
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual FGFR-targeting and pH-activatable ruthenium–peptide conjugates for targeted therapy of breast cancer†
João
Franco Machado
 ab,
Marco
Sá‡
a,
Inês
Pires‡
c,
Miguel Tarita
da Silva
b,
Fernanda
Marques
bd,
Jaime A. S.
Coelho
a,
Filipa
Mendes
bd,
M. Fátima M.
Piedade
ef,
Miguel
Machuqueiro
ce,
María Angeles
Jiménez
g,
Maria Helena
Garcia
ae,
João D. G.
Correia
*bd and
Tânia S.
Morais
*ae
ab,
Marco
Sá‡
a,
Inês
Pires‡
c,
Miguel Tarita
da Silva
b,
Fernanda
Marques
bd,
Jaime A. S.
Coelho
a,
Filipa
Mendes
bd,
M. Fátima M.
Piedade
ef,
Miguel
Machuqueiro
ce,
María Angeles
Jiménez
g,
Maria Helena
Garcia
ae,
João D. G.
Correia
*bd and
Tânia S.
Morais
*ae
aCentro de Química Estrutural, Institute of Molecular Sciences, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal. E-mail: tsmorais@fc.ul.pt
bCentro de Ciências e Tecnologias Nucleares, Instituto Superior Técnico, Universidade de Lisboa, Estrada Nacional 10 (km 139, 7), 2695-066 Bobadela LRS, Portugal. E-mail: jgalamba@ctn.tecnico.ulisboa.pt
cBioISI – Biosystems & Integrative Sciences Institute, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal
dDepartamento de Engenharia e Ciências Nucleares, Instituto Superior Técnico, Universidade de Lisboa, Estrada Nacional 10 (km 139, 7), 2695-066 Bobadela LRS, Portugal
eDepartamento de Química e Bioquímica, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal
fCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
gInstitute of Physical Chemistry Blas Cabreras (IQF-CSIC), Serrano 119, E-28006 Madrid, Spain
First published on 26th March 2024
Abstract
Dysregulation of Fibroblast Growth Factor Receptors (FGFRs) signaling has been associated with breast cancer, yet employing FGFR-targeted delivery systems to improve the efficacy of cytotoxic agents is still sparsely exploited. Herein, we report four new bi-functional ruthenium–peptide conjugates (RuPCs) with FGFR-targeting and pH-dependent releasing abilities, envisioning the selective delivery of cytotoxic Ru complexes to FGFR(+)-breast cancer cells, and controlled activation at the acidic tumoral microenvironment. The antiproliferative potential of the RuPCs and free Ru complexes was evaluated in four breast cancer cell lines with different FGFR expression levels (SKBR-3, MDA-MB-134-VI, MCF-7, and MDA-MB-231) and in human dermal fibroblasts (HDF), at pH 6.8 and pH 7.4 aimed at mimicking the tumor microenvironment and normal tissues/bloodstream pHs, respectively. The RuPCs showed higher cytotoxicity in cells with higher level of FGFR expression at acidic pH. Additionally, RuPCs showed up to 6-fold higher activity in the FGFR(+) breast cancer lines compared to the normal cell line. The release profile of Ru complexes from RuPCs corroborates the antiproliferative effects observed. Remarkably, the cytotoxicity and releasing ability of RuPCs were shown to be strongly dependent on the conjugation of the peptide position in the Ru complex. Complementary molecular dynamic simulations and computational calculations were performed to help interpret these findings at the molecular level. In summary, we identified a lead bi-functional RuPC that holds strong potential as a FGFR-targeted chemotherapeutic agent.
Introduction
Breast cancer remains the most predominantly diagnosed cancer and the leading cause of female mortality worldwide, corresponding to 15.5% of annual cancer deaths in women.1–3 Despite the great efforts on early diagnosis and treatment strategies, breast cancer recurrence and metastasis to the bones, lungs, liver, and brain renders it incurable, with this being the major reason for the current high mortality levels.4–7Dysregulation of Fibroblast Growth Factor Receptor (FGFR) signaling has been associated with the development and progression of various types of cancer, including breast cancer. Therefore, in recent years, it has emerged as a promising therapeutic target.8–10 FGFRs are a family of receptor tyrosine kinases that play key roles in cell growth, survival, angiogenesis, differentiation, and cell repair.8–12 Aberrant activation or overexpression of FGFRs has been implicated in breast cancer pathogenesis, particularly in aggressive subtypes, making it a marker of poor prognosis, as it is associated with the occurrence of metastases, early relapse, and resistance to standard therapy.8,13,14 The observation that overexpression of FGFRs by breast cancer cells comparatively to non-tumorigenic ones has paved the way for the exploration of FGFR inhibitors as potential targeted therapeutic agents.8–10,12 Although numerous ligands with high affinity and selectivity for FGFR, mainly peptides and small molecules, have reached clinical trials for several types of cancer,9,12,15 none of them have shown to be sufficiently effective for treating breast cancer.8,13,15,16 FGFRs as components of the tumor microenvironment (TME) may also be used as molecular targets for drug delivery strategies, enhancing the precision and efficacy of anticancer drugs. Nonetheless, surprisingly, the use of specific FGFR ligands as delivery vectors for targeted anticancer therapy is scarcely explored. Indeed, only one antibody–drug conjugate was reported as a promising FGFR-targeted drug for advanced cancers, such as breast, colon, esophagus, liver, and pancreas, reaching phase I clinical trials.17
Several approaches for targeted delivery of metal complexes to tumors have been developed based on exploiting the unique molecular features of tumors.18–26 In general, a targeted drug delivery system (DDS) comprises a tumor-recognizing moiety and a cytotoxic moiety, both connected directly or through a suitable linker to form a conjugate.27–30 Among the various type of targeting molecules explored in the past few years, peptides arose as one of the most promising as they can bind to their targets with high specificity and affinity, presenting low toxicity and immunogenicity which makes them good candidates for tumor-targeted DDS. Many peptide drug-conjugates have shown high potential in cancer chemotherapy,23,31–38 in particular those systems containing metal complexes (e.g. Ru, Au, Fe, Co, Ir, and Re) as cytotoxic agents.19,39–44
We have previously designed a tumor-targeting DDS based on the ruthenium–cyclopentadienyl complex [RuCp(PPh3)(2,2′-bipy)][CF3SO3] (TM34) combined with FGFR-targeting peptides. TM34 is highly potent in vitro against several cancer cell lines, in particular, breast cancer cells (MFC-7 and MDA-MB-231),45–48 and the peptide was envisaged as the vector to selectively deliver paved the way for the exploration of FGFR inhibitors as potential targeted therapeutic agents.8–10,12 Although numerous ligands TM34 to FGFR(+) breast cancer cells, thus sparing thus the non-tumorigenic tissues that have lower intrinsic levels of FGFR expression.49 These tumor-targeting ruthenium–peptide conjugates (RuPCs) were shown to be more cytotoxic in FGFR(+) breast cancer cells than in FGFR(−) cells. However, peptide conjugation also led to a considerable decrease in overall cytotoxicity compared to the parent complex TM34, limiting their further use.
Herein, to improve the ability of these RuPCs to target and kill FGFR(+) breast cancer cells, we propose a novel strategy that combines a TM34 derivative complex with an FGFR-targeting peptide connected through a pH-responsive linker. The latter allows selective and controlled release of the Ru complex in its active form only at the target, triggered by the acidic pH of TME, boosting not only its selectivity but also its therapeutic activity. A schematic representation of the design of this rationale approach is illustrated in Fig. 1.
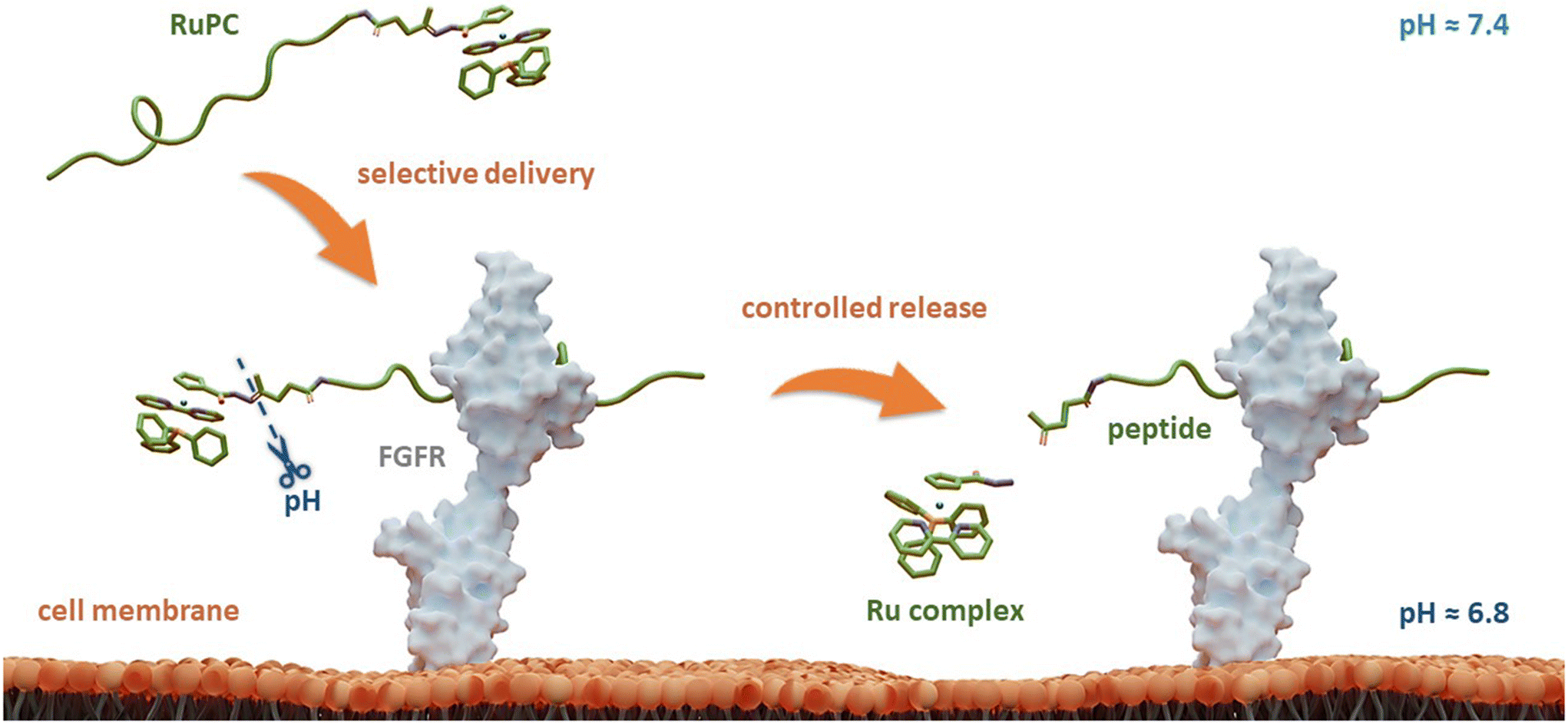 | ||
| Fig. 1 A conceptual overview of bi-functional ruthenium–peptide conjugates (RuPCs) based on targeting FGFR and a controlled release of the ruthenium active species at the acidic tumor microenvironment. | ||
The four new RuPCs proposed differ in the conjugation position and/or in the way the hydrazone, a well-known acid-labile moiety for prodrugs, was installed. In this study, four pH-responsive ruthenium–peptide conjugates, named RuPC1–RuPC4 (Fig. 2), were developed, each composed of 3 building blocks, namely a Ru complex (cytotoxic agent), an FGFR-targeting peptide (targeting agent) and a hydrazone linker (pH-sensitive linker) formed by the reaction of the two previously mentioned moieties. The hydrazone linker can be obtained by the reaction between a ketone-derivatized Ru complex (cytotoxic agent) and a hydrazide-containing peptide (delivery agent) or vice versa. Herein, we present the synthesis and characterization of new organometallic Ru complexes functionalized with ketone and hydrazide groups on the Cp or bipyridine co-ligands (1, 4, 6, and 7; Fig. 2). These two co-ligands had already been identified in our previous computational studies as the most suitable for derivatization and peptide conjugation without affecting the complex ability to interact with the cell membrane, a key process for the activity of TM34.47,49 As regards the FGFR-targeting moiety, we have developed two peptides based on the sequence VSPPLTLGQLLS, functionalized on the N-terminus with a 1,4-dioxo-pentanyl (peptide P1) or a (1-isobutyl)-4-hydrazide (peptide P2) groups, to allow the conjugation to the Ru complex. This sequence is described to bind with high affinity and specificity to the extracellular domain of FGFR and is not toxic to the tumoral cells, being used in this approach only as a carrier.49,50
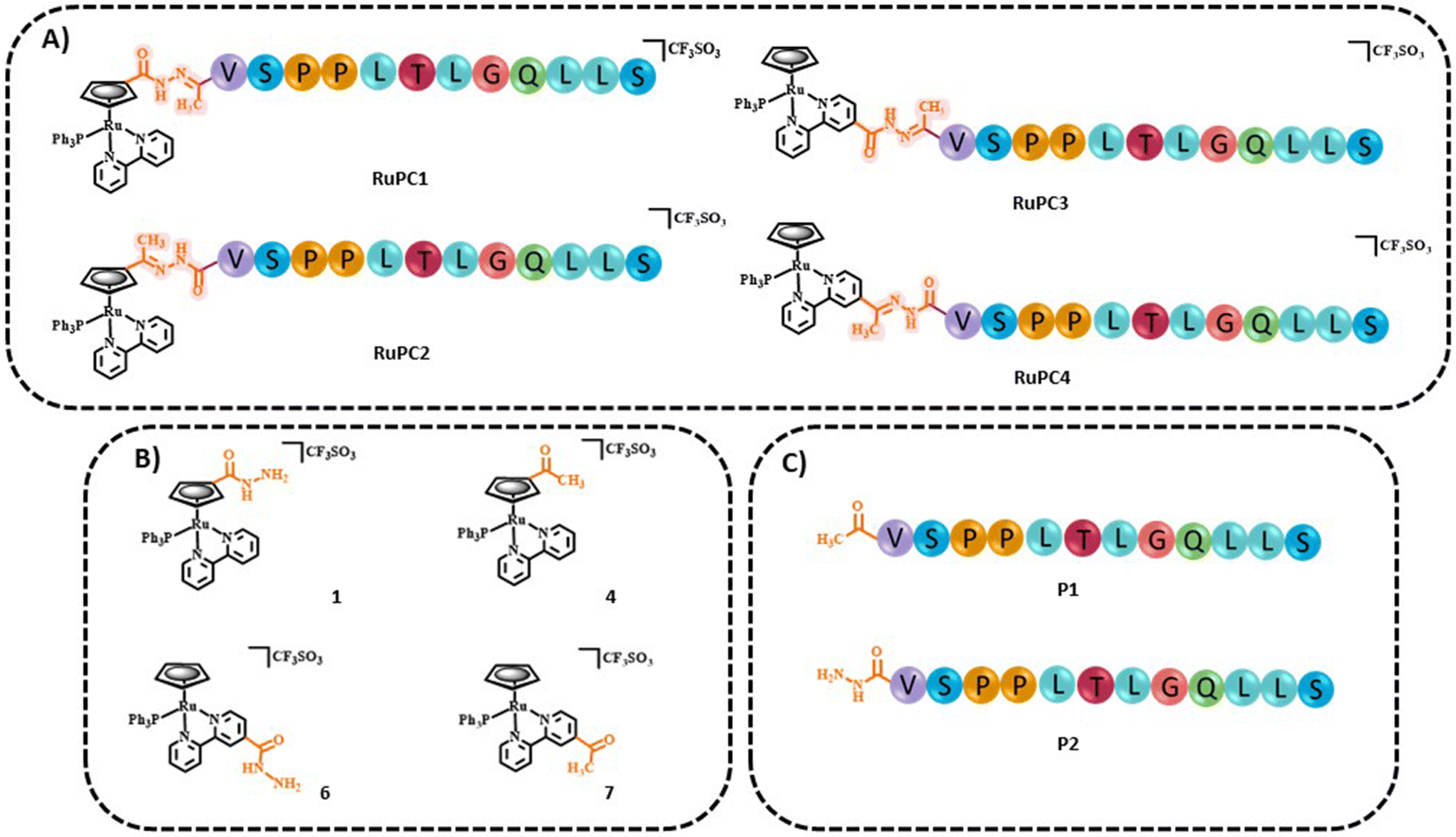 | ||
| Fig. 2 Chemical structures of (A) ruthenium–peptide conjugates (RuPC1–4), (B) active ruthenium complexes (1, 4, 6, and 7) and (C) peptides (P1 and P2). | ||
The effect of metal conjugation on the 3-dimensional structure of the peptide was evaluated by NMR spectroscopy through determination of the conformations of P1 and RuPC1 in aqueous solution. The dual effect of FGFR targeting and pH-activation of the new RuPC was evaluated in vitro in a panel of breast cancer cell lines: SKBR-3, MDA-MB-134-VI, MCF-7, and MDA-MB-231 with different FGFR expression levels, at pH 6.8 and pH 7.4 that mimic the tumor microenvironment and normal tissues/bloodstream, respectively, and in normal human dermal fibroblasts at pH 7.4. The ability of the RuPCs to release the organometallic complexes in aqueous solutions at pH 6.8 or pH 7.4 was also evaluated by RP-HPLC. Our findings suggest that the cytotoxicity and the release are strongly dependent on the position of the conjugation of the Ru complex to the peptide and the way that the hydrazone is constructed. Also, we identified a lead compound that holds the Ru complexes functionalized with ketone and hydrazide groups on the Cp or bipyridine co-ligands (1, 4, 6, and 7; Fig. 2). Also, we identified a lead compound that holds the potential to be further evaluated as a FGFR-targeted chemotherapeutic agent. The strategy presented herein holds promise for a new targeted therapeutic approach for the treatment of FGFR-related cancers.
Results and discussion
Synthesis and characterization
Two new monofunctionalized bipyridine ligands were synthesized through a Stille coupling reaction between 4-substituted- 2-(tributylstannyl)pyridine and 2-bromopyridine.The precursor [Ru(η5-C5H4COCH3)(PPh3)2Cl] was obtained by reaction of sodium acetylcyclopentadienide with [Ru(PPh3)3Cl2] in tetrahydrofuran. The final ketone/ester-derived complexes were obtained by heating, under reflux, the respective precursor complex with the corresponding bipyridine ligand in methanol. The hydrazide-derived complexes were prepared by reaction of the ester precursors with a large excess of hydrazine in ethanol under reflux. Detailed procedures, reaction schemes, and characterizations are provided in the ESI.† The structures of all complexes were confirmed by multinuclear (1H, 13C, 31P) NMR, FT-IR, and UV-vis spectroscopies and ESI-MS. The purity of all complexes was assessed by percentual elemental analysis, and HPLC (only for compounds 1, 4, and 6). Single crystals of complexes 1, 3 (used as the precursor of 4), and 4 suitable for SCXRD were obtained by diffusion of diethyl ether in a dichloromethane solution of each complex. Fig. 3 shows the molecular structures of the compounds and the selected bond lengths and angles are provided in ESI (Tables S2 and S3†).
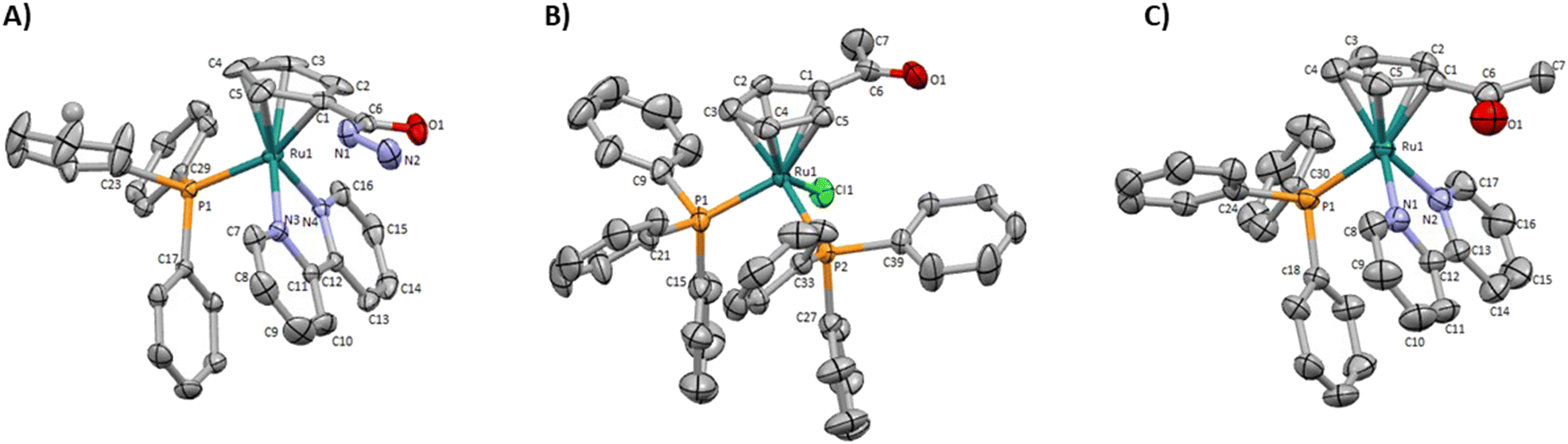 | ||
| Fig. 3 Molecular diagrams for (A) [Ru(η5-C5H4CONHNH2)(PPh3)(2,2′-bipy)]+ (1), (B) [Ru(η5-C5H4COCH3)(PPh3)2Cl] (3) and (C) [Ru(η5-C5H4COCH3)(PPh3)(2,2′-bipy)]+ (4). | ||
These monocyclopentadienyl ruthenium complexes present the usual “three-legged piano stool” geometry around the metal as confirmed by X–M–P angles close to 90° (X = N or Cl), with the remaining Cp–M–X (X = N or P) angles between 121.370(19)° and 130.16(13)°. Complexes 1 and 4 crystallize in the monoclinic crystal system in the P21/c and P21/n space groups respectively and complex 3 crystallizes the triclinic crystal system in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. For complexes 1 and 4 the asymmetric unit consists of a cationic complex and CF3SO3 anion, while for compound 3 the asymmetric unit consists of the neutral complex.
space group. For complexes 1 and 4 the asymmetric unit consists of a cationic complex and CF3SO3 anion, while for compound 3 the asymmetric unit consists of the neutral complex.
Furthermore, the stability of complexes 1, 4, 6, and 7 was evaluated in aqueous and organic solutions by UV-vis spectroscopy and NMR to assess their suitability for further conjugation to peptides and biological studies. The assays by UV-vis spectroscopy were performed in 100% DMSO (organic solution) and 5% DMSO/95% cell culture medium DMEM + GlutaMAX-I (aqueous solution) at room temperature. The NMR experiments were performed in 80% D2O/20% DMSO-d6 at room temperature by 1H NMR spectroscopy over 48 h. All complexes showed to be stable under these conditions, as the acquired electronic spectra did not display any significant variation over time regarding the number, type, shape, nor maximum absorbance of the bands (ΔAmax < 8% for π → π* and MLCT bands), or the number, chemical shift, integration, or multiplicity of the 1H resonances (Fig. S61–S71†).
The evaluation of the lipophilicity of a complex intended to be used for biomedical applications is among the primary steps of the drug development process. This important physicochemical property has a considerable impact on the pharmacokinetics and pharmacodynamics profiles of the complex, as well as a strong influence on its drug formulation. Particularly, lipophilicity influences the ability of the complex to interact with drug targets and cross-cell membranes, as well as its solubility, tissue permeability, cytotoxicity/bioactivity, and general toxicity.51 The partition coefficients in n-octanol/water of complexes 1, 4, 6, and 7 as well as the reference complex [Ru(η5-C5H5)(PPh3)(2,2′-bipy)][CF3SO3] (TM34), were estimated by the shake-flask method.52 Compared to TM34, which showed moderate lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 1.10 ± 0.05), all the derivatizations led to a slight decrease of the lipophilicity (1: logP = 0.43 ± 0.01; 4: logP = 0.25 ± 0.02 and 7: logP = 0.83 ± 0.03), with the exception for compound 6 (logP = 1.55 ± 0.06), with the bipyridine ligand functionalized with a hydrazide group, that is slightly more lipophilic than TM34.
P = 1.10 ± 0.05), all the derivatizations led to a slight decrease of the lipophilicity (1: logP = 0.43 ± 0.01; 4: logP = 0.25 ± 0.02 and 7: logP = 0.83 ± 0.03), with the exception for compound 6 (logP = 1.55 ± 0.06), with the bipyridine ligand functionalized with a hydrazide group, that is slightly more lipophilic than TM34.
The bioavailability of small metallodrugs to tumor sites is often limited due to their low efficiency in reaching selectively the tumor site, and therefore attaching specific peptides could enhance drug accumulation at the target.18,53,54 The syntheses of the novel pH-responsive Ru–peptide conjugates (RuPC1–RuPC4) comprised three main steps: (i) preparation of the organometallic complexes 1, 4, 6, and 7; (ii) synthesis and purification of the FGFR-targeting peptides derived from VSPPLTLGQLLS;55–57 where the N-terminus was functionalized with a 1,4-dioxo-pentanyl (peptide P1) or a (1-oxobutyl)-4-hydrazide (peptide P2) group; and (iii) conjugation of each organometallic complex with the respective peptide through the formation of a hydrazone bond. The peptides were prepared on a rink amide resin by ultrasound-assisted solid-phase peptide synthesis (US-SPPS), according to our previously reported methodology.50 Peptide P1 was N-functionalized with a ketone group by treating the resin with levulinic acid. Analogously, N-derivatization of P2 with the hydrazide group was performed in two steps, first by treating the resin with succinic anhydride, and then by reaction of the product with Fmoc-hydrazine, followed by final Fmoc deprotection. It is important to mention that treating the resin with an excess of succinic anhydride or for long periods, during the first step, led to the polymerization of P2 through the formation of the respective poly succinate ester. After final deprotection, peptide cleavage, and purification procedures both peptides were obtained with C-terminal amide with >98% purity. The RuPCs were obtained in high purity (>97%) upon reaction of the hydrazide-derived complex with the respective ketone-derived peptide, or vice versa (Fig. 2A). Interestingly, RuPC1, RuPC3, and RuPC4 were obtained as a 1:1 mixture of E/Z-hydrazone isomers, whereas for RuPC2 was obtained as a single isomer. P1, P2, RuPC1–RuPC4 were characterized by analytical RP-HPLC and ESI-MS, which results corroborate the proposed structures. The synthetic procedures (peptides and RuPCs), reaction schemes, and characterizations are provided in the ESI.†
The 3-dimensional conformation of a peptide can provide insights into its physicochemical and biological properties.58–60 We determine the conformation of peptide P1 and the structural impact of the conjugation to the metal (RuPC1) by NMR. The 2D 1H–1H-TOCSY spectrum of P1 exhibited a set of intense cross-peaks (Fig. S3A†), which corresponds to the major species, as well as some weak cross-peaks. This is common for Pro-containing peptides due to the cis–trans isomerization of the X-Pro bonds.61,62 Since the sequence of peptide P1 contains two Pro residues, up to four species could be present in solution, i.e., trans–trans, cis–trans, trans–cis, and cis–cis. We observed that the two Pro residues are trans in the major species since the differences between the chemical shifts of the 13Cβ and 13Cγ carbons (Δβγ = δCβ − δCγ, ppm) are small (Δβγ < 5 ppm).61,62 Sequential NOEs between the Hα proton of Ser2 and the Hδδ′ protons of Pro3 and between the Hα proton of Pro3 and the Hδδ′ protons of Pro4 confirm that the rotamer state of both X-Pro bonds is trans. Concerning the minor species, only some signals belonging to V1 and S2 of one of them could be assigned. Based on the intensities of equivalent cross-peaks, the percentages of major (trans–trans) and minor species are 90 and 10%, respectively. The NMR spectra of conjugate RuPC1 showed several sets of cross-peaks for residues Val1, Ser2, Pro3, and Pro4, two of the sets of similar intensities (their relative percentages are 47 ± 4 and 53 ± 4 according to the intensities of 19 equivalent cross-peaks). In the 2D 1H,1H-TOCSY spectrum (Fig. S3B†), four 1Hα–1HN cross-peaks are observed for Val1. According to the intensities of these Val1 cross-peaks, the percentages are 47, 40, 9 and 4%. Very likely the two major species correspond to isomers at the conjugate moieties, whereas the minor species will correspond to each of those isomers and a cis Pro rotamer. As in the free peptide, based on the small value of the difference between the chemical shifts of the 13Cb and 13Cg carbons of the two Pro residues indicates that the two X-Pro bonds in the two major species are trans. We also analyzed whether the major species of peptide P1 and conjugate RuPC1 form some preferred conformation, by examining the plots of 1Hα and 13Cα chemical shift deviations (Δδ = δobserved − δrandomcoil, ppm) as a function of peptide sequence (Fig. 4). It is noticeable that the profiles displayed by P1 and RuPC1 are almost identical, which indicates that conjugation does not affect the conformational behavior of the peptide moiety. Excluding the Pro-preceding residues, which exhibit their characteristic large values,63 most chemical shift deviations are within the random coil range, which points out to mainly disordered peptides. However, the stretch of consecutive negative ΔδHα and positive ΔδCα displayed by residues 7–10 suggests low populated helical conformations (estimated helix percentage is about 16%). The complete assignments are provided in the ESI.†
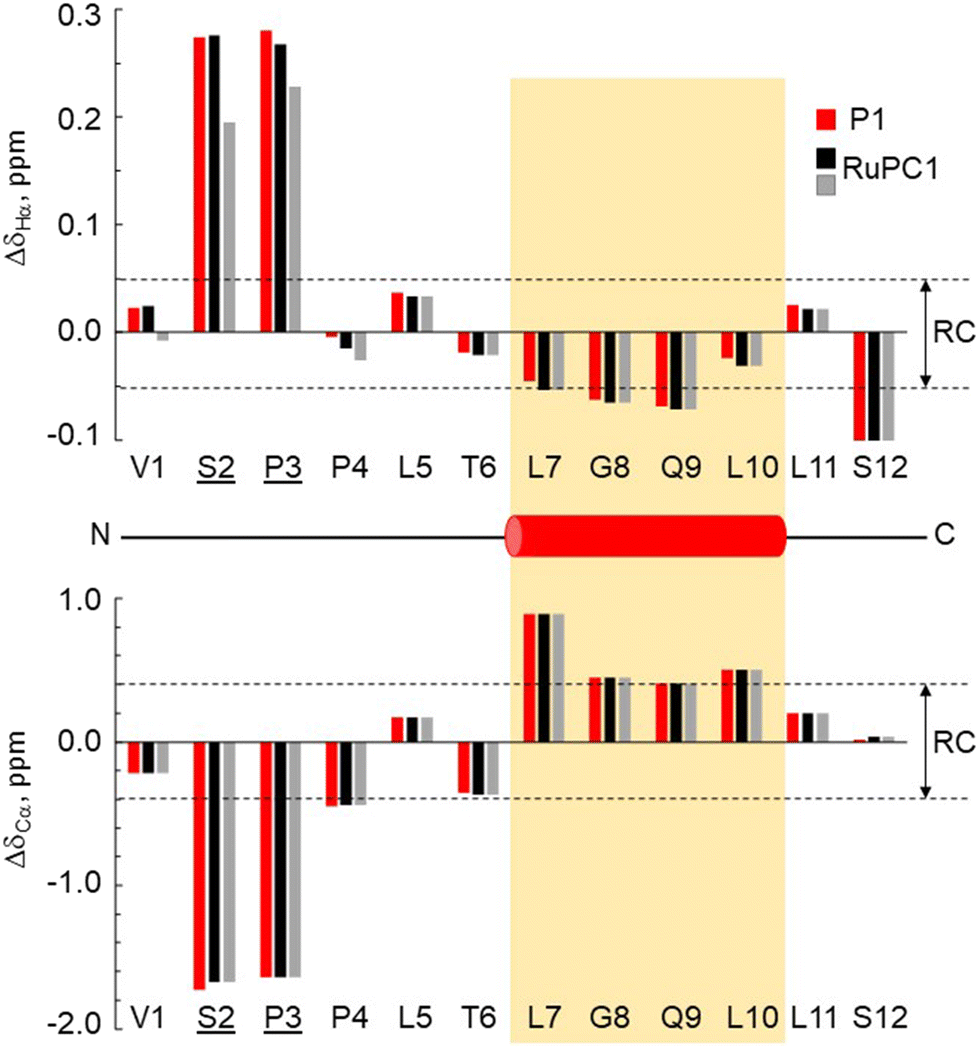 | ||
Fig. 4 Chemical shift deviations for the 1Ha protons 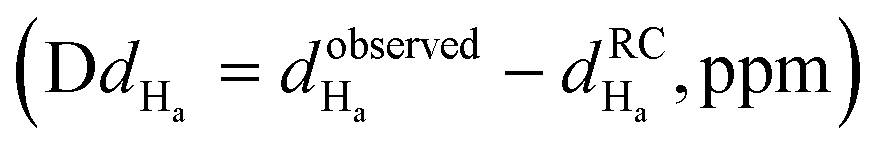 and 13Ca carbons and 13Ca carbons 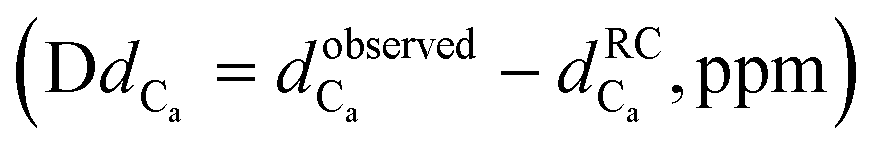 for the major species of P1 (red bars) and RuPC1 (black and grey bars) as a function of sequence. for the major species of P1 (red bars) and RuPC1 (black and grey bars) as a function of sequence. 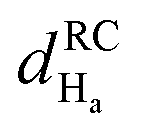 and and 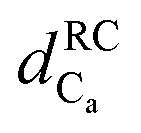 were taken from ref. 63. Pro-preceding residues are underlined. The two dashed lines indicate the random coil range (RC). Top cartoon indicates the secondary structure; helix is shown as a red cylinder. were taken from ref. 63. Pro-preceding residues are underlined. The two dashed lines indicate the random coil range (RC). Top cartoon indicates the secondary structure; helix is shown as a red cylinder. | ||
Cytotoxic activity in FGFR-positive/negative breast cancer cells
Since the RuPCs were designed as a bi-functional system with FGFR targeting and pH-activation, we envisioned that treatment of FGFR(+)-cancer cells at an acidic pH could lead to a higher inhibition of proliferation. Therefore, we evaluated the cytotoxic activities of the new conjugates, complexes, and peptides in a panel of breast cancer cell lines with different FGFR expression levels, namely SKBR-3, MDA-MB-134-VI, MCF-7, and MDA-MB-231, at pH 6.8 and pH 7.4 that mimics the tumor microenvironment and normal tissues/blood-stream, respectively.64 SKBR-3 and MDA-MB-231 cell lines are metastatic breast cancer models, while MCF-7 and MDA-MB-134-VI are hormone-dependent breast cancer models.65–67 According to the literature, SKBR-3 and MDA-MB-134-VI had the highest level of FGFR expression, followed by MCF-7, while MDA-MB-231 has no expression.68,69 For this purpose, the conjugates, and free complexes, were previously incubated in DMSO/phosphate buffer solutions at pH 6.8 and 7.4 for 48 h, before being diluted in cell medium and incubated with the cell lines at different concentrations in the range 0.1–50 μM, to determine the cellular viability after additional 48 h of incubation. An initial screening with the SKBR-3 and MDA-MB-134-VI cell lines showed that both peptides P1 and P2 are not toxic at the concentrations tested (100 μM; Table 1). In general, all conjugates and free organometallic complexes are highly cytotoxic in all the tested cell lines, with IC50 values in the micromolar range (Table 1 and Fig. S4†). The cytotoxicity is strongly dependent on the position of the conjugation of the peptide to the Ru complex. In fact, when the peptide conjugation is on the Cp ring the RuPCs are more cytotoxic than the free Ru complexes, while the opposite effect was observed for RuPCs conjugated via bipyridine ligand. As expected, except for RuPC3, the IC50 of the conjugates is dependent on the FGFR expression level and the pH value, while the activity of free complexes does not correlate with any of these factors.| Compounds | IC50 (μM) 48 h | |||||||
|---|---|---|---|---|---|---|---|---|
| MDA-MB-134-VI | SKBR-3 | MCF-7 | MDA-MB-231 | |||||
| pH 6.8 | pH 7.4 | pH 6.8 | pH 7.4 | pH 6.8 | pH 7.4 | pH 6.8 | pH 7.4 | |
| 1 | 25 ± 5.3 | 23 ± 5.0 | 45 ± 12 | 40 ± 11 | 12 ± 2.0 | 12 ± 2.0 | 38 ± 8.2 | 34 ± 7.6 |
| RuPC1 | 7.4 ± 1.4 | 17.4 ± 5.0 | 7.9 ± 1.7 | 14 ± 3.5 | 2.1 ± 0.7 | 8.5 ± 2.3 | 12.9 ± 3.1 | 26.4 ± 9.8 |
| 4 | 10.2 ± 3.8 | 12.8 ± 4.1 | 7.5 ± 1.9 | 7.5 ± 1.9 | 2.5 ± 0.6 | 3.0 ± 0.6 | 10.2 ± 1.3 | 11.3 ± 1.5 |
| RuPC2 | 6.2 ± 1.9 | 5.9 ± 1.9 | 3.3 ± 0.7 | 4.3 ± 0.8 | 1.1 ± 0.3 | 1.7 ± 0.4 | 4.0 ± 0.7 | 7.7 ± 2.5 |
| 6 | 19.8 ± 2.8 | 21.2 ± 2.9 | 7.1 ± 1.2 | 6.5 ± 1.2 | 4.3 ± 0.9 | 3.1 ± 0.5 | 20 ± 3.2 | 20.3 ± 2.3 |
| RuPC3 | 37.4 ± 6.8 | 26.7 ± 6.2 | 11.4 ± 2.1 | 10.6 ± 2.2 | 10.4 ± 3.3 | 9.5 ± 3.1 | 36.9 ± 5.8 | 23.4 ± 3.4 |
| 7 | 6.4 ± 1.4 | 6.2 ± 1.4 | 3.9 ± 1.3 | 2.7 ± 0.6 | 1.9 ± 0.4 | 2.0 ± 0.4 | 4.9 ± 1.0 | 3.9 ± 0.8 |
| RuPC4 | 17.1 ± 2.3 | 4.1 ± 1.1 | 21.8 ± 5.5 | 3.9 ± 0.8 | 9.7 ± 1.9 | 2.4 ± 0.6 | 47.7 ± 6.5 | 6.7 ± 1.2 |
| Peptides | >100 | >100 | >100 | >100 | ||||
Conjugates RuPC1, RuPC2, and RuPC4 are up to 6 times more active in FGFR-expressing cell lines than in the one that does not overexpress it. This effect is particularly evident at pH 6.8 with the higher difference observed for RuPC1. On the contrary, for the respective free complexes there was no evident correlation between the level of FGFR expression and their cytotoxic activity. Thus, these data suggest a relevant role of the peptide in the targeted delivery of the complexes to breast cancer cells overexpressing this receptor.
Regarding the effect of pH, RuPC1 was up to 4-fold more cytotoxic at pH 6.8, than at pH 7.4 in all cancer cell lines, while for RuPC2 this effect was not observed. When the peptide is conjugated to bipyridine, a different behavior is observed. Unexpectedly, RuPC3 is more cytotoxic at pH 7.4 than 6.8, with this effect being more evident in MDA-MB-134-VI and MDA-MB-231 cells. For RuPC4, at pH 7.4, this conjugate showed a cytotoxic activity similar to that of free Ru complex 7 and at pH 6.8, a significant loss of activity of this conjugate was observed in all cell lines studied, which was up to 7 times lower than its activity at pH 7.4. The respective free complexes did not show significant differences between the two pH values tested.
Regarding the free Ru organometallic complexes, it is observed that compounds functionalized with a ketone group are more cytotoxic than those containing a hydrazide group. As observed for RuPCs, also the cytotoxicity of free complexes is dependent on the position of the functionalization.
Since fibroblasts are cells present in all tissues of the human body that naturally express FGFRs,70–74 to anticipate possible adverse effects due to cytotoxicity in healthy tissues, we evaluate the in vitro toxicity of the two most promising conjugates RuPC1 and RuPC2 and their respective free Ru complexes (1 and 4) in the human dermal fibroblasts (HDF) cell line (Table 2). The free Ru complexes are more active in the normal cell line when compared with all the breast cancer cell lines (SI < 0.8), except for MCF-7. In the latter cell line, complex 4 showed a selectivity index of 2, while for complex 1 the IC50 is in the same concentration range as in the normal cell lines, which evidences the low intrinsic selectivity of free complexes. However, after conjugation of the peptide, both conjugates, as expected, are less active in normal cells than in cancer cells (1.2 < SI < 7.3). The sole exception is RuPC2 in MDA-MB-134-VI cells, where its cytotoxicity is in the same concentration range as in the normal cell (SI = 1.0). In fact, for the other three cell lines, RuPC1 was shown to increase the selectivity of complex 1 between 4 and 10 times, whereas RuPC2 was only 3 to 4 times more selective than the respective complex 4. It is also noteworthy that both conjugates show up to 6-fold higher SI values in the FGFR(+) breast cancer lines compared to the FGFR(–) normal line.
| Compounds | IC50 (μM) HDF | SI (healthy pH 7.4/tumoral pH 6.8) | |||
|---|---|---|---|---|---|
| HDF/SKBR-3 | HDF/MDA-MB-134-VI | HDF/MDA-MB-231 | HDF/MCF-7 | ||
| 1 | 10.1 ± 2 | 0.2 | 0.4 | 0.3 | 0.8 |
| RuPC1 | 15.3 ± 3.5 | 1.9 | 2.1 | 1.2 | 7.3 |
| 4 | 4.4 ± 1.3 | 0.6 | 0.4 | 0.4 | 1.8 |
| RuPC2 | 5.7 ± 1.4 | 1.7 | 0.9 | 1.4 | 5.2 |
Drug release behavior
To evaluate the ability of the RuPCs to release the active organometallic complexes in aqueous solution, RuPC1–RuPC3 were treated with phosphate buffer solutions at pH 6.8 or pH 7.4. A small amount of acetonitrile (10%) was used as a co-solvent to fully solubilize the conjugates at the working concentration (0.5 mg mL−1). The release profiles were monitored by HPLC over 50 hours. Interestingly, each of them presented a different behavior. For RuPC1, at pH 6.8, the chromatographic peak for the Ru–peptide conjugate continuously decreased as the incubation time increased (Fig. 5A). Simultaneously, a proportional increase of the signals of P1 and complex 1 was observed over the same period. After 24 h, a new peak (rt = 14.1 min) appeared, which has been assigned to the complex that results from the hydrolysis of 1 to a carboxylic acid [Ru(η5-C5H4COOH)(PPh3)(2,2′-bipy)][CF3SO3] (TM281), previously reported by us.49 Indeed, complex 1 was released from RuPC1 in an exponential-like way during the first 6 h (41%) (Fig. 5B). With a prolonged incubation period, free complex 1 was continuously released, reaching 79% after 24 h, and an almost complete release at 50 h (97%). These results suggest the successful hydrolysis of the conjugate into the respective free peptide and complex in its active form, with a small amount of it being later hydrolyzed to TM281. The hydrolysis of complex 1 to TM281, may in part explain the lower cytotoxicity of complex 1, compared to complex 4, since TM281 has already been shown to have a low cytotoxic potential.49 To be used as a potential targeted therapy the drug delivery system should be stable under non-tumoral conditions. Thus, at pH 7.4, a similar behavior for RuPC1 was observed (Fig. 5A), but at a significantly lower rate of release. As shown in Fig. 5B, at the pH of non-tumorigenic tissues, only a quarter part of complex 1 was released under a 50 h period following linear kinetics (release rate at 24 h = 14% and 50 h = 26%), suggesting that RuPC1 shows a pH-dependent controlled drug release profile compatible with the desired application. Surprisingly, RuPC2, which contains a mirrored hydrazone bond relative to that of RuPC1, showed limited drug release ability under analogous conditions. At both pH values, the chromatographic peak relative to this conjugate decreased very slowly, with a proportional slow release of P2 and complex 4 (Fig. 5C). Indeed, from Fig. 5D, we can observe that after the first 6 h only 6% of complex 4 was released from RuPC2, achieving a plateau after 24 h (15% release), which did not significantly increase with longer incubation time. Analogously, at pH 7.4, only a very small amount (9%) of complex 4 was released within 50 h of incubation. On the contrary, for RuPC3, with the peptide conjugated through the bipyridine ligand, a high release rate of complex 6 was observed (Fig. 5E), to the same extent for both pH values. For quantitative evaluation (Fig. 5F), a release of 60% was observed after 6 h, and after a prolonged incubation period, free complex 6 was continuously released reaching ca. 100% after 50 h incubation. Meanwhile, a new peak with a retention time of 11.8 min emerged and significantly intensified, which was identified by ESI-MS as the product from the hydrolysis of complex 4 to the carboxylic acid derivatized complex – [Ru(η5-C5H5)(PPh3)(2,2′-bipy-COOH)][CF3SO3] (product not isolated). Unfortunately, it was not possible to evaluate the release profile of the active complex 7 from RuPC4 since we could not find an analytical HPLC method capable of eluting and separating the conjugate RuPC4, the free peptide and complex 7. The pH-responsiveness of the conjugates can explain the differences observed in the cytotoxic profile of each RuPC and highlight the need for the complex to be released from the conjugate, i.e., to be present in its active form and increase its efficacy, as it was also reported for other drug delivery systems.19,49 Indeed, whereas RuPC2 was only slightly more active than complex 4 in the breast cancer cell lines, RuPC1 showed to be significantly more active (up to 5-fold) than the respective free complex 1 at pH 6.8. Thus, the overall results indicate that RuPC1 has a profile adequate for the controlled release of the active organometallic complex at the tumor site, whereas RuPC2 and RuPC3 do not.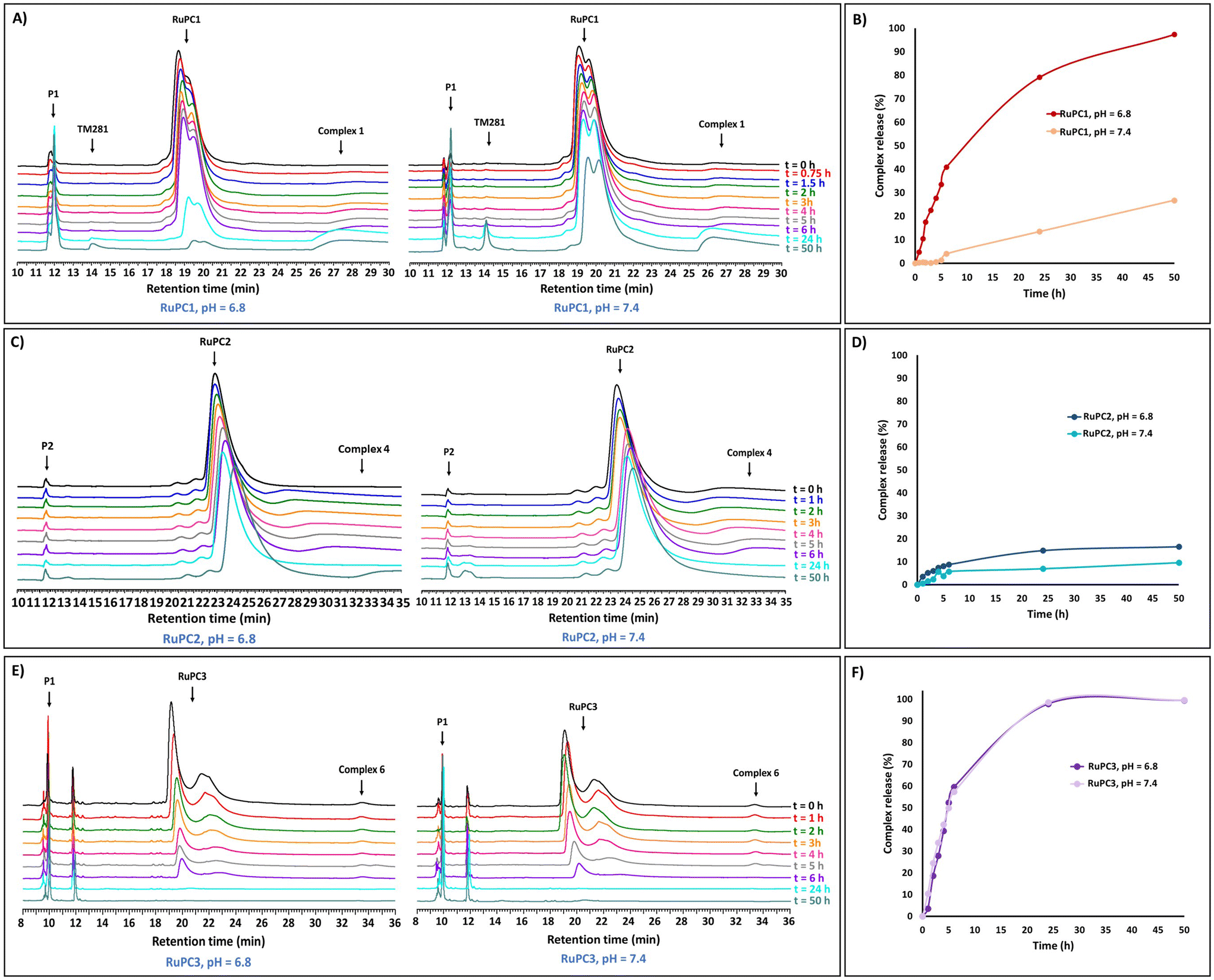 | ||
| Fig. 5 pH dependent Ru complex release profiles under pH 6.8 (tumor microenvironment) and pH 7.4 (bloodstream/healthy tissues). Time course of complexes 1, 4 and 6 release profile from RuPC1, RuPC2 and RuPC3, respectively, monitored by RP-HPLC at intervals: 0, 1, 2, 3, 4, 5,6, 24, and 48 h (A, C and E); percentage of complexes 1, 4 and 6 from RuPC1, RuPC2 and RuPC3, respectively (B, D and F). | ||
To further understand the relative stability of RuPC1 and RuPC2, we studied the hydrolysis of these hydrazones using density functional theory at M06-2X/def2-TZVPP,SDD(Ru)//M06-2X/6-31G(d,p), SDD(Ru) level of theory. We assumed that the hydrolysis rate is mainly dependent on the proton affinity (PA)75,76 and electrophilicity of the corresponding iminium ions. The calculated PA (ΔPA is 1.0 kcal mol−1) and Fukui indices f+ are greater in RuPC1 compared to RuPC2, suggesting that RuPC1 is more basic and the corresponding iminium ion more electrophilic than that of RuPC2. These results agree well with the experimental observations and, together with similar conclusions obtained for truncated model substrates (see Fig. S6–S9†), suggest that alkyl hydrazones are more easily hydrolyzed than aryl hydrazones.
Membrane interactions and permeability from MD simulations
Computational approaches, in particular molecular dynamics (MD) simulations, have been increasingly used to evaluate the interaction of drugs with the cell membrane.77–79 The interaction of lead complexes 1 and 4 with a membrane model was studied using MD simulations, to evaluate the impact of specific chemical groups, introduced in the Cp ring (hydrazide or acetyl groups, respectively), of the reference complex TM34, in its mode of interaction with the cell membrane. In particular, their effects on the membrane insertion depth, preferred orientation, and membrane permeation were studied, and a possible correlation with the cytotoxic activities was evaluated. We applied the same protocols to all compounds so that a direct comparison between TM34 and its derivatives leads to a significant error cancellation, hence providing a reliable quantification of the studied effects. These two complexes are representative of the two groups used to functionalize TM34 (hydrazide and ketone). To study the preferred partitioning region and orientation of complexes 1 and 4, we performed unrestrained MD simulations of each compound in the presence of a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer membrane. In general, all complexes showed a clear preference towards the membrane phase, similar to TM34. They reached a stable insertion position just below the average position of phosphorous atoms, within the initial 200–350 ns of simulation (Fig. 6A and S10†). Most structural properties were equilibrated after these initial time segments (Fig. S11–S13†). None of the compounds showed membrane crossing or exiting events in our MD simulations, suggesting that these average insertion values correspond to energy minima. It should be noted that the polarity and/or the hydrogen-donor ability of the substituent group influences the insertion depth, which decreases as the polarity (and hydrogen-bonding with the lipid phosphate groups) increases, following the order: TM34 (7 Å) > complex 4 (6 Å) > complex 1 (4 Å) (Fig. 6B). Although these are small membrane-insertion differences, given the size of the phospholipid bilayer thickness, they are most likely related to different membrane partition profiles, which may help interpret the compounds’ distinct cytotoxicity values. We implemented an Umbrella Sampling (US) scheme coupled to MD simulations to study the complete membrane-crossing process of TM34 and complexes 1 and 4. This approach provided detailed structural information on the compounds and the POPC membrane at all steps of the insertion pathway (Fig. S14†). The sampling obtained for the compound positions in each umbrella is very good with significant histogram overlap observed between them (Fig. S15†). The membrane deformations, observed in the US protocol, especially at more inserted umbrellas, are larger but can be easily correlated with the needed water desolvation process (Fig. S16 and S17†). The complexes’ angle profiles and rotational tumbling also seem to be stable throughout the simulations (Fig. S18–S20†). This stability is particularly important when calculating the energetics associated with the membrane-crossing process. We calculated the potential of mean force (PMF) energy profiles of all compounds (Fig. 6C) which confirmed their strong preference towards the membrane phase, with a clear energy minimum located deep below the average phosphorous region (umbrellas 1.3–1.4). The energy barrier for membrane crossing (the difference between the minimum and the maximum profiles at the membrane center) is significant, which agrees with the fact that we did not observe membrane-crossing events in the unrestrained MD simulations of these compounds. Despite having very similar PMF profiles, we still observed that complex 1 has a slightly higher energy barrier, its hydrazide substitution can establish more stabilizing interactions with the lipid headgroups. This is also in line with its smaller average insertion value, obtained from unrestrained MD. The membrane permeability coefficients (Pm) were calculated using the ISDM formalism (Fig. 6D) where the POPC bilayer is considered symmetrical so that the resulting PMF energy profiles can be duplicated. Although the absolute values of Pm cannot be directly compared with those observed in cells,80 their relative differences can provide strong hints on the membrane crossing abilities of each compound. We observed that complex 1 has a smaller Pm than TM34, which is in agreement with its structural properties and PMF energy profile. Finally, it should be noted that these complexes are cations and their calculated Pm values are still remarkably high and comparable with most hydrophobic drugs.81–83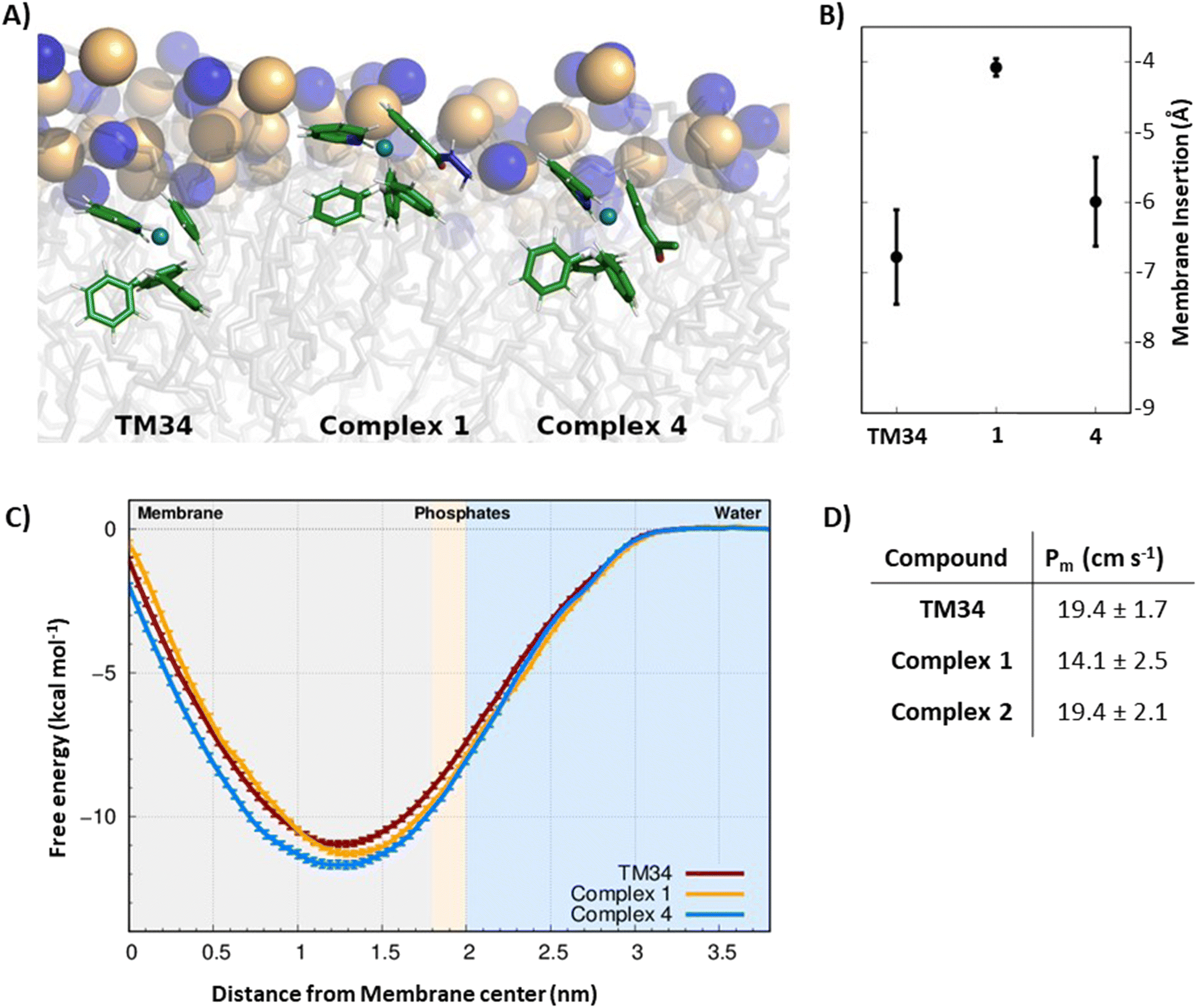 | ||
| Fig. 6 (A) Representation of the complexes at their preferred insertion depths. The structures correspond to conformations presenting each compound's preferred orientation. The POPC lipid tails are shown with transparent gray sticks with the phosphorus and nitrogen atoms represented as spheres (yellow and blue, respectively). (B) Average membrane insertion values for each compound. Insertion was calculated along the membrane normal vector, using the average position of the phosphorus atoms of the interacting monolayer as reference. Negative values correspond to the membrane-inserted positions, below the average phosphate region. The error bars show the standard deviation of the insertion. (C) Potential of mean force energy profile for the three compounds studied and using the water phase as the zero-energy reference. The light blue and light gray regions of the plot correspond to the water and membrane interior phases, respectively. The lipid phosphate group region (light pink) separates these two phases and is located at ∼1.9 nm from the membrane center. The PMF error values were calculated from the standard error of the mean between the three replicates. (D) Permeability coefficients (Pm) of the three compounds studied, calculated using the ISDM method. The error values were calculated from a jackknife leave-one-out strategy using the information from the three replicates. | ||
Conclusions
Four novel ruthenium–peptide conjugates RuPC1–RuPC4 were synthesized by combining an FGFR-targeting peptide, a cytotoxic Ru–cyclopentadienyl complex, and a pH-sensitive linker. These bi-functional RuPCs were rationally designed to selectively target FGFR-positive breast cancer cells and release in situ the cytotoxic Ru complex in a controlled way, promoting the inhibition of cancer cells with reduced side cytotoxicity to normal cells. The cytotoxicity of all RuPCs is correlated with the level of FGFR expression, and the conjugates presented selectivity for FGFR(+) breast cancer cell lines compared to normal fibroblasts. Importantly, the cytotoxicity and drug release profiles of the RuPCs were shown to be strongly dependent on the peptide conjugation position in the Ru complex and on the chemical environment around the hydrazone bond. It was possible to identify a lead bi-functional RuPC that holds the potential as an FGFR-targeted chemotherapeutic agent. This RuPC is sufficiently stable at neutral pH with a small percentage of drug release, and it achieves fast and almost complete drug release under mildly acidic conditions, such as ones found in tumor microenvironment. These promising results encourage us to further investigate these bi-functional RuPCs as a novel platform for targeted chemotherapy of FGFR-positive breast cancers. Some modifications in the structure of the conjugates, such as the use of other pH-responsive linkers and the position of conjugation of the Ru complex in the peptide sequence should be explored to obtain better therapeutic windows.Author contributions
Conceptualization: T. S. M.; funding acquisition: T. S. M., J. D. G. C., F. M., F. M., M. M., M. A. J.; investigation: J. F. M., M. S., I. P., M. T. S., F. M., J. A. S. C., F. M., M. F. M. P., M. A. J., T. S. M.; methodology: T. S. M., J. D. G. C.; validation, T. S. M., J. D. G. C.; writing – original draft preparation: J. F. M., M. S., I. P., F. M., J. A. S. C., F. M., M. F. M. P., M. M., M. A. J., J. D. G. C., T. S. M.; project administration: T. S. M.; supervision: T. S. M., J. D. G. C.; writing – review & editing: F. M., J. A. S. C., F. M., M. F. M. P., M. M., M. A. J., M. H. G., J. D. G. C., T. S. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the Fundação para a Ciência e Tecnologia (FCT), I.P./MCTES for the financial support through the projects PTDC/QUI-QIN/0146/2020, PTDC/QUI-OUT/3854/2021, UIDB/00100/2020 (CQE), LA/P/0056/2020 (IMS), UIDB/04046/2020, UIDP/04046/2020 (BioISI), UID/Multi/04349/2019 (C2TN), and for doctoral grant SFRH/BD/135915/2018 (J. F. M.), UI/BD/154814/2023 (M. S.) and 2023.01155.BD (I.P.). FCT, POPH and FSE – European Social Funds are acknowledged for the Individual Call to Scientific Employment Stimulus projects 2022/00028/CEECIND (T. S. M.), 2020/02383/CEECIND (J. A. S. C.) and CEECIND/02300/2017 (M. M.) Work at IQF-CSIC was supported by grant PID2020-112821GB-I00 from MCINN/AEI/10.13039/501100011033 to M. A. J. 3D-NMR experiments were performed at the Manuel Rico NMR Laboratory (LMR) of the Spanish National Research Council (CSIC), a node of the Spanish Large-Scale National Facility (ICTS R-LRB).Notes and references
- B. S. Chhikara and K. Parang, Chem. Biol. Lett., 2023, 10, 451–451 Search PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, CA Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed.
- S. Sauer, D. R. Reed, M. Ihnat, R. E. Hurst, D. Warshawsky and D. Barkan, Front. Oncol., 2021, 11, 659963 CrossRef CAS PubMed.
- B. Weigelt, J. L. Peterse and L. J. Van't Veer, Nat. Rev. Cancer, 2005, 5, 591–602 CrossRef CAS PubMed.
- J. Lu, P. S. Steeg, J. E. Price, S. Krishnamurthy, S. A. Mani, J. Reuben, M. Cristofanilli, G. Dontu, L. Bidaut, V. Valero, G. N. Hortobagyi and D. Yu, Cancer Res., 2009, 69, 4951–4953 CrossRef CAS PubMed.
- X. Jiang, G. Chen, L. Sun, C. Liu, Y. Zhang, M. Liu and C. Liu, Front. Oncol., 2022, 12, 977226 CrossRef CAS PubMed.
- S. Navid, C. Fan, P. O. Flores-Villanueva, D. Generali and Y. Li, Int. J. Mol. Sci., 2020, 21, 2011 CrossRef PubMed.
- M. A. Krook, J. W. Reeser, G. Ernst, H. Barker, M. Wilberding, G. Li, H. Z. Chen and S. Roychowdhury, Br. J. Cancer, 2020, 124, 880–892 CrossRef PubMed.
- I. S. Babina and N. C. Turner, Nat. Rev. Cancer, 2017, 17, 318–332 CrossRef CAS PubMed.
- E. Witsch, M. Sela and Y. Yarden, Physiology, 2010, 25, 85–101 CrossRef CAS PubMed.
- R. Porta, R. Borea, A. Coelho, S. Khan, A. Araújo, P. Reclusa, T. Franchina, N. Van Der Steen, P. Van Dam, J. Ferri, R. Sirera, A. Naing, D. Hong and C. Rolfo, Crit. Rev. Oncol. Hematol., 2017, 113, 256–267 CrossRef PubMed.
- J. Perez-Garcia, E. Muñoz-Couselo, J. Soberino, F. Racca and J. Cortes, Breast, 2018, 37, 126–133 CrossRef CAS PubMed.
- S. Wang and Z. Ding, Tumour Biol., 2017, 39, 1–10 Search PubMed.
- Y. K. Chae, K. Ranganath, P. S. Hammerman, C. Vaklavas, N. Mohindra, A. Kalyan, M. Matsangou, R. Costa, B. Carneiro, V. M. Villaflor, M. Cristofanilli and F. J. Giles, Oncotarget, 2017, 8, 16052–16074 CrossRef PubMed.
- C. Pottier, M. Fresnais, M. Gilon, G. Jérusalem, R. Longuespée and N. E. Sounni, Cancers, 2020, 12, 731 CrossRef CAS PubMed.
- S. B. Kim, F. Meric-Bernstam, A. Kalyan, A. Babich, R. Liu, T. Tanigawa, A. Sommer, M. Osada, F. Reetz, D. Laurent, S. Wittemer-Rump and J. Berlin, Targeted Oncol., 2019, 14, 591–601 CrossRef PubMed.
- Z. Xie, T. Fan, J. An, W. Choi, Y. Duo, Y. Ge, B. Zhang, G. Nie, N. Xie, T. Zheng, Y. Chen, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2020, 49, 8065–8087 RSC.
- J. F. Machado, J. D. G. Correia and T. S. Morais, Molecules, 2021, 26, 3153 CrossRef CAS PubMed.
- D. Chen, V. Milacic, M. Frezza and Q. Dou, Curr. Pharm. Des., 2009, 15, 777–791 CrossRef CAS PubMed.
- M. Mladenović, I. Morgan, N. Ilić, M. Saoud, M. V. Pergal, G. N. Kaluđerović and N. Knežević, Pharmaceutics, 2021, 13, 460 CrossRef PubMed.
- Z. Zhao, X. Tao, Y. Xie, Q. Lai, W. Lin, K. Lu, J. Wang, W. Xia and Z. W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202202855 CrossRef CAS PubMed.
- Y. Zhang, W. Zheng, Q. Luo, Y. Zhao, E. Zhang, S. Liu and F. Wang, Dalton Trans., 2015, 44, 13100–13111 RSC.
- J. Liu, X. Liao, K. Xiong, S. Kuang, C. Jin, L. Ji and H. Chao, Chem. Commun., 2020, 56, 5839–5842 RSC.
- G. Lv, L. Qiu, K. Li, Q. Liu, X. Li, Y. Peng, S. Wang and J. Lin, New J. Chem., 2019, 43, 3419–3427 RSC.
- B. Bertrand, M. A. O'Connell, Z. A. E. Waller and M. Bochmann, Chem. – Eur. J., 2018, 24, 3613–3622 CrossRef CAS PubMed.
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 951–967 CrossRef PubMed.
- O. M. Kutova, E. L. Guryev, E. A. Sokolova, R. Alzeibak and I. V. Balalaeva, Cancers, 2019, 11, 68 CrossRef CAS PubMed.
- G. Liu, L. Yang, G. Chen, F. Xu, F. Yang, H. Yu, L. Li, X. Dong, J. Han, C. Cao, J. Qi, J. Su, X. Xu, X. Li and B. Li, Front. Pharmacol., 2021, 12, 735446 CrossRef CAS PubMed.
- I. Ojima, Acc. Chem. Res., 2008, 41, 108–119 CrossRef CAS PubMed.
- J. Bhattacharyya, J. J. Bellucci, I. Weitzhandler, J. R. McDaniel, I. Spasojevic, X. Li, C. C. Lin, J. T. A. Chi and A. Chilkoti, Nat. Commun., 2015, 6, 1–12 Search PubMed.
- S. F. A. Rizvi, N. Abbas, H. Zhang and Q. Fang, J. Med. Chem., 2023, 66, 8324–8337 CrossRef CAS PubMed.
- Q. Song, X. Chuan, B. Chen, B. He, H. Zhang, W. Dai, X. Wang and Q. Zhang, Drug Delivery, 2016, 23, 1734–1746 CAS.
- L. Ayalew, J. Acuna, S. F. Urfano, C. Morfin, A. Sablan, M. Oh, A. Gamboa and K. Slowinska, ACS Med. Chem. Lett., 2017, 8, 814–819 CrossRef CAS PubMed.
- L. Hou, T. Zhong, P. Cheng, B. Long, L. Shi, X. Meng and H. Yao, Front. Bioeng. Biotechnol., 2022, 10, 938662 CrossRef PubMed.
- C. Zhang, D. Pan, K. Luo, W. She, C. Guo, Y. Yang and Z. Gu, Adv. Healthc. Mater., 2014, 3, 1299–1308 CrossRef CAS PubMed.
- L. Gong, H. Zhao, Y. Liu, H. Wu, C. Liu, S. Chang, L. Chen, M. Jin, Q. Wang, Z. Gao and W. Huang, Acta Pharm. Sin. B, 2023, 3659–3677 CrossRef CAS PubMed.
- M. Mazel, P. Clair, C. Rousselle, P. Vidal, J. M. Scherrmann, D. Mathieu and J. Temsamani, Anticancer Drugs, 2001, 12, 107–116 CrossRef CAS PubMed.
- J. F. Machado and T. S. Morais, Dalton Trans., 2022, 51, 2593–2609 RSC.
- V. P. Chavda, H. K. Solanki, M. Davidson, V. Apostolopoulos and J. Bojarska, Molecules, 2022, 27, 7232 CrossRef CAS PubMed.
- P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, Front. Chem., 2020, 8, 545283 Search PubMed.
- K. S. Gkika, D. Cullinane and T. E. Keyes, Top. Curr. Chem., 2022, 380, 1–48 CrossRef PubMed.
- L. C. C. Lee, L. Huang, P. K. K. Leung and K. K. W. Lo, Eur. J. Inorg. Chem., 2022, e202200455 CrossRef CAS.
- M. Soler, L. Feliu, M. Planas, X. Ribas and M. Costas, Dalton Trans., 2016, 45, 12970–12982 RSC.
- V. Moreno, M. Font-Bardia, T. Calvet, J. Lorenzo, F. X. Avilés, M. H. Garcia, T. S. Morais, A. Valente and M. P. Robalo, J. Inorg. Biochem., 2011, 105, 241–249 CrossRef CAS PubMed.
- A. I. Tomaz, T. Jakusch, T. S. Morais, F. Marques, R. F. M. De Almeida, F. Mendes, É. A. Enyedy, I. Santos, J. C. Pessoa, T. Kiss and M. H. Garcia, J. Inorg. Biochem., 2012, 117, 261–269 CrossRef CAS PubMed.
- L. Côrte-Real, A. P. Matos, I. Alho, T. S. Morais, A. I. Tomaz, M. H. Garcia, I. Santos, M. P. Bicho and F. Marques, Microsc. Microanal., 2013, 19, 1122–1130 CrossRef PubMed.
- L. Côrte-Real, F. Mendes, J. Coimbra, T. S. Morais, A. I. Tomaz, A. Valente, M. H. Garcia, I. Santos, M. Bicho and F. Marques, J. Biol. Inorg. Chem., 2014, 19, 853–867 CrossRef PubMed.
- J. Franco Machado, M. Machuqueiro, F. Marques, M. P. Robalo, M. F. M. Piedade, M. H. Garcia, J. D. G. Correia and T. S. Morais, Dalton Trans., 2020, 49, 5974–5987 RSC.
- R. D. M. Silva, J. F. Machado, K. Gonçalves, F. M. Lucas, S. Batista, R. Melo, T. S. Morais and J. D. G. Correia, Molecules, 2021, 26, 7349 CrossRef CAS PubMed.
- F. Hollósy, T. Lóránd, L. Örfi, D. Erös, G. Kéri and M. Idei, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2002, 768, 361–368 CrossRef PubMed.
- A. Berthod and S. Carda-Broch, J. Chromatogr. A, 2004, 1037, 3–14 CrossRef CAS PubMed.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- T. F. D. Silva, D. Vila-Viçosa and M. Machuqueiro, J. Chem. Theory Comput., 2021, 17, 3830–3840 CrossRef CAS PubMed.
- M. Jin, Y. Yu, H. Qi, Y. Xie, N. Su, X. Wang, Q. Tan, F. Luo, Y. Zhu, Q. Wang, X. Du, C. J. Xian, P. Liu, H. Huang, Y. Shen, C. X. Deng, D. Chen and L. Chen, Hum. Mol. Genet., 2012, 21, 5443–5455 CrossRef CAS PubMed.
- D. P. Perrault, G. K. Lee, S. Y. Park, S. Lee, D. Choi, E. Jung, Y. J. Seong, E. K. Park, C. Sung, R. Yu, A. Bouz, A. Pourmoussa, S. J. Kim, Y. K. Hong and A. K. Wong, Lymphatic Res. Biol., 2019, 17, 19–29 CrossRef CAS PubMed.
- B. Over, P. McCarren, P. Artursson, M. Foley, F. Giordanetto, G. Grönberg, C. Hilgendorf, M. D. Lee, P. Matsson, G. Muncipinto, M. Pellisson, M. W. D. Perry, R. Svensson, J. R. Duvall and J. Kihlberg, J. Med. Chem., 2014, 57, 2746–2754 CrossRef CAS PubMed.
- L. Doedens, F. Opperer, M. Cai, J. G. Beck, M. Dedek, E. Palmer, V. J. Hruby and H. Kessler, J. Am. Chem. Soc., 2010, 132, 8115–8128 CrossRef CAS PubMed.
- O. Demmer, A. O. Frank, F. Hagn, M. Schottelius, L. Marinelli, S. Cosconati, R. Brack-Werner, S. Kremb, H. J. Wester and H. Kessler, Angew. Chem., Int. Ed., 2012, 51, 8110–8113 CrossRef CAS PubMed.
- Y. Shen and A. Bax, J. Biomol. NMR, 2010, 46, 199–204 CrossRef CAS PubMed.
- M. Schubert, D. Labudde, H. Oschkinat and P. Schmieder, J. Biomol. NMR, 2002, 24, 149–154 CrossRef CAS PubMed.
- D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges and B. D. Sykes, J. Biomol. NMR, 1995, 5, 67–81 CrossRef CAS PubMed.
- Q. He, J. Chen, J. Yan, S. Cai, H. Xiong, Y. Liu, D. Peng, M. Mo and Z. Liu, Asian J. Pharm. Sci., 2020, 15, 416–448 CrossRef PubMed.
- X. Dai, H. Cheng, Z. Bai and J. Li, J. Cancer, 2017, 8, 3131–3141 CrossRef PubMed.
- S. E. Smith, P. Mellor, A. K. Ward, S. Kendall, M. McDonald, F. S. Vizeacoumar, F. J. Vizeacoumar, S. Napper and D. H. Anderson, Breast Cancer Res., 2017, 19, 1–12 CrossRef PubMed.
- D. L. Holliday and V. Speirs, Breast Cancer Res., 2011, 13, 1–7 CrossRef PubMed.
- Q. Zhao, A. B. Parris, E. W. Howard, M. Zhao, Z. Ma, Z. Guo, Y. Xing and X. Yang, Sci. Rep., 2017, 7, 1–14 CrossRef PubMed.
- J. Kang, Y. J. Choi, B. Y. Seo, U. Jo, S. I. Park, Y. H. Kim and K. H. Park, Sci. Rep., 2019, 9, 1–12 CrossRef PubMed.
- R. N. Gomes, F. Manuel and D. S. Nascimento, npj Regener. Med., 2021, 6, 1–12 CrossRef PubMed.
- A. Stunova and L. Vistejnova, Cytokine Growth Factor Rev., 2018, 39, 137–150 CrossRef CAS PubMed.
- R. De Araújo, M. Lôbo, K. Trindade, D. F. Silva and N. Pereira, Skin Pharmacol. Physiol., 2019, 32, 275–282 CrossRef PubMed.
- N. Abdian, P. Ghasemi-Dehkordi, M. Hashemzadeh-Chaleshtori, M. Ganji-Arjenaki, A. Doosti and B. Amiri, Cell Tissue Banking, 2015, 16, 487–495 CrossRef CAS PubMed.
- A. V. Lee, S. Oesterreich and N. E. Davidson, J. Natl. Cancer Inst., 2015, 107, 1–4 Search PubMed.
- K. Ji, C. Lee, B. G. Janesko and E. E. Simanek, Mol. Pharm., 2015, 12, 2924–2927 CrossRef CAS PubMed.
- I. Yildiz, J. Phys. Chem. A, 2016, 120, 3683–3692 CrossRef CAS PubMed.
- A. I. Bunea, S. Harloff-Helleberg, R. Taboryski and H. M. Nielsen, Adv. Colloid Interface Sci., 2020, 281, 102177 CrossRef CAS PubMed.
- J. Guo, Y. Bao, M. Li, S. Li, L. Xi, P. Xin, L. Wu, H. Liu and Y. Mu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, e1679 CAS.
- J. Sun, A. Kulandaisamy, J. Liu, K. Hu, M. M. Gromiha and Y. Zhang, Comput. Struct. Biotechnol. J., 2023, 21, 1205–1226 CrossRef CAS PubMed.
- A. M. M. Gomes, P. J. Costa and M. Machuqueiro, BBA Adv., 2023, 4, 100099 CrossRef CAS PubMed.
- D. Vila-Viçosa, B. L. Victor, J. Ramos, D. Machado, M. Viveiros, J. Switala, P. C. Loewen, R. Leitao, F. Martins and M. Machuqueiro, Mol. Pharm., 2017, 14, 4597–4605 CrossRef PubMed.
- J. B. Loureiro, R. Ribeiro, N. Nazareth, T. Ferreira, E. A. Lopes, A. Gama, M. Machuqueiro, M. G. Alves, L. Marabini, P. A. Oliveira, M. M. M. Santos and L. Saraiva, Pharmacol. Res., 2022, 175, 106026 CrossRef CAS PubMed.
- C. F. de Faria, T. Moreira, P. Lopes, H. Costa, J. R. Krewall, C. M. Barton, S. Santos, D. Goodwin, D. Machado, M. Viveiros, M. Machuqueiro and F. Martins, Biomed. Pharmacother., 2021, 144, 112362 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2332935–2332937. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00497c |
| ‡ Both authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |