DOI:
10.1039/D4DT00627E
(Paper)
Dalton Trans., 2024,
53, 11410-11416
Preparation of a high-coordinated-silicon-centered spiro-cyclic compound†
Received
1st March 2024
, Accepted 4th June 2024
First published on 10th June 2024
Abstract
Silicon compounds containing silicon–silicon bond with a variety of unusual oxidation states are quite important, because their high reactivity leads to the formation of a variety of silicon compounds. The isolation of such compounds with unusual oxidation states requires a resilient synthetic strategy. Herein, we report the synthesis of a silicon based spirocyclic compound containing a hyper-valent silicon atom and a silicon–silicon bond. The computational calculations employing natural bond orbital (NBO) analysis and energy decomposition analysis–natural orbitals for chemical valence (EDA–NOCV) reveal that the nature of bonding between the silicon atoms is of an electron sharing nature.
Introduction
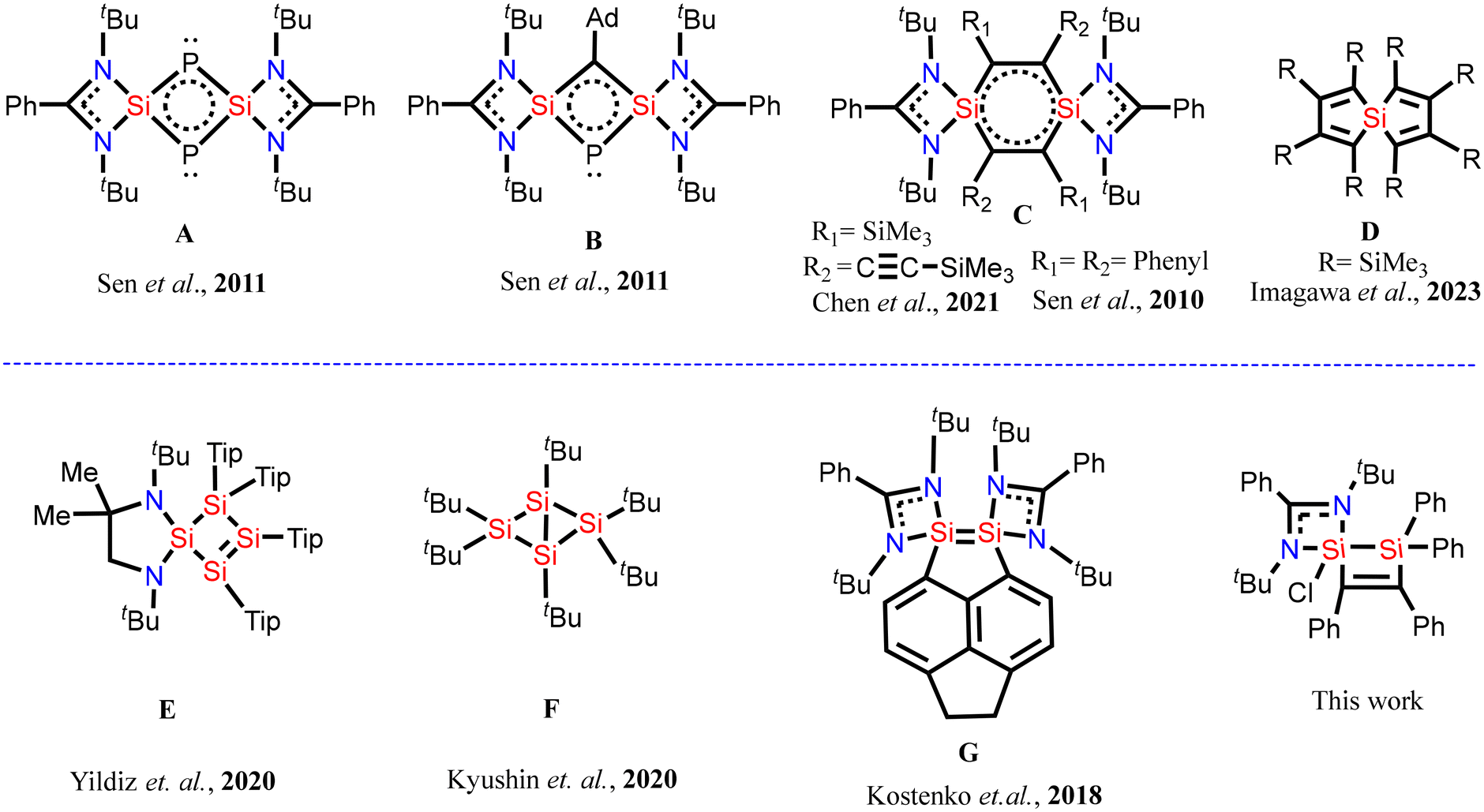
Generally, silylenes are highly reactive compounds that contain divalent silicon with a lone pair of electrons and a vacant p-orbital, similar to carbenes.1–7 The diverse reactivity of silylenes may be attributed to their unique ambiphilic nature, wherein the nucleophilic property arises from the presence of a non-bonded electron pair on the silicon atom and the electrophilic property arises due to an empty p-orbital on the silicon atom. However, donor-stabilized silylenes have also been reported wherein the electron pair present on the nitrogen atom of an amidinate moiety is donated to the empty p-orbital of the silicon atom.1 Such silylenes are more stable than the usual silylenes. Since the first report of N-heterocyclic silylene (NHSi) in 1994,4 the chemistry of this class of molecules has progressed in different directions and it has found several applications including activation of small molecules,8–10 stabilization of low-valent metal atoms11a,b and as cross-linkers in elastomers.11c Silylenes have also been used to prepare compounds with silicon–silicon bonds containing silicon atoms in unusual oxidation states.12 In addition, silylenes have also provided access to heterocyclic/spirocyclic silicon compounds that are difficult to synthesize by conventional methods. As a result, organo-silicon heterocyclic compounds and silicon centred spirocyclic compounds have been synthesised by reacting silylenes with unsaturated alkynes, alkenes, phosphorus etc.13–17 For example, Sen et al. isolated silicon-centred spirocyclic compounds, A and B, in which two silicon atoms are bridged by two-coordinate phosphorus atoms (Fig. 1).18 Similarly, Chen et al. and Sen et al. synthesized an important silicon spirocyclic compound, C, that possesses a unique stable 1,4-disilabenzene ring.13,15 Moreover, Imagawa et al. have reported a spirocyclic compound, D, wherein the spirocyclic silicon centre is bound to four carbon atoms.19 These spirocyclic compounds do not possess silicon–silicon bonds. However, Scheschkewitz and coworkers reported a spirocyclic silicon compound, E, in which one of the rings contains four silicon atoms connected to each other with silicon–silicon σ-bonds and a silicon–silicon π-bond.20 This molecule represents the first example of a main group analogue of cyclobutane-1,3-diyls, wherein no other heteroatoms are present in the ring except silicon. Similarly, Matsumoto and coworkers have synthesized a unique cyclic compound, F, in which a single silicon–silicon π-bond is present, not accompanied by a σ-bond.21 This single π-bond happens to be the longest silicon–silicon bond reported to date. Moreover, Driess and coworkers were able to stabilize a silicon–silicon double bond in a geometrically compelled disilene, G, which was accessible by inserting a suitable organic linker between two amidinato-silylene molecules.22 These spirocyclic compounds are expected to be more reactive compared to the usual silicon compounds due to ring strain and hence, reactivity studies of these compounds can give further impetus to silicon chemistry. Herein, we report preparation of a hyper-coordinated silicon-centered spirocyclic compound containing a silicon–silicon bond.
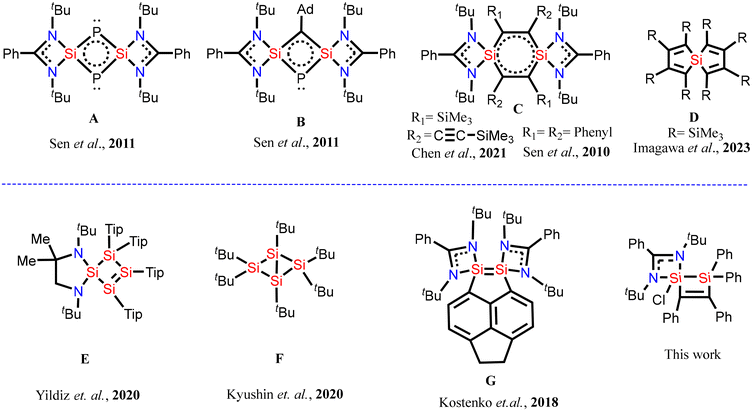 |
| | Fig. 1 Selected examples of silicon centered spirocyclic compounds: (top) spirocyclic compounds without silicon–silicon bonds and (bottom) spirocyclic compounds with silicon–silicon bonds. | |
Results and discussion
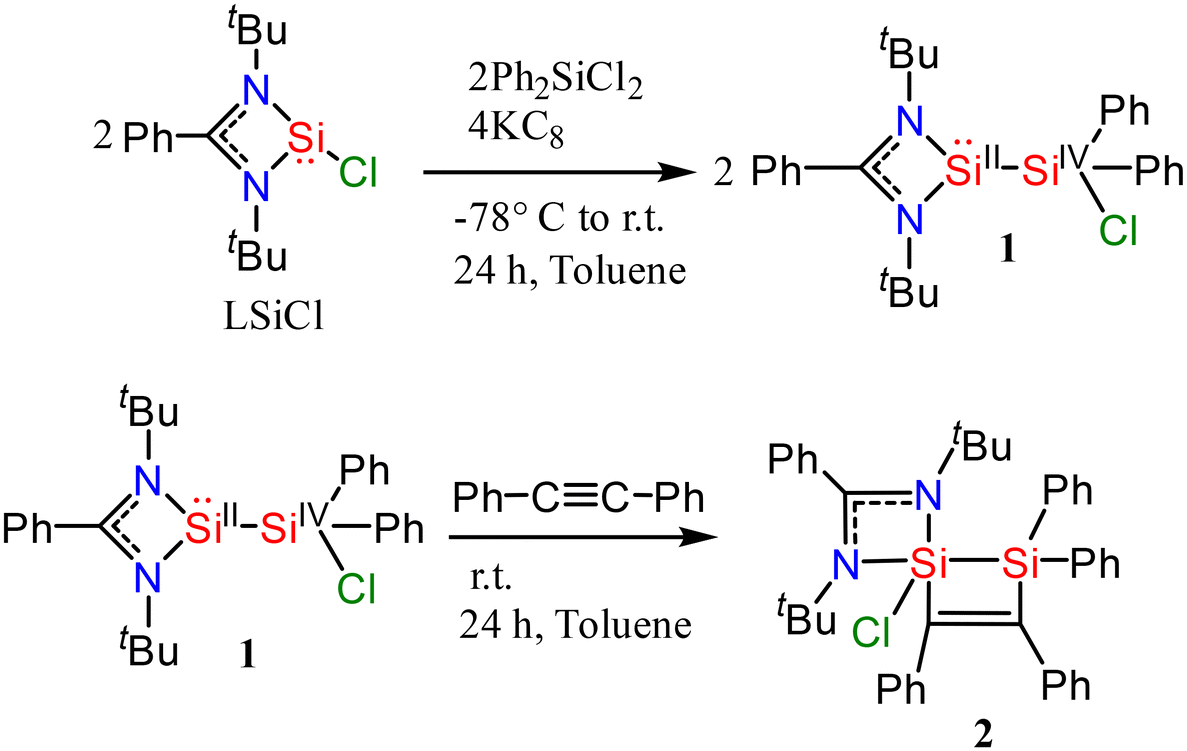
Synthesis of compound 1
Amidinato-silylene (2 mmol) and Ph2SiCl2 (2 mmol) were mixed at −77 °C in 100 ml of toluene to obtain a clear transparent solution (Scheme 1). The reaction mixture was stirred for 1 h at room temperature. Afterwards, it was cannulated to a flask containing KC8 (4 mmol) at −77 °C and was stirred overnight. Afterwards, it was filtered using a Schlenk frit under a flow of nitrogen. The clear filtrate was concentrated to about 4 ml and kept undisturbed at room temperature for two months. The block-shaped colorless crystals of compound 1 were picked from the flask with a 13% yield. Compound 1 was thoroughly characterized by single crystal X-ray diffraction (SC-XRD), and 1H, 13C and 29Si NMR spectroscopy (Fig. S1–S3 in the ESI†). The aliphatic protons of the tert-butyl group appear at 0.95 ppm while aromatic protons appear in the range of 6.86–8.16 ppm with a well resolved splitting pattern. 13C NMR also shows resonances in the expected region wherein the tert-butyl-C appears at 52.8 ppm and the methyl-C at 30.9 ppm, whereas carbon atoms from aromatic rings resonate in the range of 127–140 ppm. The 29Si NMR spectrum exhibits two resonances at 35.9 and 10.2 ppm originating from silylene-silicon and non-silylene silicon centers, respectively (Fig. S7–S9†).
 |
| | Scheme 1 Synthesis of compounds 1 and 2. | |
Synthesis of compound 2
Compound 1 (0.5 mmol) and 1,2-diphenylethyne (0.5 mmol) were mixed in toluene at room temperature (Scheme 1). Subsequently, the reaction mixture was stirred for 24 h. It was then filtered and the solvent was removed. The compound was dissolved in hexane and the concentrated solution was kept for crystallization at room temperature for one month. Block-shaped crystals of compound 2 were isolated with 37% yield in a month. 2 was thoroughly characterized by SC-XRD, and 1H, 13C and 29Si NMR spectroscopy (Fig. S4–S6 in the ESI†). The aliphatic protons of the tert-butyl group appear at 1.0 ppm while the aromatic protons resonate in the range of 8.04–6.78 ppm with well resolved splitting patterns. The 13C NMR spectrum also shows resonances in the expected region wherein tert-butyl-C appears at 54.8 ppm and methyl-C at 32.7 ppm. The carbon atoms from aromatic rings are observed in the range of 125 to 168 ppm. The 29Si NMR spectrum exhibits two resonances at 17.6 and −71.2 ppm from the two non-equivalent silicon atoms (Fig. S10 and S11†).
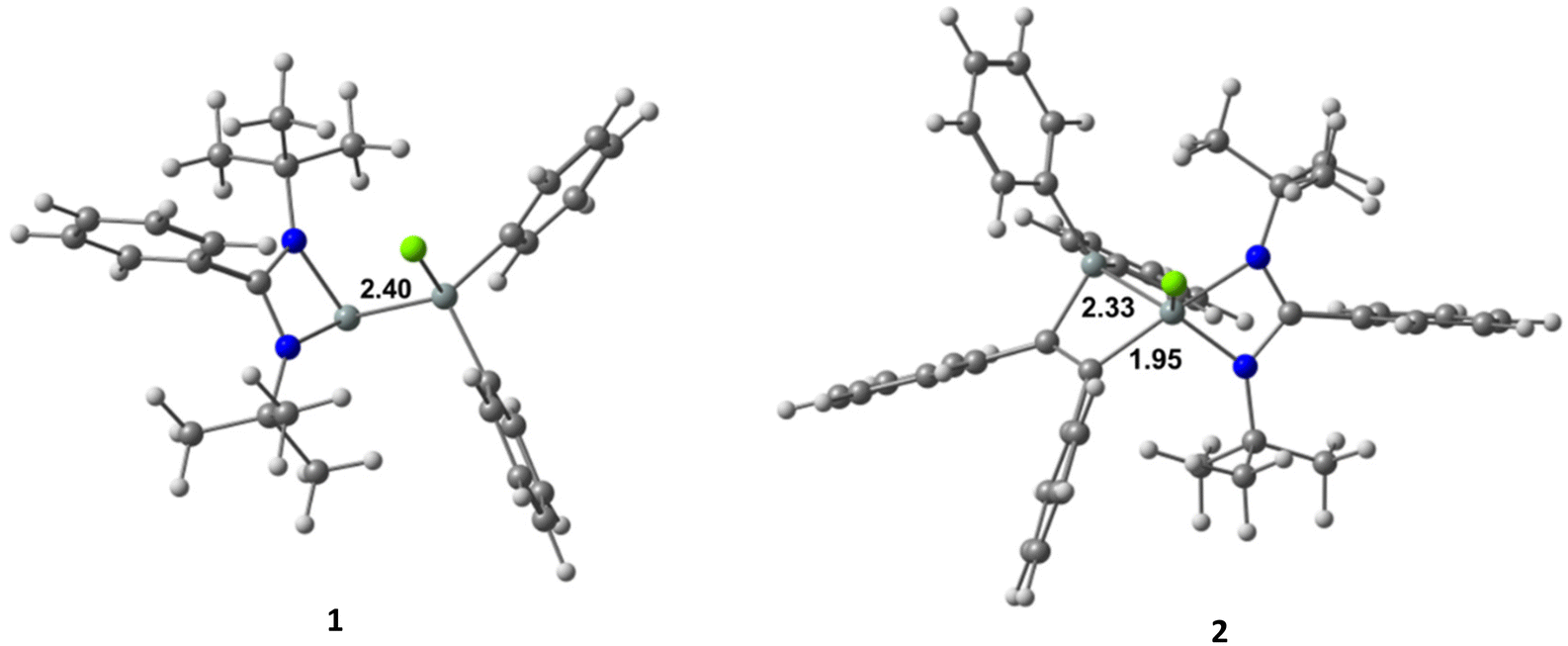
Structural description
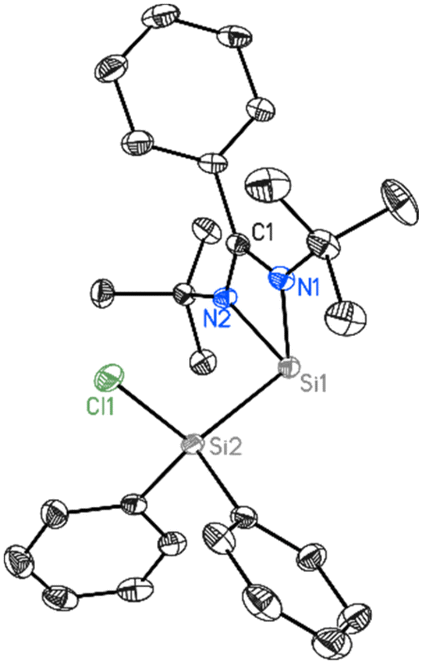
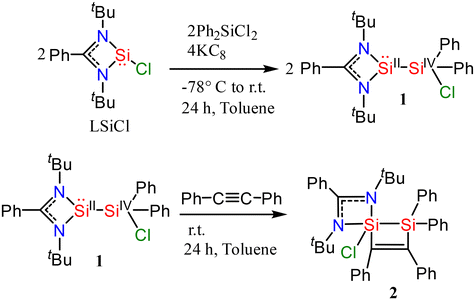
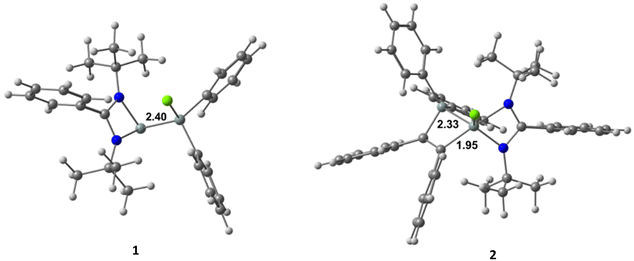
1 crystallizes in the orthorhombic space group Pna21 with two molecules in the asymmetric unit (Fig. 2). One silicon atom coordinates to the bidentate amidinato ligand. The second silicon atom Si2 is substituted by two phenyl groups and by a chlorine atom in a tetrahedral environment. The Si–Si–Cl bond angle is widened to 113.74(3)° while the Si–Si bond distance is 2.3884(8) Å and the Si–Cl bond distance is 2.0947(8) Å.
 |
| | Fig. 2 Molecular structure of 1 with anisotropic displacement parameters at the 50% probability level. Hydrogen atoms and minor positions of the disordered molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: Si1–N2 1.8563(18), Si1–N1 1.8690(18), Si1–Si2 2.3884(8), Cl1–Si2 2.0947(8), N2–Si1–N1 69.65(8), Cl1–Si2–Si1 113.73(3). | |
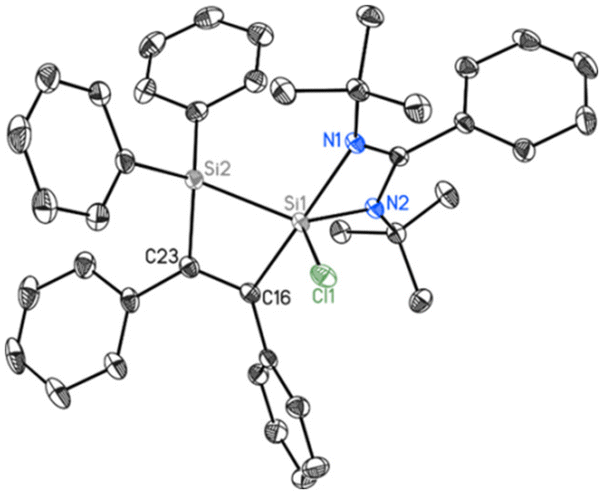
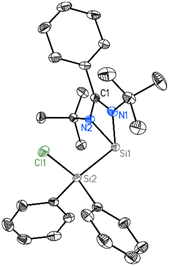
2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule in the asymmetric unit. Beside the four-membered ring formed by the silicon atom and the bidentate amidinato ligand, another four-membered ring is formed from an additional silicon atom and the two sp2-hybridized carbon atoms of the added 1,2-diphenylethyne substrate. Both spirocyclic four-membered rings are almost planar. Remarkably, in the course of the reaction the chlorine atom at the Si(IV) atom in the starting material is shifted to the former Si(II), now the penta-coordinated spiro-centre of the molecule. The bond distance between the two sp2-hybridized carbon atoms (1.3669(19) Å) confirms the double bond character (Fig. 3).
with one molecule in the asymmetric unit. Beside the four-membered ring formed by the silicon atom and the bidentate amidinato ligand, another four-membered ring is formed from an additional silicon atom and the two sp2-hybridized carbon atoms of the added 1,2-diphenylethyne substrate. Both spirocyclic four-membered rings are almost planar. Remarkably, in the course of the reaction the chlorine atom at the Si(IV) atom in the starting material is shifted to the former Si(II), now the penta-coordinated spiro-centre of the molecule. The bond distance between the two sp2-hybridized carbon atoms (1.3669(19) Å) confirms the double bond character (Fig. 3).
 |
| | Fig. 3 Molecular structure of 2 with anisotropic displacement parameters at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Cl1–Si1 2.1218(6), Si1–N2 1.8339(12), Si1–C16 1.9622(14), Si1–N1 2.0022(13), Si1–Si2 2.3378(7), Si2–C23 1.8835(15), C16–C23 1.3669(19), N2–Si1–N1 68.16(5), C16–Si1–Si2 73.79(5), C23–Si2–Si1 76.96(5). | |
Computational calculations
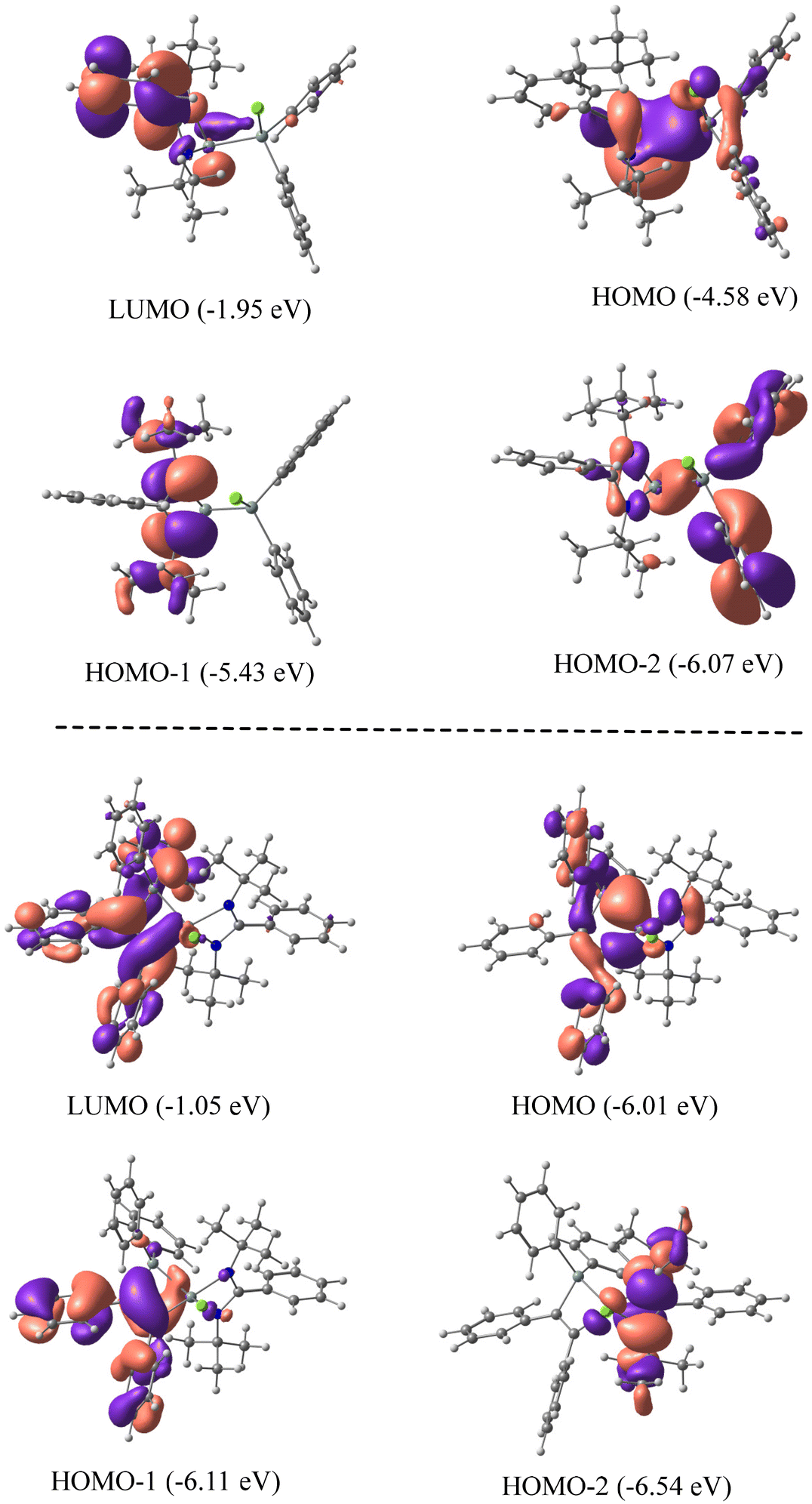
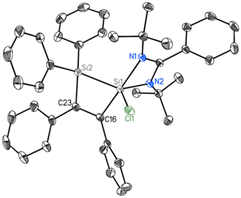
We have employed DFT methods to get insight into the electronic properties of compounds 1 and 2. Fig. 4 illustrates the optimized geometries of compounds 1 and 2 in the singlet state at the BP86-D3BJ/Def2TZVPP level. The computed SiL–Si (SiL = Si bonded to amidinato ligands) bond length of compound 1 is slightly longer than the experimental value, whereas in compound 2 the computed value matches well with the experimental one. The Wiberg bond indices of 0.86 and 0.81 and occupation numbers of 1.83–1.87 indicate a single bond nature of the SiL–Si bonds in 1 and 2. NBO analyses suggest a relatively strong polarization of charge (Table 1) towards the coordinated Si atom (57.9%) of the SiL–Si bond in 1 and a slight polarization in 2 (50.9%). The SiL–C bond shows a substantial charge concentration on the C atom (74.7%) of C2Ph2, indicating the direction of electron flow, and the negative charge on the C atom corroborates the same (Table 1). Fig. 5 shows the molecular orbital pictures of compounds 1 and 2. The HOMOs of both compounds 1 and 2 indicate the lone pair on the Si atoms. Their HOMO−1 and HOMO−2 demonstrate the lone pair on the N atoms of amidinato ligands (Fig. 5), whereas the HOMO−1 of compound 2 suggests a C–C π-bond of C2Ph2 and the HOMO−2 of compound 1 shows an SiL–Si σ type orbital interaction.
 |
| | Fig. 4 Optimized geometries of compounds 1 and 2 at the BP86-D3BJ/Def2TZVPP level. Bond lengths are in angstroms. | |
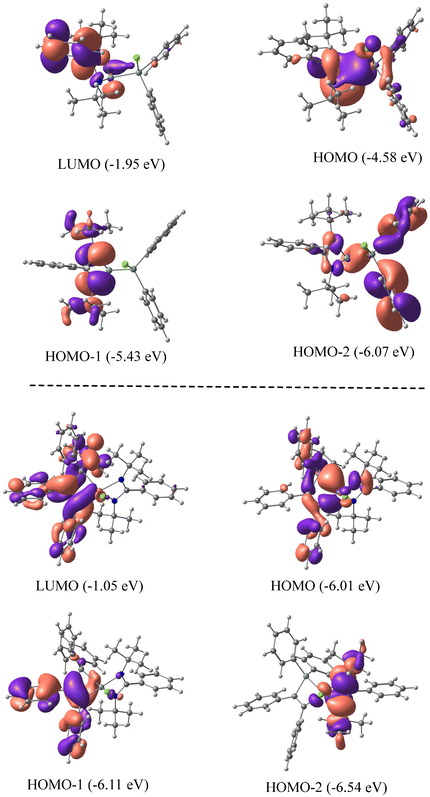 |
| | Fig. 5 Molecular orbital pictures of 1 (top) and 2 (bottom) at the BP86-D3(BJ)/def2-TZVPP level of theory. | |
Table 1 NBO results of the SiL–Si bonds (L = NHC) of LSi–Si(Cl)Ph2 (1) and compound 2 at the BP86-D3(BJ)/def2-TZVPP level of theory. Occupation number ON, polarization and hybridization of the Si–Si bonds and partial charges q
| Compound |
Bond |
ON |
Polarization and hybridization (%) |
WBI |
qSi(L) |
qSi/C |
|
1
|
SiL–Si |
1.83 |
SiL: 42.1 |
Si: 57.9 |
0.86 |
0.635 |
1.153 |
| s(10.5), p(88.5), d(1.0) |
s(27.3), p(72.3), d(0.4) |
|
2
|
SiL–Si |
1.87 |
SiL: 49.1 |
Si: 50.9 |
0.81 |
1.402 |
1.235 |
| s(33.6), p(65.7), d(0.7) |
s(22.9), p(76.5), d(0.6) |
| SiL–C |
1.90 |
SiL: 25.3 |
C: 74.7 |
0.65 |
|
−0.393 |
| s(23.9), p(75.4), d(0.7) |
s(29.6), p(70.2), d(0.2) |
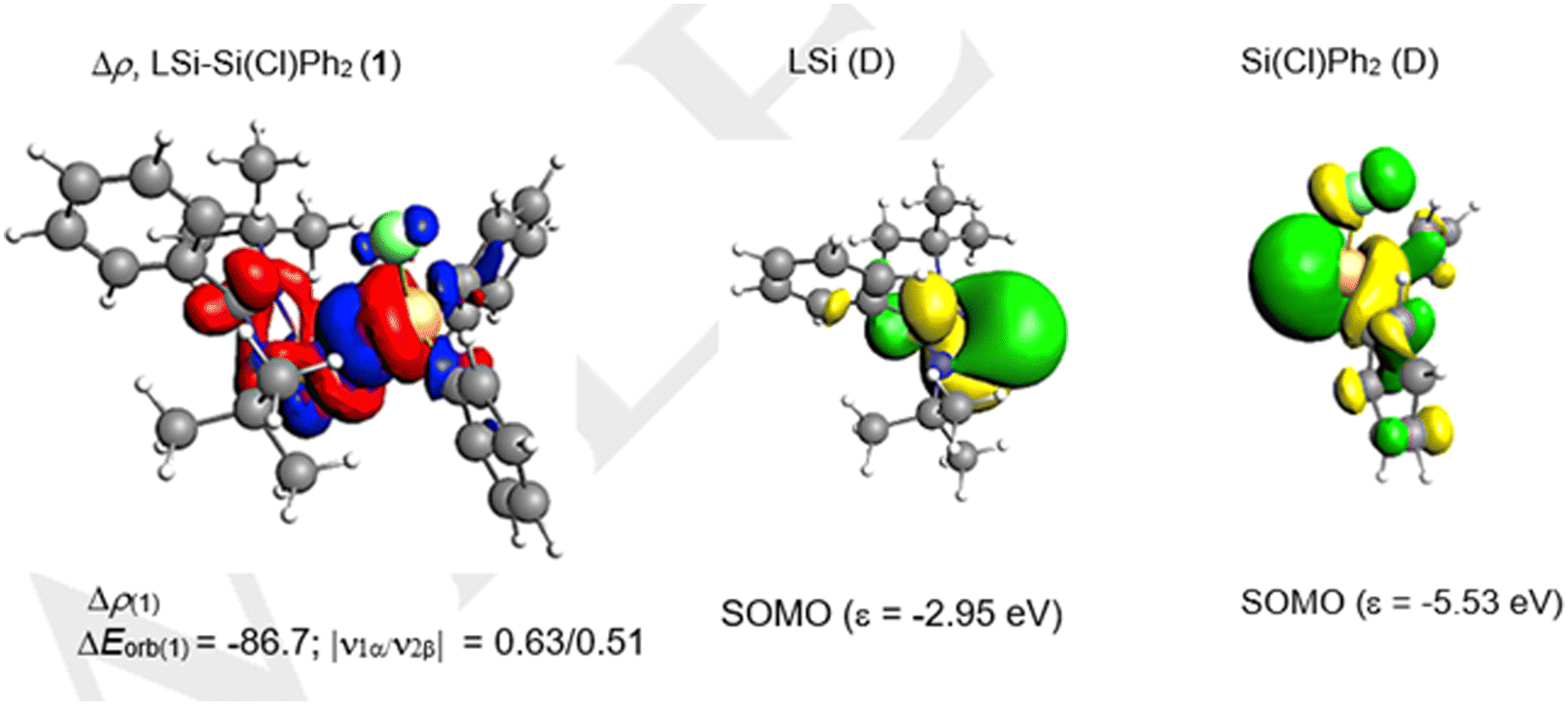
We have evaluated the nature of the SiL–Si bond of LSi–Si(Cl)Ph2 (1) using energy decomposition analysis with natural orbitals for chemical valence analysis (EDA–NOCV). To understand the best bonding scenario, we have explored three different bonding possibilities with varied charges and multiplicities, i.e., a neutral LSi and a neutral Si(Cl)Ph2 fragment in the electronic doublet state forming an electron sharing bond and a singly charged LSi (−/+) and a Si(Cl)Ph2 (+/−) fragment in the electronic singlet state forming a dative bond (Table 2). The bonding possibility with the smallest change in orbital interactions (ΔEorb) between the fragments is rated as the best bonding option.22–24 Accordingly, from Table 2, it is evident that the electron sharing interaction between neutral LSi and Si(Cl)Ph2 fragments in a doublet state with the least ΔEorb is the best bonding model.
Table 2 EDA–NOCV results of the SiL–Si bond in LSi–Si(Cl)Ph2 (1) using two different sets of fragments with different charges and electronic states (S = singlet, D = doublet) and associated bond types at the BP86-D3(BJ)/TZ2P level. Energies are in kcal mol−1. The most favourable fragmentation scheme and bond type are given by the smallest ΔEorb value written in bold. D = dative and E = electron sharing bonds
| Molecule |
Bond type |
Fragments |
ΔEint |
ΔEPauli |
ΔEelstat |
ΔEdisp |
ΔEorb |
| LSi–Si(Cl)Ph2 (1) |
E |
LSi (D) + Si(Cl)Ph2 (D) |
−75.5 |
154.3 |
−106.4 |
−20.3 |
−103.1
|
| D |
(LSi)+ (S) + (Si(Cl)Ph2)− (S) |
−163.4 |
188.1 |
−188.2 |
−20.3 |
−142.9 |
| D |
(LSi)− (S) + (Si(Cl)Ph2)+ (S) |
−237.6 |
187.7 |
−195.3 |
−20.3 |
−209.8 |
The SiL–Si bond of compound 1 is characterized by strong contributions from electrostatic and orbital interactions but with slightly dominant electrostatic characteristics (Table 3). The dispersion interactions also contribute significantly (8.8%) to the stability of the SiL–Si bond. The pair-wise breakdown of total orbital interactions (Table 3) suggests a major contribution (84.1%) from the σ electron-sharing interaction, which is depicted in the deformation density picture (Fig. 6). Additionally, a weak contribution from LSi → Si(Cl)Ph2 σ e− donation (5.8%) can also be observed (Table 3). The oxidation states of the silicon atoms were calculated by the LOBA (localized orbital bonding analysis) method25 using Multiwfn.26a The silicon atoms in 1 display the +2 and +4 oxidation states, whereas in compound 2 both silicon atoms possess the +4 oxidation state.26b
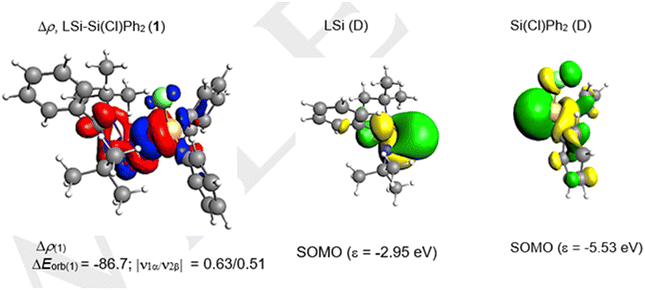 |
| | Fig. 6 The shape of the deformation density Δρ(1) that corresponds to ΔEorb(1) and the associated fragment MOs of the LSi–Si(Cl)Ph2 (1) compound at the BP86-D3(BJ)/TZ2P level at an isosurface value of 0.001 au. The eigenvalues |νn| give the size of the charge migration in e. The direction of the charge flow of the deformation densities is red → blue. | |
Table 3 The EDA–NOCV results at the BP86-D3(BJ)/TZ2P level for the Si–Si bond of the LSi–Si(Cl)Ph2 (1) compound using LSi and Si(Cl)Ph2 in the electronic doublet (D) states as interacting fragments. Energies are in kcal mol−1
| Energy |
Interaction |
LSi (D) + Si(Cl)Ph2 (D) |
|
The values in the parentheses show the contribution to the total attractive interaction ΔEelstat + ΔEorb + ΔEdis.
The values in parentheses show the contribution to the total orbital interaction ΔEorb.
|
| ΔEint |
|
−75.5 |
| ΔEPauli |
|
154.3 |
ΔEdisp![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
|
−20.3 (8.8%) |
| ΔEelstata |
|
−106.4 (46.3%) |
| ΔEorba |
|
−103.1 (44.9%) |
| ΔEorb(1)b |
LSi–Si(Cl)Ph2 σ e− sharing |
−86.7 (84.1%) |
| ΔEorb(2)b |
LSi → Si(Cl)Ph2 σ e− donation |
−6.0 (5.8%) |
| ΔEorb(rest)b |
|
−10.4 (10.1%) |
Conclusion
In conclusion, we have prepared a spirocyclic compound containing a hyper-coordinated silicon atom. This spirocycle contains a silicon–silicon bond which is rarely found in silicon-based chemistry. The theoretical studies using NBO and EDA–NOCV calculations revealed the nature of the silicon–silicon bond to be electron sharing.
Experimental section
General procedures and instrumentation
The required chemicals for the preparation of starting materials were purchased from commercial sources and were used without further purification. The required solvents were also purchased from commercial sources and were dried by stirring over Na/K alloy for four days. The dried and distilled solvents were collected immediately before use in the reactions. All manipulations were performed under inert conditions using standard Schlenk and glovebox techniques under a flow of high purity dinitrogen/argon gas. Deuterated NMR solvents (C6D6/THF-D8/toluene-D8) were dried by stirring them for several days over Na/K alloy followed by distillation under vacuum and degassing several times. 1H, 13C and 29Si NMR spectra were recorded on Bruker Avance 400/500 MHz, 125 MHz and 99 MHz NMR spectrometers, respectively, and referenced to the resonances of the solvent used. Amidinato-silylene chloride, LSiCl (L = PhC(NtBu)2), was synthesized as per the procedure reported in the literature.1
Characterization of 1.
1H NMR (400 MHz, 298K, C6D6, ppm): δ = 8.15 (d, J = 7.3, 4H, Ar–CH), 7.54–7.48 (m, 1H, Ar–CH), 7.22 (t, J = 7.4, 4H, Ar–CH), 7.18–7.12 (m, 2H, Ar–CH), 7.06 (d, J = 7.8, 1H, AR–CH), 7.02–6.94 (m, 2H, Ar–CH), 6.94–6.86 (m, 1H, Ar–CH), 0.95 (s, 18H, CH3). 13C NMR (100 MHz, 298K, C6D6): δ = 156.4 (NCN), 139.3 (Ar–C), 134.3 (Ar–C), 133.9 (Ar–C), 131.4 (Ar–C), 129.4 (Ar–C), 128.9 (Ar–C), 128.6 (Ar–C), 127.8 (Ar–C), 127.2 (Ar–C), 52.8 (tButyl-C), 30.9 (CH3). 29Si NMR (99 MHz, C6D6, ppm): δ = 35.9 and 10.2. Anal. (%) calcd for compound 1 (C27H33ClN2Si2): C, 67.96; H, 6.97; N, 5.87, Found: C, 67.82; H, 7.22; N, 5.68.
Characterization of 2.
1H NMR (500 MHz, 298K, toluene-D8, ppm): δ = 8.04–8.02 (m, 4H, Ar–CH), 7.83–7.81 (m, 2H, Ar–CH), 7.26–7.24 (m, 6H, Ar–CH), 7.17–7.10 (m, 2H, Ar–CH), 7.01–6.98 (m, 2H, Ar–CH), 6.92–6.68 (m, 7H, Ar–CH), 6.86–6.78 (m, 2H, Ar–CH), 1.00 (s, 18H, CH3). 13C NMR (125 MHz, 298K, toluene-D8, ppm): δ = 167.7 (NCN), 161.0 (Ar–C), 144.3 (Ar–C), 142.5 (Ar–C), 136.7 (Ar–C), 135.1 (Ar–C), 130.4 (Ar–C), 129.9 (Ar–CH), 129.5 (Ar–C), 128.3 (Ar–C), 126.6–126.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 54.8 (tButyl-C), 32.7 (CH3). 29Si NMR (99 MHz, 298K, toluene-D8, ppm): δ = 17.6 and −71.2.
C), 54.8 (tButyl-C), 32.7 (CH3). 29Si NMR (99 MHz, 298K, toluene-D8, ppm): δ = 17.6 and −71.2.
Crystal data.
The datasets were collected on a Bruker D8 three circle diffractometer, equipped with a SMART APEX II CCD detector and an INCOATEC microfocus source (Mo Kα radiation) with INCOATEC Quazar mirror optics. The data were integrated with SAINT27 and a multi-scan absorption correction was applied using SADABS.28 The structures were solved by SHELXT29 and refined on F2 using SHELXL30 in the graphical user interface ShelXle.31
Crystal data for 1 at 100(2) K: C27H33ClN2Si2, Mr = 477.18 g mol−1, 0.34 × 0.175 × 0.13 mm, orthorhombic, Pna21, a = 25.543(4) Å, b = 9.225(2) Å, c = 22.485(3) Å, V = 5298.2(16) Å3, Z = 8, μ(MoKα) = 0.252 mm−1, θmax = 28.393°, 200799 reflections measured, 13285 independent (Rint = 0.0488), R1 = 0.0268 [I > 2σ(I)], wR2 = 0.0668 (all data), Δρmax/Δρmin = 0.218/−0.169 e Å−3, CCDC 2292820.†
Crystal data for 2 at 100(2) K: C41H43ClN2Si2, Mr = 655.40 g mol−1, 0.183 × 0.152 × 0.124 mm, triclinic, P, a = 10.498(2) Å, b = 10.599(2) Å, c = 17.493(4) Å, α = 72.73(2)°, β = 85.07(3)°, γ = 75.62(2)°, V = 1800.3(7) Å3, Z = 2, μ(MoKα) = 0.204 mm−1, θmax = 27.219°, 80583 reflections measured, 8015 independent (Rint = 0.0409), R1 = 0.0325 [I > 2σ(I)], wR2 = 0.0849 (all data), Δρmax/Δρmin = 0.343/−0.271 e Å−3, CCDC 2326834.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. M. N. V. T. G. thanks Prof. Kathrin Hopman, Department of Chemistry, UiT for computational facilities. D. St. thanks the GRK BENCh, which is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455. Dedicated to Professor C. N. R. Rao on the occasion of his 90th birthday.
References
- S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed.
- S. Yao, Y. Xiong, A. Saddington and M. Driess, Chem. Commun., 2021, 57, 10139–10153 RSC.
- J. Li, J. Li, Y. Liu, S. Kundu, H. Keil, H. Zhu, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Inorg. Chem., 2020, 59, 7910–7914 CrossRef CAS PubMed.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- M. Kira, Chem. Commun., 2010, 46, 2893–2903 RSC.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- S. Fujimori and S. Inoue, Eur. J. Inorg. Chem., 2020, 2020, 3131–3142 CrossRef CAS PubMed.
- J. Keuter, A. Hepp, A. Massolle, J. Neugebauer, C. Mück-Lichtenfeld and F. Lips, Angew. Chem., Int. Ed., 2022, 61, e202114485 CrossRef CAS PubMed.
-
(a) Y. Wang, M. Karni, S. Yao, A. Kaushansky, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 12916–12927 CrossRef CAS PubMed;
(b) S. Du, H. Jia, H. Rong, H. Song, C. Cui and Z. Mo, Angew. Chem., Int. Ed., 2022, 61, e202115570 CrossRef CAS PubMed;
(c) F. A. D. Herz, M. Nobis, D. Wendel, P. Pahl, P. J. Altmann, J. Tillmann, R. Weidner, S. Inoue and B. Rieger, Green Chem., 2020, 22, 4489–4497 RSC.
-
(a) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568–571 CrossRef CAS PubMed;
(b) M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, Chem. Sci., 2019, 10, 6476–6648 RSC;
(c) D. Reiter, R. Holzner, A. Porzel, P. J. Altmann, P. Frisch and S. Inoue, J. Am. Chem. Soc., 2019, 141, 13536–13546 CrossRef CAS PubMed;
(d) M. K. Bisai, V. S. V. S. N. Swamy, T. Das, K. Vanka, R. G. Gonnade and S. S. Sen, Inorg. Chem., 2019, 58(16), 10536–10542 CrossRef CAS PubMed;
(e) S. K. Kushvaha, P. Kallenbach, N. V. T. S. M. Gorantla, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Chem. – Eur. J., 2023, e202303113 Search PubMed.
-
(a) Y. Chen, J. Li, Y. Zhao, L. Zhang, G. Tan, H. Zhu and H. W. Roesky, J. Am. Chem. Soc., 2021, 143, 2212–2216 CrossRef CAS PubMed;
(b) S. S. Sen, S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578–587 CrossRef CAS PubMed.
- W. H. Atwell, Organometallics, 2009, 28, 3573–3586 CrossRef CAS.
- S. S. Sen, H. W. Roesky, K. Meindl, D. Stern, J. Henn, A. C. Stückl and D. Stalke, Chem. Commun., 2010, 46, 5873–5875 RSC.
- J. Li, M. Zhong, H. Keil, H. Zhu, R. Herbst-Irmer, D. Stalke, S. De, D. Koley and H. W. Roesky, Chem. Commun., 2019, 55, 2360 RSC.
- R. Azhakar, S. Pillai Sarish, G. Tavčar, H. W. Roesky, J. Hey, D. Stalke and D. Koley, Inorg. Chem., 2011, 50(7), 3028–3036 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, H. W. Roesky, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322–2325 CrossRef CAS PubMed.
- T. Imagawa, L. Giarrana, D. M. Andrada, B. Morgenstern, M. Nakamoto and D. Scheschkewtz, J. Am. Chem. Soc., 2023, 145, 4757–4764 CrossRef CAS PubMed.
- C. B. Yildiz, K. I. Leszczynska, S. Gonzlez-Gallardo, M. Zimmer, A. Azizoglu, T. Biskup, C. W. M. Kay, V. Huch, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2020, 59, 15087–15092 CrossRef CAS PubMed.
- S. Kyushin, Y. Kurosaki, K. Otsuka, H. Imai, S. Ishida, T. Kyomen, M. Hanaya and H. Matsumoto, Nat. Commun., 2020, 11, 4009 CrossRef CAS PubMed.
- A. Kostenko and M. Driess, J. Am. Chem. Soc., 2018, 140, 16962–16966 CrossRef CAS PubMed.
-
(a) Q. Zhang, W.-L. Li, C. Xu, M. Chen, M. Zhou, J. Li, D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 11078 CrossRef CAS PubMed;
(b) D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 12319 CrossRef CAS PubMed;
(c) R. Saha, S. Pan, G. Merino and P. K. Chattaraj, Angew. Chem., Int. Ed., 2019, 58, 8372 CrossRef CAS PubMed;
(d) Q. Wang, S. Pan, Y. Wu, G. Deng, G. Wang, L. Zhao, M. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2019, 58, 17365 CrossRef CAS PubMed;
(e) S. M. N. V. T. Gorantla, P. Parameswaran and K. C. Mondal, J. Comput. Chem., 2021, 42, 1159 CrossRef CAS PubMed;
(f) S. K. Kushvaha, S. M. N. V. T. Gorantla and K. C. Mondal, J. Phys. Chem. A, 2022, 126, 845–858 CrossRef CAS PubMed;
(g) G. Frenking, S. M. Gorantla, S. Pan and K. C. Mondal, Chem. – Eur. J., 2020, 26, 14211 CrossRef PubMed.
- F. Engelhardt, C. Maaß, D. M. Andrada, R. Herbst-Irmer and D. Stalke, Chem. Sci., 2018, 9, 3111–3121 RSC.
- A. J. W. Thom, E. J. Sundstrom and M. Head-Gordon, Phys. Chem. Chem. Phys., 2009, 11, 11297–11304 RSC.
-
(a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed;
(b) S. K. Kushvaha, P. Kallenbach, N. V. T. S. M. Gorantla, R. Herbst-Irmer, D. Stalke, P. Parameswaran and H. W. Roesky, Chem. – Eur. J., 2024, 30, e202303113 CrossRef CAS PubMed.
-
Bruker AXS Inc., in Bruker Apex CCD, SAINT v8.40B, Bruker AXS Inst. Inc., WI, USA, Madison, 2019 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Sai Manoj N. V. T.
Gorantla
a,
Sai Manoj N. V. T.
Gorantla