
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defluorination of HFCs by a magnesium reagent†
Daniel J.
Sheldon
 ,
Joseph M.
Parr
and
Mark R.
Crimmin
*
,
Joseph M.
Parr
and
Mark R.
Crimmin
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 28th March 2024
Abstract
Reaction of a series of HFCs with a main group reagent containing a Mg–Mg bond results in defluorination to form the corresponding magnesium fluoride complex. In the case of 1,1,1,2-tetrafluoroethane (HFC-134a) generation of the fluoride occurs alongside selective formation of 1,1-difluoroethene. DFT calculations have been carried out to better understand the selectivity and compare the barriers for sp3 C–F bond activation with sp3 C–H bond activation in this system.
Due to their unique chemical and physical properties hydrofluorocarbons (HFCs) have been widely used as 3rd generation refrigerants. The largest use HFC for this purpose is 1,1,1,2-tetrafluoroethane (HFC-134a). Perhaps as a result, HFC-134a has become one of the most abundant fluorinated gases in the atmosphere. HFC-134a has a 100-year global warming potential (GWP100) of 1530 and is contributing to climate change. Legislation is now in place to limit the use of HFCs globally.1–5 The planned phase-down of HFCs is however a gradual process; developed countries have committed to phase-down by 85% by 2036, with other countries committing to 80% by the late 2040s.6 Consequently, HFCs may continue to be emitted for a long time. There is a need to develop processes to repurpose HFCs, ideally with destruction and removal of the fluorine content of these molecules.
Reactions of 1,1,1,2-tetrafluoroethane nearly always involve pathways that result in the formal elimination of an equivalent of HF (Fig. 1). Strong bases, organometallic reagents, or heterogeneous catalysts react with HFC-134a to form 1,1,2-trifluoroethene.7–9 This chemoselectivity is driven by the strong sp3 C–F bond strengths of both fluorinated sites, and the relative acidity of the adjacent sp3 C–H bonds. In the case of reactions with organometallic compounds, in situ deprotonation of 1,1,2-trifluoroethane to form a trifluorovinyl moiety is common. For example, it has been reported that the reaction of HFC-134a with 2 equiv. n-BuLi at −78 °C forms trifluorovinyllithium. This species, which decomposes if warmed to room temperature, can react at −78 °C with a wide range of electrophiles including metal halides, main group halides, CO2, aldehydes and epoxides.7,8,10–26 Other reports include the transfer of the trifluorovinyl group onto zinc chlorides for application in palladium-catalysed Negishi cross-coupling reactions.19,27–31
 | ||
| Fig. 1 Known HF elimination pathway for HFC-134a and this work. | ||
To the best of our knowledge there are no examples of reactions of HFC-134a which involve solely sp3 C–F bond activation. In this communication, we show that reaction of HFC-134a with a main group reagent containing a Mg–Mg bond leads to exclusive formation of 1,1-difluoroethene due to the formal 1,2-elimination of two F atoms from the substrate. This is a rare example of selective sp3 C–F bond activation of this industrially important HFC.
HFC-134a (1 bar, 25 °C, approx. 7 equiv.) was added to a degassed C6D6 solution of 1 and 4-dimethylaminopyridine (DMAP, 2 equiv.). The reaction mixture was agitated and left to proceed for 16 h at 25 °C, during which time the dark red solution turned yellow. 1H and 19F NMR spectroscopy of the resultant reaction mixture reveal the formation of 2 (86% yield) and 1,1-difluoroethene (Scheme 1). 2 has been previously reported and characterised by our group, and shows a diagnostic resonance at δ = −183.9 ppm in the 19F NMR spectrum.32 1,1-Difluoroethene has a resonance at δ = −81.8 ppm, in accordance with data in the literature.33 Due to its volatility and partitioning between solution and reaction headspace, the amount of 1,1-difluoroethene was not quantified, subsequent onwards reaction however, support generation in reasonable yields (vide infra).
 | ||
| Scheme 1 Defluorination of HFC-134a by a magnesium reagent (1) and DMAP. | ||
Jones and co-workers have shown that the magnesium nucleophile used in this reaction, 1,34,35 displays enhanced reactivity on the addition of a Lewis base,36,37 while we have recently demonstrated its use in the defluorination of PTFE.32 Defluorination of HFC-134a with 1 can also occur in the absence of DMAP, but requires heating to 80 °C, and results in 30% yield of an analogue of 2 which does not have DMAP coordinated.
To gain further insight into this unique reactivity, the mechanism of defluorination of HFC-134a was studied using computational techniques (DFT). Binding of DMAP to 1 was assumed to be fast and reversible under the reaction conditions, reinforced by observed fluxionality in 1H NMR spectroscopic data for 1·DMAP suggesting that the DMAP can move rapidly between Mg centres.32,36,37
The reaction is initiated by attack of 1·DMAP at the sp3 C–F bond of the –CH2F group of HFC-134a, viaTS-1 (ΔG‡298 K = 19.5 kcal mol−1), to form Int-1. This reactivity mode for 1·DMAP has been established previously in the defluorination of poly(tetrafluoroethene) (PTFE).32TS-1 is asymmetric, the three-coordinate Mg centre of 1·DMAP inserts into the C–F bond, with the fluoride ion migrating into a bridging position between both Mg sites. One way to conceptualise this reaction is as a bimetallic oxidative addition of the C–F bond of the HFC to 1·DMAP. Calculated NPA charges in 1·DMAP reveal the 3-coordinate Mg atom has a less positive charge (+0.84) compared to the four-coordinate Mg atom (+1.07), and hence attack originates from the three-coordinate Mg atom. It has been proposed that polarisation and stretching of the Mg–Mg bond in 1·DMAP leads to enhanced reactivity compared to the symmetric species 1 or 1·DMAP2.32,37 NBO calculations reveal a flow of charge from 1·DMAP to the fluorine atom of HFC-134a as TS-1 is traversed (see ESI†). Int-1 undergoes fluoride elimination viaTS-2 (ΔG‡298 K = 6.6 kcal mol−1) to form 1,1-difluoroethene, and reaction of the magnesium fluoride intermediate with a second equivalent of DMAP forms 2. This second step effectively breaks one of the C–F bonds of the CF3 group of HFC-134a completing the formal 1,2-defluorination (Fig. 2).
 | ||
| Fig. 2 Calculated reaction pathway for defluorination of HFC-134a by 1 + DMAP. Gibbs energies in kcal mol−1. G09:B3PW91-GD3BJ/6-311+G*(C,H,N,F)/SDDAll(Mg)//B3PW91-GD3BJ/6-31G**(C,H)/6-311+G*(N,F)/SDDAll(Mg). Inset, representation of TS-1 annotated with NPA charges on key atomic sites. | ||
The mechanism proposed based on DFT calculations is perhaps intuitive. The fluorophilicity of 1 likely results in selective sp3 C–F bond activation over sp3 C–H bond activation, while the trends in bonds strengths would favour reaction at the isolated sp3 C–F bond.38 Alternative mechanisms were calculated and found to lead to transition states that were less energetically accessible than TS-1. Deprotonation of the sp3 C–H bond of HFC-134a with 1·DMAP was calculated to occur viaTS-3 with ΔG‡298 K = 27.1 kcal mol−1. Attack at a fluorine atom of the CF3 group of HFC-134a was calculated to occur viaTS-4, ΔG‡298 K = 26.5 kcal mol−1. A concerted pathway involving simultaneous breaking of two sp3 C–F bonds was also considered but a suitable transition state could be not be located.
Consideration of selectivity in the possible bond breaking events for HFC-134a raises the question as to how small changes to structure might affect the reaction outcome. A range of hydrofluoroethanes including 1,1,1-trifluoroethane (HFC-143a, GWP100 = 5810), 1,1-difluoroethane (HFC-143a, GWP100 = 164) and 1,1,1,2,2-pentafluoroethane (HFC-125, GWP100 = 3740) are widely available and used in the refrigeration sector.9,39 Along with HFC-134a, this series of substrates represents a group with systematic changes in number and position of fluorine atoms.
The computational model was extended to examine the activation barriers for sp3 C–F activation and sp3 C–H activation of this range of HFCs. Several trends are consistent across the series: (i) in general activation barriers increase with lower fluorine content of the substrate, (ii) in cases where multiple C–F bonds are present there is preference for CFH2 > CF3 and CF2H > CF3 groups – reflecting the known trends in sp3 C–F bond strengths,38 and (iii) sp3 C–F activation is consistently a more facile process than sp3 C–H bond activation (Fig. 3). For example, the activation barriers for sp3 C–F bond activation (TS-1) increase in the order 1,1,1,2,2-pentafluoroethane (HFC-125, ΔG‡298 K = 20.5, 25.8 kcal mol−1), 1,1,1-trifluoroethane (HFC-143a, ΔG‡298 K = 29.6 kcal mol−1), 1,1-difluoroethane (HFC-152a, ΔG‡298 K = 32.7 kcal mol−1). For 1,1,1,2,2-pentafluoroethane, there are two possible isomeric transition states for C–F bond activation, with attack of 1·DMAP occurring at either the CF2H or CF3 bond of the substrate, the lowest energy pathway involves defluorination of the difluoromethyl site.
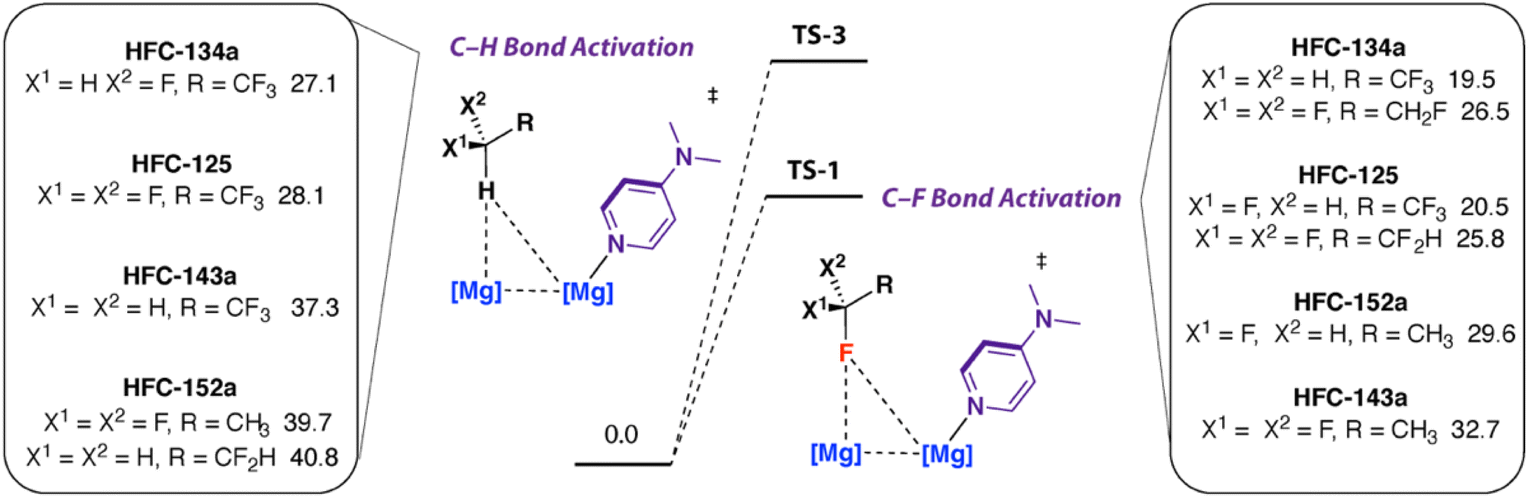 | ||
| Fig. 3 Calculated barriers for C–F and C–H activation of HFCs with 1 + DMAP. Gibbs energies in kcal mol−1. G09:B3PW91-GD3BJ/6-311+G*(CH,N,F)/SDDAll(Mg)//B3PW91-GD3BJ/6-31G**(C,H)/6-311+G*(N,F)/SDDAll(Mg). | ||
The calculations suggest that these HFCs should also undergo chemoselective defluorination under accessible conditions with 1 + DMAP. Reaction of 1,1,1,2,2-pentafluoroethane with 1 + DMAP at 25 °C in C6D6 for 5 days led to the formation of 2 in a 59% yield. Reaction of 1 + DMAP with 1,1-difluoroethane in C6D6 occurred slowly at 25 °C, but was complete within 16 h at 60 °C forming 2 in 41% yield. 1 + DMAP also reacts with 1,1,1-trifluoroethane in C6D6 to form 2 after 16 h at 60 °C (Scheme 2). The elevated temperatures required for these reactions are consistent with the trends in calculated activation energies. In all these cases, magnesium hydride side-products derived from sp3 C–H activation are not observed, suggesting selective reaction at the sp3 C–F bond.
 | ||
| Scheme 2 Defluorination of HFC-125, HFC-143a, HFC-152a by 1 + DMAP. | ||
A minor fluoroalkene containing product could be identified from the reaction of 1,1,1,2,2-pentafluoroethane with 1 and DMAP, identified as α,β,β-trifluorostyrene-d5, formed in a 5–10% yield (based on 1 equivalent of 1). α,β,β-Trifluorostyrene-d5 likely derives from a defluorinative coupling of the HFC and the benzene-d6 reaction solvent. While the pathway for its formation remains unclear, generation of a fluoroethene intermediate cannot be ruled out at this stage.
In the case of both 1,1-difluoroethane and 1,1,1-trifluoroethane, we could not identify the organic products of these reactions. No obvious fluorine containing species were present following vacuum transfer of the volatiles as evidenced by 19F NMR spectroscopy. No products beyond 2 could be isolated from the residue and as such, the precise fate of the organic fragment in these reactions remains an open question. Nevertheless, the lack of clear formation of ethene products implies is that, without fluorine substitution at both carbon atoms of the ethane, the 1,2-elimination pathway may be switched off. In these cases, sp3 C–F bond activation would generate intermediate organomagnesium complexes likely to decompose through α-elimination pathways forming 2 along with unstable carbene fragments.
In terms of further use of the products from the defluorination of HFCs, we have previously shown that 2 can act as a nucleophilic source of fluoride, capably of fluorinating a small array of highly activated electrophiles.32 In the case of HFC-134a, the volatile product 1,1-difluoroethene could be separated from 2 by vacuum transfer, allowing further derivatisation by nucleophilic substitution (Scheme 3). 1,1-Difluoroethene was defluorosilylated upon reaction with the lithium silanide 4 to form the organosilicon compound 5 in 40% (NMR yield, over two steps based on 1 as the limiting reagent). 5, a known compound, was characterised by a diagnostic resonance in the 19F NMR spectrum at δ = −103.0 ppm.40 Related defluorosilylation reactions of fluoroalkenes including difluoroethene have been previously reported.41–43
 | ||
| Scheme 3 Two-step derivatisation of HFC-134a via 1,2-defluorination and nucleophilic substitution. Yield determined by 19F NMR spectroscopy. | ||
In summary, we report the defluorination of a range of HFC refrigerants using a magnesium reagent. In the case of 1,1,1,2-tetrafluoroethene (HFC-134a), the largest commercial HFC, a highly unusual pathway for 1,2-defluorination to form 1,1-difluoroethene was observed. This pathway complements established reaction patterns which nearly always result in formation of 1,1,2-trifluoroethene through elimination of an equivalent of HF. DFT calculations have been used to compare the chemoselectivity in these systems. sp3 C–F bond activation universally occurs with lower barriers than sp3 C–H bond activation, likely due to participation of fluorophilic magnesium sites in the key transition state for bond breaking. The development of mechanistic understanding in reactions that defluorinate HFCs has the potential to underpin future approaches to repurpose these potent greenhouse gases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the ERC for generous funding (Fluorocycle: 101001071) and Imperial College London and the EPSRC for DTP studentship funding (DJS). We thank Apollo Scientific for donation of HFCs.References
- F. Polonara, L. J. Kuijpers and R. Peixoto, Int. J. Heat Technol., 2017, 35, S1–S8 CrossRef.
- A. J. Manning, A. L. Redington, D. Say, S. O'Doherty, D. Young, P. G. Simmonds, M. K. Vollmer, J. Mühle, J. Arduini and G. Spain, et al. , Atmos. Chem. Phys., 2021, 21, 12739–12755 CrossRef CAS.
- H. Flerlage, G. J. M. Velders and J. de Boer, Chemosphere, 2021, 283, 131208 CrossRef CAS PubMed.
- G. J. M. Velders, J. S. Daniel, S. A. Montzka, I. Vimont, M. Rigby, P. B. Krummel, J. Muhle, S. O'Doherty, R. G. Prinn and R. F. Weiss, et al. , Atmos. Chem. Phys., 2022, 22, 6087–6101 CrossRef CAS.
- J. M. Calm, Int. J. Refrig., 2008, 31, 1123–1133 CrossRef CAS.
- E. A. Heath, Int. Leg. Mater., 2017, 56, 193–205 CrossRef.
- J. Burdon, P. L. Coe, I. B. Haslock and R. L. Powell, Chem. Commun., 1996, 49–50 RSC.
- K. K. Banger, A. K. Brisdon and A. Gupta, Chem. Commun., 1997, 139–140 RSC.
- D. J. Sheldon and M. R. Crimmin, Chem. Soc. Rev., 2022, 51, 4977–4995 RSC.
- S. Duric, B. M. Schmidt, N. M. Ninnemann, D. Lentz and C. C. Tzschucke, Chem. – Eur. J., 2012, 18, 437–441 CrossRef CAS PubMed.
- K. K. Banger and A. K. Brisdon, J. Organomet. Chem., 1999, 582, 301–309 CrossRef CAS.
- A. K. Brisdon, I. R. Crossley, R. G. Pritchard and G. Sadiq, J. Organomet. Chem., 2007, 692, 2125–2130 CrossRef CAS.
- N. A. Barnes, A. K. Brisdon, W. I. Cross, J. G. Fay, J. A. Greenall, R. G. Pritchard and J. Sherrington, J. Organomet. Chem., 2000, 616, 96–105 CrossRef CAS.
- P. L. Coe, J. Burdon and I. B. Haslock, J. Fluor. Chem., 2000, 102, 43–50 CrossRef CAS.
- J. Burdon, P. L. Coe, I. B. Haslock and R. L. Powell, J. Fluor. Chem., 1997, 85, 151–153 CrossRef CAS.
- K. K. Banger, R. P. Banham, A. K. Brisdon, W. I. Cross, G. Damant, S. Parsons, R. G. Pritchard and A. Sousa-Pedrares, J. Chem. Soc., Dalton Trans., 1999, 427–434 RSC.
- J. E. Phelps, S. B. Frawley and R. G. Peters, Heteroat. Chem., 2009, 20, 393–397 CrossRef CAS.
- K. Wu and Q.-Y. Chen, Tetrahedron, 2002, 58, 4077–4084 CrossRef CAS.
- J. Burdon, P. L. Coe, I. B. Haslock and R. L. Powell, J. Fluor. Chem., 1999, 99, 127–131 CrossRef CAS.
- K. Nickisch, W. Elger, J. Cessac, N. Kesavaram, B. Das, R. Garfield, S.-Q. Shi, O. Amelkina and R. Meister, Steroids, 2013, 78, 255–267 CrossRef CAS PubMed.
- N. A. Barnes, A. K. Brisdon, F. R. W. Brown, W. I. Cross, I. R. Crossley, C. Fish, J. V. Morey, R. G. Pritchard and L. Sekhri, New J. Chem., 2004, 28, 828–837 RSC.
- K. K. Banger, A. K. Brisdon, C. J. Herbert, H. A. Ghaba and I. S. Tidmarsh, J. Fluor. Chem., 2009, 130, 1117–1129 CrossRef CAS.
- G. DiMartino, M. B. Hursthouse, M. E. Light, J. M. Percy, N. S. Spencer and M. Tolley, Org. Biomol. Chem., 2003, 1, 4423–4434 RSC.
- D. J. Brauer and G. Pawelke, J. Organomet. Chem., 2000, 604, 43–51 CrossRef CAS.
- K. Wu and Q.-Y. Chen, J. Fluor. Chem., 2003, 122, 171–174 CrossRef CAS.
- T. Okano, M. Chokai, M. Hiraishi, M. Yoshizawa, T. Kusukawa and M. Fujita, Tetrahedron, 2004, 60, 4031–4035 CrossRef CAS.
- A. Raghavanpillai and D. J. Burton, J. Org. Chem., 2004, 69, 7083–7091 CrossRef CAS PubMed.
- K. Fuchibe, K. Shigeno, N. Zhao, H. Aihara, R. Akisaka, T. Morikawa, T. Fujita, K. Yamakawa, T. Shimada and J. Ichikawa, J. Fluor. Chem., 2017, 203, 173–184 CrossRef CAS.
- R. Anilkumar and D. J. Burton, Tetrahedron Lett., 2002, 43, 2731–2733 CrossRef CAS.
- H. Miyauchi, N. Kondo and N. Nagasaki, Substituted bis(trifluorovinylbenzene) compound, US Pat, US2020017429, 2020 Search PubMed.
- P. V. Ramachandran and G. V. Reddy, J. Fluor. Chem., 2008, 129, 443–446 CrossRef CAS.
- D. J. Sheldon, J. M. Parr and M. R. Crimmin, J. Am. Chem. Soc., 2023, 145, 10486–10490 CrossRef CAS PubMed.
- N. O. Andrella, N. Xu, B. M. Gabidullin, C. Ehm and R. T. Baker, J. Am. Chem. Soc., 2019, 141, 11506–11521 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- C. Jones, Nat. Rev. Chem., 2017, 1, 59 CrossRef CAS.
- K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS PubMed.
- K. Yuvaraj, I. Douair, D. D. L. Jones, L. Maron and C. Jones, Chem. Sci., 2020, 11, 3516–3522 RSC.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb and M. I. Gomis, et al., Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Oxford, UK, 2021 Search PubMed.
- H. Sakaguchi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2018, 57, 328–332 CrossRef CAS PubMed.
- G. Coates, H. Y. Tan, C. Kalff, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2019, 58, 12514–12518 CrossRef CAS PubMed.
- T. Hanamoto, S. Harada, K. Shindo and M. Kondo, Chem. Commun., 1999, 2397–2398 RSC.
- Unreacted HFC-134a is also present in this reaction mixture, and if left for 16 hours some deprotonation by 4 is observed, leading to a more complicated product mixture.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00636d |
| This journal is © The Royal Society of Chemistry 2024 |