-toxic in the dark. Nevertheless, these two classes of treatments present many differences.6 PDT agents are photostable, whereas PACT agents change their chemical formula upon irradiation, and it is worth noting that the major advantage of PACT over PDT is the oxygen-independent mechanism of action of the drugs that enhances their efficiency in hypoxic environments such as those of tumours.
Transition metal containing complexes, owing to their rich photophysics and photochemistry that can also be tuned properly by varying the metal centre and/or ligands, are considered very promising compounds to be used in PACT given their capacity to modify their structure upon light irradiation. The ideal candidate should be a prodrug stable and nontoxic to cells in the dark, but activatable within tumour tissues by light of relatively long wavelengths, and able to both penetrate in depth and minimize photodamage of endogenous compounds. Metal complexes including Pt(IV),7 Re(I),8 and Ir(III)9 together with Rh(III)10 and Ru(II)11,12 have all been investigated extensively for this purpose. Photoactivatable Ru(II)-centred complexes, in particular polypyridyl ruthenium compounds, emerged as promising candidates owing to their low ligand exchange propensity observed in the dark, together with their high photoreactivity. Various studies have reported that they may undergo photosubstitution reactions, mediated by solvent molecules, forming photoproducts having significantly higher cytotoxicity compared to the precursor complex.13–16 These resultant “uncaged” metal Ru(II) complexes can bind DNA in a manner similar to cisplatin, thus inducing cell death.17
Bonnet and co-workers have recently presented a new ruthenium complex, [Ru(dpp)(bpy)(mtmp)]2+ (Scheme 1), bearing three different bidentate chelating ligands: dpp (4,7-diphenyl-1,10-phenanthroline) and bpy (2,2′-bipyridine), which are non-photocleavable spectator groups, and an mtmp ligand (2-methylthiomethylpyridine).18 This complex will be called Ru complex from now on. The advantage of using thioether ligands is that their softness allows efficient coordination to Ru(II) in the ground state, while their photosubstitution efficiency is greater than that of pyridines as a consequence of the relative weakness of the Ru–S bond compared to the Ru–N one in the excited state.19 The mtmp ligand, indeed, acts as a photolabile group, which is photosubstituted by solvent molecules under green light irradiation generating the active DNA photobinding agent.18
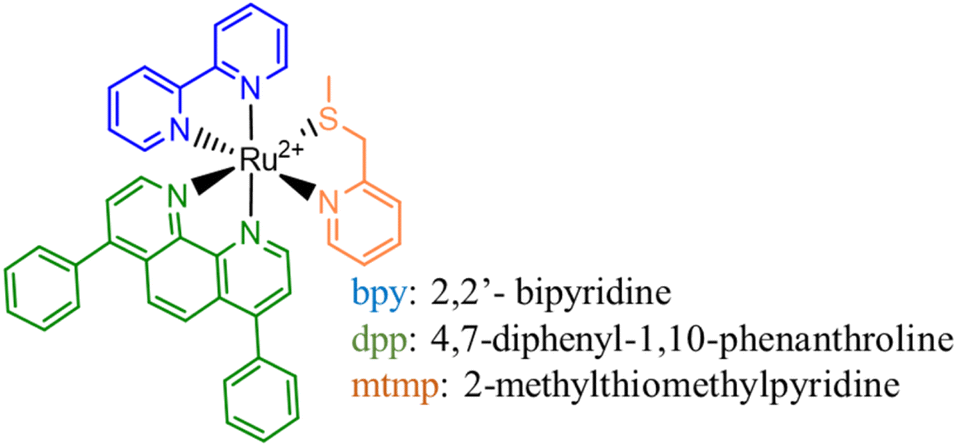 | ||
| Scheme 1 Schematic representation of the structure of the Ru(II) complex under investigation. | ||
Stimulated by the experimentally reported efficacy of such a novel synthesized PACT agent, a detailed computational investigation of the photophysical properties of the complex has been carried out by means of density functional theory (DFT). Over the course of years, new information has been added through the use of computational tools to the description of the mechanism of photosubstitution for the detachment of both mono- and bi-dentate ligands and to the rationalization of the observed behaviours in terms of the involved triplet excited states.20–24 Here, the outcomes of our computational analysis are reported aiming at helping the comprehension of the described phenomena and the synthesis of new efficacious PACT agents. All the mechanistic aspects of the photodissociation process have been explored in detail and all the involved singlet and triplet states have been fully described with the aim to contribute to addressing two of the unresolved issues that need clarification in order to ultimately design efficacious Ru complexes suitable for PACT application. One aim is general and concerns the involvement of solvent molecules in photosubstitution and the other is specific about the excellent role played by pyridyl–thioether chelates as caging groups.
Results and discussion
The fully optimized ground state (GS) structure of the Ru complex, in water solvent and without imposing any symmetry constraint, is shown in Fig. S1 of the ESI† along with the main structural parameters. The trisheteroleptic complex adopts a distorted octahedral geometry. The average Ru–N(bpy) and Ru–N(dpp) bond lengths are 2.102 and 2.120 Å, respectively, whereas the Ru–N(mtmp) bond length is 2.150 Å. The Ru–S bond distance is significantly longer by about 0.25 Å. The bpy and dpp bite angles are 77.8 degrees, while that of the mtmp ligand is 81.1 degrees.Photophysical properties of the Ru complex
In order to properly describe the photophysical processes accounting for PACT action, a preliminary benchmarking of the exchange and correlation functionals has been carried out. A superposition of the Ru complex simulated spectra and the experimental one and a comparison of the calculated maximum absorption wavelengths with those experimentally available, obtained in acetonitrile solvent, are provided in Fig. S2 and Table S1 of the ESI,† respectively. From the reported data, the M05 functional emerges as the functional that better simulates the experimental feature and it has been employed, hence, for its complete characterization: both the examination of the photochemical characteristics and the light-induced reaction mechanisms.The UV-vis spectrum of the Ru complex has been thus recorded in the solvent that could better mimic the physiological environment, that is water, and is reported in Fig. 1, where the most important transitions are included.
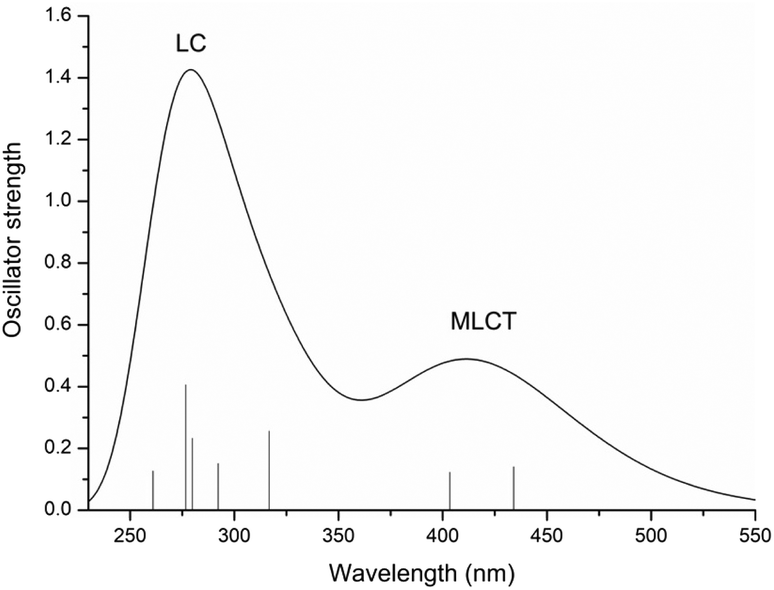 | ||
| Fig. 1 Calculated UV-vis absorption spectrum of the Ru complex in water at the M05/6-31G** (SDD for Ru) level of theory. | ||
The main data describing the absorption spectrum of Ru are collected in Table S2 of the ESI.† Only the electronic transitions with an oscillator strength (f) higher than 0.01, considered to be essential for determining the different absorption bands, are reported. The only exception is the reported first transition, for the S1 state. The composition percentage of molecular orbitals, for each of the listed transitions, is also included. Natural Transition Orbital (NTO) plots25 for the most important transitions of the spectrum are shown in Fig. S3.† For the Ru complex, the calculated absorption spectrum displays essentially two bands. A first weaker absorption band, of appreciable intensity, is observed in the range of 350–500 nm and centred at 434 nm, a value that reproduces very well the maximum absorption wavelength of 430 nm experimentally detected.15 TDDFT calculations assign this transition to the S5 singlet excited state and the analysis carried out with the TheoDORE (theoretical density, orbital relaxation and exciton analysis) package26 confirms a band with the MLCT character. Indeed, all the electronic transitions originating from such a band involve a charge transfer from a molecular orbital primarily located on the metal (i.e. HOMO (H) and H-2) to an orbital outspread on one of the two N^N ligands, either bpy or dpp. In particular, the excitation to the S1 state, calculated at 486 nm (Tr1), originates from the mixed transitions H → L + 1 (61%) and H → L (33%) that mainly involve the bpy ligand. The most intense peak at higher energy, between 250 and 350 nm, is found at 275 nm. Despite a little contribution of the type MLCT, the band is characterized by electronic transitions that are mainly centred on the ligand, as confirmed by the NTOs and TheoDORE analysis.
Excited states study
Considering that the lifetime of singlet excited states is approximately five times shorter than that of triplet states (10−8vs. 10−3 s), it seems reasonable to suggest that the pathway for converting the system under investigation into a diaquo species may entail an excited triplet state instead of a singlet one. Specifically, for Ru-based PACT agents, it is generally accepted that photoactivated dissociation and solvolysis occur due to the population of a dissociative triplet metal-centred 3MC state from a triplet metal–ligand charge transfer 3MLCT state that lies close enough in energy. Such an 3MLCT state, in turn, is generated by a metal-to-ligand charge-transfer singlet excited state (1MLCT) that efficiently spin–flips to a triplet one.27–301MLCT to 3MLCT transitions are usually very fast and thermally non-activated, whereas 3MLCT to 3MC conversions can entail the overcoming of an activation barrier and take time. With Kasha's rules in mind,31 it seems reasonable to hypothesize that when the S5 state, identified as the bright state, is populated by photon absorption, an internal conversion can occur before the intersystem crossing (ISC) process with consequent population of the first singlet excited state S1. From here a spin–flip is required to achieve the PACT active state, a triplet state. In order to properly identify and characterize the triplet states responsible for the photodissociation process and then describe the photoactivity of Ru, a primary investigation, within the vertical approximation, of the photophysical features of the triplet states below the S1 and T1–T6 has been performed. The energies and the characteristics of the selected six triplet states are reported in Table S3 of the ESI,† whereas the NTO plots for these triplet excited states are shown in Fig. S4.† While the first four triplet states share the mixed characteristics of LC/MLCT, the last two exhibit a certain contribution of the MC characteristic together with the CT from the metal to the ligand. To establish the feasibility of ISC processes from S1 to the triplet manifold, the spin–orbit matrix elements and the energy difference between the singlet and triplet states lying below have been calculated and are reported in Table 1. Looking at these data, it is worth noting that the triplet states show a common behaviour, independent of their characteristics, and that the influence of the metal is evident from the values of the SOC elements computed for all the S1 → Tn (n = 1–6) radiationless transitions. Therefore, based on the high SOC values, it is plausible to suggest that the transition to the lowest singlet excited state (S1) is followed by an efficient ISC to the triplet manifold.Once the feasibility of ISC processes is established, the structures of the involved singlet and triplet excited states have been optimized within the TDDFT approximation in order to properly characterize the triplet states and compute the effective energy splitting between the coupled states. The structures obtained for the triplet states have been subsequently used as starting points for the optimization within the unrestricted Kohn–Sham formalism (UKS),34,35 as it is reported to help in reducing the instability of excited triplet states involving charge transfer. The analysed triplet excited states converged into four distinct states denoted as Ta, Tb, Tc and Td. Their optimized structures, together with the corresponding spin density distributions, were used to determine their characteristics and are reported in Fig. 2, where the spin density values on the Ru atom and the relative energies calculated with respect to the ground state GS are also included. Furthermore, a comparison of the main bond distances of the calculated triplet states with those of the GS is reported in Table S4.† Cartesian coordinates of GS and Ta, Tb, Tc and Td triplet states are collected in Table S5.† As it clearly appears, in all the intercepted triplet structures, the bonds with the mtmp group are lengthened, supporting the dissociative characteristic of these triplets leading to the detachment of the S,N-donor ligand. In particular, the structures of the triplet states Ta and Tb show modest differences compared to that of the ground state (see Table S4†), while the optimized triplet excited states Tc and Td have been found to be dissociative with respect to the mtmp ligand and are responsible for the photoreactivity of this complex.
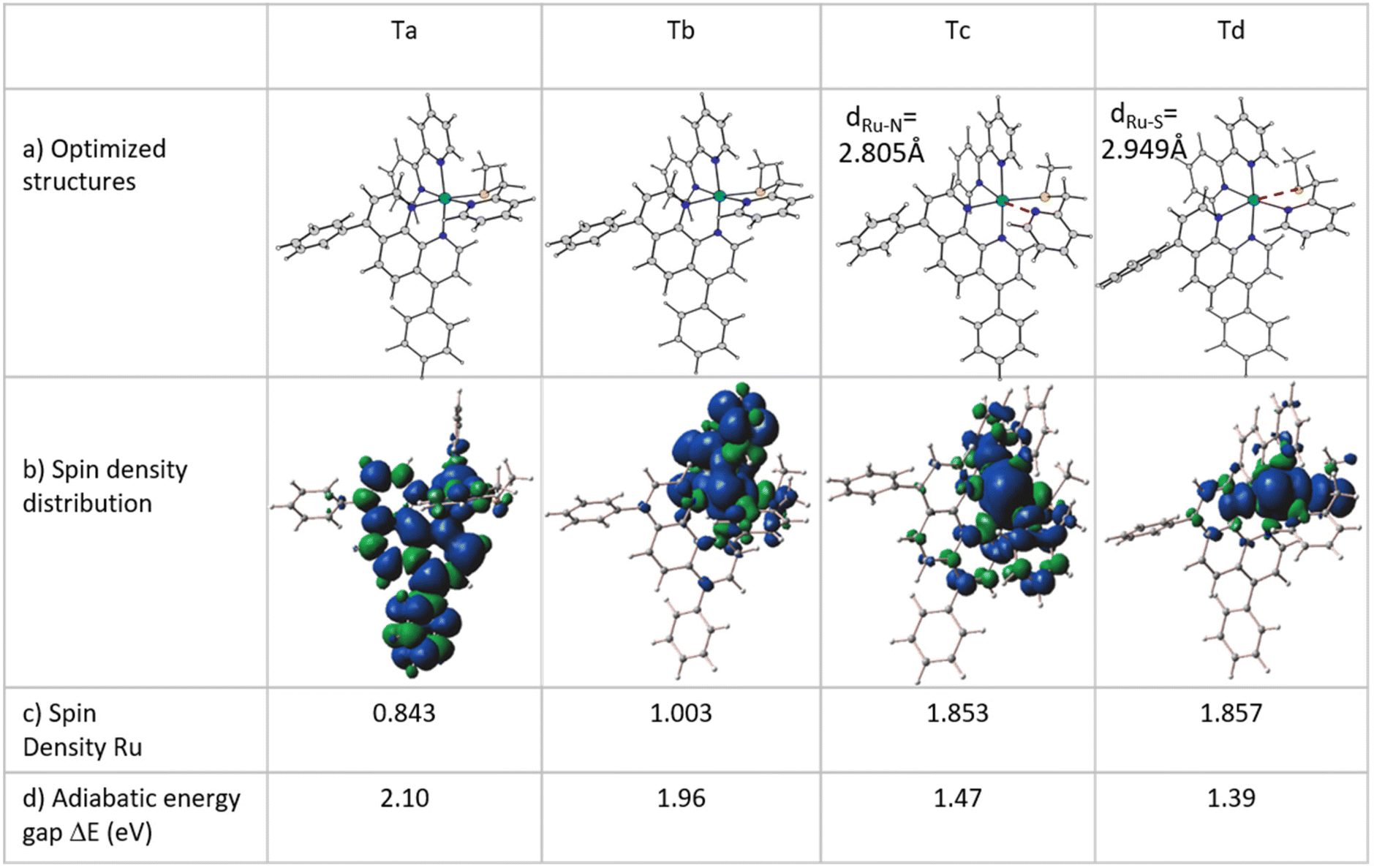 | ||
| Fig. 2 (a) Structure of optimized triplet states Ta, Tb, Tc and Td within the unrestricted DFT formalism. (b) Computed spin density distributions of the intercepted triplet states. (c) Spin density values (SD in a.u.) on the ruthenium metal center. (d) Energy gap between the optimized triplet states and the S0 ground state. | ||
Tc and Td triplet states appear to be characterized by a significant reorganization in the coordination sphere of the Ru centre. In Tc, the detachment of the pyridyl arm of the mtmp ligand takes place with an increase in the Ru–N bond distance of about 0.7 Å and a deviation of 67.9 degrees with respect to the plane containing the metal. This rearrangement is accompanied also by an increase in the length of the Ru–N bond in the trans position by about 0.3 Å. The lowest energy triplet state Td is characterized, instead, by a clear elongation of the Ru–S bond and shows a large deviation from the GS structure with a strong distortion of the metal coordination sphere. The Ru–S distance increases by approximately 0.6 Å and the pendant arm containing the sulphur donor atom lies at 51.1 degrees with respect to the plane containing the metal centre, while the trans Ru–N bond lengthens by more than 0.3 Å. All the obtained triplet states have been properly characterized and all the possible photophysical processes examined. Looking at Fig. 2b, the spin density distribution plot confirms the MLCT characteristic of the Ta and Tb triplets.
The Mulliken spin density near unity on the Ru(II) centre is indicative of one unpaired electron on the metal centre in the excited state as it should be expected for the 3MLCT state. In contrast, the spin density values of about 1.9 a.u. for both Tc and Td triplet states show that the unpaired electrons are essentially localized at the Ru centre as for a triplet with 3MC nature. This nature of both Tc and Td states facilitates ligand photodissociation. The relative energies of the photoactivated excited states follow the order S1 (1MLCT) > Ta (3MLCT) > Tb (3MLCT) > Tc (3MC) > Td (3MC). The Ru complex shows more stable metal-centred excited states, 3MC states, than the 3MLCT triplet state, a characteristic that enhances the probability of an internal conversion (3MLCT → 3MC state). Relaxed scans have been performed on the triplet potential energy surfaces to determine the barriers for the conversion from the Tb state to the Tc and Td ones. The corresponding relaxed potential energy surfaces are shown in Fig. 3 as obtained by varying the S1–C2–C3–N4 and N4–C3–C2–S1 dihedral angles by −2.0 degrees for Tc and Td states, respectively. In both cases, an abrupt transition from the 3MLCT state to the 3MC one is observed for an estimated barrier of 7.0 kcal mol−1 along the path for the Tc state formation and of 4.5 kcal mol−1 along the path for the Td state generation. This behaviour indicates that the electron density transition does not occur smoothly. Therefore, we can assume that when the dihedral angles change and the complex geometry is distorted, the involved orbitals don't mix.36
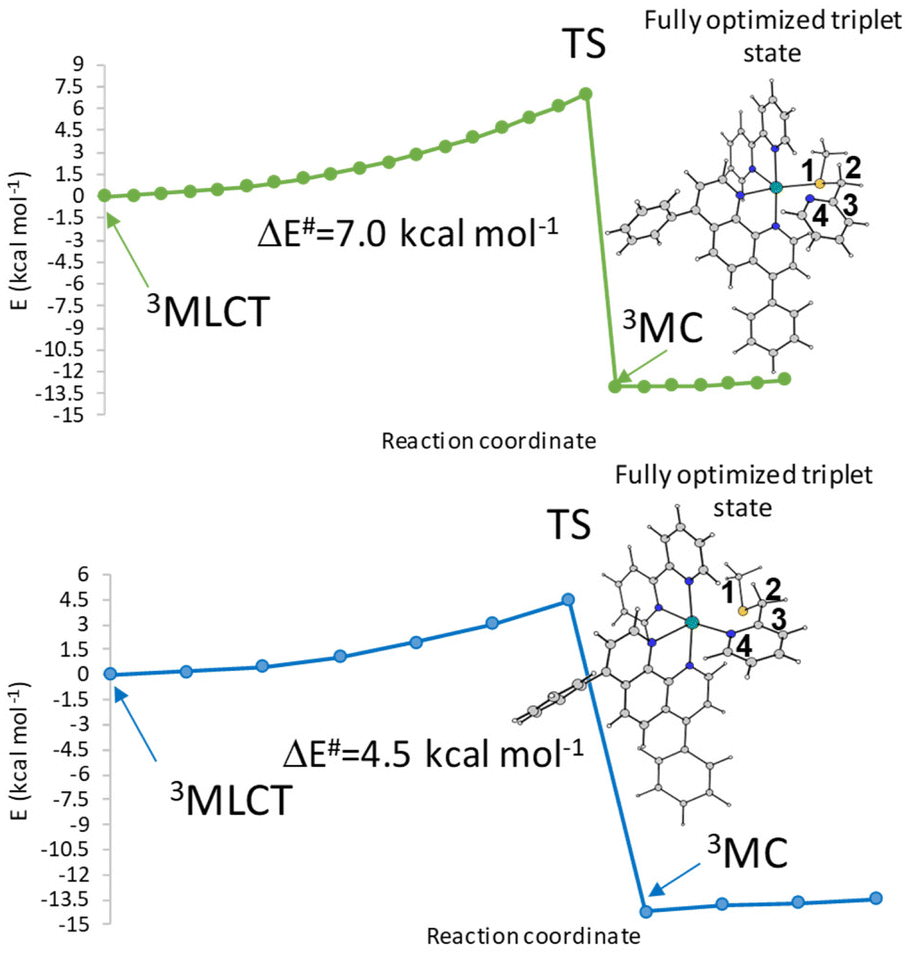 | ||
| Fig. 3 Relaxed potential energy scans from the 3MLCT state to 3MC (a) Tc and (b) Td of the Ru complex calculated by varying the S1–C2–C3–N4 and N4–C3–C2–S1 dihedral angles (°). | ||
The simplified Jablonski diagram that represents the key photophysical processes in the Ru complex is reported in Fig. 4. Looking at the energy gaps S1–Tn, the triplet state Ta (3MLCT) that lies at an energy close to the S1 state (1MLCT) could be populated through an ISC mechanism. The intersystem crossing process can be followed by a fast internal conversion and the triplet Tb can be populated. The state Tb (3MLCT) can either return to the ground state by radiative deactivation or it can evolve into a nonradiative triplet metal-centred state (3MC) Tc or Td, lying below the Tb (3MLCT) state, leading to ligand dissociation.
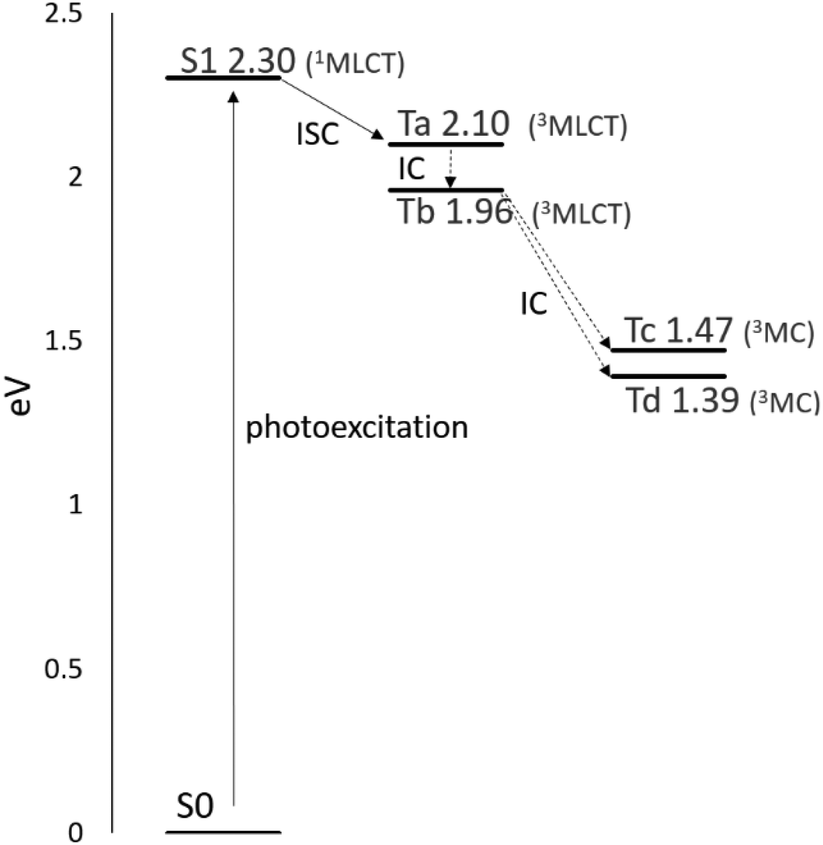 | ||
| Fig. 4 Simplified Jablonski diagram depicting the key photophysical processes for the Ru complex: photoexcitation, intersystem crossing (ISC), and internal conversion (IC). The diagram highlights that 3MC states fall below the 3MLCT states. | ||
A strategy commonly used to enhance the photosubstitution efficiency that decreases the 3MLCT-3MC energy gap or even inverts their energy order to favor the population of the 3MC states consists of introducing steric strain on the dissociating ligand.37–39 Alternatively, electronic effects can elicit photosubstitution, as assumed in the present case, and for some ligands, this effect is due to the 3MLCT state destabilization.40 Since for pyridyl–thioether chelates no indication has been given about their influence on photosubstitution, the analysis of the molecular orbitals for the Ru complex has been compared with that of other three complexes, which are [Ru(bpy)3]2+, [Ru(phen)(bpy)2]2+ with phen = 1,10-phenanthroline, and [Ru(mtmp)(bpy)2]2+ (Fig. 5). The three highest occupied molecular orbitals (HOMOs) are, as expected, metallic t2g orbitals (dxy, dxz, dyz) and are separated from the lowest unoccupied molecular orbital (LUMO) by an energy gap of 3.82 eV. The LUMOs are composed mainly of ligand centered π* orbitals. Specifically, the π* orbitals of the bpy and dpp ligands are involved. Higher in energy lie the metal-centered (eg*) vacant MOs (dσ*) showing σ-antibonding interactions between the dx2−y2 and dz2 orbitals of the metal and the S and N atoms of the mtmp ligand. On going from the reference [Ru(bpy)3]2+ to [Ru(phen)(bpy)2]2+, it is possible to deduce that the presence of the phen ligand does not introduce any significant change, except for a further degeneration removal of the HOMO d orbitals. The comparison between [Ru(phen)(bpy)2]2+ and [Ru(mtmp)(bpy)2]2+, instead, shows clearly that substitution of the bpy ligand with mtmp causes a slight increase of the HOMO–LUMO energy gap due to slight stabilization of the HOMO and, very importantly, stabilization of the vacant MOs (dσ*) that are primarily dx2−y2 and dz2-like orbitals responsible for the generation of the 3MC states. A very similar diagram is obtained when the Ru MOs are examined involving stabilization of the 3MC states, which accelerates the photosubstitution reaction.
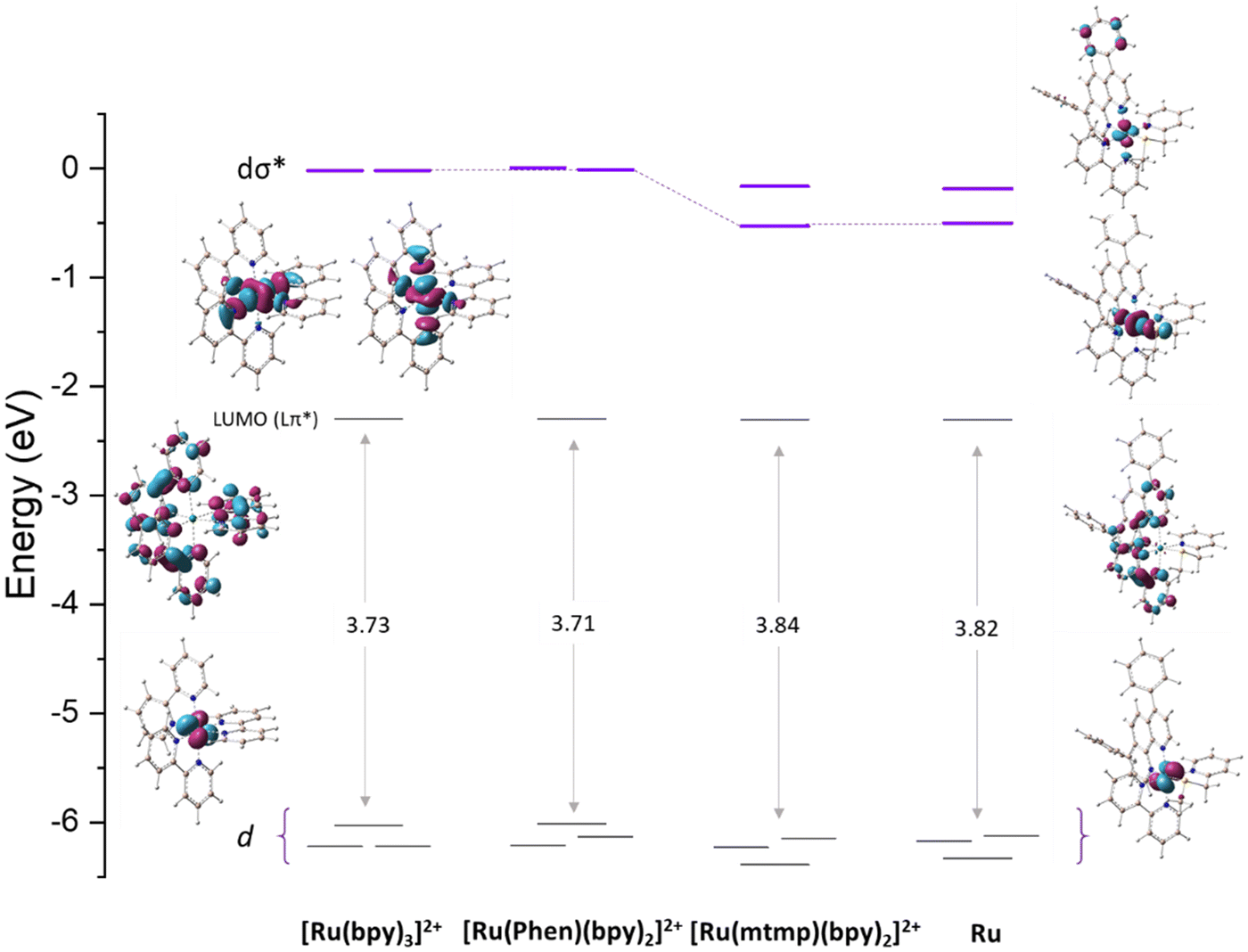 | ||
| Fig. 5 Energy diagram of the molecular orbitals of the Ru complex compared with those of [Ru(bpy)3]2+, [Ru(phen)(bpy)2]2+ (phen = phenanthroline), and [Ru(mtmp)(bpy)2]2+ with representative plots of HOMOs, LUMOs and dσ* orbitals. | ||
Mechanism of Ru complex photodissociation
When exposed to visible light, ruthenium complexes undergo photochemical substitution reactions, where one of the ligands in the coordination sphere of the metal centre is replaced by solvent molecules. Over the years, the intricate nature of these systems has limited the exploration of their photoactivation mechanism. Particularly, from a theoretical perspective, the investigation of excited triplet states involves their challenging optimization.Here, we report the detailed computational analysis of all the steps of the photoactivation mechanism of a ruthenium complex, including those leading to the formation of the final active solvated complexes, simulated with water molecules used to mimic the physiological environment. It is worth underlining that the triplet pathway has been built up starting from the optimized structures of the triplet states lying below the bright state and the transition states located and checked for their connection with the appropriate minima. Explicit water molecules have been included in the model and the transition states for their coordination have been intercepted. Based on all the data described above and the experimental results,18 two viable pathways for the activation by light of the Ru complex, shown in Scheme 2, can be proposed involving the previously described 3MLCT (Ta, Tb) and 3MC (Tc, Td) states.
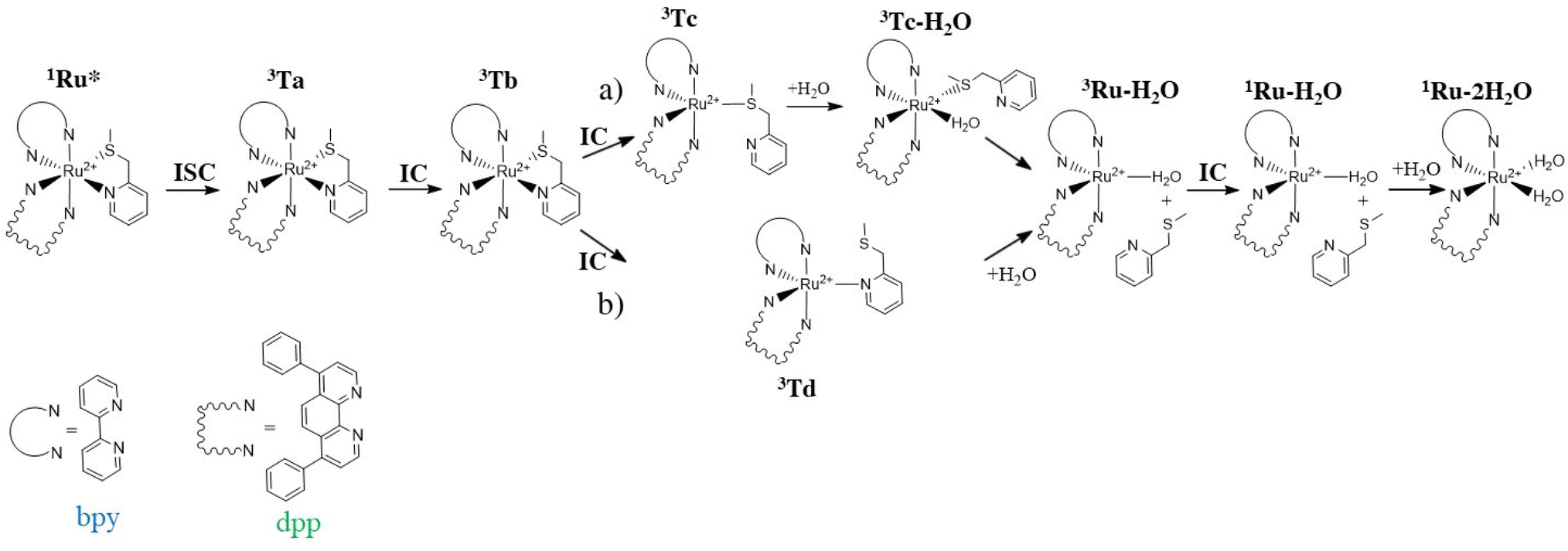 | ||
| Scheme 2 Proposed activation pathways of the Ru complex involving both 3MC (a) Tc and (b) Td states as intermediates for the formation of biaquated photoproducts having cytotoxic activity. | ||
Scheme 2 describes the whole process by which the biaquated photoproducts, the real cytotoxic species, are generated through photoinduced ligand dissociation. The excitation, taking place when the Ru complex is exposed to light of appropriate wavelength, leads to the population of the 1MLCT state (1Ru*), followed by an efficient ISC process that allows the formation of the excited 3MLCT state Ta which could contribute to populate the Tb state by internal conversion, which represents a key intermediate in the complex photoactivation reaction. The second step consists of ligand dissociation. The triplet Tb can be deactivated by the population of the nearby 3MC states with dissociative characteristics.
As stated above, two distinct 3MC states, named Tc and Td, have been identified for the Ru complex. It is interesting to note that in both 3MC states, one of the arms of the mtmp ligand is almost detached, resulting in the formation of an intermediate to be considered five-coordinated. From here, the reaction path bifurcates for the formation of a monoaquated complex that occurs in the next step. Along the pathway named (a), after the formation of the intermediate triplet Tc, the reaction proceeds with a stepwise ligand substitution. In the first step, a water molecule enters the coordination sphere of the metal, thereby restoring hexa-coordination to the ruthenium atom (3Tc–H2O). In the subsequent second step, the ruthenium mono-aquated complex named 3Ru–H2O is generated with the definitive detachment of the mtmp ligand. Along the triplet pathway named (b), from the intermediate triplet Td state, the formation of the mono-aquated ruthenium complex 3Ru–H2O occurs in one step through an associative ligand substitution of the mtmp ligand by a water molecule. Both triplet states, Tc and Td, produce the same intermediate, 3Ru–H2O. The last step of the process, that is the formation of the biaquated complex, takes place along a ground state singlet path. Indeed, after the formation of the mono-aquated species, the coordination complex may return to its ground state (1Ru–H2O) and the coordination of the second water molecule can occur forming the final photoproduct 1Ru–2H2O, the desired cytotoxic species. The calculated energy profiles for both processes represented in Scheme 2 are shown in Fig. 6. The intercepted structures of transition states are reported in the same figure, while the Cartesian coordinates of all the stationary points are reported in Table S6.† As depicted in Fig. 6, the hydrolysis process for the triplet state Tc proceeds in two distinct steps. The initial step of the nucleophilic substitution mechanism involves the rapid entry of a water molecule overcoming a very low activation energy barrier of 1.1 kcal mol−1 (TS1c). The water molecule saturates the metal coordination sphere, resulting in the formation of an intermediate with octahedral geometry (Tc–H2O). In the second step, the mtmp ligand leaves the metal coordination sphere and the pentacoordinated ruthenium complex denoted as the 3Ru–H2O complex is formed, overcoming an energy barrier of 6.5 kcal mol−1 (TS2c). The hydrolysis process for the triplet state Td, instead, takes place in a single step in which a water molecule approaches the pentacoordinated complex, and going through the transition state TSd, it replaces the mtmp ligand in an associative manner. As anticipated, the formation of the mono-aquated complex occurs, overcoming an energy barrier of 8.4 kcal mol−1 (TSd).
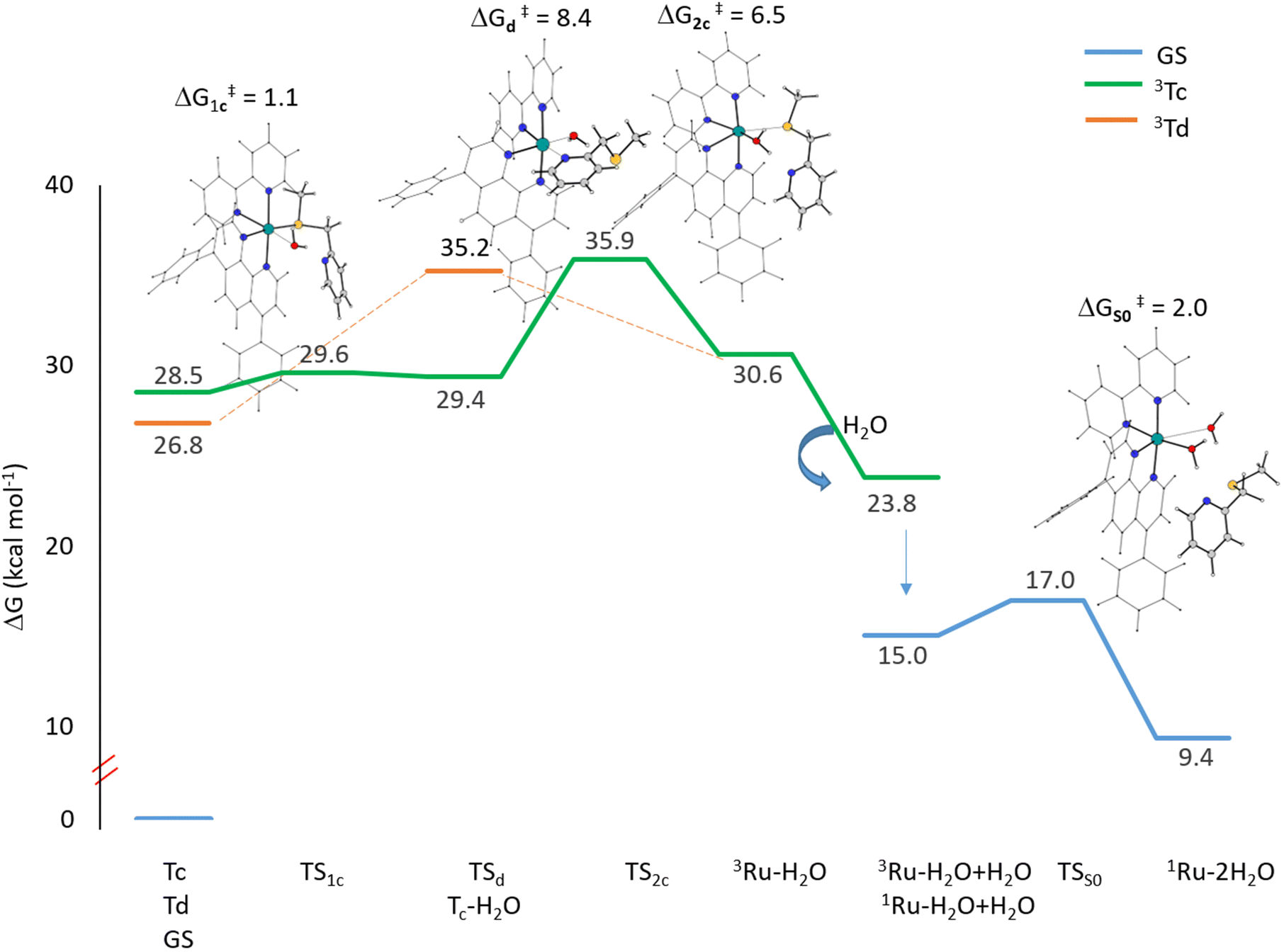 | ||
| Fig. 6 Calculated free energy profiles in water describing the light-induced substitution mechanism of the Ru complex. Relative energies are in kcal mol−1 and calculated with respect to the ground state singlet energy of the complex. | ||
The definitive release of the mtmp ligand and the formation of the Ru–H2O complex are calculated to be endothermic along both triplet pathways. When a second water molecule comes in close proximity to the 3Ru–H2O complex, it stabilizes the system by approximately 7 kcal mol−1. Several explorations for the formation of the final biaquated product have been conducted in the triplet state, but all of them failed. This suggests that the formation of the final product, Ru–2H2O, occurs directly in the ground singlet state. Indeed, the solvent adduct of the Ru–H2O complex in the triplet state undergoes a transition to the corresponding singlet state that lies 15.0 kcal mol−1 above the energy of the Ru complex. Along the singlet path, a second water molecule binds to the mono-aquated Ru complex, overcoming a very low activation energy barrier of only 2.0 kcal mol−1. The final product formation is calculated to be endothermic of 9.4 kcal mol−1. Overall, the results show that for the hydrolysis process to occur, leading to the formation of the cytotoxic agent in PACT, very low energy barriers have to be overcome along calculated pathways, confirming the experimentally detected high efficacy of the studied complex.
Computational details
All molecular geometry optimizations have been carried out with the Gaussian16 software package,41 at the density functional level of theory, employing the M05 functional.42 For the Ru atom, the relativistic compact Stuttgart–Dresden effective core potential has been used43 in conjunction with the corresponding split valence basis set. The standard double-ζ 6-31G** basis sets have been used for all the rest of the atoms. The impact of the aqueous solvent has been taken into account by performing optimization of the solvent considering a dielectric constant of 78.4, within the Tomasi implicit polarizable continuum model44,45 as implemented in Gaussian16. The electronic spectra have been obtained, within the nonequilibrium time-dependent TDDFT approach, as vertical electronic excitation on the ground-state structure, at the same level of theory used for other calculations. Such computational protocol has been selected on the basis of the outcomes of a preliminary benchmark study, considering the performance of several exchange–correlation functionals, on the absorption spectrum of the complex compared with that experimentally detected (Fig. S2 and Table S1†). The optimized structures of all the excited states potentially involved in the complex photoactivation have been explored using the TDDFT approach in a condensed phase. In the case of triplet states, the obtained guesses have then been used as starting points for the optimization within the unrestricted Kohn–Sham formalism.34,35 Stationary points along the examined reaction pathways have been identified as minima or transition states by the number of imaginary vibrational frequencies (0 or 1, respectively). These calculations have been also used for calculating zero-point energy (ZPE) corrections. Intercepted transition states have been carefully checked to be properly connected to the correct minima by intrinsic reaction coordinate (IRC) analysis.46,47 To ascertain the probability that a triplet state could be populated, spin–orbit matrix elements have been computed using the quadratic-response TD-DFT approach, as implemented in the ORCA code.48,49 Relativistic corrections have been obtained by the zeroth order regular approximation (ZORA). ZORA-DEF2-SVP and SARC-ZORA-SVP basis sets have been used for the main and metal atoms, respectively. The RIJCOSX approximations have been introduced to speed up the calculation time, as suggested in the ORCA manual.Conclusions
Here, a detailed computational investigation of the photophysical properties of a trisheteroleptic ruthenium complex, named the Ru complex, having a mtmp ligand that serves as a protecting group towards the coordination of biomolecules present in cells has been carried out by means of DFT and its time-dependent extension. The Ru complex is a highly efficacious PACT agent since, upon light irradiation, the “uncaged” complex is released that, as aquated cisplatin, acts as an activated drug able to bind to DNA inducing cell death. All the mechanistic aspects of the photodissociation process have been explored in depth and all the involved states have been fully described. The results of such a detailed investigation provide important information about both the reasons why pyridyl–thioether chelates form excellent caging groups for ruthenium-based PACT compounds and the mechanism by which solvent molecules are involved in the photosubstitution mechanism. First of all, through a comparison of the energies and plots of MOs of Ru with those of the other three polypyridyl complexes, it has been demonstrated that the presence of the mtmp ligand causes stabilization of the vacant MOs responsible for the generation of the 3MC states facilitating the population of such dissociative triplet metal-centred states from a triplet with the metal to ligand charge transfer, 3MLCT, character. Once the most appropriate protocol has been chosen, through a benchmark, the experimental spectrum has been reproduced and all the characteristics of singlet and triplet excited states have been explored. Very importantly, calculation of SOC elements for all the triplet states lying below the S1 state shows that they can be populated through efficient ISC. Optimization, using unrestricted DFT, of triplet states identified by TDDFT calculations converged into four distinct triplets denoted as Ta, Tb, Tc and Td, with the former two having the 3MLCT character and the latter two having the 3MC character. Tc and Td triplet states appear to be characterized by a significant reorganization in the coordination sphere of the Ru centre. In particular, Tc shows the detachment of the pyridyl arm of mtmp and Td is characterized, instead, by a clear elongation of the Ru–S bond in comparison with the complex ground state structure. Starting from such triplet states, the mechanism of the mtmp ligand substitution by water leading to the formation of the real cytotoxic species has been elucidated including, for the first time, the role played by the water solvent molecules. The hydrolysis process for the Tc triplet proceeds in two steps consisting of the rapid coordination of a water molecule followed by the mtmp ligand loss due to the definitive detachment of the pyridyl arm. As a result, the pentacoordinated ruthenium complex denoted as 3Ru–H2O is formed. The same complex is obtained in one step starting from the Td state through an associative substitution reaction causing the definitive breaking of the Ru–S bond. The second water molecule attack leading to the formation of the biaquated Ru–2H2O complex occurs after the transition to the corresponding singlet state, overcoming a very low energy barrier. The outcomes of our computational analysis confirm that electronic effects can play a very important role in promoting photosubstitution, as it happens in the present case, due to the 3MC state stabilization.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Italian Association for Cancer Research, AIRC (ID 25578). The authors gratefully acknowledge the computing time granted by the CINECA (project IsCa3_MoAPACT).References
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Stimuli-Responsive Therapeutic Metallodrugs, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- N. J. Farrer, L. Salassa and P. J. Sadler, Photoactivated chemotherapy (PACT): the potential of excited-state d-block metals in medicine, Dalton Trans., 2009, 10690–10701 RSC.
- U. Schatzschneider, Photoactivated Biological Activity of Transition-Metal Complexes, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef CAS.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Molecular mechanisms of cisplatin resistance, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- E. Wexselblatt, E. Yavin and D. Gibson, Cellular interactions of platinum drugs, Inorg. Chim. Acta, 2012, 393, 75–83 CrossRef CAS.
- S. Bonnet, Why develop photoactivated chemotherapy?, Dalton Trans., 2018, 47, 10330–10343 RSC.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, A Potent Trans-Diimine Platinum Anticancer Complex Photoactivated by Visible Light, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed.
- I. Chakraborty, J. Jimenez and P. K. Mascharak, CO-Induced apoptotic death of colorectal cancer cells by a luminescent photoCORM grafted on biocompatible carboxymethyl chitosan, Chem. Commun., 2017, 53, 5519–5522 RSC.
- A. Kastl, A. Wilbuer, A. L. Merkel, L. Feng, P. D. Fazio, M. Ocker and E. Meggers, Dual anticancer activity in a single compound: visible-light-induced apoptosis by an antiangiogenic iridium complex, Chem. Commun., 2012, 48, 1863–1865 RSC.
- M. R. Kim, H. Morrison and S. I. Mohammed, Effect of a photoactivated rhodium complex in melanoma, Anticancer Drugs, 2011, 22, 896–904 CrossRef CAS PubMed.
- Z. Li, A. David, B. A. Albani, J.-P. Pellois, C. Turro and K. R. Dunbar, Optimizing the Electronic Properties of Photoactive Anticancer Oxypyridine-Bridged Dirhodium(II,II) Complexes, J. Am. Chem. Soc., 2014, 136, 17058–17070 CrossRef CAS PubMed.
- D. A. Lutterman, P. K.-L. Fu and C. Turro, cis-[Rh2(μ-O2CCH3)2(CH3CN)6]2+ as a Photoactivated Cisplatin Analog, J. Am. Chem. Soc., 2006, 128, 738–739 CrossRef CAS PubMed.
- L. Zhang, P. Wang, X.-Q. Zhou, L. Bretin, X. Zeng, Y. Husiev, E. A. Polanco, G. Zhao, L. S. Wijaya, T. Biver, S. E. Le Dévédec, W. Sun and S. Bonnet, Cyclic Ruthenium-Peptide Conjugates as Integrin-Targeting Phototherapeutic Prodrugs for the Treatment of Brain Tumors, J. Am. Chem. Soc., 2023, 145, 14963–14980 CrossRef CAS PubMed.
- D. Havrylyuk, D. K. Heidary, Y. Sun, S. Parkin and E. C. Glazer, Photochemical and Photobiological Properties of Pyridyl-pyrazol(in)e-Based Ruthenium(II) Complexes with Sub-micromolar Cytotoxicity for Phototherapy, ACS Omega, 2020, 5, 18894–18906 CrossRef CAS PubMed.
- S. Bonnet, Ruthenium-Based Photoactivated Chemotherapy, J. Am. Chem. Soc., 2023, 145, 23397–23415 CrossRef CAS PubMed.
- L. Conti, E. Macedi, C. Giorgi, B. Valtancoli and V. Fusi, Combination of light and Ru(II) polypyridyl complexes: Recent advances in the development of new anticancer drugs, Coord. Chem. Rev., 2022, 469, 214656 CrossRef CAS.
- T. N. Singh and C. Turro, Photoinitiated DNA Binding by cis-[Ru(bpy)2(NH3)2]2+, Inorg. Chem., 2004, 43, 7260–7262 CrossRef CAS PubMed.
- Q. Chen, J.-A. Cuello-Garibo, L. Bretin, L. Zhang, V. Ramu, Y. Aydar, Y. Batsiun, S. Bronkhorst, Y. Husiev, N. Beztsinna, L. Chen, X.-Q. Zhou, C. Schmidt, I. Ott, M. J. Jager, A. M. Brouwer, B. E. Snaar-Jagalska and S. Bonnet, Photosubstitution in a trisheteroleptic ruthenium complex inhibits conjunctival melanoma growth in a zebrafish orthotopic xenograft model, Chem. Sci., 2022, 13, 6899–6919 RSC.
- R. N. Garner, L. E. Joyce and C. Turro, Effect of Electronic Structure on the Photoinduced Ligand Exchange of Ru(II) Polypyridine Complexes, Inorg. Chem., 2011, 50, 4384–4391 CrossRef CAS PubMed.
- J. D. Knoll, B. A. Albani and C. Turro, New Ru(II) Complexes for Dual Photoreactivity: Ligand Exchange and 1O2 Generation, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS PubMed.
- C. Kreitner and K. Heinze, Excited state decay of cyclometalated polypyridine ruthenium complexes: insight from theory and experiment, Dalton Trans., 2016, 45, 13631–13647 RSC.
- M. Kayanuma, Photosubstitution reaction of a bidentate ligand in a Ru(II) complex in aqueous solution, Comput. Theor. Chem., 2022, 1213, 113745 CrossRef CAS.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Theoretical Study of the Full Photosolvolysis Mechanism of [Ru(bpy)3]2+: Providing a General Mechanistic Roadmap for the Photochemistry of [Ru(N^N)3]2+-Type Complexes toward Both Cis and Trans Photoproducts, Inorg. Chem., 2020, 59, 14679–14695 CrossRef CAS PubMed.
- M. L. A. Hakkennes, M. S. Meijer, J. P. Menzel, A.-C. Goetz, R. Van Duijn, M. A. Siegler, F. Buda and S. Bonnet, Ligand Rigidity Steers the Selectivity and Efficiency of the Photosubstitution Reaction of Strained Ruthenium Polypyridyl Complexes, J. Am. Chem. Soc., 2023, 145, 13420–13434 CrossRef CAS PubMed.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- F. Plasser, TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- J. S. García, F. Alary, M. Boggio-Pasqua, I. M. Dixon and J.-L. Heully, Is photoisomerization required for NO photorelease in ruthenium nitrosyl complexes?, J. Mol. Model, 2016, 22, 284 CrossRef PubMed.
- S. E. Greenough, G. M. Roberts, N. A. Smith, M. D. Horbury, R. G. McKinlay, J. M. Żurek, M. J. Paterson, P. J. Sadler and V. G. Stavros, Ultrafast photo-induced ligand solvolysis of cis-[Ru(bipyridine)2(nicotinamide)2]2+: experimental and theoretical insight into its photoactivation mechanism, Phys. Chem. Chem. Phys., 2014, 16, 19141–19155 RSC.
- J. D. Knoll, B. A. Albani and C. Turro, New Ru(II) Complexes for Dual Photoreactivity: Ligand Exchange and 1O2 Generation, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS PubMed.
- C.-W. Yin, M.-K. Tsai and Y. J. Chen, Low-Temperature Observation of the Excited-State Decay of Ruthenium-(Mono-2,2′:6′,2′′-Terpyridine) Ions with Innocent Ligands: DFT Modeling of an 3MLCT–3MC Intersystem Crossing Pathway, ACS Omega, 2023, 8, 11623–11633 CrossRef CAS PubMed.
- M. Kasha, Characterization of electronic transitions in complex molecules, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- F. Ponte, D. M. Scopelliti, N. Sanna, E. Sicilia and G. Mazzone, How Computations Can Assist the Rational Design of Drugs for Photodynamic Therapy: Photosensitizing Activity Assessment of a Ru(II)-BODIPY Assembly, Molecules, 2022, 27, 5635 CrossRef CAS PubMed.
- F. Ponte, S. Scoditti, P. Barretta and G. Mazzone, Computational Assessment of a Dual-Action Ru(II)-Based Complex: Photosensitizer in Photodynamic Therapy and Intercalating Agent for Inducing DNA Damage, Inorg. Chem., 2023, 62, 8948–8959 CrossRef CAS PubMed.
- D. Escudero, E. Heuser, R. J. Meier, M. Schäferling, W. Thiel and E. Holder, Unveiling Photodeactivation Pathways for a New Iridium(III) Cyclometalated Complex, Chem. – Eur. J., 2013, 19, 15639–15644 CrossRef CAS PubMed.
- D. Escudero, W. Thiel and B. Champagne, Spectroscopic and second-order nonlinear optical properties of Ruthenium(II) complexes: a DFT/MRCI and ADC(2) study, Phys. Chem. Chem. Phys., 2015, 17, 18908–18912 RSC.
- Y.-J. Tu, S. Mazumder, J. F. Endicott, C. Turro, J. J. Kodanko and H. B. Schlegel, Selective Photodissociation of Acetonitrile Ligands in Ruthenium Polypyridyl Complexes Studied by Density Functional Theory, Inorg. Chem., 2015, 54, 8003–8011 CrossRef CAS PubMed.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Light-activated ruthenium complexes photobind DNA and are cytotoxic in the photodynamic therapy window, Chem. Commun., 2012, 48, 9649–9651 RSC.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- J. Roque III, D. Havrylyuk, P. C. Barrett, T. Sainuddin, J. McCain, K. Colón, W. T. Sparks, E. Bradner, S. Monro, D. Heidary, C. G. Cameron, E. C. Glazer and S. A. McFarland, Strained, Photoejecting Ru(II) Complexes that are Cytotoxic Under Hypoxic Conditions, Photochem. Photobiol., 2020, 96, 327–339 CrossRef PubMed.
- C. E. Welby, G. K. Armitage, H. Bartley, A. Sinopoli, B. S. Uppal and P. I. P. Elliott, Photochemical ligand ejection from non-sterically promoted Ru(ii)bis(diimine) 4,4′-bi-1,2,3-triazolyl complexes, Photochem. Photobiol. Sci., 2014, 13, 735–738 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016. GaussView 5.0.Wallingford, E.U.A Search PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Exchange-correlation functional with broad accuracy for metallic and nonmetallic compounds, kinetics, and noncovalent interactions, J. Chem. Phys., 2005, 123, 161103 CrossRef PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Energy-adjusted ab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Miertuš and J. Tomasi, Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes, Chem. Phys., 1982, 65, 239–245 CrossRef.
- K. Fukui, Formulation of the reaction coordinate, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, An improved algorithm for reaction path following, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, e1327 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometry of the Ru complex, TDDFT benchmark, singlet and triplet transitions, NTOs, selected bond lengths of Ta, Tb, Tc and Td triplet states, and relaxed potential energy scans. See DOI: https://doi.org/10.1039/d4dt00834k |
| This journal is © The Royal Society of Chemistry 2024 |