
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is bismuth(III) able to inhibit the activity of urease? Puzzling results in the quest for soluble urease complexes for agrochemical and medicinal applications†
Laura
Contini
a,
Arundhati
Paul
 b,
Luca
Mazzei
b,
Stefano
Ciurli
*b,
Davide
Roncarati
*c,
Dario
Braga
a and
Fabrizia
Grepioni
*a
b,
Luca
Mazzei
b,
Stefano
Ciurli
*b,
Davide
Roncarati
*c,
Dario
Braga
a and
Fabrizia
Grepioni
*a
aDepartment of Chemistry “G. Ciamician”, University of Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: fabrizia.grepioni@unibo.it
bLaboratory of Bioinorganic Chemistry, Department of Pharmacy and Biotechnology (FaBiT), University of Bologna, Viale Giuseppe Fanin 40, Bologna I-40127, Italy. E-mail: stefano.ciurli@unibo.it
cDepartment of Pharmacy and Biotechnology (FaBiT), University of Bologna, Via Selmi 3, 40126 Bologna, Italy. E-mail: davide.roncarati@unibo.it
First published on 4th June 2024
Abstract
Bismuth(III) complexes have been reported to act as inhibitors of the enzyme urease, ubiquitously present in soils and implicated in the pathogenesis of several microorganisms. The general insolubility of Bi(III) complexes in water at neutral pH, however, is an obstacle to their utilization. In our quest to improve the solubility of Bi(III) complexes, we selected a compound reported to inhibit urease, namely [Bi(HEDTA)]·2H2O, and co-crystallized it with (i) racemic DL-histidine to obtain the conglomerate [Bi2(HEDTA)2(μ-D-His)2]·6H2O + [Bi2(HEDTA)2(μ-L-His)2]·6H2O, (ii) enantiopure L-histidine to yield [Bi2(HEDTA)2(μ-L-His)2]·6H2O, and (iii) cytosine to obtain [Bi(HEDTA)]·Cyt·2H2O. All compounds, synthesised by mechanochemical methods and by slurry, were characterized in the solid state by calorimetric (DSC and TGA) and spectroscopic (IR) methods, and their structures were determined using powder X-ray diffraction (PXRD) data. All compounds show an appreciable solubility in water, with values ranging from 6.8 mg mL−1 for the starting compound [Bi(HEDTA)]·2H2O to 36 mg mL−1 for [Bi2(HEDTA)2(μ-L-His)2]·6H2O. The three synthesized compounds as well as [Bi(HEDTA)]·2H2O were then tested for inhibition activity against urease. Surprisingly, no enzymatic inhibition was observed during in vitro assays using Canavalia ensiformis urease and in vivo assays using cultures of Helicobacter pylori, raising questions on the efficacy of Bi(III) compounds to counteract the negative effects of urease activity in the agro-environment and in human health.
Introduction
Urease (urea amidohydrolase; EC 3.5.1.5) is a nickel-dependent non-redox metalloenzyme that is prevalent in nature.1–7 It is found in fungi, algae, bacteria, plants, and soil, operating within the global N cycle transformations. Urease catalyses the hydrolysis of urea, leading to the formation of aqueous ammonium hydrogen carbonate and to a pH increase from neutrality to ca. 9.0–9.5. It has a high catalytic efficiency, accelerating the hydrolysis of urea by ca. 15 orders of magnitude (see Fig. 1).8–10 | ||
| Fig. 1 Urea hydrolysis catalysed by urease. | ||
This reaction is crucial for the agrochemical field, as urea is the most used nitrogen-based fertiliser and its hydrolysis is required to generate forms of nitrogen that serve as plant nutrients.11 However, it is also the leading cause of several agronomic,12 environmental,13–15 and economic problems16 and strongly affects the global N cycle.17,18 In particular, urea hydrolysis causes a rapid increase in the soil pH, which can lead to the loss of urea nitrogen as gaseous ammonia. Ammonia is toxic to plants and is reported to contribute to the formation of fine particulate matter (PM2.5),19 which is a significant health hazard. Additionally, ammonia can be converted by other soil enzymes, such as ammonia monooxygenase (AMO), to several nitrogen oxides, especially N2O, a greenhouse gas with 300 times the heat-trapping capacity of CO2.20–22
Due to its ubiquitous presence in nature, the enzyme plays a crucial role in other aspects of life. In addition to the agrochemical field, it is also central to the medical field, where it is implicated in the pathogenesis of several microorganisms,23 including Klebsiella aerogenes,24Proteus mirabilis,25,26 and Helicobacter pylori.27,28 The role of urease in the pathogenesis of H. pylori is crucial: by exploiting the increase in pH generated by urea hydrolysis, the bacterium can colonise the environment of the gastric mucosa, which would otherwise be too acidic for its survival.29–32 Interest in novel ways to eradicate H. pylori is high as it reportedly infects nearly half of the world's population and has been classified by the World Health Organization (WHO) as well as by the International Agency for Research on Cancer, as a class I carcinogen, associated with the onset of gastric cancer.33,34 Additionally, the WHO recently added it to the list of 12 priority pathogens that pose the greatest threat to human health and for which new antibiotics are urgently needed.35
The quest for novel urease inhibitors is thus central to various fields36,37 and several compounds have been found to inhibit urease.7,38–56 Another strategy to inhibit urease is the use of coordination polymers (CPs) and complexes,57,58 formed by the self-assembly of metal ions and organic ligands and representing one of the most prolific research areas of crystal engineering and medicinal inorganic chemistry in the last 15 years. Interest in such compounds comes from their double organic–inorganic nature and the possibility of simultaneously exhibiting the properties of both classes. They are of great interest in the field of enzyme inhibition, as the inhibition efficiency and mechanism can be tuned by proper selection of the oxidation state of the metal centre, coordination environment, and chemical nature of the ligand.59–62
Bismuth-based complexes are of particular interest in the field of urease inhibitors.10,63 Although bismuth has no known natural biological role, some preparations based on Bi(III), such as bismuth subsalicylate (BSS) and bismuth subcitrate (CBS), have been used to treat H. pylori in combination with antibiotics such as tetracycline, clarithromycin, and amoxicillin.64–66 Despite the efficacy of these bismuth formulations, the mechanism of action of bismuth compounds is still not fully understood.10 In any case, urease inhibition by bismuth compounds such as [Bi(HEDTA)]·2H2O, [Bi(Cys)3] and ranitidine bismuth citrate (RBC) has been reported.‡63
The use of bismuth-based compounds as inhibitors of soil urease for agrochemical applications is hindered by their low solubility in water at soil pH (6.5–7.5).67 Overcoming solubility issues is also relevant from a synthetic point of view, as most Bi(III) precursors are either insoluble (Bi2O3) or unstable in water at neutral pH values (BiCl3 and Bi(NO3)3). Mechanochemistry provides a promising solution to this issue,68 as it is a synthetic technique that requires little to no solvent. Additionally, Friščić et al.69 proved it to be a very versatile technique for synthesizing bismuth complexes as they investigated the synthesis of the API bismuth subsalicylate via ion- and liquid-assisted grinding.
Our approach is based on the application of crystal engineering strategies70–76 to the preparation of (i) a novel complex and (ii) a co-crystal of Bi(III), both characterized by improved solubility properties in water with respect to bismuth subsalicylate (<1 mg mL−1) and bismuth subcitrate (insoluble in water). The starting material [Bi(HEDTA)2]·2H2O (1),77,78 for which only solution synthesis and crystal data had been reported, and its anhydrous form [Bi(HEDTA)2] (2),79 for which only crystallographic information are available, were also prepared by solid-state methods, and their solubility determined. The synthesized compounds were tested for their efficacy to inhibit urease in vitro using a plant enzyme as well as in vivo using cultures of H. pylori.
Experimental
Materials and methods
All reagents (see Chart 1) were purchased from Sigma-Aldrich or Alfa Aesar and used without further purification. | ||
| Chart 1 The reagents employed in this study. | ||
Synthesis
All compounds were synthesized by solid state and slurry methods, as summarized in Table 1.| Compound | Synthetic method |
|---|---|
| a CSD Refcode HAYPEC.77 b CSD Refcode SOXXOS.79 c LAG = liquid assisted grinding80 (here the solvent is water). | |
| (1) [Bi(HEDTA)]·2H2O β-forma | Slurry/LAGc |
| (2) [Bi(HEDTA)] anhydrousb | [Bi(HEDTA)]·2H2O at 150 °C for 4 h |
| (3) [Bi2(HEDTA)2(μ-L-His)2]·6H2O + [Bi2(HEDTA)2(μ-D-His)2]·6H2O | Slurry/LAG |
| (4) [Bi2(HEDTA)2(μ-L-His)2]·6H2O | Slurry/LAG |
| (5) [Bi(HEDTA)]·Cyt·2H2O | Slurry/LAG |
The synthesis of [Bi(HEDTA)]·2H2O (1) (see Table 1) was carried out by reacting 0.5 mmol of H4EDTA and 0.25 mmol of Bi2O3 either via slurry in 1 mL of water or mechanochemically via liquid assisted grinding (LAG).80 For the LAG synthesis, the reagents were added to a 5 mL agate jar in the presence of 100 μL of water and two 5 mm agate balls; a Retsch MM200 ball miller was employed, operated at a frequency of 20 Hz for 90 min. In both cases the product obtained, characterized via powder X-ray diffraction (PXRD), was the β polymorph, i.e., the form previously obtained from solution synthesis.77,79 The compound was then used as a reagent for the syntheses of the new complexes. Heating of (1) in an oven at 150 °C for 4 hours yielded the known anhydrous form of the complex, [Bi(HEDTA)] (2).79 The conglomerate [Bi2(HEDTA)2(μ-L-His)2]·6H2O + [Bi2(HEDTA)2(μ-D-His)2]·6H2O (3), the complex [Bi2(HEDTA)2(μ-L-His)2]·6H2O (4) and the co-crystal [Bi(HEDTA)]·Cyt·2H2O (5) were obtained by reaction of (1) (0.5 mmol) with a stoichiometric amount of histidine (DL-his or L-His) and cytosine (Cyt), either via slurry or liquid assisted grinding (LAG). LAG syntheses were carried out in the same conditions used for the synthesis of (1). No change in the product formation was observed by varying the amount of water used for the LAG procedure. Compound (5) could also be obtained by slurry of stoichiometric amounts of the three separate reagents, i.e., Bi2O3, H4EDTA and cytosine.
Powder X-ray diffraction
X-ray diffraction patterns were collected in Bragg–Brentano geometry on a PANalytical X'Pert Pro automated diffractometer, equipped with an X'celerator detector, using Cu-Kα radiation (λ = 1.5418 Å) without monochromator in the 3–40° 2θ range (step size 0.033°; time per step: 20 s; Soller slit 0.04 rad, antiscatter slit: 1/2, divergence slit: 1/4; 40 mA·40 kV).For structural solution from powder, data in the 2θ range 3–70° 2θ range were collected on a Panalytical X'Pert PRO automated diffractometer equipped with a PIXcel detector in transmission geometry (capillary spinner), using Cu-Kα radiation (λ = 1.5418 Å) without monochromator in the 2θ range between 3° and 70° (continuous scan mode, step size 0.0130°, counting time 170.6 s, Soller slit 0.02, antiscatter slit ¼, divergence slit ¼, 40 mA 40 kV). Six patterns were recorded and summed to enhance the signal to noise ratio. To solve the structures from powder data, the software EXPO201481 was used. Selected peaks in the 3–65°2θ range were chosen and used to index the unit cell of the compound using the algorithm N-TREOR09.82 The structures were solved by simulated annealing using fragments retrieved from the Cambridge Structural Database. Ten runs for simulated annealing trials were set, and a cooling rate (defined as the ratio Tn/Tn − 1) of 0.95 was used. In all cases the best solution was chosen for Rietveld refinement, which was performed using the software EXPO 2014 A Chebyshev function with 20 parameters was used to fit background. All the hydrogen atoms were fixed in calculated positions with the software Mercury. Crystal data tables and Rietveld refinements are collected in the ESI.† CCDC 2340273 and 2340274.†
Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) traces were recorded using a PerkinElmer Diamond apparatus. The samples (3–8 mg range) were placed in open aluminium pans. All measurements were conducted in the 30–170 °C range at a heating rate of 10 °C min−1. DSC traces are reported in the ESI.†Thermogravimetric analysis (TGA)
TGA measurements were performed with a PerkinElmer TGA8000 in the 25–700 °C temperature range, under N2 gas flow at a heating rate of 10 °C min−1. TGA traces are reported in the ESI.†Solubility
The solubility of the synthesized complexes was measured by placing 500 mg of powder and 1 mL of water in a closed vessel. The mixture was left stirring at room temperature and after 5 h the undissolved solid was filtered and weighed, allowing to determine the change in solubility with respect to Bi2O3. To verify that the procedure was not altering the compound, the undissolved powder was analysed via X-ray powder diffraction.Enzymatic assays
Stock solution (50 mM) of (1) were prepared from the corresponding dihydrated solid in 2 mM HEPES buffer at pH 7.0 and 7.5, or in 3 mM phosphate buffer at pH 7.0. A working solution of (1) was prepared in the 0.12–8.0 mM concentration range by diluting the stock in the same buffers. A stock solution of Canavalia ensiformis (jack bean) urease (JBU, type C-3 powder, ≥ 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per g, Merck Milan, Italy) to a final concentration of 50 μg mL−1 (100 nM) was prepared in 20 mM HEPES buffer at pH 7.5. The JBU stock solution (0.100 mL) was added to 9.775 mL of the working solution of (1) and, after one-hour incubation, the reaction was started by the addition of 0.125 mL of a 8 M urea water solution to reach a final concentration of 100 mM substrate and 1 nM enzyme. Urease activity was determined in triplicate using the pH-STAT method, as originally described.83 In particular, a T1 pH-meter equipped with a 50–14 T electrode (Crison Instruments, SA), was used to record, every 0.5 min, the volume of a 50 mM HCl solution necessary to maintain the pH at a determined and constant value. The measurement started 0.5 min after urea addition to allow time to reach uniform substrate concentration in the sample volume and continued for the first 3 minutes after substrate addition, under constant magnetic stirring. The reaction rates were averaged, normalized with respect to the rate of the control samples in the absence of (1), and the obtained residual activity values (%) were plotted as a mean percentage ± standard deviation (SD) as a function of the concentration of the tested compound on a semi-log graph. Prism v. 10.1.1 software was used to plot the results.
000 units per g, Merck Milan, Italy) to a final concentration of 50 μg mL−1 (100 nM) was prepared in 20 mM HEPES buffer at pH 7.5. The JBU stock solution (0.100 mL) was added to 9.775 mL of the working solution of (1) and, after one-hour incubation, the reaction was started by the addition of 0.125 mL of a 8 M urea water solution to reach a final concentration of 100 mM substrate and 1 nM enzyme. Urease activity was determined in triplicate using the pH-STAT method, as originally described.83 In particular, a T1 pH-meter equipped with a 50–14 T electrode (Crison Instruments, SA), was used to record, every 0.5 min, the volume of a 50 mM HCl solution necessary to maintain the pH at a determined and constant value. The measurement started 0.5 min after urea addition to allow time to reach uniform substrate concentration in the sample volume and continued for the first 3 minutes after substrate addition, under constant magnetic stirring. The reaction rates were averaged, normalized with respect to the rate of the control samples in the absence of (1), and the obtained residual activity values (%) were plotted as a mean percentage ± standard deviation (SD) as a function of the concentration of the tested compound on a semi-log graph. Prism v. 10.1.1 software was used to plot the results.
Antimicrobial activity tests against Helicobacter pylori
The compounds (1), (2), (3), and (5) together with [Bi(Citrate)], and H4EDTA were used to carry out antimicrobial assay against H. pylori [H. pylori G27 wild-type strain was gifted in 1988 by Chiron corporation, now part of the GlaxoSmithKline company]. The antimicrobial activity was assessed by the broth microdilution method, according to the Clinical and Laboratory Standards Institute (CLSI) and the European Committee for Antimicrobial Susceptibility Testing (EUCAST) guidelines. All solutions were tested in parallel, employing the H. pylori G27 strain, to determine their minimal inhibitory concentrations (MICs). [Bi(Citrate)] was dissolved (125 mM) in 10% (w/v) ammonia water solution at a concentration of 125 mM; all other Bi-containing compounds were dissolved (125 mM) in ultrapure water or 10% ammonia water solution. Freshly prepared solutions were used for the assay in 11 progressive 2-fold dilution series ranging from 125.0 μM to 0.1 μM in a 96-well microtiter plate. Exponentially growing H. pylori liquid culture (optical density at 600 nm = 0.6–0.7) was diluted and inoculated into each well to a final concentration of 5 × 105 CFU mL−1. All experiments and dilutions were performed in Brucella broth supplemented with 5% fetal bovine serum. Bacterial cultures were incubated in a microaerophilic environment with constant agitation at 37 °C and examined visually after 72 h. MIC values were defined as the lowest concentration of compound that inhibited the visible growth of bacteria. Negative controls were implemented to check for the sterility of the solutions, while a positive control was added to assess bacterial growth and fitness. In addition, to rule out any effect of the solvent, bacterial growth was also monitored for H. pylori G27 cells exposed to the vehicle solution in which bismuth-containing solutions were prepared (10% ammonia or water, but without adding bismuth compounds). Each experiment was performed in triplicate.Results and discussion
Preparation and characterization of [Bi(HEDTA)]·2H2O (1) and [Bi(HEDTA)] (2)
Several papers and reviews highlight that Bi(III) forms stable complexes with H4EDTA in aqueous solution.78,84 Bhat and Krishna Iyer85 assessed that the deprotonated complex [Bi(EDTA)]− is stable at pH 1.5–10, with a stability constant of ca. 26. At higher pH, bismuth hydroxide is formed via hydrolysis. We further verified the stability of (1) in water by performing 1H NMR on an aqueous solution of the complex (see Fig. ESI 14†). In the solid state Bi(III) and H4EDTA form both neutral [Bi(HEDTA)] and anionic [Bi(EDTA)]− species. We concentrated our efforts on the synthesis of the neutral form. According to the literature, the complex can be obtained from solution as the dihydrate (1) in two polymorphic forms, or as the anhydrous (2), depending on the reaction conditions (temperature and rate of precipitation).77–79 In order to explore faster and scalable techniques, we focused our efforts on the preparation of the complex with solid-state techniques, such as mechanochemistry and slurry. We choose bismuth oxide as an accessible source of bismuth(III), despite it being utterly insoluble in water.69 The results of neat grinding, liquid assisted grinding (LAG) and slurry of a physical mixture of H4EDTA and Bi2O3 in a 1:2 stoichiometric ratio are represented in Scheme 1.
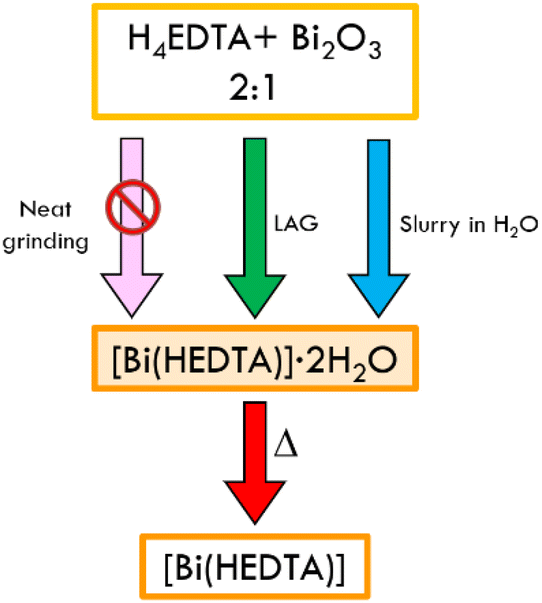 | ||
| Scheme 1 Details of the neat grinding, LAG, slurry and crystallization processes for the synthesis of [Bi(HEDTA)]·2H2O (1), and subsequent thermal dehydration to obtain the anhydrous complex. | ||
Neat grinding of the reagents resulted in a physical mixture of the starting materials, while LAG and slurry in water resulted in the formation of the known β polymorph of (1)77 (see Fig. 2) as a crystalline powder. Recrystallization from water, in which the solid is slightly soluble, yielded single crystals of the same polymorph. Structural characterization of the LAG and slurry product was done by comparing the experimental powder diffraction patterns with the calculated ones for the α and β forms of (1) (see Fig. ESI 1†).
 | ||
| Fig. 2 Packing of [Bi(HEDTA)]·2H2O (1) viewed down the a-axis, highlighting the presence of water layers (pale blue) in the crystal. H atoms not shown for clarity. | ||
Thermal stability of the coordination polymer (1) was assessed via TGA and DSC measurements (see Fig. ESI 9†). TGA was performed in the 25–700 °C range; the trace displays an initial weight loss of about 7%, attributed to the loss of two H2O molecules per formula unit. The complex is then stable up to 250 °C, where a multistep degradation occurs. To confirm the hypothesis that the initial weight loss observed in TGA is due to dehydration of the complex, DSC was performed in the 25–200 °C range. The trace shows a broad endothermic peak at 133 °C (peak temperature), corresponding to the first weight loss observed in TGA, and is thus attributed to dehydration. No other thermal events were observed in the analysed range. In agreement with the results obtained with DSC and TGA, heating of (1) above 130 °C yields the anhydrous (2) (see Fig. 3), as confirmed by comparison of the experimental powder diffraction pattern with the calculated one for the anhydrous form present in the CSD (refcode SOXXOS, see Fig. ESI 2†).
 | ||
| Fig. 3 Coordination environment around bismuth in the complex [Bi(HEDTA)] (2). | ||
Thermal stability of the coordination polymer (2) was assessed via TGA and DSC measurements in the 25–700 °C and in the 25–200 °C ranges, respectively. Both resulting traces (see Fig. ESI 10†) are consistent with those for (1) following the dehydration process.
Preparation and characterization of the conglomerate [Bi2(HEDTA)2(μ-L-His)2]·6H2O + [Bi2(HEDTA)2(μ-D-His)2]·6H2O (3) and of [Bi2(HEDTA)2(μ-L-His)2]·6H2O (4)
The results of ball milling, liquid assisted grinding (LAG) and slurry of a physical mixture of (1) and either DL- or L-Histidine (DL-His and L-His respectively) in a 1:1 stoichiometric ratio are presented in Scheme 2.
 | ||
| Scheme 2 Details of the neat grinding, LAG and slurry processes for the reaction of [Bi(HEDTA)]·2H2O with L- or DL-histidine. | ||
While neat grinding of the reagents resulted in a physical mixture of unreacted starting materials, LAG and slurry in water yielded the complexes crystalline powders with superimposable patterns (see ESI†), therefore, upon reaction of DL-histidine with (1), spontaneous resolution of DL-histidine occurs, and the conglomerate (3) is formed, i.e., a 50:50 mixture of crystals of enantiopure [Bi2(HEDTA)2(μ-L-His)2]·6H2O and of enantiopure [Bi2(HEDTA)2(μ-D-His)2]·6H2O [where the enantiopure [Bi2(HEDTA)2(μ-D-His)2]·6H2O is exactly the complex (4), see below]. Despite numerous attempts and techniques tested, single crystals could not be obtained. The structure was thus solved from powder data, collected on the product of the reaction of DL-histidine with (1), with the help of information derived from thermogravimetric analysis (TGA), which allowed to determine the stoichiometry of the complex and confirm the presence of six water molecules per formula unit (See Fig. ESI 11†). Fig. 4 shows the structure of the complex (4) obtained as pure compound upon reaction with L-histidine, or as a part of the conglomerate (3) if DL-histidine is used.
 | ||
| Fig. 4 The dimeric complex obtained by reaction of [Bi(HEDTA)]·2H2O with DL- or L-histidine [conglomerate (3) and L-histidine complex (4), respectively] and a projection of the packing down the crystallographic b-axis (with purple coordination polyhedra around the bismuth ions). Histidine C atoms in orange, water O atoms in blue. H atoms not shown for clarity. | ||
Crystalline (4) is a discrete complex where the each HEDTA3− anion acts as a multidentate ligand coordinating one bismuth(III) cation, and the L-histidine molecule asymmetrically bridges the two Bi3+ cations [Bi-NL-his 2.440(4) and 2.826(4) Å]. The two bismuth cations, the two L-histidine and the two HEDTA3− ligands are related by a crystallographic two-fold axis; six water molecules are present per dimeric unit. Fig. ESI 3† show a comparison of the calculated powder diffraction pattern of (4), calculated on the basis of the structural resolution via simulated annealing, and the experimental patterns of the bulk products obtained with LAG and slurry, confirming that the structure obtained is representative of the products of both procedures.
Thermal stability of the conglomerate (3) and of the pure L-histidine complex (4) was assessed via TGA and DSC measurements (see Fig. ESI 12†). TGA was performed in the 25–700 °C range; the trace displays an initial weight loss of about 8%, which has been attributed to the loss of the six H2O molecules present per unit formula. The complex is then stable up to 250 °C, where a multistep degradation occurs. To confirm the hypothesis that the initial weight loss observed in TGA was due to dehydration of the complex, DSC was performed in the 25–200 °C range. The trace shows one broad endothermic peak at 72 °C corresponding to the first weight loss observed in TGA and thus attributed to dehydration. No other thermal events have been observed in the analysed range.
Preparation and structural characterization of [Bi(HEDTA)]·Cyt·2H2O (5)
The results of ball milling, Liquid Assisted Grinding (LAG) and slurry of a physical mixture of (1) and cytosine (Cyt) in a 1:1 stoichiometric ratio are presented in Scheme 3. While neat grinding of the reagents resulted in a physical mixture of unreacted starting materials, LAG and slurry in water resulted in the formation of the cocrystal (5) as a crystalline powder.
 | ||
| Scheme 3 Details of the slurry, ball milling and crystallization processes for the synthesis of the co-crystal [Bi(HEDTA)]·Cyt·2H2O (5). | ||
Despite numerous attempts and crystallization techniques tested, single crystals of the co-crystal could not be obtained. The structure was thus solved from powder data, with the help of information derived from thermogravimetric analysis (TGA), which allowed to determine the stoichiometry of the complex and confirm the presence of two water molecules per formula unit (see Fig. ESI 13†).
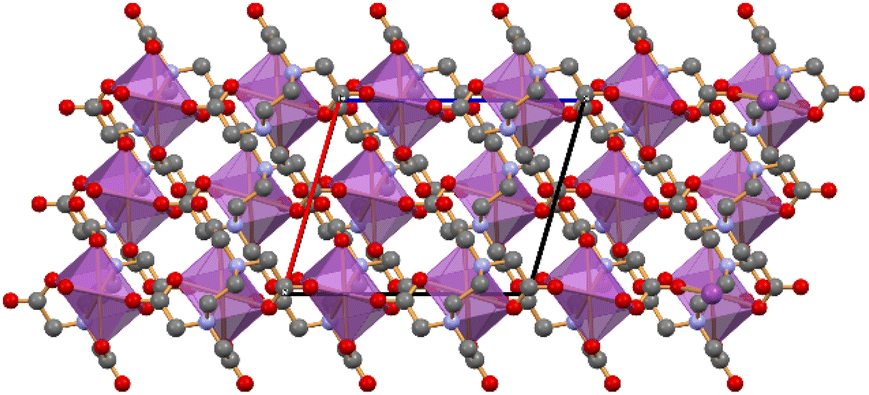
In crystalline (5), the HEDTA3− anion acts as a multidentate ligand and simultaneously binds three Bi(III) ions, each octacoordinated by six O and two N atoms. Fig. 5 shows the 1D ribbon formed by the neutral [Bi(HEDTA)] units, extending parallel to the crystallographic c-axis; cytosine and water molecules fill in the channels between the 1D ribbons (Fig. 5b).
 | ||
| Fig. 5 (Top) The 1D ribbon of [Bi(HEDTA)] units(coordination polyhedra around the bismuth ions in purple) extending parallel to the c-axis in crystalline [Bi(HEDTA)]·Cyt·2H2O (5). (Bottom) A side-view, down the c-axis, shows how cytosine (large caps) and water molecules fill the cavities between neighbouring 1D-ribbons (water oxygens in blue; H atoms not shown for clarity). | ||
Although our original intent was the preparation of a novel mixed ligand Bi(III) ethylenediaminetetraacetate complex with cytosine as an additional ligand, in the crystalline structure the cytosine does not act as an additional ligand to Bi(III) but remains in the lattice as an independent molecule. The compound can thus be described as a hydrated co-crystal formed by cytosine and by a 1D coordination polymer of [Bi(HEDTA)].
Fig. ESI 8† shows a comparison of the calculated powder diffraction pattern of (5), calculated on the basis of the structural resolution via simulated annealing, and the experimental pattern of the bulk products obtained with LAG and slurry, and confirms that the structure obtained is representative of the products of both crystallization techniques.
Thermal stability of the coordination polymer (5) was assessed via TGA and DSC measurement (Fig. ESI 13†). TGA was performed in the 25–700 °C range; the trace displays an initial weight loss of about 5%, which has been attributed to the loss of the two water molecules present per unit formula. The complex is then stable up to 250 °C, where a multistep degradation occurs. To confirm the hypothesis that the initial weight loss observed in TGA was due to dehydration of the complex, DSC was performed in the 25–200 °C range. The trace shows two broad and overlapped endothermic peaks at 114 and 130 °C respectively, corresponding to the first weight loss observed in TGA and thus attributed to dehydration. No other thermal events have been observed in the analysed range.
Solubility tests
The thermodynamic solubility at 30 °C for both (1) and (2) was determined, and results are shown in Table 2. As usually observed when dealing with anhydrous and hydrated forms of the same molecule or complex, the solubility in water of anhydrous (2) is about twice the solubility observed for the hydrated form (1); this is in keeping with the general observation that molecules or complexes are usually more soluble in their anhydrous or less hydrated form. Water solubility of (1) makes it interesting for applications in the agrochemical field. Additionally, the complex can be exploited to prepare other bismuth-based derivatives. The presence of histidine in compounds (3) and (4) determines an increase in thermodynamic solubility compared to that of (1) and (2), as can be seen in Table 2. On the other hand, the solubility of (5) is decreased as compared to (3) and (4), but it is still significantly higher than that observed for (1).| Compound | Solubility (mg mL−1) | Solubilitya (mol L−1) |
|---|---|---|
| a Molar concentration of Bi(III). | ||
| (1) [Bi(HEDTA)]·2H2O | 6.84 | 12.8 |
| (2) [Bi(HEDTA)] | 11.5 | 21.5 |
| (3) [Bi2(HEDTA)2(μ-D-His)2]·6H2O + [Bi2(HEDTA)2(μ-L-His)2]·6H2O conglomerate | 25.7 | 36 |
| (4) [Bi2(HEDTA)2(μ-L-His)2]·6H2O | 25.7 | 36 |
| (5) [Bi(HEDTA)]·Cyt·2H2O | 12.5 | 19.4 |
Kinetic studies of urease catalysis in the presence of [Bi(HEDTA)]·2H2O (1)
We examined the prototypical Bi(III) hydrated complex (1), devoid of any co-crystallized additional moieties, because this same compound had been previously reported to inhibit Klebsiella aerogenes urease (KAU) with a competitive mechanism that allegedly involved the binding of Bi(III) to a catalytically essential cysteine residue.63 In addition, the co-crystallized D/L histidine or cytosine moieties present in compounds (3), (4) and (5) are not expected to contribute to enzyme inhibition. The inhibition studies involved the determination of the activity of Canavalia ensiformis (jack bean) urease (JBU) in the absence and presence of (1) at the optimal pH for the enzyme activity (pH = 7.5) and at the pH value (7.0) at which this compound was previously reported to inhibit KAU.63 The integrity of (1) in solution was ascertained and confirmed using 1H NMR spectroscopy (See Fig. ESI 14†). One hour incubation of JBU with different (0.12–8.0 mM) concentrations of (1) at pH 7.0 or 7.5 and in the presence of either HEPES or phosphate buffer (the latter used in the previous study63) showed a substantial invariance of the enzymatic activity at concentrations well above the reported Ki = 1.74 mM63 (Fig. 6).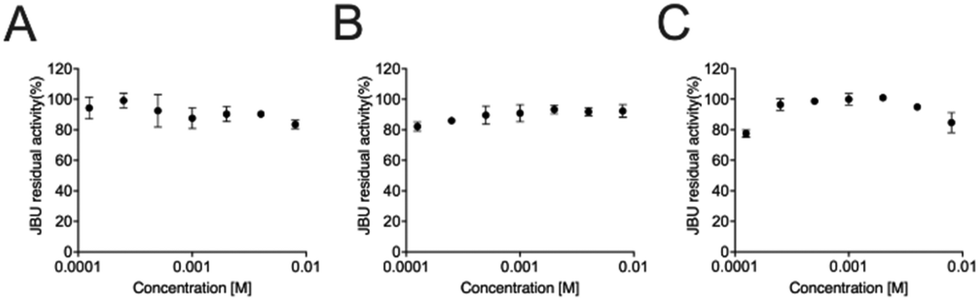 | ||
| Fig. 6 Dose–response semi-log plot for the activity of JBU as a function of the concentration of (1) at different pH values and buffering systems: (A) HEPES at pH 7.5; (B) HEPES at pH 7.0; (C) phosphate at pH 7.0. | ||
Antimicrobial activity tests
In parallel to the in vitro studies on the simplest of the synthesized compounds reported above, which highlighted the absence of urease inhibition at concentrations up to one order of magnitude higher than the reported inhibition constant, the possible antimicrobial activity of the Bi(III)-containing compounds obtained in this work was assessed in vivo on the H. pylori G27 strain. Freshly prepared solutions of (1), (2), (3) and (5) were added to H. pylori G27 liquid culture and their minimal inhibitory concentration (MIC) was determined. As a control, Bi(citrate) was tested in parallel. In our experimental conditions, we consistently measured MIC = 15 μM for Bi(citrate), in agreement with previous reports on the same strain.86 However, all other compounds yielded much higher MIC values (62.5 μM, Fig. ESI 15†). Focusing on the prototypical compound (1), we compared its antimicrobial activity to H4EDTA, as control, obtaining the same MIC value (62.5 μM, Fig. ESI 16†). The observed inhibitory effect of H4EDTA on H. pylori growth is consistent with previous studies that indicated a concentration range of 25–100 μM for the MIC value.87Conclusions
Urease is a nickel-dependent enzyme that is responsible for several pathogenic states in humans, as well as increasing the environmental impact of agriculture. Therefore, studies aimed to modulate the enzymatic ureolytic activity could provide means to improve treatment of infectious diseases as well as more sustainable primary food production. In this study, a crystal engineering approach was taken to prepare via co-crystallization, compounds based on bismuth(III). Bi(III) compounds have been reported to act as effective urease inhibitors for agrochemical and medicinal applications, even though their use is limited by their very low water solubility. Furthermore, the functional mechanism of clinically use bismuth-based drugs on H. pylori infections remains obscure as it differs in variety of treatment regimens.88 All compounds were characterized by solid state methods and their structures determined by diffraction methods. The aim of this work was twofold: on the one hand, we wanted to ascertain if co-crystallization of [Bi(HEDTA)]·2H2O with coformers such as histidine and cytosine could lead to more soluble compounds, while, on the other hand, we wanted to verify the urease inhibition activity in vitro and in vivo by these compounds. The outcomes of these studies were both satisfying and frustrating. On the one hand, the first goal of this project was achieved, as the compounds (3), (4) and (5) appear to be significantly more soluble than the precursor (1). On the other hand, the investigation of the in vitro inhibition of a plant urease by (1), as well as the measurement of the in vivo control of growth of H. pylori G27 strain cells, indicated no significant concentration-dependent inactivation, in contrast to previous reports indicating this compound acting as a competitive inhibitor of urease.63 These results suggest that the action of the [Bi(HEDTA)]·2H2O complex is not significantly different from that of the H4EDTA ligand itself, which is well known to remove the two essential Ni(II) ions from the urease active site at low pH and high concentration.89,90Bi(III) behaves similarly to Zn(II), sharing high affinity for N- and O- as well as S-based ligands. The active site of several metallo-hydrolases contain Zn(II) as an essential cofactor, while other enzymes, not containing metal ions as cofactors, use histidine imidazole –N, cysteine –SH, or serine –OH groups for catalysis. Therefore, the use of Bi(III)-based drugs could impact a large number of metabolic pathways. To the best of our knowledge, only one study has been reported that shows the inhibition of metallo-β-lactamases, a specific class of hydrolases, by Bi(III)-containing drugs, whereby Bi(III) replaces the Zn(II) ions in the active site.91 In the case of urease, a unique Ni(II)-dependent hydrolase that also uses a conserved cysteine thiol as an essential residue, it was expected that Bi(III) could either substitute the Ni(II) ions in the active site or bind to the essential cysteine blocking catalysis, as previously suggested.63 The absence of these effects suggests that in the case of H. pylori, whose growth is inhibited by Bi(III) complexes,92 the action of Bi(III)-based drugs must not specifically target urease but rather affect other metabolic processes such as energy production and/or cell wall disruption. The results of the present study highlights the need to investigate the action of Bi(III) coordination compounds on other classes of bimetallic hydrolases such as, for example, arginase or agmatinase, functioning in the urea cycle.93
Author contributions
Laura Contini, Arundhati Paul: investigation, formal analysis, methodology, writing – original draft; Dario Braga: resources, writing; Luca Mazzei, Stefano Ciurli, Davide Roncarati, Fabrizia Grepioni: conceptualization, supervision, investigation, formal analysis, methodology, writing, resources.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
L. M., A. P. and S. C. acknowledge financial support from the Consorzio Interuniversitario di Risonanze Magnetiche di Metallo-Proteine (CIRMMP). F. G., D. B., A. P., S. C. and L. M. acknowledge the project “NICE – Nature Inspired Crystal Engineering” (PRIN2020). A PNRR DM 351/2022 PhD project (LC) is acknowledged. All authors acknowledge financial support (RFO) from the University of Bologna.References
- H. L. Mobley, M. D. Island and R. P. Hausinger, Microbiol. Rev., 1995, 59, 451–480 CrossRef CAS PubMed
.
- A. Sirko and R. Brodzik, Acta Biochim. Pol., 2000, 47, 1189–1195 CrossRef CAS PubMed
- M. J. Maroney and S. Ciurli, Chem. Rev., 2014, 114, 4206–4228 CrossRef CAS PubMed
- L. Mazzei, F. Musiani and S. Ciurli, J. Biol. Inorg. Chem., 2020, 25, 829–845 CrossRef CAS PubMed
- R. P. Hausinger, Microbiol. Rev., 1987, 51, 22–42 CrossRef CAS PubMed
- H. L. Mobley and R. P. Hausinger, Microbiol. Rev., 1989, 53, 85–108 CrossRef CAS PubMed
-
L. Mazzei, F. Musiani and S. Ciurli, in The Biological Chemistry of Nickel, ed. D. Zamble, M. Rowińska-Żyrek and H. Kozlowski, The Royal Society of Chemistry, 2017, pp. 60–97 Search PubMed
- B. P. Callahan, Y. Yuan and R. Wolfenden, J. Am. Chem. Soc., 2005, 127, 10828–10829 CrossRef CAS PubMed
- Y. Qin and J. M. S. Cabral, Biocatal. Biotransform., 2002, 20(1), 1–14 CrossRef CAS
- K. Kappaun, A. R. Piovesan, C. R. Carlini and R. Ligabue-Braun, J. Adv. Res., 2018, 13, 3–17 CrossRef CAS PubMed
- Fertilizer Outlook 2017–2021, https://www.fertilizer.org/resource/fertilizer-outlook-2017-2021/, (accessed 1 March 2024).
- C. N. Hewitt, A. R. MacKenzie, P. Di Carlo, C. F. Di Marco, J. R. Dorsey, M. Evans, D. Fowler, M. W. Gallagher, J. R. Hopkins, C. E. Jones, B. Langford, J. D. Lee, A. C. Lewis, S. F. Lim, J. McQuaid, P. Misztal, S. J. Moller, P. S. Monks, E. Nemitz, D. E. Oram, S. M. Owen, G. J. Phillips, T. A. M. Pugh, J. A. Pyle, C. E. Reeves, J. Ryder, J. Siong, U. Skiba and D. J. Stewart, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18447–18451 CrossRef CAS PubMed
- R. A. Duce, J. LaRoche, K. Altieri, K. R. Arrigo, A. R. Baker, D. G. Capone, S. Cornell, F. Dentener, J. Galloway, R. S. Ganeshram, R. J. Geider, T. Jickells, M. M. Kuypers, R. Langlois, P. S. Liss, S. M. Liu, J. J. Middelburg, C. M. Moore, S. Nickovic, A. Oschlies, T. Pedersen, J. Prospero, R. Schlitzer, S. Seitzinger, L. L. Sorensen, M. Uematsu, O. Ulloa, M. Voss, B. Ward and L. Zamora, Science, 2008, 320, 893–897 CrossRef CAS PubMed
- R. J. Diaz and R. Rosenberg, Science, 2008, 321, 926–929 CrossRef CAS PubMed
- P. Y. Oikawa, C. Ge, J. Wang, J. R. Eberwein, L. L. Liang, L. A. Allsman, D. A. Grantz and G. D. Jenerette, Nat. Commun., 2015, 6, 8753 CrossRef CAS PubMed
- J. N. Galloway and E. B. Cowling, AMBIO: J. Hum. Environ., 2002, 31, 64–71 CrossRef PubMed
- D. Coskun, D. T. Britto, W. Shi and H. J. Kronzucker, Nat. Plants, 2017, 3, 1–10 Search PubMed
- X. Zhang, E. A. Davidson, D. L. Mauzerall, T. D. Searchinger, P. Dumas and Y. Shen, Nature, 2015, 528, 51–59 CrossRef CAS PubMed
- S. N. Behera and M. Sharma, Sci. Total Environ., 2010, 408, 3569–3575 CrossRef CAS PubMed
- H. Tian, R. Xu, J. G. Canadell, R. L. Thompson, W. Winiwarter, P. Suntharalingam, E. A. Davidson, P. Ciais, R. B. Jackson, G. Janssens-Maenhout, M. J. Prather, P. Regnier, N. Pan, S. Pan, G. P. Peters, H. Shi, F. N. Tubiello, S. Zaehle, F. Zhou, A. Arneth, G. Battaglia, S. Berthet, L. Bopp, A. F. Bouwman, E. T. Buitenhuis, J. Chang, M. P. Chipperfield, S. R. S. Dangal, E. Dlugokencky, J. W. Elkins, B. D. Eyre, B. Fu, B. Hall, A. Ito, F. Joos, P. B. Krummel, A. Landolfi, G. G. Laruelle, R. Lauerwald, W. Li, S. Lienert, T. Maavara, M. MacLeod, D. B. Millet, S. Olin, P. K. Patra, R. G. Prinn, P. A. Raymond, D. J. Ruiz, G. R. van der Werf, N. Vuichard, J. Wang, R. F. Weiss, K. C. Wells, C. Wilson, J. Yang and Y. Yao, Nature, 2020, 586, 248–256 CrossRef CAS PubMed
- X. Zhu, M. Burger, T. A. Doane and W. R. Horwath, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6328–6333 CrossRef CAS PubMed
- J. N. Galloway, F. J. Dentener, D. G. Capone, E. W. Boyer, R. W. Howarth, S. P. Seitzinger, G. P. Asner, C. C. Cleveland, P. A. Green, E. A. Holland, D. M. Karl, A. F. Michaels, J. H. Porter, A. R. Townsend and C. J. Vöosmarty, Biogeochemistry, 2004, 70, 153–226 CrossRef CAS
- R. A. Burne and Y.-Y. M. Chen, Microbes Infect., 2000, 2, 533–542 CrossRef CAS PubMed
- A. Palusiak, Front. Cell. Infect. Microbiol., 2022, 12, 991657 CrossRef CAS PubMed
- M. V. C. Grahl, A. F. Uberti, V. Broll, P. Bacaicoa-Caruso, E. F. Meirelles and C. R. Carlini, Int. J. Mol. Sci., 2021, 22, 7205 CrossRef CAS PubMed
- C. Coker, C. A. Poore, X. Li and H. L. Mobley, Microbes Infect., 2000, 2, 1497–1505 CrossRef CAS PubMed
- H. Mobley, Aliment. Pharmacol. Ther., 1996, 10, 57–64 CrossRef CAS PubMed
- J. Baj, A. Forma, M. Sitarz, P. Portincasa, G. Garruti, D. Krasowska and R. Maciejewski, Cells, 2020, 10, 27 CrossRef PubMed
- P. Malfertheiner, M. C. Camargo, E. El-Omar, J.-M. Liou, R. Peek, C. Schulz, S. I. Smith and S. Suerbaum, Nat. Rev. Dis. Primers, 2023, 9, 1–24 CrossRef PubMed
- S. Diaconu, A. Predescu, A. Moldoveanu, C. S. Pop and C. Fierbințeanu-Braticevici, J. Med. Life, 2017, 10, 112–117 CAS
- M. Naumann, Int. J. Med. Microbiol., 2001, 291, 299–305 CrossRef CAS PubMed
- B. E. Dunn and S. H. Phadnis, Yale J. Biol. Med., 1998, 71, 63–73 CAS
-
IARC, Schistosomes, Liver Flukes and Helicobacter pylori, IARC, Lyon, France, 1994, vol. 61 Search PubMed
- S. Ishaq and L. Nunn, Gastroenterol. Hepatol. Bed Bench, 2015, 8, S6–S14 Search PubMed
- Antimicrobial Resistance Division (AMR), Global Coordination and Partnership (GCP), Medicines Selection, IP and Affordability (MIA), 2017.
-
S. Kiss and M. Simihăian, Improving Efficiency of Urea Fertilizers by Inhibition of Soil Urease Activity, Springer Netherlands, Dordrecht, 2002 Search PubMed
- P. Kafarski and M. Talma, J. Adv. Res., 2018, 13, 101–112 CrossRef CAS PubMed
- L. Mazzei, A. Paul, M. Cianci, M. Devodier, D. Mandelli, P. Carloni and S. Ciurli, J. Inorg. Biochem., 2024, 250, 112398 CrossRef CAS PubMed
- K. Macegoniuk, W. Tabor, L. Mazzei, M. Cianci, M. Giurg, K. Olech, M. Burda-Grabowska, R. Kaleta, A. Grabowiecka, A. Mucha, S. Ciurli and Ł. Berlicki, J. Med. Chem., 2023, 66, 2054–2063 CrossRef CAS PubMed
- L. Mazzei, M. Cianci and S. Ciurli, Chem. – Eur. J., 2022, 28, e202201770 CrossRef CAS PubMed
- L. Mazzei, D. Cirri, M. Cianci, L. Messori and S. Ciurli, J. Inorg. Biochem., 2021, 218, 111375 CrossRef CAS PubMed
- L. Mazzei, L. Massai, M. Cianci, L. Messori and S. Ciurli, Dalton Trans., 2021, 50, 14444–14452 RSC
- L. Mazzei, U. Contaldo, F. Musiani, M. Cianci, G. Bagnolini, M. Roberti and S. Ciurli, Angew. Chem., 2021, 133, 6094–6100 CrossRef
-
L. Mazzei and S. Ciurli, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2021, pp. 1–11 Search PubMed
- L. Mazzei, M. N. Wenzel, M. Cianci, M. Palombo, A. Casini and S. Ciurli, ACS Med. Chem. Lett., 2019, 10, 564–570 CrossRef CAS PubMed
- L. Mazzei, M. Cianci, U. Contaldo and S. Ciurli, J. Agric. Food Chem., 2019, 67, 2127–2138 CrossRef CAS PubMed
- L. Mazzei, M. Cianci, A. G. Vara and S. Ciurli, Dalton Trans., 2018, 47, 8240–8247 RSC
- L. Mazzei, M. Cianci, F. Musiani, G. Lente, M. Palombo and S. Ciurli, J. Inorg. Biochem., 2017, 166, 182–189 CrossRef CAS PubMed
- L. Mazzei, M. Cianci, U. Contaldo, F. Musiani and S. Ciurli, Biochemistry, 2017, 56, 5391–5404 CrossRef CAS PubMed
- L. Mazzei, M. Cianci, F. Musiani and S. Ciurli, Dalton Trans., 2016, 45, 5455–5459 RSC
- L. Mazzei, M. Cianci, S. Benini, L. Bertini, F. Musiani and S. Ciurli, J. Inorg. Biochem., 2016, 154, 42–49 CrossRef CAS PubMed
- S. Benini, M. Cianci, L. Mazzei and S. Ciurli, J. Biol. Inorg. Chem., 2014, 19, 1243–1261 CrossRef CAS PubMed
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Mangani and S. Ciurli, J. Am. Chem. Soc., 2004, 126, 3714–3715 CrossRef CAS PubMed
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, J. Biol. Inorg. Chem., 2000, 5, 110–118 CrossRef CAS PubMed
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, Structure, 1999, 7, 205–216 CrossRef CAS PubMed
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Ciurli and S. Mangani, J. Biol. Inorg. Chem., 1998, 3, 268–273 CrossRef CAS
- Â. de Fátima, C. de P. Pereira, C. R. S. D. G. Olímpio, B. G. de Freitas Oliveira, L. L. Franco and P. H. C. da Silva, J. Adv. Res., 2018, 13, 113–126 CrossRef PubMed
- L. Habala, F. Devínsky and A. E. Egger, J. Coord. Chem., 2018, 71, 907–940 CrossRef CAS
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC
- Z. Chen, Z. Wang, J. Ren and X. Qu, Acc. Chem. Res., 2018, 51, 789–799 CrossRef CAS PubMed
- C.-M. Che and F.-M. Siu, Curr. Opin. Chem. Biol., 2010, 14, 255–261 CrossRef CAS PubMed
- W. Yang, Z. Peng and G. Wang, J. Enzyme Inhib. Med. Chem., 2023, 38, 361–375 CrossRef CAS PubMed
- L. Zhang, S. B. Mulrooney, A. F. K. Leung, Y. Zeng, B. B. C. Ko, R. P. Hausinger and H. Sun, Biometals, 2006, 19, 503–511 CrossRef CAS PubMed
- Y. Cao, J. Zhang, Y. Liu, L. Zhang, L. Wang, J. Wang, Y. Qi, H. Lv, J. Liu, L. Huo, X. Wei and Y. Shi, Medicine, 2021, 100, e27923 CrossRef CAS PubMed
- M. P. Dore, H. Lu and D. Y. Graham, Gut, 2016, 65, 870–878 CrossRef CAS PubMed
- R. M. Zagari, S. Rabitti, L. H. Eusebi and F. Bazzoli, Eur. J. Clin. Invest., 2018, 48, e12857 CrossRef PubMed
- J. d. S. Rosário, F. H. Moreira, L. H. F. Rosa, W. Guerra and P. P. Silva-Caldeira, Molecules, 2023, 28, 5921 CrossRef PubMed
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed
- V. André, A. Hardeman, I. Halasz, R. S. Stein, G. J. Jackson, D. G. Reid, M. J. Duer, C. Curfs, M. T. Duarte and T. Friščić, Angew. Chem., Int. Ed., 2011, 50, 7858–7861 CrossRef PubMed
- F. Grepioni, L. Casali, C. Fiore, L. Mazzei, R. Sun, O. Shemchuk and D. Braga, Dalton Trans., 2022, 51, 7390–7400 RSC
- L. Casali, L. Mazzei, R. Sun, M. R. Chierotti, R. Gobetto, D. Braga, F. Grepioni and S. Ciurli, Cryst. Growth Des., 2022, 22, 4528–4537 CrossRef CAS
- L. Casali, V. Broll, S. Ciurli, F. Emmerling, D. Braga and F. Grepioni, Cryst. Growth Des., 2021, 21, 5792–5799 CrossRef CAS
- D. Braga, Chem. Commun., 2023, 59, 14052–14062 RSC
- C. Fiore, A. Lekhan, S. Bordignon, M. R. Chierotti, R. Gobetto, F. Grepioni, R. J. Turner and D. Braga, Int. J. Mol. Sci., 2023, 24, 5180 CrossRef CAS PubMed
- R. Sun, L. Casali, R. J. Turner, D. Braga and F. Grepioni, Molecules, 2023, 28, 1244 CrossRef CAS PubMed
- L. Casali, L. Mazzei, O. Shemchuk, L. Sharma, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2019, 7, 2852–2859 CrossRef CAS
- S. P. Summers, K. A. Abboud, S. R. Farrah and G. J. Palenik, Inorg. Chem., 1994, 33, 88–92 CrossRef CAS
- V. Stavila, R. L. Davidovich, A. Gulea and K. H. Whitmire, Coord. Chem. Rev., 2006, 250, 2782–2810 CrossRef CAS
- L. M. Shkol’nikova, R. L. Davidovich, V. S. Fundamenskii, K. D. Suyarov and R. L. Davidovich, Koord. Khim., 1991, 17, 253–261 Search PubMed
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, J. Appl. Crystallogr., 2013, 46, 1231–1235 CrossRef CAS
- A. Altomare, G. Campi, C. Cuocci, L. Eriksson, C. Giacovazzo, A. Moliterni, R. Rizzi and P.-E. Werner, J. Appl. Crystallogr., 2009, 42, 768–775 CrossRef CAS
- R. L. Blakeley, E. C. Webb and B. Zerner, Biochemistry, 1969, 8, 1984–1990 CrossRef CAS PubMed
-
A.E Martell, R.M Smith and R.J Motekaitis, NIST standard reference database, 1997 Search PubMed
- T. R. Bhat and R. Krishna Iyer, J. Inorg. Nucl. Chem., 1967, 29, 179–185 CrossRef CAS
- S. Kumar, C. Schmitt, O. Gorgette, M. Marbouty, M. Duchateau, Q. Giai Gianetto, M. Matondo, J.-M. Guigner and H. De Reuse, mBio, 2022, 13, e01633-22 CrossRef PubMed
- T. Nagai and S. Oita, J. Gen. Appl. Microbiol., 2004, 50, 115–118 CrossRef CAS PubMed
- P. Malfertheiner and M. Selgrad, Curr. Opin. Gastroenterol., 2014, 30, 589–595 CrossRef CAS PubMed
- N. E. Dixon, C. Gazzola, C. J. Asher, D. S. W. Lee, R. L. Blakeley and B. Zerner, Can. J. Biochem., 1980, 58, 474–480 CrossRef CAS PubMed
- L. Mazzei, V. Broll and S. Ciurli, Soil Sci. Soc. Am. J., 2018, 82, 994–1003 CrossRef CAS
- R. Wang, T.-P. Lai, P. Gao, H. Zhang, P.-L. Ho, P. C.-Y. Woo, G. Ma, R. Y.-T. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439–450 CrossRef PubMed
- R. Wang, H. Li, T. K.-Y. Ip and H. Sun, Adv. Inorg. Chem., 2020, 75, 183–205 CrossRef CAS
- E. Uribe, M.-B. Reyes, I. Martínez, K. Mella, M. Salas, E. Tarifeño-Saldivia, V. López, M. García-Robles, J. Martínez-Oyanedel, M. Figueroa, N. Carvajal and G. Schenk, J. Inorg. Biochem., 2020, 202, 110802–110808 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction patterns, crystal data, Rietveld refinements plots, DSCand TGA traces, 1H NMR, antimicrobial activity tests. CCDC 2340273 and 2340274. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00778f |
| ‡ Ref. 63 reports measurements conducted on Bi(EDTA) synthesized as described previously by Summers et al. (see ref. 77 in this article). In Summers's paper the synthesis yields complex (2), which is labelled as Bi(HEDTA); the single crystal structure of complex (2), however, with coordinates deposited in the CSD (refcode HAYPEC), is that of the dihydrate Bi(HEDTA)·2H2O. Therefore, the complex studied in ref. 63 is the dihydrated complex Bi(HEDTA)·2H2O. For this reason, our enzymatic assays have also been conducted on the dihydrated complex. |
| This journal is © The Royal Society of Chemistry 2024 |