
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrate and nitroarene hydrogenations catalyzed by alkaline-earth nickel phosphide clathrates†
Marquix A. S.
Adamson
a,
Lin
Wei
a,
Philip
Yox
ab,
Fatema H. B.
Hafiz
a and
Javier
Vela
 *ab
*ab
aDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: vela@iastate.edu
bAmes National Laboratory, U.S. Department of Energy, Ames, Iowa 50011, USA
First published on 27th February 2024
Abstract
The alkaline-earth-containing nickel phosphide clathrates AeNi2P4 (Ae = Ba, Sr) are investigated as catalysts for the reduction of nitrate or nitroarenes in aqueous or ethanolic solution, respectively. While AeNi2P4 clathrates are inactive in their bulk polycrystalline form, they become active in nitrate hydrogenation after size reduction by either grinding or ball milling. However, while the clathrate structure remains intact after manual grinding, ball milling is of limited use as it results in significant clathrate degradation. Ground AeNi2P4 catalysts are also active in nitroarene hydrogenation. Condensation products such as azoxy- and azo-benzenes form early (4 h) but anilines accumulate after long reaction times (24 h). Unexpectedly, BaNi2P4 partially devinylates nitrostyrene to nitrobenzene. Overall, BaNi2P4 is more active than SrNi2P4 in both nitrate and nitroarene hydrogenation. These results showcase the potential utility of clathrates in a growing number of catalytic transformations.
Introduction
Green and sustainable chemistries benefit from catalysts that can efficiently remove pollutants or selectively generate value-added compounds.1,2 For example, the reduction of nitrogen oxides and nitroarenes is key to addressing outstanding challenges posed by some of the multiple anthropogenic effects on the global nitrogen cycle as well as in the production of organic dyes for the fine chemicals and electronics industries. Specifically, high concentrations of nitrate (NO3−) in groundwater produced by the degradation of excess fertilizer has been linked to a higher incidence of cases of blue baby syndrome and certain forms of cancer.3,4 Similarly, because of their mutagenicity or bioaccumulation, multiple nitroarenes including nitrobenzene are considered priority pollutants by the U.S. Environmental Protection Agency (EPA).5–7 Therefore, new catalysts that can facilitate the removal of these contaminants by transforming them into less toxic and—ideally—more valuable products are highly desirable.A potent method for reducing nitrate and nitroarenes to useful or relatively benign compounds is catalytic hydrogenation with H2 or NaBH4 (Scheme 1). Recent developments in this area include versatile bimetallic and intermetallic catalysts which,8,9 in addition to reducing nitroarenes,10–13 can in some cases tolerate a variety of functional groups.14,15 Intermetallic nanocatalysts prepared from single-source heterobimetallic precursors have also exhibited a strong correlation between catalyst structure and product selectivity.10,16,17 For example, depending on the crystalline phase of the nanocatalyst used, nitrobenzene is selectively reduced either directly to aniline or to azo(xy) condensation products formed by condensation of nitroso and hydroxylamine intermediates (Scheme 1b).10,18
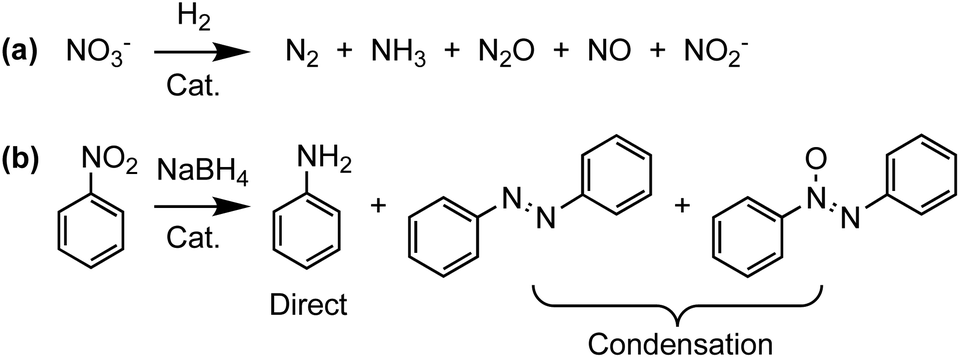 | ||
| Scheme 1 Catalytic hydrogenations of nitrate and nitrobenzene showing some of their possible products. | ||
In comparison to noble metal-containing catalysts, there are fewer base metal catalysts that permit the reduction of NO3− or nitroarenes at or near ambient conditions.18–21 Among selected examples, we previously demonstrated that nanoscale Ni2P catalyzes the full hydrogenation of NO3− with very high selectivity toward ammonia (NH3).22 Computations showed that the Ni2P(001) surface exhibits partial H• saturation coverage, which enables coadsorption and thus activation and reduction of NO3−. Interestingly, this led to the development of a self-sustainable photocatalytic NO3− reduction system using Ni2P-modified, nanostructured semiconductors. Specifically, starting with 2 mM aqueous NO3−, heterostructured Ni2P/Ta3N5 and Ni2P/TaON displayed NO3− conversions as high as 79% and 61%, respectively, within 12 h under 419 nm light, which corresponded to apparent quantum yields of 3–4%.23 Similar photocatalytic schemes have been reported for nitrobenzene.24,25
Because multiple first row, transition metal phosphides and related materials in addition to Ni2P are also capable of catalyzing the hydrogen evolution reaction (HER)26–28 as well as—its reverse—hydrogen activation, we wondered whether a more complex framework based on nickel phosphide could serve as a noble metal-free catalyst for nitrogen-centered hydrogenation. For example, transition metal-based clathrates exhibit high flexibility in their 3D frameworks. This prompted our interest in nickel phosphide clathrates such as BaNi2P4 and SrNi2P4, which are comprised of Ni8P16 cages encapsulating alkaline-earth (Ae) guest atoms (Fig. 1).29 Like Ni2P and other transition metal phosphides, AM2P4 clathrates display metallic character, as determined through transport measurements and computations.30 Moreover, the guest atom (Ba or Sr) affects the electron density and overall reactivity of the clathrate, which could open a new avenue for fine tuning the activity or selectivity of new families of catalysts.
 | ||
| Fig. 1 Unit cells of the crystalline nickel phosphide clathrates (a) BaNi2P4 and (b) SrNi2P4 (green: Ba or Sr, grey: Ni, orange: P).29,30 | ||
In this work, we demonstrate that the ternary nickel phosphide clathrates BaNi2P4 and SrNi2P4 are indeed capable of catalyzing the hydrogenation of NO3− and nitroarenes at or near ambient temperature and atmospheric pressure. In the case of NO3− hydrogenation, AeNi2P4 clathrates are selective toward NH3. Because this mimics the behavior of binary Ni2P, we infer there may be mechanistic similarities between the two systems. We also find that, under the same conditions, BaNi2P4 achieves up to three times higher activity than SrNi2P4 in NO3− hydrogenation, which may be partly attributed to the exact nature of the guest atom. In the case of nitroarenes, these clathrates enable the formation of condensation products like azobenzene and azoxybenzene at early reaction times, although direct aniline products accumulate at longer times. From our previous work on nanoscale binary nickel phosphides, we believe that dimensional and electronic fine-tuning within the AeNi2P4 clathrate family and other similar frameworks may be used to further enhance their catalytic activity and selectivity.
Results and discussion
Catalyst dimensionality and activation
AeNi2P4 clathrates are typically made by a traditional, high-temperature, solid-state reaction where the elements are mixed, sealed in a silica ampule, and heated to 850 °C.29 The resultant mixture is ground, resealed, and further annealed until phase-pure clathrates form, as determined by powder XRD (Fig. 2). As-prepared, polycrystalline or “bulk” BaNi2P4 and SrNi2P4—with approximate particle sizes of 0.20–0.37 mm—are inactive toward NO3− hydrogenation (Table 1, entries 4 and 12). Previously, annealing under 1 atm of H2 at 400 °C for 1 h was used to successfully activate nanocrystalline metal phosphides by removing capping ligands.31 In addition, surface reconstruction induced by H• adsorption is known to enhance the activity of Ni2P nanocatalysts.22 Thus, even though the clathrates in this work lack surface ligands, we decided to follow a similar H2 annealing and activation protocol across all of the different catalysts studied. Indeed, H2 annealing partially activates bulk BaNi2P4 toward NO3− hydrogenation, achieving 10% reduction when starting from 2.0 mM aqueous NO3− at 60 °C and pH = 2 (using 10 mg catalyst, see Table 1 entry 5).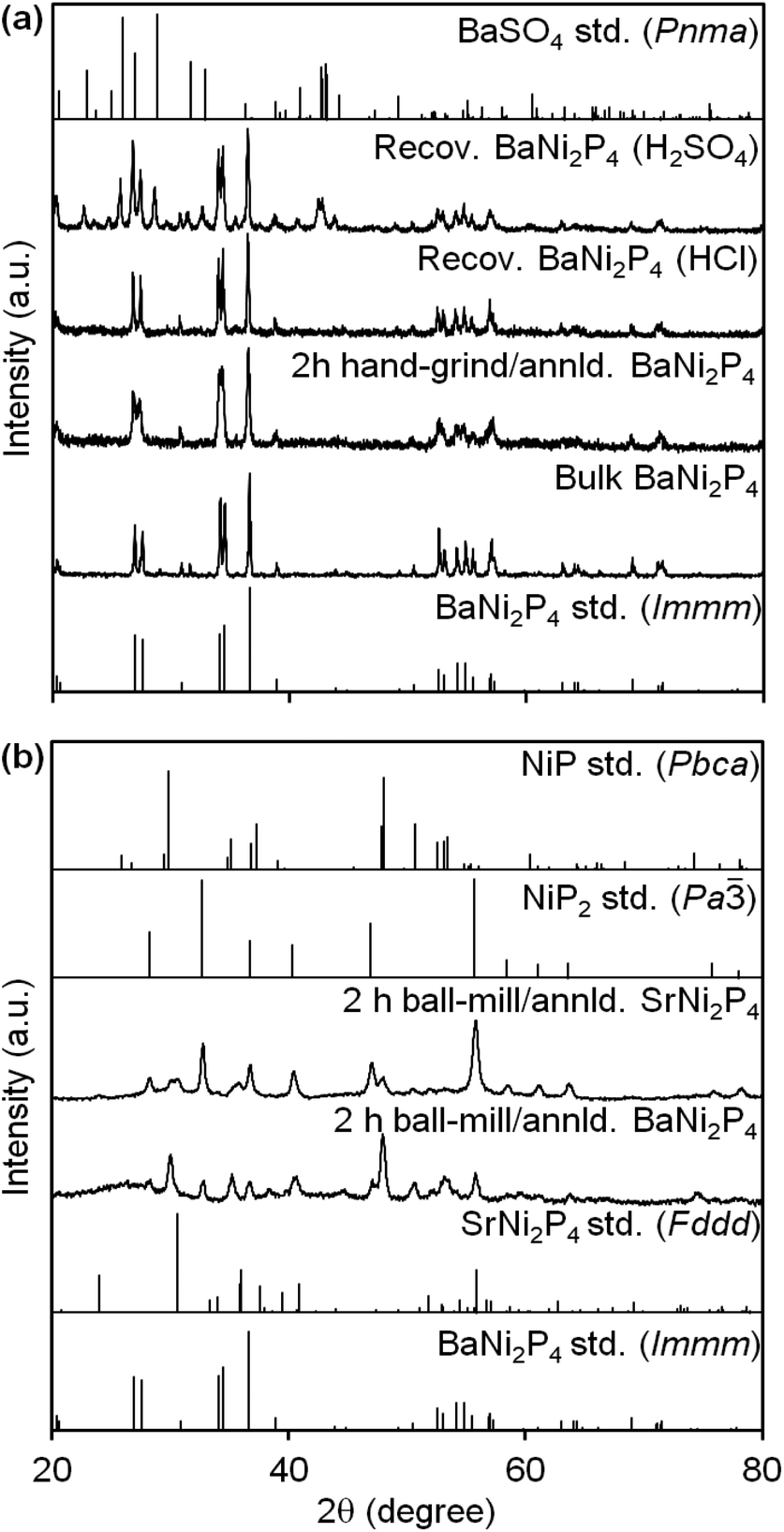 | ||
| Fig. 2 Powder XRD of AeNi2P4 (Ae = Ba or Sr) clathrates before and after (a) hand-grinding or (b) ball-milling. XRD of two BaNi2P4 samples recovered after catalysis with different acids are included in (a). Reference XRD patterns are shown for comparison (BaNi2P4, ICSD79104; SrNi2P4, ICSD429359; NiP, ICSD27159; NiP2, ICSD22221; BaSO4, ICSD33730). | ||
| Catalyst | Pretreatmenta | Pretreated XRD (nm)b | Reactantc | t (h) | Conversion (%) | Product(s) (selec./%) | Recovered XRD (nm)b | TONd (TOFe h−1) | |
|---|---|---|---|---|---|---|---|---|---|
| a 10 mg cat. b Estimated from XRD peak widths using the Scherrer equation when <100 nm; reported in nm unless specified otherwise. c 2.0 mM NO3− or NO2−, 60 °C, pH = 2/H2O; or 50 mM nitroarene, RT/EtOH. d TON = moles of converted reactant/moles of surface-active catalyst sites (calculated for select cases, only). e TOF = TON/catalysis time. f 1 atm H2, 1 h, 400 °C. Adjusted pH with: g H2SO4. h HNO3, or i HCl. j Products with functional group (see ESI†). k Multiple Ph–vinyl (C–C) breaking products. | |||||||||
| 1 | None | — | — | NO3− | 12 | 0 | — | — | — |
| 2 | Ni2Pg | Annld.f | NO3− | 4 | 98 | NH4+ (100) | — | 31 (8) | |
| 3 | Ni5P4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g g |
Annld.f | NO3− | 4 | 81 | NH4+ (100) | — | 1149 (287) | |
| 4 | BaNi2P4g |
— | BaNi2P4 (0.2 mm) | NO3− | 12 | 0 | — | — | — |
| 5 | BaNi2P4g |
Annld.f | BaNi2P4 (0.2 mm) | NO3− | 12 | 10 | NH4+ (100) | — | — |
| 6 | BaNi2P4g |
h/2 Hand-grind, annld.f | BaNi2P4 (0.35 μm) | NO3− | 12 | 25 | NH4+ (100) | — | — |
| 7 | BaNi2P4g |
2 h Hand-grind, annld.f | BaNi2P4 (44 ± 5) | NO3− | 12 | 79 | NH4+ (94) | 65% BaNi2P4 (59 ± 15), 35% BaSO4 (40 ± 3) | 1188 (99) |
| 8 | BaNi2P4h |
2 h Hand-grind, annld.f | BaNi2P4 (44 ± 5) | NO3− | 12 | 15 | NH4+ (100) | 69% BaNi2P4 (44 ± 13), 31% Ni2P (32 ± 9) | — |
| 9 | BaNi2P4i |
2 h Hand-grind, annld.f | BaNi2P4 (44 ± 5) | NO3− | 12 | 6.5 | NH4+ (100) | BaNi2P4 (82 ± 10) | — |
| 10 | BaNi2P4g |
h/2 Ball-mill, annld.f | 73% NiP2 (52 ± 39), 27% ZrO2? (>100). | NO3− | 12 | 30 | NH4+ (100) | — | |
| 11 | BaNi2P4g |
2 h Ball-mill, annld.f | 73% NiP (20 ± 6), 27% NiP2 (27 ± 1) | NO3− | 12 | 43 | NH4+ (100) | 50% BaSO4 (55 ± 33), 37% NiP2 (38 ± 27), 13% ZrO2? (>100) | — |
| 12 | SrNi2P4g |
— | SrNi2P4 (0.37 mm) | NO3− | 6 | 0 | — | — | — |
| 13 | SrNi2P4g |
2 h Hand-grind, annld.f | SrNi2P4 (40 ± 15) | NO3− | 12 | 28 | NH4+ (91) | SrNi2P4 (51 ± 1) | 324 (27) |
| 14 | SrNi2P4h |
2 h Hand-grind, annld.f | SrNi2P4 (40 ± 15) | NO3− | 12 | 50 | NH4+ (100) | 62% SrNi2P4 (63 ± 3), 38% Ni2P (32 ± 6) | — |
| 15 | SrNi2P4 | 2 h Ball-mill, annld.f | 29% SrNi2P4 (9.6 ± 2.1), 51% NiP2 (16 ± 6), 20% NiP (14 ± 4) | — | — | — | — | — | |
| 16 | None | — | — | PhNO2 | 4 | 9 | — | — | — |
| 17 | BaNi2P4 | 2 h Hand-grind, annld.f | PhNO2 | 4 | 47 | Aniline (4), azo (7), azoxy (89) | — | — | |
| 18 | BaNi2P4 | 2 h Hand-grind, annld.f | PhNO2 | 24 | 98 | Aniline (48), azo (49), azoxy (3) | — | 2456 (102) | |
| 19 | SrNi2P4 | 2 h Hand-grind, annld.f | PhNO2 | 4 | 45 | Aniline (4), azo (2), azoxy (94) | — | — | |
| 20 | SrNi2P4 | 2 h Hand-grind, annld.f | PhNO2 | 24 | 58 | Aniline (16), azo (19), azoxy (65) | — | 1114 (46) | |
| 21 | BaNi2P4 | 2 h Hand-grind, annld.f | 3-Nitrostyrene | 4 | 95 | Anilinej (16), azoj (8), azoxyj (69), devinyl.k (1), ethylj (26) | — | — | |
| 22 | BaNi2P4 | 2 h Hand-grind, annld.f | 3-Nitrostyrene | 24 | 99 | Anilinej (20), azoj (0), azoxyj (54), devinyl. (39)k, ethylj (38) | — | 2481 (103) | |
Smaller, nanoscale catalysts are known to have a much larger surface area and contain a higher concentration of active sites and defects that enhance catalytic performance.32 In fact, nanocrystalline clathrates were recently prepared by ball milling or through ionic liquid-assisted approaches.33,34 Therefore, we decided to explore whether a similar, top-down approach based on hand-grinding or ball-milling could be used to increase the surface area and, with it, enhance the catalytic activity of AeNi2P4 clathrates (Fig. 3).
 | ||
Fig. 3 Catalytic activity (●) and selectivity ( ) of BaNi2P4 against NO3− hydrogenation as a function of (a) hand-grinding time (prior to catalysis) and (b) particle size (12 h reaction data shown). (c) Catalytic activity (black) and selectivity (red) of nanocrystalline binary phosphides vs. 2 h hand-ground clathrates (Table 1, entries 1–3, 7 and 13). (General conditions: 2 mM aqueous NO3−, 60 °C, pH = 2—adjusted with H2SO4—see Experimental). ) of BaNi2P4 against NO3− hydrogenation as a function of (a) hand-grinding time (prior to catalysis) and (b) particle size (12 h reaction data shown). (c) Catalytic activity (black) and selectivity (red) of nanocrystalline binary phosphides vs. 2 h hand-ground clathrates (Table 1, entries 1–3, 7 and 13). (General conditions: 2 mM aqueous NO3−, 60 °C, pH = 2—adjusted with H2SO4—see Experimental). | ||
Hand-grinding results in noticeable broadening of the reflections in the powder XRD of these materials, indicating a decrease in apparent particle size (Fig. 2a). Using the Scherrer equation, the average single crystalline domain size after 2 h of hand-grinding is as small as 44 ± 5 nm for BaNi2P4 and 40 ± 15 nm for SrNi2P4 (Table 1, entries 7 and 13). Scanning electron microscopy (SEM) images of the hand-ground clathrate powders confirm a substantial decrease in particle size—see ESI.†
In contrast to hand-grinding, ball-milling the clathrates for 2 h results in pronounced decay (Fig. 2b). After 2 h of ball-milling, BaNi2P4 completely decomposes into a mixture of nanocrystalline (<30 nm) NiP2 (73%) and NiP (27%) (Table 1, entry 11). The strontium clathrate is only marginally more stable under these conditions: after 2 h of ball-milling, 29% of nanocrystalline SrNi2P4 remains (9.6 ± 2.1 nm; Table 1, entry 15). Thus, while both hand-grinding and ball-milling are effective at decreasing particle size, only the former can keep the clathrate phase intact.
Particle size, binary comparison, and effect of guest atom
Because the localized high pressure and excessive shear produced by ball-milling results in significant clathrate decomposition, we focused on hand-grinding for a majority of the catalysis experiments in this study. Fig. 3a shows that there is a fairly linear relationship between NO3− conversion with BaNi2P4 and the length of time the material is hand-ground prior to catalysis. With the aforementioned standard conditions—2.0 mM aq. NO3− at 60 °C, pH = 2, 10 mg catalyst—conversion reaches 25% and 79% after 0.5 h and 2 h of hand-grinding, respectively (Table 1, entries 5–7). Fig. 3b further demonstrates the effect that grinding has in decreasing average particle size and increasing NO3− conversion. Interestingly, while ball-milling for 0.5 h also results in a modest 30% conversion, this effect tapers off after ball-milling for 2 h, which results in only 43% conversion (about half of the maximum overall activity observed after hand-grinding the catalyst; entries 7–11). In addition, the activity of the latter likely stems from the presence of nickel phosphide binaries produced by the decomposition of the clathrate under ball milling conditions.Based on their turnover numbers (TONs) and turnover frequencies (TOFs), the clathrate catalysts used here have comparable or superior activity relative to the better-known nanoscale binary nickel phosphides Ni5P4 and Ni2P (Table 1, entries 2–3, 7).22 The clathrates are also highly selective to ammonium (NH4+; produced by protonation of NH3 under the acidic conditions used). For example, under the same conditions above, nanocrystalline Ni2P and Ni5P4 reduce 98% and 81% of NO3− within 4 h (Fig. 3c). In comparison, hand-ground BaNi2P4 only reaches similar NO3− conversion and NH4+ selectivity after 12 h (entry 7). As in the case of the bulk clathrates above, bulk Ni2P and Ni5P4 are also inactive. Therefore, clear opportunities exist to further control the dimensionality and, with it, the activity of AeNi2P4 clathrate nanocatalysts.
Multiple factors inherent to the catalyst may contribute to differences in catalytic performance, such as the electronegativities and ionic sizes of the guest atoms, and the size and shape of the clathrate cages. The electronegativity of the guest atom—0.89 for Ba and 0.95 for Sr—may affect the electron density on the catalyst surface,35 consequently influencing the ability to bind and activate NO3− and H2. Similarly, the different nickel phosphide frameworks may show different nitrate and hydrogen adsorption energetics. The larger, truncated octahedral cages encapsulate Ba2+ (156 pm ionic radius) cations, whereas the smaller, twisted Kelvin cells encapsulate Sr2+ (140 pm) cations.36 As measured by the TON and TOF values achieved by each catalyst, hand-ground BaNi2P4 (1188, 99 h−1) outperforms hand-ground SrNi2P4 (324, 27 h−1) in our NO3− hydrogenation experiments (Table 1). This suggests that more electropositive metals and larger cages could enhance the clathrate's nitrate reduction ability.
Acid effect and identity of recovered catalysts
Similar to the hydrogen evolution reaction (HER),22,23,37 NO3− hydrogenation with metal phosphides is pH dependent, becoming faster with the increased acidity of the medium. In the case of the clathrates here, initial acidification with H2SO4, HNO3, or HCl has some impact in catalytic behavior. More specifically, NO3− conversions range depending on the acid and catalyst combination used (see below). For example, there is a drastic rise in conversion over hand-ground BaNi2P4 from 6.5% to 79% in going from HCl to HNO3 to H2SO4 (Table 1, entries 7–9). In most cases, at least 25% of the initial NO3− is reduced within 12 h at 60 °C and pH = 2, and always with a high selectivity for NH4+ (2.0 mM initial aq. NO3−, 10 mg of catalyst; see ESI†).To further probe the reasons behind these differences, we recovered the solids left after select catalytic experiments and re-measured their powder XRD. As shown in Fig. 2, BaNi2P4 is unaffected when HCl is used, and the catalyst retains the same crystalline phase without any byproducts or impurities (Table 1, entry 9, see ESI†). In contrast, changing the proton source to H2SO4 leads to the formation of a small amount of insoluble BaSO4; however, a majority of the recovered solids are still made of crystalline BaNi2P4 (Table 1, entries 7). Across all experiments, there is a slight decrease in the Scherrer size of the clathrates after catalysis, suggesting that some catalyst etching and dissolution occur.
Nitroarene reduction and chemoselectivity
We next turned our attention to the ability of the clathrates to mediate the reduction of nitroarenes in ethanol (Scheme 1b). Previous reports showed that nickel phosphide nanoparticles—like Ni2P and Ni12P5—are highly active catalysts for reducing nitrobenzene,38 benzonitrile,39 and other nitroarenes.40,41 The reduction of nitroarenes normally occurs with high selectivity for the direct aniline products over a period of a few hours.Benchtop hydrogenation of nitrobenzene is catalyzed by the hand-ground clathrates BaNi2P4 and SrNi2P4 (Fig. 4). BaNi2P4 fully reduces nitrobenzene within 24 h under ambient conditions—when starting with 50 mM nitroarene and using 10 mg of catalyst (see Experimental). However, unlike regular nitroarene reduction, quantification by GCMS shows clathrate catalysis follows an indirect pathway to aniline by means of forming condensation products like azobenzene and azoxybenzene at first (Table 1, entry 18). The condensation pathway is similarly followed when using SrNi2P4, yet less than 60% of the starting material is converted within the same timeframe under the same conditions (Table 1, entry 20). As a direct comparison between catalysts, BaNi2P4 reaches TON and TOF values of 2456 and 102 h−1, respectively, while SrNi2P4 is ca. half as active, with TON and TOF values of 1114 and 46 h−1, respectively. These results are consistent with the NO3− hydrogenation experiments discussed above, where the barium clathrate is also the more active catalyst. In other words, the sterically bulkier nitrobenzene molecules are more rapidly reduced over catalysts containing the more electropositive metal (Ba) and the larger cage clathrate (BaNi2P4).
 | ||
| Fig. 4 Catalytic activity (black) and selectivity (red) of (a) BaNi2P4 and (b) SrNi2P4 against nitrobenzene reduction. (Conditions: 50 mM PhNO2, R.T.; catalysts hand-ground for 2 h and annealed under H2—see Experimental). | ||
We further probed the activity and chemoselectivity of the activated AeNi2P4 clathrates against 3-nitrostyrene, containing both nitro and vinyl groups, both of which are susceptible to chemical reduction (Scheme 2). Nitrostyrene is a reactant often employed in catalytic reductions to study the chemoselectivity against different functional groups.42–44 Under ambient conditions and starting with 50 mM of 3-nitrostyrene and using 10 mg catalyst, both BaNi2P4 and SrNi2P4 show 95% conversion—TONs and TOFs of 2481–1882 and 103–78 h−1—within 4 h (Fig. 5; Table 1, entry 21, see Experimental). Interestingly, the clathrate catalysts consistently show a strong preference for the activation of nitro over vinyl groups, with marked selectivity for azo(xy) condensation products over anilines, ethyl benzenes, or other reduction products.
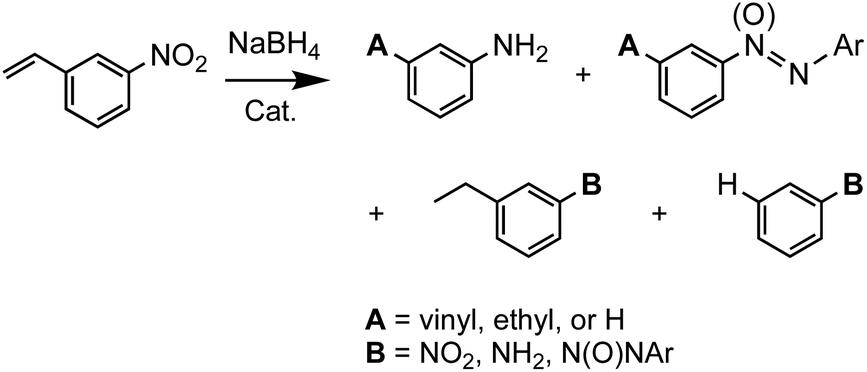 | ||
| Scheme 2 Catalytic hydrogenation of 3-nitrostyrene showing some of its possible products. | ||
 | ||
| Fig. 5 Catalytic activity (black) and selectivity (red) of BaNi2P4 against 3-nitrobenzene hydrogenation. (Conditions: 50 mM 3-NO2-styrene, R.T.; catalyst hand-ground for 2 h and annealed under H2, see Experimental and ESI†). | ||
Unexpected devinylation and surface analysis
Interestingly, catalytic runs over 24 h using hand-ground BaNi2P4 result in a substantial amount of a different and unexpected product, nitrobenzene (Table 1, entry 22). Unlike ethyl reduction products formed by the selective reduction of the styrene double bond, devinylated products like nitrobenzene are formed by the activation and reduction of the aromatic (Ar)C–aliphatic(C) bond in 3-nitrostyrene, leading to loss of the vinyl group. Notably, devinylation is specific to the more active BaNi2P4 clathrate after hand grinding and H2-annealing and is absent with the other catalysts tested. After 24 h with this catalyst, devinylated aromatics are the second-most prominent class of reduction products behind azo(xy) compounds.To our knowledge, this represents one of the first reports of barium- or clathrate-based devinylation of a styrene. Previous examples of styrene devinylation and related carbon–carbon bond activation require similarly long reaction times (12–24 h) but significantly higher temperatures (80–180 °C)—whereas this work takes place at ambient temperature.45,46 Similar to our results, reported styrene devinylation yields remain moderate (<50%).47 Additional investigations are ongoing to probe the nature of the active species, the reaction mechanism, and possible optimization of this unique, clathrate-mediated transformation.
Finally, because surface chemistry plays a key role in determining catalytic activity, we studied one of the clathrates at different stages of catalysis by X-ray photoelectron spectroscopy (XPS) (Fig. 6). Specifically, we find that the spectra of BaNi2P4 before and after the hydrogenation of 3-nitrostyrene show XPS peaks corresponding to Ba, Ni, and P species (in addition to C and O, see ESI†). In all samples, the Ba 3d5/2 region shows two main peaks at 780.3 eV and 795.6 eV, which can be attributed to Ba2+.48 Also, the Ni 2p3/2 and Ni 2p1/2 regions exhibit peaks at 852.6 eV and 870.3 eV, which are reminiscent of the low valent ‘Niδ+’ sites present in Ni2P;22 however, unlike the XPS of Ni2P, the Ni region in BaNi2P4 lacks a Ni2+ impurity.22 In all cases, the P 2p region contains two peaks: a more prominent peak at a binding energy of 134.3 eV, corresponding to the oxidized form P5+—surface PO43− impurity—and a smaller peak at 129.1 eV corresponding to P3−—phosphide. Because the XPS spectra lack any evidence of other species that could be catalytically active, such as Ni2+ or Ni0, and because the XPS spectra are equivalent before and after catalysis, we conclude that the active species under an atmosphere of H2 exists on the surface of the clathrate crystals.
 | ||
| Fig. 6 Ba 3d, Ni 2p, and P 2p XPS regions of BaNi2P4 before (black) and after (red) catalytic hydrogenation of 3-nitrostyrene. (Conditions: 50 mM 3-NO2-styrene, R.T.; catalyst hand-ground for 2 h and annealed under H2, see Experimental and ESI†). | ||
Conclusions
In summary, top-down approaches to reducing the particle size of bulk nickel phosphide clathrates AeNi2P4 (A = Ba, Sr) such as hand-grinding or ball-milling, followed by thermal annealing under H2 results in their activation toward the hydrogenation of aqueous NO3− or the reduction of nitroarenes. While ball milling is a well-known technique for conveniently reducing particle size, our studies show that ball milling leads to significant degradation of the clathrate phases into binary phosphides. This underscores how careful characterization of the catalyst after activation as well as before and after catalysis is key to identifying the actual active species. Fortunately, hand grinding is a milder way to decrease particle size and increase the surface area and catalytic activity, while at the same time preserving the integrity of the clathrate phase.Without size reduction, the bulk clathrates lack catalytic activity. After hand-grinding and H2 annealing, BaNi2P4 converts 79% NO3− under near-ambient conditions (10 mg catalyst, 1 atm H2, 60 °C, pH = 2, 12 h) starting from 2.0 mM NO3− and is highly selective toward NH4+. Longer hand-grinding times result in higher NO3− conversions in a nearly linear fashion. We also observe that lowering the pH of the initial solution with different acids can have a direct impact on the overall efficiency of the catalysis. In all cases, turnover number and frequency calculations show that the BaNi2P4 clathrate is the more active catalyst against both NO3− and nitrobenzene reduction. BaNi2P4 fully reduces nitrobenzene and nitrostyrene within 24 h under ambient conditions using 10 mg catalyst and 50 mM nitroarene. Within the same timeframe and under the same conditions, SrNi2P4 partially reduces nitrobenzene and fully reduces nitrostyrene. Interestingly, a previously unreported devinylation of nitrostyrene to nitrobenzene occurs when using the more active BaNi2P4 clathrate catalyst. Grinding appears to have an immense effect on the ability of the catalyst to devinylate, as this reaction is absent when bulk BaNi2P4 is used.
Given that a small particle size and high surface area are key to successful catalytic reduction with AeNi2P4 clathrates, additional synthesis of nanosized AeNi2P4 and similar phases may enable additional catalytic studies and applications. Furthermore, other nickel phosphide clathrates containing one or more guest atoms (e.g., EuNi2P4) may be used to further enhance or fine tune the catalytic activity and selectivity of nitrogen-based and similar reductions.
Experimental
Materials
Metallic barium (Ba, 99.9%), strontium (Sr, 99.9%), copper powder (Cu, 99.9%), and 3-nitrostyrene (96%) were purchased from Sigma-Aldrich; nitrobenzene (99%) from Oakwood Chemical; sodium borohydride (NaBH4, 99%) from Acros Organics; ethanol (EtOH, 200 proof) from Decon Labs; nickel powder (Ni, 99.996%) and red phosphorus (P4, 99%) from Alfa Aesar; sulfuric acid (H2SO4, certified ACS plus), hydrochloric acid (HCl, certified ACS plus), nitric acid (HNO3, certified ACS plus), and sodium nitrate (NaNO3, certified ACS) from Fisher; hydrogen gas (H2) and argon gas (Ar) from Airgas. Colorimetric kits (0.10–25.0 mg L−1 NO3–N, 0.002–1.00 mg L−1 NO2–N, 2.0–150 mg L−1 NH4–N) were purchased from Merck. All chemicals were used as received. MilliQ water was used over all experiments.Sample preparation
We prepared nanostructured Ni2P and Ni5P4 using previously reported methods. In all cases, annealing under 1 atm of H2 at 400 °C for 1 h was performed before catalysis.22,49 Nickel phosphide clathrates AeNi2P4 (Ae = Ba, Sr) were synthesized as previously reported.29,30 BaNi2P4 and SrNi2P4 were synthesized by stoichiometric solid-state reactions between the elements in glassy carbon crucibles sealed in evacuated silica ampules. Samples were heated to 850 °C over a period of 17 h, and then kept at this temperature for 140 h. Samples were opened in an Ar glovebox, ground, resealed, and reannealed in a similar manner. This grind-seal-anneal process was repeated 2–3 times until phase-pure samples were obtained. As prepared clathrates were ground in an agate mortar for 0.5–2 h, then annealed under 1 atm H2 (5–10% H2 in Ar) at 400 °C for 1 h.Characterization
UV−Vis absorption spectra were collected with a photodiode-array Agilent 8453 UV−Vis spectrometer. Powder X-ray diffraction (XRD) was recorded using a Rigaku Ultima IV diffractometer with a Cu Kα radiation (40 kV, 44 mA). X-ray photoelectron spectroscopy (XPS) was conducted with a Kratos Amicus/ESCA 3400 instrument. The samples were irradiated with 240 W unmonochromated Mg Kα X-rays, and photoelectrons emitted at 0° from the surface were energy analyzed using a DuPont type analyzer. The pass energy was set at 150 eV. CasaXPS was used to process the raw data files. The binding energy of C 1s at 284.8 eV was used for reference.NO3− catalytic hydrogenation
Approximately 10 mg of dry catalyst was placed in an H2-filled 3-neck round bottom flask. 30 mL of an aqueous solution of NaNO3 or HNO3 (2.0 mM), previously sparged with Ar for 15–20 min was added to the flask and the pH was adjusted with H2SO4 or HCl. 1 atm of H2 (5–10% in Ar) was introduced into the reactor at a rate of 20 mL min−1 while vigorously stirring at 700 RPM at 60 °C. Aliquots were taken at different times for colorimetric quantification of the different aqueous NO3−, NO2− and NH4+ products.Nitroarene catalysis
Nitrobenzene (0.1 mmol, 10 μL) or 3-nitrostyrene (0.1 mmol, 14 μL) and ethanol (2 mL) were placed in a 5 mL vial. After stirring to ensure homogeneity, an aliquot was taken out, diluted, and analyzed by GCMS. 10 mg of a ground and annealed clathrate sample was then added and stirred at 300 RPM, followed by NaBH4 (23 mg, 0.6 mmol) while stirring continued. Aliquots were taken at certain times and filtered using 0.2 μm PTFE syringe filters. Part of the filtered aliquots were then diluted for use in GCMS analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the U.S. National Science Foundation, Division of Chemistry, Macromolecular, Supramolecular, and Nanochemistry Program (2305062) for funding of this work. We thank Dapeng Jing for assistance with XPS spectroscopy.References
- J. Wang, W. S. Cheon, J.-Y. Lee, W. Yan, S. Jung, H. W. Jang and M. Shokouhimehr, Dalton Trans., 2023, 52, 3567–3574 RSC.
- R. Rameshan, A. Tiwari, S. Kanungo and S. Roy, Inorg. Chem., 2023, 62, 9934–9944 CrossRef CAS PubMed.
- A. J. Burgin and S. K. Hamilton, Front. Ecol. Environ., 2007, 5, 89–96 CrossRef.
- S. Seitzinger, J. A. Harrison, J. K. Böhlke, A. F. Bouwman, R. Lowrance, B. Peterson, C. Tobias and G. Van Drecht, Ecol. Appl., 2006, 16, 2064–2090 CrossRef CAS PubMed.
- United States Environmental Protection Agency. Priority Pollutant List, 2014. https://www.epa.gov/sites/default/files/2015-09/documents/priority-pollutant-list-epa.pdf (Accessed 2023-1-8).
- M. Bilal, A. R. Bagheri, P. Bhatt and S. J. Chen, J. Environ. Manage., 2021, 291, 112685-1–112685-13 Search PubMed.
- J. Tiwari, P. Tarale, S. Sivanesan and A. Bafana, Environ. Sci. Pollut. Res., 2019, 26, 28650–28667 CrossRef CAS PubMed.
- I. Sanchis, E. Diaz, A. H. Pizarro, J. J. Rodriguez and A. F. Mohedano, Sep. Purif. Technol., 2022, 290, 120750-1–120750-10 CrossRef.
- M. Armbrüster, Sci. Technol. Adv. Mater., 2020, 21, 303–322 CrossRef PubMed.
- C. L. Daniels, D.-J. Liu, M. A. S. Adamson, M. Knobeloch and J. Vela, J. Phys. Chem. C, 2021, 125, 24440–24450 CrossRef CAS.
- S. Guo, H. Li, K. N. Heck, X. Luan, W. Guo, G. Henkelman and M. S. Wong, Appl. Catal., B, 2022, 305, 121048-1–121048-9 CrossRef.
- Z. Shen, G. Peng, Y. Gao and J. Shi, Environ. Sci.: Water Res. Technol., 2021, 7, 1078–1089 RSC.
- M. Gholinejad, F. Khosravi, M. Afrasi, J. M. Sansano and C. Nájera, Catal. Sci. Technol., 2021, 11, 2652–2702 RSC.
- M. Chen, C. R. Bowers and W. Huang, Acc. Mater. Res., 2021, 2, 1190–1202 CrossRef CAS.
- L. Zhang, J. Pan, L. Liu, S. Zhang, X. Wang, S. Song and H. Zhang, Small, 2022, 2201271-1–2201271-6 Search PubMed.
- C. L. Daniels, D. L. Mendivelso-Perez, B. A. Rosales, D. You, S. Sahu, J. S. Jones, E. A. Smith, F. P. Gabbaï and J. Vela, ACS Omega, 2019, 4, 5197–5203 CrossRef CAS PubMed.
- C. L. Daniels, M. Knobeloch, P. Yox, M. A. S. Adamson, Y. Chen, R. W. Dorn, H. Wu, G. Zhou, H. Fan, A. Rossini and J. Vela, Organometallics, 2020, 39, 1092–1104 CrossRef CAS.
- M. A. S. Adamson, P. Yox, T. Hernandez, F. Wang and J. Vela, Chem. Mater., 2022, 34, 746–755 CrossRef CAS.
- Y. Sheng, X. Lin, S. Yue, Y. Liu, X. Zou, X. Wang and X. Lu, Mater. Adv., 2021, 2, 6722–6730 RSC.
- A. L. Schio, M. R. F. Soares, G. Machado and T. Barcellos, ACS Sustainable Chem. Eng., 2021, 9, 9661–9670 CrossRef.
- D.-Y. Kuo, E. Nishiwaki, R. A. Rivera-Maldonado and B. M. Cossairt, ACS Catal., 2023, 13, 287–295 CrossRef CAS.
- L. Wei, D.-J. Liu, B. A. Rosales, J. W. Evans and J. Vela, ACS Catal., 2020, 10, 3618–3628 CrossRef CAS.
- L. Wei, M. A. S. Adamson and J. Vela, ChemNanoMat, 2020, 6, 1179–1185 CrossRef CAS.
- Y. Dai, C. Li, Y. Shen, T. Lim, J. Xu, Y. Li, H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, Nat. Commun., 2018, 9, 60 CrossRef PubMed.
- L. Pei, H. Tan, M. Liu, R. Wang, X. Gu, X. Ke, J. Jia and Z. Zheng, Green Chem., 2021, 23, 3612–3622 RSC.
- J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- A. T. Landers, M. Fields, D. A. Torelli, J. Xiao, T. R. Hellstern, S. A. Francis, C. Tsai, J. Kibsgaard, N. S. Lewis, K. Chan, C. Hahn and T. F. Jaramillo, ACS Energy Lett., 2018, 3, 1450–1457 CrossRef CAS.
- M. F. Delley, Z. Wu, M. E. Mundy, D. Ung, B. M. Cossairt, H. Wang and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 15390–15402 CrossRef CAS PubMed.
- J.-A. Dolyniuk, J. Wang, K. Lee and K. Kovnir, Chem. Mater., 2015, 27, 4476–4484 CrossRef CAS.
- J. Wang, J.-A. Dolyniuk, E. H. Krenkel, J. L. Niedziela, M. A. Tanatar, E. I. Timmons, T. Lanigan-Atkins, H. Zhou, Y. Cheng, A. J. Ramirez-Cuesta, D. L. Schlagel, U. S. Kaluarachchi, L.-L. Wang, S. L. Bud'ko, P. C. Canfield, R. Prozorov, O. Delaire and K. Kovnir, Chem. Mater., 2020, 32, 7932–7940 CrossRef CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- C. Xie, D. Yan, H. Li, S. Du, W. Chen, Y. Wang, Y. Zou, R. Chen and S. Wang, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- R. Shirataki, M. Hokazono, T. Nakabayashi and H. Anno, IOP Conf. Ser.: Mater. Sci. Eng., 2011, 18, 142012 Search PubMed.
- P. Simon, Z. Tang, W. Carrillo-Cabrera, K. Chiong, B. Böhme, M. Baitinger, H. Lichte, Y. Grin and A. M. Guloy, J. Am. Chem. Soc., 2011, 133, 7596–7601 CrossRef CAS PubMed.
- J. Mullay, Estimation of Atomic and Group Electronegativities. In Structure and Bonding, ed. K. D. Sen and C. K. Jørgensen, Springer, Berlin, Heidelberg, 1987, pp 1–25 Search PubMed.
- J.-A. Dolyniuk, B. Owens-Baird, J. Wang, J. V. Zaikina and K. Kovnir, Mater. Sci. Eng., 2016, 108, 1–46 CrossRef.
- J. F. Callejas, J. M. McEnaney, C. G. Read, J. C. Crompton, A. J. Biacchi, E. J. Popczun, T. R. Gordon, N. S. Lewis and R. E. Schaak, ACS Nano, 2014, 8, 11101–11107 CrossRef CAS PubMed.
- P. Liu, Z.-X. Zhang, S. W. Jun, Y.-L. Zhu and Y.-X. Li, React. Kinet. Mech. Catal., 2019, 126, 453–461 CrossRef CAS.
- Y.-F. Zen, Z.-C. Fu, F. Liang, Y. Xu, D.-D. Yang, Z. Yang, X. Gan, Z.-S. Lin, Y. Chen and W.-F. Fu, Asian J. Org. Chem., 2017, 6, 1589–1593 CrossRef CAS.
- T. Geng, H. Wang, H. Wu and S. Zhang, J. Nanopart. Res., 2020, 22, 237 CrossRef CAS.
- R. Gao, L. Pan, W. Wang, X. Zhang, L. Wang and J. J. Zou, ACS Catal., 2018, 8, 8420–8429 CrossRef CAS.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- Y. Pei, Z. Qi, T. W. Goh, L.-L. Wang, R. V. Maligal-Ganesh, H. L. MacMurdo, S. Zhang, C. Xiao, X. Li, F. F. Tao, D. D. Johnson and W. Huang, J. Catal., 2017, 356, 307–314 CrossRef CAS.
- S. Furukawa, Y. Yoshida and T. Komatsu, ACS Catal., 2014, 4, 1441–1450 CrossRef CAS.
- S. B. Bharate, R. Mudududdla, R. Sharma and R. A. Vishwakarma, Tetrahedron Lett., 2013, 54, 2913–2915 CrossRef CAS.
- R. K. DiNello and D. H. Dolphin, J. Org. Chem., 1981, 46, 3498–3502 CrossRef CAS.
- T. Hisayasu, N. Hara and H. Tamiaki, Bull. Chem. Soc. Jpn., 2022, 95, 1553–1560 CrossRef CAS.
- The international XPS database for XPS reference spectra. (https://xpsdatabase.net/). (Accessed 2023-11-16).
- K. Aso, A. Hayashi and M. Tatsumisago, Inorg. Chem., 2011, 50, 10820–10824 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Hydrogenation results using different binary and ternary nickel phosphides, and powder XRD patterns of recovered clathrates. See DOI: https://doi.org/10.1039/d4dt00332b |
| This journal is © The Royal Society of Chemistry 2024 |