
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DLPNO-CCSD(T) and DFT study of the acetate-assisted C–H activation of benzaldimine at [RuCl2(p-cymene)]2: the relevance of ligand exchange processes at ruthenium(II) complexes in polar protic media†
Vicente
Ojea
 * and
María
Ruiz
* and
María
Ruiz
Departamento de Química, Facultade de Ciencias, Universidade da Coruña, E-15078 A Coruña, Spain. E-mail: vicente.ojea@udc.es
First published on 25th April 2024
Abstract
To gain mechanistic insights into the acetate-assisted cyclometallations of arylimines promoted by [RuCl2(p-cymene)]2 in polar protic media, DFT geometry optimizations (with M06 and ωB97X-D3 functionals and the cc-pVDZ-PP[Ru] basis set) followed by DLPNO-CCSD(T)/CBS energy evaluations were performed using benzaldimine as a model substrate and methanol as the solvent (with CPCM or SMD models). The calculation results show that coordination of the imine to an acetate ruthenium precursor is followed by anion (chloride or acetate) dissociation as the rate-determining step of the process. H-Bonding of two explicit MeOH to the anion reduces the calculated activation energy to ca. 23 kcal mol−1, in good agreement with the experimental half-life at room temperature. Subsequent AMLA/CMD C–H activation of the intermediate cationic complexes is a faster, reversible process. Alternative reaction pathways involving neutral diacetate ruthenium complexes offer AMLA/CMD transition state structures of lower energy but are precluded due to higher energy barriers for the initial ligand exchange processes at ruthenium. Solvent assistance accelerates the final chloride/acetate exchange processes on the cycloruthenate intermediates, particularly when compression in the condensed phase is taken into consideration. The performance of six DFT functionals (with the aug-pVTZ-PP[Ru] basis set) was assessed using the DLPNO-CCSD(T)/CBS reference energies. Neutral diacetate ruthenium complexes were incorrectly predicted as being kinetically relevant when using hybrid DFT methods (PBE0-D3(BJ), M06-2X or ωB97M-V). Good agreement between the calculated barrier heights and our benchmark energy results was obtained by using double-hybrid DFT methods. PWPB95 with D3(BJ) or D4 dispersion energy corrections was found to be the most accurate (ΔG≠ MUE of ca. 1 kcal mol−1). This study may aid our understanding of and help with further experimental investigations of synthetically useful carboxylate-assisted C–H bond functionalizations involving (N,C)-cyclometallated (p-cymene)Ru(II) intermediate complexes in sustainable polar protic solvents.
1. Introduction
Transition-metal-catalyzed C–H bond functionalizations have emerged as essential tools in organic synthesis. Among widely used transition metals, ruthenium has powerful advantages for C–H activations, including high versatility, broad functional group tolerance, and relatively low cost.1 In particular, carboxylate-assisted ruthenium(II) catalysis constitutes a reliable tool for either directed or nondirected C–H activations.2 These features have enabled the employment of non-hazardous, renewable, and environmentally benign solvents for the development of sustainable ruthenium(II)-catalyzed C–H functionalizations.3Chelation assistance has proved instrumental for enhancing the site-selectivity of ruthenium catalyzed C–H activation and functionalization. The transformation requires binding of a directing group (DG) to the metal center, formation of a key ruthenacyclic intermediate and subsequent functionalization or annulation. Heterocycles with a strongly coordinating nitrogen atom (pyridine, oxazoline, pyrazole…) have been successfully utilized as DGs in these aromatic C–H bond transformations (see Scheme 1). In many of these processes [RuCl2(p-cymene)]2 was revealed as an optimal, bench-stable, commercial catalyst precursor.4 Moreover, many cyclometallated (p-cymene)Ru(II)-complexes formed in this way could be isolated5 and were shown to be key intermediates and catalytic species6 for the functionalization of arenes. Since many of the N-containing heterocycles, such as aryl substituted pyridines, may be difficult to modify or remove, the use of imines constitutes a more efficient synthetic alternative. Imines are readily accessible, inexpensive, can undergo a variety of chemical transformations and prove to be one of the most efficient transient DGs reported to date in transition-metal-catalyzed functionalizations.7 Thus, following on from an early report by Oi and Inoue,8a efficient arylations,5c,8b,c alkylations,7d,9 annulations10 and amidations11 of arylimines by carboxylate-assisted (arene)ruthenium(II) catalysis have been developed. Moreover, recent examples reveal the convenience of employing polar protic solvents, such as alcohols7e,10b,c or water8c in such functionalizations to achieve efficiency and sustainability improvements.
 | ||
| Scheme 1 Ruthenium(II) catalyzed C–H functionalization or annulation of arenes equipped with N-containing DGs. L stands for η6-arene co-ligand and X for carboxylate or chloride. | ||
Different mechanistic possibilities have been considered for the activation of C–H bonds by transition metal complexes.12 The generally accepted proposal for carboxylate-assisted C–H activations involves coordination of the substrate to the electrophilic metal to form a complex with increased acidity that facilitates subsequent deprotonation by a carboxylate group in either an intra- or intermolecular13 fashion (see Chart 1). When cleavage of the C–H bond and formation of the carbon–metal bond take place in a concerted fashion, frequently involving a 6-membered transition state (TS), the process is known as “concerted metalation–deprotonation”14 (CMD) or “ambiphilic metal-ligand assistance”12a,15 (AMLA).16
 | ||
| Chart 1 AMLA/CMD TS structures for carboxylate-assisted ruthenation of C–H bonds. DG stands for directing group, L stands for η6-arene co-ligand and X for carboxylate or chloride. | ||
Most of the current knowledge of the mechanism of carboxylate-assisted C–H activations by ruthenium(II) complexes is based on computational evidence.17 DFT studies covering the reactions of phenylpyrazoles,18 benzylamines,19 phenylpyridines,20N-aryl-oxazolidinones,21 naphthols,22 aryl carboxylic acids,23 arylphosphonates,24 arylacetamides,16d,25 benzamides,26 pyridylindoles,27 phenylketones,28 hydroxy-chromones29 or phenylimidazoles30 promoted by [RuCl2(p-cymene)]2 and acetate anions (or preformed Ru(OAc)2(p-cymene)) support initial substrate (A) binding to the ruthenium precursor (B) followed by dissociation of a ligand (from C) and creation of a key intermediate with a vacant site at which the M–C bond can form (see Scheme 2). Most of these electrophilic intermediates are cationic in nature (D+)31 but alternative routes involving neutral intermediates equipped with two acetate ligands bound to the ruthenium (FL) and showing η6–η2 slippage32 or substitution of the arene co-ligand by solvent or a second substrate molecule33 have been also reported. From either cationic or neutral intermediates the C–H activation event (to E+ or GL, respectively) is followed by final HOAc exchange to form a stable cycloruthenate complex (H) or enable a subsequent functionalization process. In some of these DFT studies the C–H activation events were claimed to be rate determining5g,18,20b,21,23e,24,26d although transition states for initial anion dissociation, ligand substitution or final HOAc exchange at ruthenium were not located and the corresponding energy barriers were assumed to be diffusion-controlled.34 Thus, the potential kinetic relevance of ligand exchange processes at ruthenium has been overlooked and further quantum mechanical calculations on carboxylate-assisted C–H activations by ruthenium(II) complexes are required.
 | ||
| Scheme 2 Routes involving cationic and neutral intermediates for C–H activation at Ru(II)(arene) complexes. DG stands for directing group, X stands for chloro or acetate, L stands for arene, solvent or substrate as η2-ligands. | ||
Although a number of examples of stoichiometric or catalytic C–H bond activations and functionalizations of arenes equipped with N-containing DGs by ruthenium(II) are present in the literature,4–9 to the best of our knowledge, no computational studies have been disclosed for the cyclometallation of arylimines or for the involvement of discrete molecules of polar protic solvents in directed C–H bond activation events promoted by Ru(II)(p-cymene) complexes.35,36 Thus, herein we introduce a detailed computational study of the cyclometallation of N-phenyl benzaldimine (A) promoted by [RuCl2(p-cymene)]2 (B0) and acetate anions in methanol. The main aims of the study are directed to the evaluation of reaction pathways involving cationic or neutral electrophilic ruthenium species in the C–H activation process, the identification of the rate-determining step (RDS) and the establishment of the prevalence of kinetic or thermodynamic control for the global process. For these purposes the effect of the polar protic solvent on geometry optimizations and energy evaluations has also been considered. In addition, selected hybrid and double-hybrid DFT methods among the top performers in the MOR41 and MOBH35 databases37 were evaluated against our benchmark results to localize an accurate scheme of reduced computational cost for the study of the thermodynamics and kinetics of these cyclometallation processes. Our calculations in methanol as a solvent show that the C–H activation event is reversible and kinetically irrelevant and that the rate of cyclometallation should be determined by ligand exchange processes at the ruthenium intermediate complexes.
2. Results and discussion
Calculations reported in this paper were performed for the model system shown in Scheme 3, that is, the reaction of A with stoichiometric B0 in the presence of 2 equivalents of acetate anions (sodium cations were excluded) leading to chloro ruthenacycle H. The computational protocol involved geometry optimizations using M0638 and ωB97X-D339 functionals in combination with a polarized double-ζ basis set and an effective core potential for ruthenium (cc-pVDZ-PP[Ru]) followed by highly accurate yet computationally efficient energy evaluations with an approximate local coupled-cluster method40 and extrapolation to the complete basis set limit (DLPNO-CCSD(T)/CBS level).41 The influence of the polar protic solvent on the geometry optimization process and final energy evaluations have been considered with a hybrid explicit–implicit solvent model,42 by using polarizable continuum solvation models (CPCM and SMD)43 but also including explicit molecules of MeOH. The discussion of the theoretical calculations is presented in terms of the quasi-harmonic44 Gibbs energies in methanol at 298 K corrected for the change from the gas phase (1 atm) to two different standard states in solution (with either a solute concentration of 1.0 M or a liquid methanol concentration of 24.6 M) to better reflect the experimental conditions (see Rationale for the selection of methods and Computational methods in the ESI† for further details). To enable a comparison of the competitive reaction channels the ruthenium dimeric precursor B0, imine A, acetate and an adequate number of methanol molecules were taken separately as zero for the quasi-harmonic Gibbs energies. Throughout the whole article, the difference in Gibbs energy between an intermediate or TS structure and the global reference is abbreviated to ΔGr. | ||
| Scheme 3 Model reaction system under study. | ||
This work is divided into four sections. The first one is related to the pre-activation of ruthenium precursors and the formation of key electrophilic intermediates. The second section describes the C–H activation event and the third one the final ligand exchange processes at the most significant ruthenacyclic intermediate. In the fourth section the competence between reaction pathways is discussed and the lowest energy profile and the RDS are uncovered. The fourth section is divided into three subsections that include separate analyses of the results obtained using the CPCM or SMD solvation models and a final comparison with the experimental results that supports the computational results. In the first, second and third sections, energy barriers and relative Gibbs energies calculated at the DLPNO-CCSD(T)/CBS(CPCM) level after geometry optimizations with the M06 functional are reported first, followed by the corresponding values, between brackets, calculated at the same level on the ωB97X-D3 geometries. For comparison purposes, a third set of energy values calculated at the DLPNO-CCSD(T)/CBS(SMD) level on the ωB97X-D3 geometries are reported last, in brackets and italics.
2.1. Pre-activation by anion dissociation and ligand exchange processes at ruthenium
The reaction of the dimeric dichloro(p-cymene)ruthenium precursor (B0) with acetate in methanol was studied first. Kinetic evidence reported by Bassetti et al.45 indicates that this process should be initiated by ruthenium–chloro bond breaking, release of chloride and the formation of a cationic intermediate (see B0+ in Scheme S2 of the ESI†). Such a dissociation process was calculated to be endergonic by 2.7 [0.9] kcal mol−1 and showed an activation energy of 20.2 [18.9] kcal mol−1. Next, acetate/chloride exchange processes also take place easily on ruthenium at room temperature, to give chloro–acetate derivative B1 and diacetate derivative B2 as key cyclometallation promoters (see Chart 2). | ||
| Chart 2 Acetate derivatives from [RuCl2(p-cymene)2]2 dimer opening. Relative quasi-harmonic free energies (in kcal mol−1) calculated at the DLPNO-CCSD(T)/CBS(CPCM) level in methanol (298 K, 1 M) using geometries optimized at the M06/cc-pVDZ-PP[Ru](IEF-PCM) level are shown in blue. The corresponding values calculated at the same level by using geometries optimized at the ωB97X-D3/cc-pVDZ-PP[Ru](CPCM) level are shown between brackets in red. Energy values calculated at the DLPNO-CCSD(T)/CBS(SMD) level in methanol (298 K, 1 M) are shown in italics. | ||
Formation of chloro–acetate derivative B1 from ruthenium dimeric precursor B0 was calculated to be exergonic by 8.3 [9.0] [2.8] kcal mol−1 while diacetate derivative B2 lies at −11.9 [−12.0] [2.1] kcal mol−1. Solvent-assisted dissociation of B1 or B2 led to the solvated (B+S) and the unsolvated (B+) cationic precursors, which were characterized to be higher in energy than the starting compounds, at 5.6 [5.8] and 21.7 [19.1] kcal mol−1, respectively. Solvent-assisted dissociation of B2 to form free p-cymene and mono-, di- or tri-solvated Ru(OAc)2 complexes was calculated to be endergonic by more than 25 kcal mol−1 (see ESI, Scheme S3†). These results are consistent with the intermediate species identified by 1H NMR in the cyclometallations of 2-arylquinazolines with [RuCl2(p-cymene)]2 and KOAc in MeOH as the solvent.5e
Binding of ligand A to monomeric precursors B1 or B2 takes place by coordination of the nitrogen atom to ruthenium (see Scheme 4, entry a). Such coordination processes involve TS structures TS(B1-C1) or TS(B2-C2) located at 8.5 and 6.4 [9.7 and 6.6] [19.3 and 16.9] kcal mol−1 and give rise to the chloro–acetate complex C1 or diacetate complex C2 which were found at 0.2 and −5.8 [−4.1 and −4.8] [2.4 and 4.7] kcal mol−1, respectively. The loss of an anion from the tetracoordinate ruthenium intermediates gives rise to the key cationic intermediate, with a vacant coordination site available at the metal. Dissociation of the chloride at Ru(II) from C1 led to cationic intermediate D+ showing a bidentate acetate ligand and lying at −4.4 [−3.4] [4.0] kcal mol−1. Going through TS(C1-D+), such dissociation required surpassing a ΔGr of 17.1 [16.4] [23.7] kcal mol−1. Nevertheless, D+ can also be formed by dissociation of acetate from diacetate complex C2, viaTS(C2-D+), which showed a slightly higher ΔGr of 18.0 [18.4] [26.7] kcal mol−1 (see ESI, Schemes S4a and S4b†). Alternative reaction pathways to cationic intermediate D+ by coordination of ligand A to cationic precursors B+ or B+S resulted in a higher energy by more than 5 kcal mol−1 (see ESI, Scheme S4c†).
 | ||
| Scheme 4 (a) Formation of cationic intermediate D+ by the reaction of imine (A) with monomeric precursors B1 or B2 followed by anion dissociation; (b) equilibria between diacetate complex C2 and neutral intermediates of the FL series by p-cymene slippage or decoordination and substitution by solvent or a second imine molecule. Relative quasi-harmonic free energies (in kcal mol−1) calculated at the DLPNO-CCSD(T)/CBS(CPCM) level in methanol (298 K, 1 M) using geometries optimized at M06/cc-pVDZ-PP[Ru](IEF-PCM) are shown in blue. The corresponding values calculated at the same level by using geometries optimized at the ωB97X-D3/cc-pVDZ-PP[Ru](CPCM) level are shown between brackets in red. Energy values calculated at the DLPNO-CCSD(T)/CBS(SMD) level in methanol (298 K, 1 M) are shown in italics. | ||
From C2, equilibria involving slippage of the p-cymene moiety (from η6- to η2-ligand) and its decoordination or substitution by solvent or a second imine molecule give rise to key neutral intermediates Fc, F, FS, and FA which are found at 6.6, 16.1, 2.9, and −6.1 [8.2, 18.8, 5.8, and −2.8] [20.3, 31.5, 17.9 and 9.4] kcal mol−1, respectively (see Scheme 4, entry b). Most stable TS structures for the ligand exchange processes at ruthenium were located on the dissociative pathways. Thus, from Fc, decoordination of the p-cymene moiety through TS(Fc-F) required surpassing a ΔGr of 18.7 [>26.9] [>39.3]46 kcal mol−1 and gave rise to the 16-electron species F. Addition of a solvent molecule to F, viaTS(F-FS), led to the solvated diacetate complex FS. The lowest energy pathway for the addition of a second imine molecule to F involved coordination of the N-phenyl moiety to ruthenium(II), acting as an η2-ligand, to form the F·A complex, followed by rearrangement to the N,N-coordinated bis-imine ruthenium complex FAviaTS(F-FA) which was characterized by a ΔGr of 19.5 [19.1] [30.0] kcal mol−1 (see ESI, Schemes S5a and S5b†). Competing TS structures in the solvent-assisted interchange pathways from Fc to FS or to FA were also located but resulted in higher energies (see ESI, Scheme S5c†).
2.2. C–H activation events
 | ||
| Scheme 5 (a) C–H activation of the cationic precursor (D+ or D+*), involving the cationic agostic intermediates (cis-INT(D+-E+) or trans-INT(D+-E)) and intramolecular or intermolecular routes for proton abstraction and formation of the acetate–ruthenacycles (E+ and E). (b) C–H activation of the bis-imine diacetate neutral intermediate FA, involving the agostic intermediate INT(FA-GA) and intramolecular proton abstraction leading to ruthenacycle GA. Relative quasi-harmonic free energies (in kcal mol−1) calculated at the DLPNO-CCSD(T)/CBS(CPCM) level in methanol (298 K, 1 M) using the geometries optimized at M06/cc-pVDZ-PP[Ru](IEF-PCM) are shown in blue. The corresponding values calculated at the same level by using the geometries optimized at the ωB97X-D3/cc-pVDZ-PP[Ru](CPCM) level are shown between brackets in red. Energy values calculated at the DLPNO-CCSD(T)/CBS(SMD) level in methanol (298 K, 1 M) are shown in italics. | ||
Two different pathways for the acetate-assisted C–H activation event have been considered, involving deprotonation of the cationic agostic intermediates by either a complexed acetate or a free one, as depicted in Scheme 5(a). Both deprotonation pathways, denoted as intramolecular and intermolecular, respectively, may lead to the same acetate–ruthenacycle E after a molecule of acetic acid is released. In the intramolecular pathway, starting from cis-INT(D+-E+), the κ1-acetate ligand abstracts the proton using the unbonded oxygen atom and gives rise to cationic intermediate E+, which was found at −1.0 [−1.5] [6.7] kcal mol−1. The intramolecular pathway involves the classical AMLA/CMD process, which, going through the six-membered cyclic TS structure cis-TS(D+-E+)2, must overcome a ΔGr of 10.9 [10.5] [18.8] kcal mol−1. Alternatively, the association of a free molecule of acetate to the activated C–H bond in trans-INT(D+-E) facilitates subsequent intermolecular deprotonation leading to intermediate E and HOAc. After the association of HOAc with chloride to form AcOH·Cl−, the acetate–ruthenacycle E was calculated to be 18.6 [17.8] [7.1] kcal mol−1 more stable than the starting compounds. Going through TS structure trans-TS(D+-E)2 intermolecular deprotonation (also denoted as the external-CMD pathway) required surpassing a ΔGr of 12.4 [13.9] [25.5] kcal mol−1 and was disfavoured over the intramolecular one by 1.5 [3.4] [6.7] kcal mol−1.48
2.3. Final ligand exchange processes at ruthenacycles
Cyclometallated chloro(p-cymene)Ru(II) complexes have interesting biological6p–r and catalytic6c,d,e,f activities. The replacement of the chloride leaving group by a water molecule is thought to be essential for their mechanism of action in biological systems, while the chloro/carboxylate ligand exchange processes are mandatory for their entrance into the catalytic cycles of carboxylate-assisted C–H bond functionalizations. As neither the detailed mechanism nor the kinetic relevance of the chloro/carboxylate ligand exchange processes at cyclometallated (p-cymene)Ru(II) complexes are yet clear, we decided to undertake modelling in methanol solution by using the acetate cycloruthenate E as a model system. At our benchmark level, the lowest energy route from E to chloro cycloruthenate H follows an associative pathway with solvent assistance, as depicted in Scheme 6. Initial acetate/methanol exchange at ruthenium through TS structure TS(E-H)1·S required surpassing a ΔGr of 9.3 [10.7] [21.4] kcal mol−1 and led to the monosolvated intermediate INT(E-H)+·S (see ESI, Scheme S10a†). Subsequent methanol/chloride exchange on the cationic intermediate takes place through TS(E-H)2·S (ΔGr of 8.8 [9.5] [17.4] kcal mol−1) to afford the chloro cycloruthenate H (see ESI, Scheme S10b†), which was found to be 19.3 [18.3] [10.6] kcal mol−1 lower in energy than the starting compounds. Thus, final acetate/chloride exchange on the ruthenacycle is calculated to be exergonic by 0.7 [0.5] [3.5] kcal mol−1. Dissociative pathways for the acetate/chloride exchange, with or without solvent assistance, and direct acetic acid/chloride exchange on intermediate E+ were also considered but were more than 4 kcal mol−1 higher in energy (see ESI, Scheme S10c†). | ||
| Scheme 6 Acetate/chloride exchange and formation of the chloro–ruthenacycle (H). Relative quasi-harmonic free energies (in kcal mol−1) calculated at the DLPNO-CCSD(T)/CBS(CPCM) level in methanol (298 K, 1 M) using the geometries optimized at M06/cc-pVDZ-PP[Ru](IEF-PCM) are shown in blue. The corresponding values calculated at the same level by using the geometries optimized at the ωB97X-D3/cc-pVDZ-PP[Ru](CPCM) level are shown between brackets in red. Energy values calculated at the DLPNO-CCSD(T)/CBS(SMD) level in methanol (298 K, 1 M) are shown in italics. Hydrogen bonds are represented by dotted lines. | ||
2.4. Competence between reaction pathways
2.4.1.1. Standard gas-phase thermochemical analysis. 3D representations50 of the optimized geometries and energy plots at the DLPNO-CCSD(T)/CBS(CPCM) level for the most significant stationary points located on the competitive pathways are shown in Fig. 1. Fig. 1(a) plots the energy results evaluated under standard conditions (1.0 M) after geometry optimizations (in methanol solution) with the M06 functional. The corresponding plot calculated at the same level by using the ωB97X-D3 geometries is included in the ESI,† see Fig. S2(a).† In these plots, relative energies for the pre-activation and C–H activation processes (from B1 to E) are referenced to A+½B0 + 2AcO− while for the final ligand exchange processes on the ruthenacycle (from E to H) A + ½B0 + S + 2AcO− is set to zero. Analysis of both plots allows us to conclude the following. First, chemical equilibria between chloro and acetate ruthenium(p-cymene) precursors are shifted towards monomeric diacetate complex B2, which constitutes the lowest energy point of the profiles and serves as the reference for calculating the barrier heights in the forward reaction. From B2 (located in Fig. 1 under the precursors A+½B0, which serve as the reference for the reaction coordinate) the lowest energy pathway to cycloruthenates involves cationic intermediates D+, INT(D+-E+) and E+. In this pathway the imine/chloride ligand exchange process on ruthenium leading to key cationic intermediate D+ constitutes the slower, RDS for the global process. Thus, an activation energy of 29.0 [28.4] kcal mol−1 is calculated for the forward reaction. Subsequent steps from cationic intermediate D+ to acetate–cycloruthenate E are faster, as the CMD/AMLA C–H activation must surpass a barrier of 22.8 [22.5] kcal mol−1. Formation of acetate–cycloruthenate E from precursor B2 is found to be exergonic by 6.7 [5.8] kcal mol−1. The final solvent-assisted chloride/acetate ligand exchange on ruthenacycle E must overcome an activation free energy of 27.9 [28.5] kcal mol−1 and should take place faster than [or at comparable rate to] anion dissociation leading to key cationic intermediate D+. Formation of chloro–ruthenacycle H from acetate precursor E is calculated to be exergonic by only 0.7 [0.5] kcal mol−1 and may constitute an equilibrium process, which is mainly driven by dissociation of AcOH·Cl− and formation of AcOH·AcO− (see ESI, Scheme S11†). As the energy barrier for the forward reaction (29.0 [28.4] kcal mol−1viaTS(C1-D+)) is almost the same as the barrier required for the reversion of cyclometallation (from E to D+ through cis-TS(D+-E+)2, 29.5 [28.3] kcal mol−1), calculations show that the cyclometallation of benzaldimines in methanol should be reversible, as previously reported for ruthenium-catalyzed hydroarylation7e or deuteration7h of aromatic carbonyl compounds using imines as TDGs in apolar solvents and also in accordance with deuterium exchange experiments reported for many C–H bond ruthenations of related arenes equipped with N-containing DGs in polar protic solvents such as water, methanol, acetic acid or HFIP.51
 | ||
| Fig. 1 Relative quasi-harmonic Gibbs energy plots calculated at the DLPNO-CCSD(T)/CBS(CPCM) level in methanol (in kcal mol−1). (a) At 298 K and 1 M after geometry optimizations at the M06/cc-pVDZ-PP[Ru](IEF-PCM) level. (b) At 298 K and 24.6 M after geometry optimizations at the ωB97X-D3/cc-pVDZ-PP[Ru](CPCM) level. Pre-activation by anion dissociation and ligand exchange processes at ruthenium are shown in black, C–H activations involving cationic intermediates are shown towards the right in blue and the corresponding processes involving neutral intermediates are shown towards the left in brown and green. The final ligand exchange processes at ruthenacycles are shown towards the right in purple. | ||
On the basis of the results obtained at the DLPNO-CCSD(T)/CBS(CPCM) level, participation of the reaction pathways involving neutral intermediates should be excluded. Neutral intermediates are shown in Fig. 1 towards the left of the A+½B0 reference. From Fc, the neutral intermediate with the p-cymene moiety acting as the η2-ligand, the slowest step corresponds to the formation of agostic intermediate INT(Fc-Gc)via TS structure TS(Fc-Gc)1. Thus, the progress of the C–H activation event through the pathway involving neutral intermediate Fc will show an activation energy of 30.6 [32.9] kcal mol−1, 1.6 [4.5] kcal mol−1 higher than that offered by the pathway involving cationic intermediate D+. Alternative reaction pathways involving solvated or bis-imine ligated neutral intermediates (FS and FA) are also precluded due to higher activation energies for the exchange processes of the p-cymene ligand with methanol or a second imine molecule. Nevertheless, in a related fashion to that reported by Ackermann et al. on the grounds of their computational investigation of the acetate-assisted ruthenium-catalyzed C–H activations of one of two N-coordinated arylpyridine motifs,33c the bis-imine N-ligated ruthenium intermediates FA, INT(FA-GA) and GA could offer the lowest energy pathway for the cyclometalation of benzaldimines in apolar solvents at higher reaction temperatures or at room temperature under visible-light irradiation conditions, which are known to facilitate decomplexation of the p-cymene ligand from the neutral ruthenium intermediates.52 For the system under study the energy barrier required for C–H activation via the neutral bis-imine ligated intermediate INT(FA-GA) was calculated to be ca. 2 [2] kcal mol−1 lower than the corresponding barrier in the pathway involving the cationic intermediate cis-INT(D+-E+).
A comparison of the energy plots obtained by performing the geometry optimizations with the M06 or ωB97X-D3 functionals (Fig. 1(a) and Fig. S2(a),† respectively) reveals only small differences. Activation energies calculated at the DLPNO-CCSD(T)/CBS(CPCM) level for the geometries optimized with the ωB97X-D3 functional for anion dissociation and formation of key cationic intermediate D+, the C–H activation event from D+ to E+ and the final solvent-assisted dissociative ligand exchange at ruthenacycles differs by less than 1 kcal mol−1 from the corresponding values obtained by using the M06 geometries. Nevertheless, for the competing pathways involving neutral intermediates F–G the calculated activation energies were found to be between 2 and 9 kcal mol−1 higher when the geometry optimizations were performed with the ωB97X-D3 functional instead of the M06 one. Thus, inclusion of dispersion energy corrections and range-separated treatment for the exchange contributions in the DFT geometry optimizations appears to improve the discrimination between the pathways involving the cationic and neutral intermediates.
2.4.1.2. Condensed-phase thermochemical analysis. Since entropic contributions calculated within the ideal gas approximation at 1.0 M are likely to exaggerate the expected values in the condensed phase, a second thermochemical analysis was performed to model the reduction of the translational degrees of freedom at the concentration of the liquid solvent. When a pressure parameter to adjust the concentration to that of the liquid solvent53 (24.6 M for methanol) is taken into consideration, monosolvation of chloride anions, association of chloride to acetic acid and the formation of hydrogen diacetate (by association of acetate to acetic acid) are more favoured processes (by an additional term of ca. 1.9 [1.9] kcal mol−1), as depicted in Scheme S11 of the ESI.† Relative quasi-harmonic Gibbs energies calculated at the DLPNO-CCSD(T)/CBS(CPCM) level in the condensed phase (298 K, 24.6 M) using geometries optimized at the ωB97X-D3/cc-pVDZ-PP[Ru](CPCM) level for the most significant stationary points are depicted in Fig. 1(b). Due to the exergonic monosolvation of the chloride anions under condensed-phase conditions, in Fig. 1(b) the relative energies are referenced to A + ½B0 + 2S + 2AcO−.
By comparison of Fig. 1(b) and Fig. S2(a) in the ESI† some differences can be observed for the progress of cyclometallation in the condensed phase with respect to standard conditions. (1) In the forward direction the Gibbs energies of activation remain unchanged for the formation of key intermediate D+ and for the subsequent C–H activation involving cationic intermediates (at ca. [28] and [22] kcal mol−1, respectively) but are reduced for the final ligand exchange process at cycloruthenates (by ca. [1] kcal mol−1); (2) the energy barrier for C–H activation in the reverse direction (from E to D+) is also conserved at ca. [28] kcal mol−1. Thus, under condensed-phase conditions the pathway involving cationic intermediates also offers the lowest energy profile and the reversibility of the C–H activation event should not be compromised. It should be noted that the solvent compression effect significantly reduces the energy gap between TS(C1-D+) and TS(C2-D+) and between cis-TS(D+-E+)2 and trans-TS(D+-E)2 (by ca. [1.9] kcal mol−1). Thus, the contribution of the pathways involving acetate anion dissociation in the RDS or intermolecular deprotonation for the C–H activation event may be increased in polar protic solvents.48 The same conclusions can be drawn by comparing the energy plots calculated under standard and condensed-phase conditions by using the M06 geometries (depicted in Fig. 1(a) and Fig. S2(b) in the ESI†).
2.4.1.3. Performance of DFT methods. Single point energy evaluations with top hybrid DFT performers in the MOR41 and MOBH35 databases37 using an augmented polarized triple-ζ basis set (denoted as aug-cc-pVTZ-PP[Ru], see Computational methods in the ESI† for further details) did not allow to reproduce the DLPNO-CCSD(T)/CBS(CPCM) energy results (see Table S2 in the ESI†). In a different manner, single point energy evaluations with double-hybrid DFT methods were found to be in good agreement with our benchmark calculations at the DLPNO-CCSD(T)/CBS(CPCM) level. According to the results obtained with the B2GP-PLYP,54 B2K-PLYP55 or PWPB9556 methods, the formation of key cationic intermediate D+ was the RDS and the reaction channels involving neutral intermediates were all disfavoured by more than 5 [3] kcal mol−1. Nevertheless, energy evaluations with double-hybrid DFT methods underestimated the exergonic character of the global transformation by more than 3 [2] kcal mol−1 and overestimated the Gibbs activation energy for the RDS by more than 9 [4] kcal mol−1. Better correlation with our benchmark results was obtained by applying Grimme's D3(BJ)57 or D458 dispersion energy corrections to the energy evaluations with the PWPB95 method, as the overestimation of the activation energies was reduced to 3 [2] kcal mol−1. Moreover, the lowest MUEs for the relative Gibbs energies of precursors and cyclometallated intermediates (B1, B2, E and H) and for the whole set of barrier heights in Table S2† were obtained when the energy evaluations were performed with the PWPB95 method adding the D3(BJ) or D4 dispersion energy corrections and using the ωB97X-D3 geometries (see entries 5 and 20). In addition, by using this computational scheme (PWPB95 + D3(BJ)//ωB97X-D3) the MUE for the barrier heights along the most favourable pathway and involving the cationic intermediates was only 0.3 kcal mol−1 (see entry 21).
2.4.2.1. Pure implicit (SMD) solvation model. As the SMD model provides a more realistic description of the non-electrostatic solute–solvent interactions and is recommended for the estimation of the solvation energies of charged species, following the suggestion of one referee, we have performed energy evaluations by employing the SMD model for the most significant stationary points located after geometry optimizations with the ωB97X-D3 functional. At our benchmark level with the SMD solvation model, monosolvation of chloride anions and their association to acetic acid are favoured processes (by −1 and −2 kcal mol−1, respectively), while the formation of hydrogen diacetate is disfavoured (by 2 kcal mol−1), even under standard conditions (see Scheme S11 of the ESI†). Thus, using the SMD solvation model, to describe the most stable aggregation states and relative energies for all the intermediates represented in Schemes 4–6, the chloride anions have been replaced by those that are monosolvated or associated with acetic acid. Plots of the energy results at the DLPNO-CCSD(T)/CBS(SMD) level evaluated under standard conditions (1.0 M) and under condensed-phase conditions (24.6 M) are included in the ESI† (see parts (a) and (b) of Fig. S3†). According to these SMD profiles (1) the chemical equilibria between chloro and acetate ruthenium(p-cymene) precursors are shifted towards monomeric diacetate complex B1, which sets the reference for calculating the barrier height for cycloruthenation in the forward direction; (2) the lowest energy pathway for C–H bond activation also involves cationic intermediates, (3) an activation energy of [26.9] kcal mol−1 is calculated for chloride dissociation leading to cationic intermediate D+, (4) subsequent CMD/AMLA C–H activation events from D+ to E+ are faster and require an activation energy of [21.2] kcal mol−1, (5) formation of the acetate–cycloruthenate E from precursor B1 is found to be exergonic by [4.7] kcal mol−1 and (6) the C–H activation event is reversible and must surpass an activation barrier of [25.9] kcal mol−1 in the reverse direction (from E to D+). Finally, according to the SMD profile, the progress of C–H bond activation through the pathways involving neutral diacetate ruthenium intermediates is also precluded in methanol as solvent, as it would require activation energies more than [9] kcal mol−1 higher than that offered by the pathway involving cationic intermediate D+.
Analysis of the energy plots calculated at the benchmark level with the SMD model under standard conditions and condensed-phase conditions (see parts (a) and (b) of Fig. S3 in the ESI†) reveals some differences and similarities to those aspects described above for the CPCM profiles. The switch from standard to condensed-phase conditions in the SMD profiles (a) reduces the energy barriers for anion dissociation leading to key intermediate D+ and for subsequent C–H activation involving cationic intermediate INT(D+-E+) by 1.9 kcal mol−1 (from [26.9] to [25.0] and from [21.2] to [19.3] kcal mol−1, respectively), while the energy barrier for the reverse reaction (from E to D+) is maintained (at ca. [26] kcal mol−1); (b) the energy gap between TS(C1-D+) and TS(C2-D+) and between cis-TS(D+-E+)2 and trans-TS(D+-E)2 is also reduced by ca. [1.8] kcal mol−1.
In this manner, energy plots calculated for our benchmark level using either the SMD or CPCM solvation models describe the C–H bond activation of benzaldimine in MeOH as a reversible and kinetically irrelevant process. Nevertheless, a remarkable difference of 1.5/3.2 kcal mol−1 is observed for the activation energies leading to cycloruthenate formation, as the values calculated using the CPCM model under standard and condensed-phase conditions are [28.4] and [28.2] kcal mol−1 and are reduced to [26.9] and [25.0] kcal mol−1 with the use of the SMD model. All these values are in line with the activation energies reported in previous computational studies on the C–H bond activation processes for related arenes equipped with heterocyclic DGs in alcoholic solvents, which have been described as system-dependent and generally ranging from 25 to 28 kcal mol−1.5g,18,19,23e,29a,b
2.4.2.2. Hybrid explicit–implicit (SMD) solvation model. Continuum solvation models are computationally efficient and are widely used in computational homogeneous catalysis. In particular, PCM and SMD solvation models have been successfully employed to investigate many ruthenium(II)-catalyzed C–H activation and functionalization processes in almost all kinds of solvents (as 2-MeTHF, dioxane, toluene, DCE, acetonitrile, MeOH, TFE, tert-butanol, HFIP, or AcOH).12–33 Nevertheless, the suitability of implicit solvent models for the study of ionic reactions that are performed in protic solvents is still controversial, as strong, specific, solvent–solute H-bonding interactions are seemingly ignored. These kinds of directed interactions are not captured by the CPCM and SMD models and this deficiency could compromise the accuracy of our results. To overcome these limitations, we considered the application of a hybrid explicit–implicit (cluster/continuum) solvation model,42 by adding a limited number of solvent molecules to the reaction system inside the continuum model cavity. Nevertheless, it is not clear how many solvent molecules need to be added to describe the solvent effects with the desired accuracy, while the addition of solvent molecules to selected binding sites or regions around the solute is guided by assumptions and may change from one intermediate to another along the reaction coordinate. In any case, the static ab initio evaluation of a small set of low-lying configurations for the solvent–solute clusters can lead to meaningful improvements in the description of the “averaged” solvent effects by pure implicit solvation models, thus providing insights into the relevance of the solvent–solute H-bonding interactions and more “realistic” energetic estimations.
Taking into consideration experimental measurements and the results of molecular dynamics simulations that are consistent with a coordination shell of chloride or acetate anions in methanol solutions essentially made up to 3 or 4 solvent molecules,59 and trying to get a glance of the relevance of specific solvent–solute interactions in the acetate-assisted C–H activation of benzaldimine at [RuCl2(p-cymene)]2 in MeOH, we studied the addition of up to 3 or 4 explicit MeOH molecules to the most relevant stationary points in the C–H bond activation pathway involving cationic intermediates, from B1 to E+. We considered it reasonable to assume that one, two or three solvent molecules should hydrogen bond to the chloride and acetate moieties. After geometry optimizations at the ωB97X-D3/cc-pVDZ-PP[Ru](CPCM) level in methanol solution, the relative quasi-harmonic Gibbs energies of the solvated species were evaluated at our benchmark level (DLPNO-CCSD(T)/CBS(SMD)) using separate reagents (A + ½B0 + 2AcO−) and an adequate number of explicit solvent molecules (to ensure the stoichiometric balance) as a common energetic reference. As described below (in the energy profile using the SMD solvation model) for the non-solvated reaction channel, aside from the MeOH molecules maintaining specific interactions near the reaction center, one explicit MeOH molecule is needed to accomplish the exergonic monosolvation of the chloride that is freed through the formation of the B1 precursors. Thus, in Fig. 2 (and in Fig. S4, S5 and S6 of the ESI†) the active MeOH molecules (surrounding the reaction center of any intermediate or TS) are highlighted in yellow while the solvent molecule associated with the stoichiometric chloride is highlighted in blue. The non-solvated and solvated reaction channels, considering specific interactions of any intermediate with zero, one, two or three (yellowish) MeOH molecules, are denoted as the S0-, S1-, S2- and S3-reaction channels, respectively. The (yellowish) methanol molecules bound the chloride atoms are denoted as S, and those associated with the acetate moiety by H-bonding the oxygen atom bound to ruthenium(II) or one in the pendant free arm are denoted as Sk and Sp, respectively. Solvent molecules bound to an external acetate are designated as Se and those involved in proton-relay mechanisms are designated as Sr.
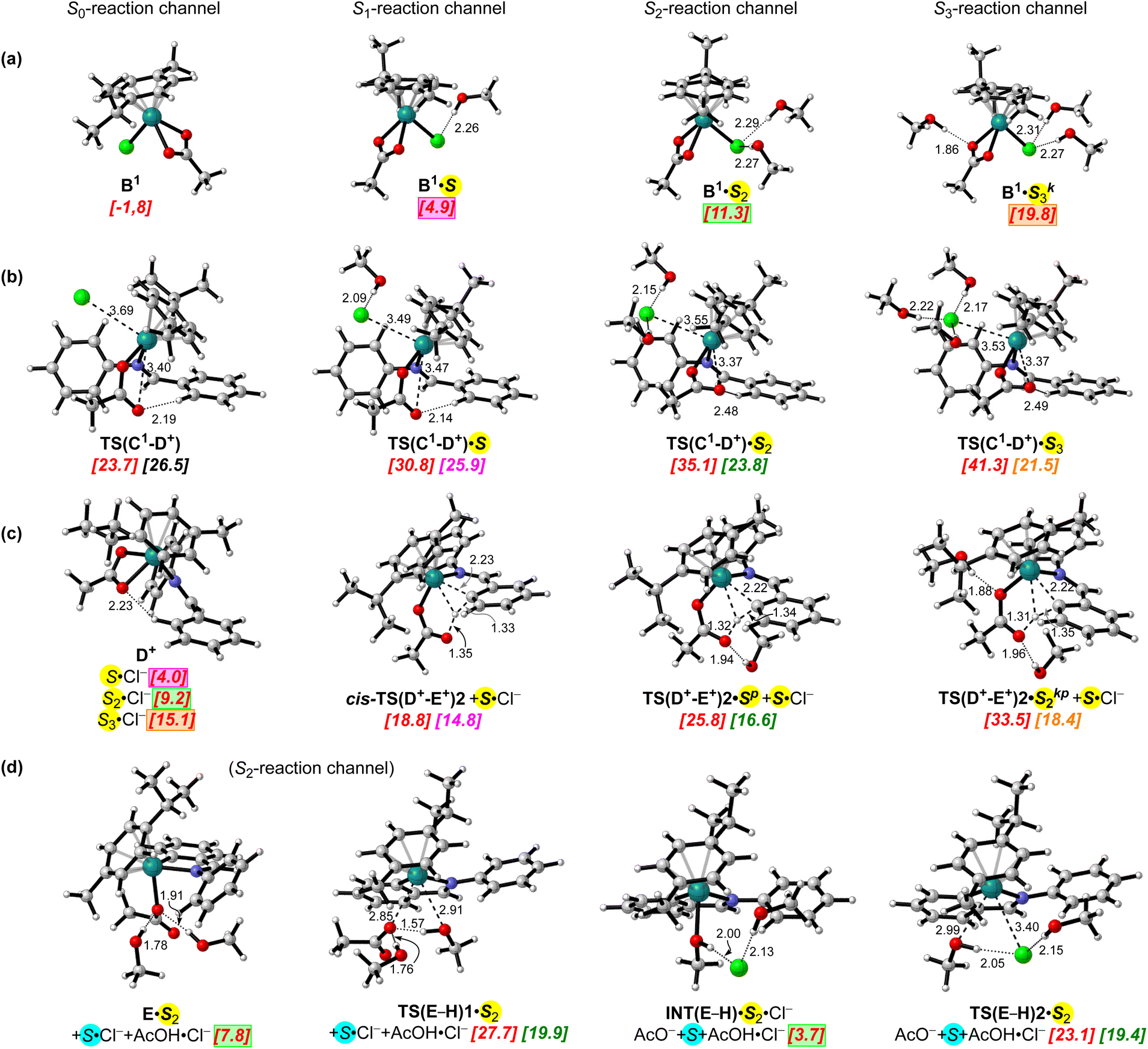 | ||
| Fig. 2 3D representations of the most significant stationary points located in the S0-, S1-, S2- and S3-reaction channels. (a) Key chloro acetate (p-cymene)ruthenium complexes; (b) TS structures for chloride dissociation from intermediates C1·Sn leading to cationic intermediate D+; (c) relevant TS structures for the C–H bond activation process from D+ to cycloruthenate E+, (d) TS structures and intermediates for acetate/exchange processes between cycloruthenates E and H. Quasi-harmonic Gibbs energies calculated at the DLPNO-CCSD(T)/CBS(SMD) level in methanol (298 K, 1 M) after geometry optimizations at the ωB97X-D3/cc-pVDZ-PP[Ru](cPCM) level are shown between brackets, in italics and red, relative to separated reagents (A + ½B0 + 2AcO−) and an adequate number of explicit solvent molecules to ensure stoichiometric balance. Activation barriers are shown in magenta, green, or orange with respect to the corresponding lowest energy precursors in the S1-, S2- or S3-reaction channels, which are highlighted using the same colours. The hydrogen bonds are represented by dotted lines. | ||
Based on the results described in previous sections, we focused our analysis of the specific solvent–solute interactions in chloride dissociation from intermediates C1·Sn leading to D+viaTS(C1-D+)·Sn and C–H bond activation process from D+ to cycloruthenate E+viaTS(D+-E+)1·Sn, INT(D+-E+)1·Sn and TS(D+-E+)2·Sn. 3D representations of the most significant stationary points located in the S0-, S1-, S2- and S3-reaction channels are depicted in Fig. 2, including selected distances and relative energies. The complete energy profiles for the S1-, S2- and S3-reaction channels are included in the ESI,† see Fig. S4, S5 and S6,† respectively.
Most stable TS structures located in the S1-, S2- and S3-reaction channels leading to cationic intermediate D+ showed the chloride leaving group associated with one, two and three MeOH molecules, respectively, with distances and angles falling in the ranges usually chosen to characterize hydrogen bonds (r(H⋯Cl) < 2.2 Å, r(O⋯Cl) < 3.2 Å, and θ(OH⋯Cl) < 14°). The solvated TS structures TS(C1-D+)·Sn were also characterized by slightly shorter distances between ruthenium and chloride (ca. 3.50 A) than the non-solvated one (ca. 3.70), indicating a higher early character of these TS structures in the solvated reaction channels. TS structures TS(C1-D+)·S1, TS(C1-D+)·S2 and TS(C1-D+)·S3 were characterized at 30.8, 35.1 and 41.3 kcal mol−1, respectively, showing activation energies of 26.5, 25.9 and 21.5 kcal mol−1 relative to the corresponding B1·S1, B1·S2 and B1·S3 precursors (see parts (a) and (b) of Fig. 2 and Schemes S12 and S13a–c in the ESI†). Thus, mono-, di- and tri-solvation of the chloride leaving group in TS(C1-D+)·Sn led to reductions of 1.0, 3.1 and 5.4 kcal mol−1 in the calculated energy barriers from the value of 26.9 kcal mol−1 previously estimated for the S0-reaction channel, viaTS(C1-D+). Alternative transition structures for the chloride dissociation step in the S2- and S3-reaction channels, showing one MeOH molecule bound to the oxygen atom of the pendant free arm of the acetate moiety were found to be higher in energy (see TS(C1-D+)·S2p and TS(C1-D+)·S3p in Fig. S5, S6 and S13c in the ESI†).
For the analysis of the C–H activation process we found it reasonable to include in the corresponding Sn-reaction channels all possible intermediates and transition states with “n” (yellowish) MeOH molecules, either H-bonding to chloride that was freed during their formation (Cl−·Sn) or bound to the oxygen atoms of the acetate moieties (in TS(D+-E+)1·S(3−n), INT(D+-E+)·S(3−n) and TS(D+-E+)2·S(3−n)). In this manner, the lowest energy pathways for C–H bond activation in the S1-, S2- or S3-reaction channels involve the non-solvated TS structures cis-TS(D+-E+)1 and cis-TS(D+-E+)2 accompanied by the corresponding mono-, di- or tri-solvated chloride anions (Cl−·S, Cl−·S2 or Cl−·S3, see Fig. 2(c)). Reaction partners cis-TS(D+-E+)1 and cis-TS(D+-E+)2 plus Cl−·S were found at 15.5 and 18.8 kcal mol−1, respectively, in the S1-reaction channel, while for the S2- and the S3-reaction channels the same TS structures were found at 20.7 and 24.0 and 26.6 and 29.9 kcal mol−1, combined with Cl−·S2 and Cl−·S3, respectively (see Scheme S14 and Fig. S4, S5 and S6 in the ESI†). These three combinations enable the progress of C–H bond activation from the lowest-lying, non-solvated ruthenium precursor D+ with an activation barrier of only 14.8 kcal mol−1, 6.4 kcal mol−1 lower than that calculated by using the pure implicit solvation model. Such a reduction of the activation barrier for the progress of C–H activation via the same TS structures is mainly due to the energy balance between non-solvated and solvated precursors and by-products that are introduced into the corresponding continuum model cavities to achieve a realistic description of the system. The lowest-lying TS structures arising from any solvated cationic intermediate D+·Sn in the S1-, S2- or S3-reaction channels were TS(D+-E+)2·Sp, with one MeOH molecule bound to the oxygen atom at the pendant free arm of the acetate moiety, and TS(D+-E+)2·S2kp, with two MeOH molecules H-bonding to both oxygens of the acetate moiety. These transition structures were found in the S2- and S3-reaction channels at 25.8 and 33.5 kcal mol−1, respectively, and showed activation energies of 16.6 and 18.4 kcal mol−1 from their respective precursors, D+·S1 and D+·S2 (see Fig. 2(c) and Schemes S15a and S15b in the ESI†). Other TS structures in the S2- and S3-reaction channels involving C–H bond activation by proton-relay mechanisms or deprotonation by an external acetate molecule were found to be higher in energy (see Fig. S5, S6 and S7 in the ESI†).
We have also employed the hybrid explicit/implicit (SMD) solvation model to study the acetate/chloride exchange processes between cycloruthenates E and H. The energy profiles calculated at our benchmark level for the S1- and S2-reaction channels are included in Fig. S8a and S8b in the ESI.† The most relevant stationary points located in the S2-reaction channel are depicted in Fig. 2(d). At the DLPNO-CCSD(T)/CBS(SMD) level, the energy barriers for the acetate/chloride exchange processes in the S1- and S2-reaction channels differ by less than 1 kcal mol−1. H-Bonding of the second MeOH molecule to the acetate acting as a leaving group increases the activation energy for the methanolysis of the acetate cycloruthenate by ca. 1.0 kcal mol−1, from a value of 19.0 kcal mol−1 in the S1-reaction channel viaTS(E-H)1·S to a value of 19.9 kcal mol−1 in the S2-reaction channel viaTS(E-H)1·S2 (see Fig. S8a and S8b in the ESI†). Association of two MeOH molecules to the chloride anion reduces its nucleophilicity, and methanol/chloride exchange in the S2-reaction channel viaTS(E-H)2·S2 showed an activation barrier 0.6 kcal mol−1 higher than that calculated viaTS(E-H)2·S in the S1-reaction channel. The energy barriers for the acetate/chloride exchange processes in the solvated pathways are estimated to be ca. 19.5 and 24.5 kcal mol−1 in the forward and reverse directions, respectively. In this manner, mono- or di-solvation of the acetate leaving group led to a reduction of the energy barrier by ca. 8.5 kcal mol−1, from the value of 28.5 kcal mol−1 previously estimated for the non-solvated ligand exchange process, viaTS(E-H)1 (see Fig. S3†). This reduction of the energy barrier is mainly due to the removal of the entropy cost associated with the binding of methanol molecule(s) and cycloruthenate, which is considered to be an integral part of the barrier for the non-solvated ligand exchange processes (from E to TS(E-H)1) but not applicable to the solvated reaction channels (from E·Sn to TS(E-H)1·Sn).
It should be noted that the activation barrier calculated for the progress of chloride/acetate ligand exchange in the reverse direction from INT(E-H)·S2·Cl− to E·S2viaTS(E-H)1·S2 (24.0 kcal mol−1) is almost the same as that required for the RDS in the forward direction, from precursor B1·S2 to key cationic intermediate D+ + Cl−·S2viaTS(C1-D+)·S2 (23.8 kcal mol−1). Thus, acetate/chloride exchange on cycloruthenates and C–H bond activation on cationic intermediates are calculated to be reversible and kinetically irrelevant processes in MeOH at room temperature.
In conclusion, calculations employing a hybrid explicit/implicit solvation model do not change the main results that were reached using the pure implicit CPCM or SMD solvent models. In any of the Sn-reaction channels the energy barriers for the C–H bond activation process were calculated to be lower than those for the previous anion dissociation step. Thus, chloride dissociation leading to cationic intermediate D+viaTS(C1-D+)·Sn is the RDS and subsequent C–H bond activation events in the CMD pathways are faster and reversible processes. In addition, calculations with the hybrid explicit–implicit solvation model indicate that 2 or 3 MeOH molecules H-bonding to the chlorine atom increase the aptitude of the solvated chloride to act as a leaving group in the anion dissociation step, in a sort of acid catalysis by the polar protic solvent that accelerates cycloruthenation.
As indicated by one of the referees, Ru(II)-catalyzed C–H bond activations and functionalizations of arenes equipped with imines and other nonprotic DG that are promoted by [RuCl2(p-cymene)]2 are also frequently conducted in the presence of silver salts of weakly coordinating anions and stoichiometric amounts of base, generally carbonate salts. A comparison of our computational results with experiments conducted with these protocols is also possible. The most common mechanistic proposal for (p-cymene)Ru(II)-catalyzed C–H activations that are conducted in the presence of silver salts assumes that the initial chloride/acetate exchange processes on ruthenium precursors (like B0 and B1) should take place more easily and that diacetate ruthenium complexes (like B2 and C2) should become the key active intermediates (see ref. 7e as an example). In this situation the involvement of TS structures like TS(C1-D+) is precluded but, according to our computational results, TS(C2-D+) would offer the lowest energy pathway for acetate dissociation. Thus, subsequent AMLA/CMD C–H activation on the corresponding intermediate cationic complexes would also be faster and reversible processes. Several studies have postulated that the role played by stoichiometric bases in experimental catalytic protocols is to deprotonate the AcOH moiety of the first cycloruthenate intermediate (like E+) to form a more stable OAc adduct (like E). Thus, for processes that are conducted in the presence of silver salts or with an excess of base, the thermodynamics of the acetate/ligand exchange at the ruthenacycle intermediates could be affected. In particular, association of chloride to acetic acid should be suppressed but, in our opinion, comparison with or inference from our main computational results (regarding (a) binding of a substrate with a nonprotic DG to ruthenium, (b) anion dissociation accelerated by H-bonding to protic polar solvent molecules and leading to key cationic intermediate D+ as the RDS and (c) subsequent reversible AMLA/CMD C–H activation events) should not be compromised.
3. Conclusions
The reaction mechanisms for the acetate-assisted C–H activation of N-phenyl benzaldimine at Ru(p-cymene)2Cl2 in methanol have been computationally studied at the DLPNO-CCSD(T)/CBS(CPCM and SMD) level. The most favourable reaction pathway involves the coordination of the imine to the ruthenium precursor followed by ligand dissociation and formation of a cationic ruthenium intermediate as the RDS of the process. Subsequent C–H bond activation events are faster and reversible processes. Two or three MeOH molecules H-bonding to chlorine in the anion dissociation step may speed up the cycloruthenation process, in a sort of acid catalysis by the polar protic solvent. Solvent assistance accelerates the final chloride/acetate ligand exchange process at the cationic cycloruthenate intermediate, particularly when solvent compression in the condensed phase is taken into consideration. Neutral diacetate ruthenium intermediates lead to TS structures of lower energy for the AMLA/CMD C–H bond activation, which result not productive due to higher energy barriers for the ligand exchange processes at acetate ruthenium precursors in MeOH solution. Energy evaluations with hybrid DFT methods (PBE0-D3(BJ), M06-2X or ωB97M-V) failed to reproduce the energy gap between the pathways involving cationic and neutral ruthenium intermediates. In this manner, the use of double-hybrid functionals in combination with dispersion energy corrections was found to be necessary for an accurate evaluation of the kinetics of this acetate-assisted cycloruthenation at the DFT level. Energy evaluations at the PWPB95 + D3(BJ)/aug-cc-pVTZ-PP[Ru](CPCM) level reproduce the DLPNO-CCSD(T)/CBS(CPCM) results at a small fraction of their computational cost and should be reliable for the study of new synthetically useful C–H bond activations/functionalizations involving (N,C)-cyclometallated (p-cymene)Ru(II) complexes in polar protic media. We are currently working on the application of this computational scheme to understand the regioselectivity shown by arenes equipped with N-containing DGs in their acetate-assisted cyclometallations at [RuCl2(p-cymene)]2.Author contributions
V. O. performed the computational calculations. The manuscript was written through contributions of both authors. Both authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Centro de Supercomputación de Galicia (CESGA) for providing computer facilities. Funding for open access charge from Universidade da Coruña/CISUG is acknowledged.References
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- N. Kaplaneris, M. Vilches-Herrera, J. Wu and L. Ackermann, ACS Sustainable Chem. Eng., 2022, 10(21), 6871–6888 CrossRef CAS.
- For reviews on. (a) Annulations of alkynes, see L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (b) Arylations, see P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721–5745 CrossRef CAS; (c) Olefinations, see R. Manikandana and M. Jeganmohan, Chem. Commun., 2017, 53, 8931–8947 RSC. For a relevant sulfonation, see: (d) P. Marcé, A. J. Paterson, M. F. Mahon and C. G. Frost, Catal. Sci. Technol., 2016, 6, 7068–7076 RSC. For recent examples on annulation by 1,3-diyne insertion, see: (e) G. S. Sontakke, C. Ghosh, K. Pal and C. M. R. Volla, J. Org. Chem., 2022, 87, 14103–14114 CrossRef CAS PubMed; (f) Y. Luo and L. Dong, J. Org. Chem., 2022, 87, 5577–5591 CrossRef CAS PubMed. For a recent alkenylation, see: (g) C. N. Shambhavi and M. Jeganmohan, Org. Lett., 2023, 25, 358–363 CrossRef CAS PubMed.
- (a) D. L. Davies, O. Al-Duaij, J. Fawcett, M. Giardiello, S. T. Hilton and D. R. Russell, Dalton Trans., 2003, 4132–4138 RSC; (b) B. Li, T. Roisnel, C. Darcel and P. H. Dixneuf, Dalton Trans., 2012, 41, 10934–10937 RSC; (c) B. Li, C. Darcel, T. Roisnel and P. H. Dixneuf, J. Organomet. Chem., 2015, 793, 200–209 CrossRef CAS; (d) W.-G. Jia, T. Zhang, D. Xie, Q.-T. Xu, S. Ling and Q. Zhang, Dalton Trans., 2016, 45, 14230–14237 RSC; (e) P. Kuzman, F. Pozgan, A. Meden, J. Svete and B. Stefane, ChemCatChem, 2017, 9, 3380–3387 CrossRef CAS; (f) C. A. Riedl, L. S. Flocke, M. Hejl, A. Roller, M. H. M. Klose, M. A. Jakupec, W. Kandioller and B. K. Keppler, Inorg. Chem., 2017, 56, 528–541 CrossRef CAS PubMed; (g) R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, Faraday Discuss., 2019, 220, 386–403 RSC.
- (a) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed; (b) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032–3035 CrossRef CAS PubMed; (c) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (d) J. Li, S. Warratz, D. Zell, S. D. Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed; (e) C. Yuan, L. Zhu, R. Zeng, Y. Lan and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1277–1281 CrossRef CAS PubMed; (f) L. Zhang, K. Deng, G. Wu, J. Yang, S. Tang, X. Fu, C. Xia and Y. Ji, J. Org. Chem., 2020, 85, 12670–12681 CrossRef CAS PubMed; (g) P. Piehl, R. Amuso, A. Spannenberg, B. Gabriele, H. Neumann and M. Beller, Catal. Sci. Technol., 2021, 11, 2512–2517 RSC; (h) X. Tan, X. Hou, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2021, 60, 4619–4624 CrossRef CAS PubMed; (i) Y.-X. Xu, Y.-Q. Liang, Z.-J. Cai and S.-J. Ji, Org. Lett., 2022, 24, 2601–2606 CrossRef CAS PubMed; (j) P. M. Illam and A. Rit, Catal. Sci. Technol., 2022, 12, 67–74 RSC; (k) J. M. Zakis, T. Smejkal and J. Wencel-Delord, Chem. Commun., 2022, 58, 483–490 RSC and references cited therein. (l) M. T. Findlay, P. Domingo-Legarda, G. McArthur, A. Yen and I. Larrosa, Chem. Sci., 2022, 13, 3335–3362 RSC and references cited therein. (m) M. T. Findlay, A. S. Hogg, J. J. Douglas and I. Larrosa, Green Chem., 2023, 25, 2394–2400 RSC; (n) C.-Y. Shi, X.-Q. Chen, R. Zeng, H. Lu, Z. Fan and A. Zhang, Org. Lett., 2023, 25, 3870–3875 CrossRef CAS PubMed; (o) D. M. N. Udayanga, N. Le, E. N. Schwirian, B. Donnadieu, K. Nash, W. Collier, C. E. Webster and X. Cui, Org. Lett., 2023, 25, 8745–8750 CrossRef CAS PubMed. For recent articles on the biological activity of chloro(p-cymene)cycloruthenates, see: (p) C. A. Riedl, L. S. Flocke, M. Hejl, A. Roller, M. H. M. Klose, M. A. Jakupec, W. Kandioller and B. K. Keppler, Inorg. Chem., 2017, 56, 528–541 CrossRef CAS PubMed; (q) C. A. Riedl, M. Hejl, M. H. M. Klose, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2018, 47, 4625–4638 RSC; (r) P. Getreuer, L. Marretta, E. Toyoglu, O. Dömötör, M. Hejl, A. Prado-Roller, K. Cseh, A. A. Legin, M. A. Jakupec, G. Barone, A. Terenzi, B. K. Keppler and W. Kandioller, Dalton Trans., 2024, 53, 5567–5579 RSC.
- For recent reviews, see: (a) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456–19464 RSC; (b) J. I. Higham and J. A. Bull, Org. Biomol. Chem., 2020, 18, 7291–7315 RSC, and. (c) C. Jacob, B. U. W. Maes and G. Evano, Chem. – Eur. J., 2021, 27, 13899–13952 CrossRef CAS PubMed. For significative examples of imines as TDGs in C–H bond functionalizations catalyzed by (p-cymene)ruthenium(II) complexes, see: (d) F. Li, Y. Zhou, H. Yang, D. Liu, B. Sun and F.-L. Zhang, Org. Lett., 2018, 20, 146–149 CrossRef CAS PubMed; (e) Z.-Y. Li, H. H. C. Lakmal, X. Qian, Z. Zhu, B. Donnadieu, S. J. McClain, X. Xu and X. Cui, J. Am. Chem. Soc., 2019, 141, 15730–15736 CrossRef CAS PubMed; (f) G. Li, Q. Liu, L. Vasamsetty, W. Guo and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 3475–3479 CrossRef CAS PubMed; (g) Y. Wu, N. Liu, M. Qi, H. Qiao, X. Lu, L. Ma, Y. Zhou and F.-L. Zhang, Org. Lett., 2021, 23, 3923–3927 CrossRef CAS PubMed; (h) S. Kopf, F. Ye, H. Neumann and M. Beller, Chem. – Eur. J., 2021, 27, 9768–9773 CrossRef CAS PubMed ; see also ref. 6o.
- (a) S. Oi, Y. Ogino, S. Fukita and Y. Inoue, Org. Lett., 2002, 4, 1783–1785 CrossRef CAS PubMed; (b) B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, ACS Catal., 2011, 1, 1221–1224 CrossRef CAS; (c) B. Li, K. Devaraj, C. Darcel and P. H. Dixneuf, Tetrahedron, 2012, 68, 5179–5184 CrossRef CAS.
- (a) L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Angew. Chem., Int. Ed., 2009, 48, 6045–6048 CrossRef CAS PubMed; (b) L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875–1877 CrossRef CAS PubMed; (c) J. Li, K. Korvorapun, S. De Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Nat. Commun., 2017, 8, 15430 CrossRef PubMed.
- For representative annulations by alkene hydroarylation of imines see ref. 7e and 7f. For representative annulations by cycloaddition on imines see: (a) M. Tamizmani, B. Ramesh and M. Jeganmohan, J. Org. Chem., 2018, 83, 3746–3755 CrossRef CAS PubMed. For representative annulations by alkyne insertions on arylimines, see: (b) C.-H. Hung, P. Gandeepan and C.-H. Cheng, ChemCatChem, 2014, 6, 2692–2697 CrossRef CAS; (c) J. Li and L. Ackermann, Tetrahedron, 2014, 70, 3342–3348 CrossRef CAS.
- R. Manoharan and M. Jeganmohan, Eur. J. Org. Chem., 2016, 4013–4019 CrossRef CAS.
- For reviews on computational studies on C–H bond activation by metal species, see: (a) Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820–5831 RSC; (b) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed; (c) D. Balcells and O. Eisenstein, Theoretical Studies on the Reaction Mechanism of Metal-Assisted C−H Activation, in Comprehensive Inorganic Chemistry II, ed. J. R. Poeppelmeier, Elsevier, Amsterdam, 2013, vol. 9, pp. 695–726 Search PubMed.
- I. Özdemir, S. Demir, B. Çetinkaya, C. Gourlaouen, F. Maseras, C. Bruneau and P. H. Dixneuf, J. Am. Chem. Soc., 2008, 130, 1156–1157 CrossRef PubMed.
- (a) M. Lafrance, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 14570–14571 CrossRef CAS PubMed; (b) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed.
- For systems in which the more electron-rich substrates show increased reactivity and the dominant process should be of electrophilic character, related terms such as “base-assisted internal electrophilic substitution” (BIES, see ref. 16a and 16d) or “electrophilic-CMD” (eCMD, see ref. 16b and 16c) have been coined (a) W. Ma, R. Mei, G. Tenti and L. Ackermann, Chem. – Eur. J., 2014, 20, 15248–15251 CrossRef CAS PubMed; (b) E. Tan, O. Quinonero, M. Elena de Orbe and A. M. Echavarren, ACS Catal., 2018, 8, 2166–2172 CrossRef CAS PubMed; (c) L. Wang and B. P. Darrow, ACS Catal., 2019, 9, 6821–6836 CrossRef CAS PubMed; (d) T. Rogge, J. C. A. Oliveira, R. Kuniyil, L. Hu and L. Ackermann, ACS Catal., 2020, 10, 10551–10558 CrossRef CAS.
- Computational studies on carboxylate-assisted C–H activation and functionalization by ruthenium(II) complexes have been reviewed, see: (a) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed; (b) C. Shan, L. Zhu, L.-B. Qu, R. Bai and Y. Lan, Chem. Soc. Rev., 2018, 47, 7552–7576 RSC.
- A. G. Algarra, W. B. Cross, D. L. Davies, Q. Khamker, S. A. Macgregor, C. L. McMullin and K. Singh, J. Org. Chem., 2014, 79, 1954–1970 CrossRef CAS PubMed . See also ref. 5g.
- S. Ruiz, P. Villuendas, M. A. Ortuño, A. Lledós and E. P. Urriolabeitia, Chem. – Eur. J., 2015, 21, 8626–8636 CrossRef CAS PubMed.
- (a) A. Gray, A. Tsybizova and J. Roithova, Chem. Sci., 2015, 6, 5544–5553 RSC; (b) L. Zhang, L. Yu, J. Zhou and Y. Chen, Eur. J. Org. Chem., 2018, 5268–5277 CrossRef CAS.
- J. A. Leitch, P. B. Wilson, C. L. McMullin, M. F. Mahon, Y. Bhonoah, I. H. Williams and C. G. Frost, ACS Catal., 2016, 6, 5520–5529 CrossRef CAS.
- M. Zhang and G. Huang, Chem. – Eur. J., 2016, 22, 9356–9365 CrossRef CAS PubMed.
- (a) J.-L. Yu, S.-Q. Zhang and X. Hong, J. Am. Chem. Soc., 2017, 139, 7224–7243 CrossRef CAS PubMed; (b) S. Jambu, M. Tamizmani and M. Jeganmohan, Org. Lett., 2018, 20, 1982–1986 CrossRef CAS PubMed. For benzoate-assisted process, see: (c) N. Y. P. Kumar, T. Rogge, S. R. Yetra, A. Bechtoldt, E. Clot and L. Ackermann, Chem. – Eur. J., 2017, 23, 17449–17453 CrossRef CAS PubMed; (d) M. Simonetti, R. Kuniyil, S. A. Macgregor and I. Larrosa, J. Am. Chem. Soc., 2018, 140, 11836–11847 CrossRef CAS PubMed; (e) For a trifluoroacetate-assisted process, see: L. Zhang, L.-L. Wang and D.-C. Fang, ACS Omega, 2022, 7, 6133–6141 CrossRef CAS PubMed.
- P. Chen, Y. Sun, Y. Wu, L. L. Liu, J. Zhu and Y. Zhao, Org. Chem. Front., 2017, 4, 1482–1492 RSC.
- (a) Q. Bu, T. Rogge, V. Kotek and L. Ackermann, Angew. Chem., 2018, 130, 773–776 CrossRef; (b) R. Sivasakthikumaran, S. Jambu and M. Jeganmohan, J. Org. Chem., 2019, 84, 3977–3989 CrossRef CAS PubMed.
- (a) B. Ling, Y. Liu, Y.-Y. Jiang, P. Liu and S. Bi, Organometallics, 2019, 38, 1877–1886 CrossRef CAS; (b) B. Lian, L. Zhang and D.-C. Fang, Org. Chem. Front., 2019, 6, 2600–2606 RSC; (c) W. Yan, Q. Cheng, Y. Jiao, C.-T. Cao and Z. Tang, J. Org. Chem., 2023, 88, 14945–14952 CrossRef CAS PubMed; (d) S. Sau, K. Mukherjee, K. Kondalarao, V. Gandon and A. K. Sahoo, Org. Lett., 2023, 25, 7667–7672 CrossRef CAS PubMed; (e) See also the ESI for ref. 25a. For related DFT studies on benzimidates and phenylisoindolinone see: ; (f) A. Anukumar, M. Tamizmani and M. Jeganmohan, J. Org. Chem., 2018, 83, 8567–8580 CrossRef CAS PubMed; (g) Y.-C. Yuan, Q.-L. Lu, X.-T. Zhu, S. Posada-Pérez, M. Solà, A. Poater, T. Roisnel and R. Gramage-Doria, Org. Chem. Front., 2023, 10, 42–53 RSC.
- (a) A. Schischko, N. Kaplaneris, T. Rogge, G. Sirvinskaite, J. Son and L. Ackermann, Nat. Commun., 2019, 10, 3553 CrossRef PubMed; (b) C. Deng, P. Hu, Y. Wang, S. Wang and W. Zhang, Dalton Trans., 2019, 48, 9181–9186 RSC.
- M. Zhang, J. Zhang, Z. Teng, J. Chen and Y. Xia, Front. Chem., 2020, 8, 648, DOI:10.3389/fchem.2020.00648.
- (a) B. Ling, J. Wang, Y. Liu, Y.-Y. Jiang, P. Liu, J. Feng and S. Bi, Eur. J. Org. Chem., 2021, 266–273 CrossRef CAS; (b) H.-J. Long, L. Zhang, B. Lian and D.-C. Fang, Org. Chem. Front., 2022, 9, 1056–1064 RSC.
- L. A. Machado, E. R. S. Paz, M. H. Araujo, L. D. Almeida, I. A. O. Bozzi, G. G. Dias, C. L. M. Pereira, L. F. Pedrosa, F. Fantuzzi, F. T. Martins, L. A. Cury and E. N. da Silva, Eur. J. Org. Chem., 2022, e202200590 CrossRef CAS.
- For substrates featuring a protic DG, anion dissociation may be facilitated by proton transfer and HX departure leading to neutral intermediates (contact ion-pairs) with only one acetate ligand, see Scheme S1 at the ESI† and ref. 18, 22, 23a, 24, 26a–c and 29.
- K. R. Bettadapur, V. Lanke and K. R. Prabhu, Org. Lett., 2015, 17, 4658–4661 CrossRef PubMed.
- (a) J. McIntyre, I. Mayoral-Soler, P. Salvador, A. Poater and D. J. Nelson, Catal. Sci. Technol., 2018, 8, 3174–3182 RSC; (b) C. L. McMullin, N. A. Rajabi and J. S. Hammerton, Org. Biomol. Chem., 2019, 17, 6678–6686 RSC; (c) T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 15640–15645 CrossRef CAS PubMed; (d) X. Chen, H. C. Gülen, J. Wu, Z.-J. Zhang, X. Hong and L. Ackermann, Angew. Chem., Int. Ed., 2023, 62, e202302021 CrossRef CAS PubMed.
- DFT studies covering the C–H bond activation/functionalization processes of arenes featuring a protic directing group (derived from pyrazol, naphtol, benzoic acid, hydroxychromone, or benzamide) reported lower barriers for the initial ligand dissociation at ruthenium (termed as O–H or N–H activation processes) than those calculated for the subsequent C–H activation events (see ref 18, 22, 23e, 26b, 26c, 26d, 27b, 29a and 29b). For the ruthenium-catalyzed annulation of benzamide and propargyl alcohol the opposite was true and the initial N–H activation (accompanied with release of HOAc) was calculated higher in energy than the subsequent C–H activation event (see ref. 26a). In addition, concerted ligand dissociation and C–H activation has been reported for phenylpyridines or hydroxychromones (see ref. 20b, 26b, 29a and 29b). Conversely, in previous computational studies on the C–H activation/functionalization processes of arenes equipped with a non-protic DG the kinetic relevance of the dissociation step leading to the key cationic ruthenium intermediates was overlooked (see ref. 5g, 21, 24, and 26d). In reference 5g, the activation energies of the final acetate/chloride ligand exchange processes on ruthenacyclic intermediates were also not evaluated. For other studies in which the C–H activation event was reported as kinetically irrelevant and subsequent functionalization was found as the turnover-limiting step in the catalytic cycle, the computational assessment of the initial ligand exchange processes leading to the key neutral or cationic ruthenium intermediates was also overlooked (see ref. 23a, 23b, 23c, 25a, 25b, 27a and 28).
- During the preparation of this manuscript an experimental and DFT study on ruthenium(II)-catalyzed C–H activation and intramolecular hydroarylation of transient imines of indole derivatives has been published, see ref. 6o. For previous geometry optimizations and thermochemical evaluations on cycloruthenates derived from N-sulfonyl-benzaldimines, see ref. 10a. For previous computational studies of acetate-assisted C–H activations of imines at rhodium, iridium, or palladium, see: (a) D. L. Davies, S. M. A. Donald, O. Al-Duaij, J. Fawcett, C. Little and S. A. Macgregor, Organometallics, 2006, 25, 5976–5978 CrossRef CAS; (b) L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492–3500 CrossRef CAS; (c) K. J. T. Carr, D. L. Davies, S. A. Macgregor, K. Singh and B. Villa-Marcos, Chem. Sci., 2014, 5, 2340–2346 RSC; (d) J. Li, W. Hu, Y. Peng, Y. Zhang, J. Li and W. Zheng, Organometallics, 2014, 33, 2150–2159 CrossRef CAS; (e) L. Zhang and D.-C. Fang, J. Org. Chem., 2016, 81, 7400–7410 CrossRef CAS PubMed.
- For detailed kinetic studies on acetate-assisted C–H activations of arenes equipped with N-containing DGs, see: (a) E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161–10170 CrossRef PubMed; (b) I. Fabre, N. von Wolff, G. Le Duc, E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, Chem. – Eur. J., 2013, 19, 7595–7604 CrossRef CAS PubMed . See also ref. 5e, 6i, 10b, 18.
- (a) S. Dohm, A. Hansen, M. Steinmetz, S. Grimme and M. P. Checinski, J. Chem. Theory Comput., 2018, 14, 2596–2608 CrossRef CAS PubMed; (b) J. M. A. Iron and T. Janes, J. Phys. Chem. A, 2019, 123, 3761–3781 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- (a) C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed; (b) C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef PubMed . The choice of an inexpensive double-ζ basis set for DFT geometry optimizations has a relatively small impact on the DLPNO-CCSD(T) reaction energies and barriers, with differences that are generally much smaller than 1 kcal mol−1, see: M. Wang, X. He, M. Taylor, W. Lorpaiboon, H. Mun and J. Ho, J. Chem. Theory Comput., 2023, 19, 5036–5046 Search PubMed.
- Calculations with the M06 functional were performed with Gaussian 16 Rev C.01 program (see ESI† for a full citation) while those made with the ωB97X-D3 functional and all the refined single-point energy calculations were performed by using ORCA (version 5.0.3), see. F. Neese, WIREs Comput. Mol. Sci., 2022, 12, e1606, DOI:10.1002/wcms.1606.
- For recent reviews on hybrid explicit–implicit solvation models, see: (a) J. R. Pliego Jr. and J. M. Riveros, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, 10 Search PubMed; (b) M. Das, A. R. Gogoi and R. B. Sunoj, J. Org. Chem., 2022, 87, 1630–1640 CrossRef CAS PubMed.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS; (b) For a review, see: J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed; (c) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- R. Salvio, F. Juliá-Hernández, L. Pisciottani, R. Mendoza-Meroño, S. García-Granda and M. Bassetti, Organometallics, 2017, 36, 3830–3840 CrossRef CAS.
- TS(Fc-F) could not be located at the (B97X-D3/cc-pVDZ-PP[Ru](CPCM) hypersurface. Chart S1 in the ESI† includes a constrained energy profile for the dissociation of the p-cymene moiety from Fc leading to F.
- Carbonate has been previously located trans to the agostic interaction for the C(sp3)–H activation of 2-bromoalkylbenzenes promoted by Pd(OAc)2 and PtBu3 as catalysts, see: M. Chaumontet, R. Piccardi, N. Audic, J. Hitce, J.-L. Peglion, E. Clot and O. Baudoin, J. Am. Chem. Soc., 2008, 130, 15157–15166 CrossRef CAS PubMed.
- Intermolecular deprotonation processes have been found to be favoured over the intramolecular ones in previous computational studies on cycloruthenation of benzylamines in MeOH (see ref. 19) and of 2-phenylpyridines in NMP or dioxane (see ref. 33a and 20b).
- Alternative pathway involving neutral bis-imine intermediates and transition structures characterized by a cis disposition between the two imine moieties at the ruthenium resulted higher in energy, see ESI,† Scheme S6.† Analogous structures with either a cis or a trans disposition between the two molecules of the substrate ligated to the ruthenium have been reported for the cyclometallation of 2-phenylpyridines, see ref. 33b and 33c.
- CYLview, 1.0b; C. Y. Legault, Université de Sherbrooke, 2009. https://www.cylview.org (accessed January 2024).
- (a) Phenylpyridines in D2O: M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977–3980 CrossRef CAS PubMed; (b) Phenylbenzimidazoles in TFE (CD3OD): S. Nunewar, S. Kumar, A. W. Meshram and V. Kanchupalli, J. Org. Chem., 2022, 87, 13757–13762 CrossRef CAS PubMed; (c) Phenylisoxazoles in HFIP(D2O): P. Kumar and M. Kapur, Chem. Commun., 2022, 58, 4476–4479 RSC; (d) Indole-carboxamides in TFE: G. K. Das Adhikari, S. R. Mohanty, S. K. Banjare, N. Prusty, G. Murmu and P. C. Ravikumar, J. Org. Chem., 2023, 88, 952–959 CrossRef CAS PubMed; (e) Phenyl-imidazo[1,2-a]pyridines in AcOH(D2O): J. A. Tali and R. Shankar, Org. Lett., 2023, 25, 3200–3205 CrossRef CAS PubMed; (f) Phenylpyrazolidinones in CD3OD: see ref. 4f.
- (a) A. Sagadevan, A. Charitou, F. Wang, M. Ivanova, M. Vuagnat and M. F. Greaney, Chem. Sci., 2020, 11, 4439–4443 RSC; (b) K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 18103–18109 CrossRef CAS PubMed.
- R. L. Martin, P. J. Hay and L. R. Pratt, J. Phys. Chem. A, 1998, 102, 3565–3573 CrossRef CAS.
- A. Karton, A. Tarnopolsky, J.-F. Lamére, G. C. Schatz and J. M. L. Martin, J. Phys. Chem. A, 2008, 112, 12868–12886 CrossRef CAS PubMed.
- A. Tarnopolsky, A. Karton, R. Sertchook, D. Vuzman and J. M. L. Martin, J. Phys. Chem. A, 2008, 112, 3–8 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122, DOI:10.26434/chemrxiv.7430216.v2 (and ChemRxiv 2019).
- (a) H. L. Robinson and M. C. R. Symon, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2131–2144 RSC; (b) O. M. Cabarcos, C. J. Weinheimer, T. J. Martínez and J. M. Lisy, J. Chem. Phys., 1999, 110, 9516–9526 CrossRef CAS; (c) T. Megyes, T. Radnai and A. Wakisaka, J. Phys. Chem. A, 2002, 106, 8059–8065 CrossRef CAS; (d) T. Megyes, T. Radnay, T. Grózsc and G. Pálinkás, J. Mol. Liq., 2002, 101, 3–18 CrossRef CAS; (e) K. Ozutsumi and H. Ohtaki, Pure Appl. Chem., 2004, 76, 91–96 CrossRef CAS; (f) M. Pagliai, G. Cardini and V. Schettino, J. Phys. Chem. B, 2005, 109, 7475–7481 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional Chart, Figures, and Tables; Rationale for the selection of methods; Computational methods; Computed cartesian coordinates, lowest frequencies, and energies. See DOI: https://doi.org/10.1039/d4dt00380b |
| This journal is © The Royal Society of Chemistry 2024 |