
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination chemistry of alkali metal dimesityl-thio- and dimesityl-selenophosphinites [(L)2A-EPMes2]2 (A = Li, Na, K; E = S, Se; L = THF, THP) and [(18C6)K-SPMes2]†
Richard C. C.
Dorow
,
Phil
Liebing
 ,
Helmar
Görls
and
Matthias
Westerhausen
*
,
Helmar
Görls
and
Matthias
Westerhausen
*
Friedrich Schiller University Jena, Institute of Inorganic and Analytical Chemistry, Humboldtstraße 8, D-07743 Jena, Germany. E-mail: m.we@uni-jena.de
First published on 29th February 2024
Abstract
The reactions of dimesitylphosphane oxide Mes2P(O)H with Lawessons reagent and dimesitylphoshane with selenium yield Mes2P(E)H with E = S (1a) and E = Se (1b), respectively, with moderate yields. Metalation of dimesitylphosphane sulfide 1a with n-butyllithium, sodium hydride or potassium hydride in THF allows the isolation of dinuclear dimesityl-thiophosphinites of the type [(thf)2A-S-PMes2]2 [A = Li (4), Na (5), K (2a)] with central four-membered A2S2 rings. The weaker base THP leads to the very similar aggregate [(thp)2K-S-PMes2]2 (3a) as has also been observed for the homologous potassium dimesityl-selenophosphinites of the type [(L)2K-Se-PMes2]2 [L = thf (2b), thp (3b)]. Addition of 18-crown-6 ether leads to deaggregation and expectedly to formation of mononuclear [(18C6)K-S-PMes2] (6). Moderate yields have been obtained due to dismutation reactions that yield the corresponding phosphinates AE2PMes2 and phosphanides APMes2, a degradation process which has been observed earlier also for Li-O-PMes2. This side reaction hampers the application of these thio- and selenophosphinites as catalysts in the addition of phosphane sulfides and selenides across alkynes.
Introduction
Diarylphosphane oxides Ar2P(O)H are an important substance class for the formation of P–C, P–O and P–N bonds yielding tertiary phosphane oxides of the type Ar2P(O)R (Ar = aryl, R = alkyl, aryl, H).1 Often the P–H functionality lacks reactivity and drastic reaction conditions and/or activation of these secondary phosphane oxides by substitution of the P-bound hydrogen atom by an electropositive metal are required for the addition of the P–H bond onto alkenes and alkynes. Metalation of these phosphane oxides yields diarylphosphinites of the type M-O-PAr2; electropositive metals such as alkali and alkaline-earth metals are bound at the oxygen atom and the P atom is in a trigonal-pyramidal environment. Decreasing electronegativity and increasing softness of the s-block metal ions are beneficial for catalytic addition of Ar2P(O)H across alkynes.2 Thus, heavy alkali metal phosphinite catalysts Ar2P-O-A (especially of the softer alkali metals A like potassium, rubidium and cesium) are the preferred choice for quantitative conversion of diarylphosphane oxides and alkynes into alkenyl-diarylphosphane oxides (Scheme 1) whereas small and hard alkaline-earth metal congeners (especially of magnesium and calcium) show no catalytic activity. In addition, heterobimetallic alkali metal/magnesium phosphinite complexes proved to be very reactive in this hydrophosphorylation reaction, too.3 | ||
| Scheme 1 Catalytic hydrophosphorylation of alkynes with diarylphosphane oxides in the presence of catalytic amounts of AO-PAr2 (Ar = aryl). | ||
The instability in solution of some alkali metal phosphinites has been recognized; thus, lithium dimesitylphosphinite Li-OPMes2 dismutates into lithium phosphinate Li(O2PMes2) and lithium phosphanide LiPMes2 (Scheme 2).4 This degradation process can be prevented by coordination of soft cadmium species to the free electron pairs of the phosphorus atoms. Contrary to the hydrogen atom, the alkali metal ions are always bonded to the hard oxygen base and at the phosphorus site, a soft free electron pair is available for coordination chemistry to soft (transition) metals.5 Despite the reactivity, tetranuclear [(thf)Li-OPPh2]4 and dinuclear [(dme)Li-OPPh2]2 with tetra-coordinate lithium atoms have been isolated and structurally authenticated.6,7
 | ||
| Scheme 2 Dismutation of dissolved lithium dimesitylphosphinite in solvent L yielding lithium dimesitylphosphinate and lithium dimesitylphosphanide. Possible aggregation and deaggregation equilibria in solution are neglected. | ||
Potassium diarylphosphinites crystallize as tetranuclear complexes with central K4O4 heterocubane cages, the tetra-coordination of the oxygen bases decelerates dismutation processes which have been observed for the lithium derivatives.8 Despite these challenges, alkali metal phosphinites have been known for a long time.9 NMR studies verify that in solution the phosphinite ions bind via the oxygen atom to the alkali metals.10–12 A similar coordination behavior has been observed for the homologous diphenyl-thiophosphinites of lithium, sodium and potassium.11 Heavier diaryl-chalcogenophosphinites have also been studied by NMR spectroscopy.10,12 In general, these alkali metal phosphinites are accessible by two different strategies: (i) reduction of chloro-diarylphosphane oxides with s-block metals and (ii) deprotonation of diarylphosphane oxides with alkali metal hydrides or organometallics.
Diphenylphosphane oxide (P–O 148.81(11) pm),13 sulfide (P–S 195.60(7) pm)8 and selenide (P–Se 211.27(9) pm)14 have very similar molecular structures due to lack of steric pressure. Slight differences arise from a significantly larger electronegativity difference for the oxide (Allred-Rochow electronegativity values: P 2.06, O 3.50, S 2.44, Se 2.48)15 making the P atom more positive. The C–P–C bond angle of 108.40(7)° and the P–C bond lengths (179.82(15) and 180.15(14) pm)13 for Ph2P(O)H differ only slightly from the values for Ph2P(S)H (C–P–C 105.97(9)°, P–C 180.7(2) and 181.1(2) pm)8 and Ph2P(Se)H (C–P–C 107.56(14)°, P–C 180.4(3) and 181.0(3) pm).14 Deprotonation and formation of diphenylphosphinites6,7 and diphenyl-thiophosphinites8 lead to significant elongation of the P–O and P–S bonds. In addition, [(thp)K-SPPh2]∞ crystallizes with a unique wavy strand structure.8
The properties of diarylphosphinites strongly depend on the chalcogen (electronegativity, radius) and on the alkali metal (electronegativity, radius, surface charge density, hardness) and therefore, we started our investigation with K-SPMes2 to compare this complex with well-studied K-OPMes2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and K-SPPh28 and varied the chalcogen as well as alkali metal atom to elucidate their influence on NMR and structural parameters.
8 and K-SPPh28 and varied the chalcogen as well as alkali metal atom to elucidate their influence on NMR and structural parameters.
Results and discussion
Synthetic procedures
We chose mesityl-substituted chalcogenophosphinites due to beneficial crystallization behavior and enhanced stability with respect to dismutation into phosphinates and phosphanides as had been observed for Li-OPMes2 yielding LiPMes2 and LiO2PMes2.4 Thus, the structure of [(18C6)K-OPPh2] was impaired by cocrystallization with the phosphinate [(18C6)K-O2PPh2] whereas an analytically pure, crystalline sample of [(18C6)K-OPMes2] was studied for the derivative with the bulkier mesityl substituents.16An advantageous straightforward synthesis of potassium dimesityl-chalcogenophosphinites was the deprotonation of the dimesitylphosphane chalcogenides with potassium hydride as depicted in Scheme 3.
 | ||
| Scheme 3 Synthesis of thf (2) and thp (3) adducts of potassium dimesityl-chalcogenophosphinites via metalation of dimesitylphosphane chalcogenides (E = S (a), Se (b)) with potassium hydride in an ethereal solvent L (L = THF, THP). | ||
For the elucidation of the structural influence of the alkali metal, lithium and sodium dimesityl-thiophosphinites were included in this investigation. Metalation of Mes2P(S)H (1a) succeeded smoothly in THF with nBuLi or NaH yielding lithium (4) and sodium dimesityl-thiophosphinites (5), respectively, as shown in Scheme 4.
 | ||
| Scheme 4 Synthesis of thf adducts of lithium and sodium dimesityl-thiophosphinites (4, 5) via metalation of dimesitylphosphane sulfide (1a) with nBuLi and NaH in THF. | ||
All these dimesityl-chalcogenophosphinites crystallized as dinuclear molecules. The bulky mesityl groups hinder aggregation and formation of coordination polymers. The dinuclear complexes deaggregated after addition of a stoichiometric amount of [18]crown-6 yielding yellow mononuclear [(18C6)K-SPMes2] (6) as depicted in Scheme 5.
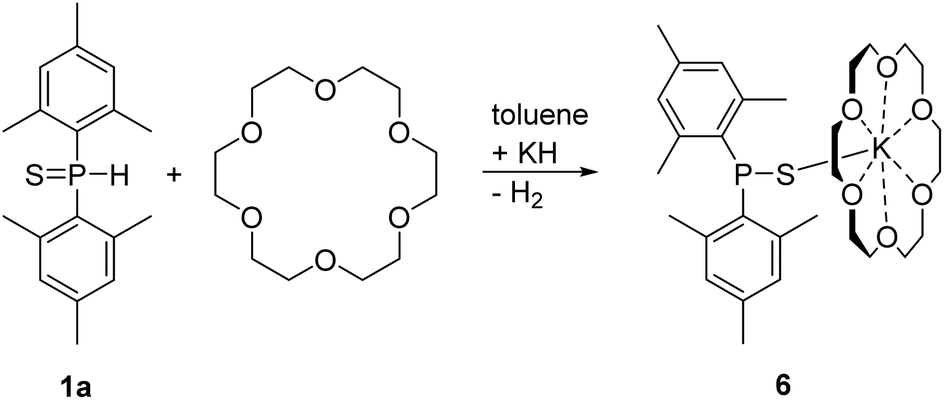 | ||
| Scheme 5 Synthesis of mononuclear [(18C6)K-SPMes2] (6) via metalation of dimesitylphosphane sulfide (1a) with KH in toluene in the presence of [18]crown-6. | ||
The above described thio- and selenophosphinites could be obtained as analytically pure compounds. However, excess of phosphane sulfides and selenides as well as other proton sources had to be avoided to suppress dismutation into the corresponding dimesitylphosphinates and dimesitylphosphanides.
NMR spectroscopy
Selected NMR parameters of compounds of the type Ar2P(E)H and Ar2PE-A(L)n (Ar = Ph, Mes; E = O, S, Se, A = Li, Na, K, L = ether base) are compared in Table 1. Increasing radius (and hence softness) of the chalcogen atom leads to higher frequency shifted 31P NMR resonances. Substitution of the phenyl substituents by bulkier mesityl groups causes a high field shift of the signals. Deprotonation of diarylphosphane oxides and hence formation of the alkali metal diarylphosphinites shifts the 31P resonances to lower field by approx. 60 ppm whereas chemical 31P shift differences are significantly smaller for the sulfur and selenium congeners. The influence of the aryl groups and the chalcogen atoms on the 1J(31P,1H) coupling constants of Ar2P(E)H is rather small and values around 465 Hz are observed. The 1J(31P,77Se) coupling does also not depend on the substitution pattern of the aryl group. However, metalation of the diarylphosphane selenides and formation of the alkali metal selenophosphinites halves the values of these coupling constants. This finding can be expected because the P–Se bond lengths elongate upon deprotonation from 211.28(5) pm for 1b to 223.29(8) and 223.88(6) pm for 2b and 3b, respectively. Generally, also 1J(31P,1H) coupling constants in PIII compounds with phosphorus atoms in pyramidal environments are much smaller than those values of PV congeners with tetrahedrally coordinated phosphorus atoms.| E | O | S | Sea |
|---|---|---|---|
| a 1 J(31P,77Se) coupling constants are given in brackets, 1J(31P,1H) values in parentheses. b Chemical shift of the tetrahydropyran adduct [(thp)KSPPh2]∞. c No NMR data given due to decomposition according to Scheme 2. | |||
| Ph2P(E)Ha | 21.717 |
20.9 (465)18 | 5.64 (461) [−740]19 |
| Mes2P(E)Ha | 11.4 (481) | −1.5 (467) | −27.7 (461) [−726] |
| Li-EPPh2 | 88.910 |
20.720 |
5.1 [−316]21 |
| Na-EPPh2 | 90.510 |
22.320 |
6.8 [−360] |
| K-EPPh2 | 86.810 |
25,20 26.8b8 |
9.7 [−377] |
| Li-EPMes2 | —c4 |
16.4 | −11.9 [−316] |
| Na-EPMes2 | 101.518 |
16.9 | −13.9 [−320] |
| K-EPMes2 | 95.98 |
21.0 | −8.9 [−351] |
Molecular structures
For comparison reasons, the molecular structures of dimesitylphosphane sulfide (1a) and selenide (1b) have been determined. The molecular structure and atom labelling scheme of 1a are depicted in Fig. 1, compound 1b is shown in the ESI.† Selected bond lengths and angles are listed in Table 2. Due to a diminished electronegativity difference between P and E for the heavier chalcogens S and Se, the P–E bond becomes less heteropolar with decreasing positive character of the phosphorus atom. | ||
| Fig. 1 Solid state molecular structure and atom labelling scheme of Mes2P(S)H (1a). The ellipsoids represent a probability of 30%, H atoms bound to C atoms are neglected for the sake of clarity. Selected bonding parameters are listed in Table 2. | ||
| E | O | S (1a) | Se (1b) |
|---|---|---|---|
| P1–E1 | 148.54(13) | 195.68(6) | 211.28(5) |
| P1–C1 | 181.51(18) | 182.34(17) | 182.91(13) |
| P1–C10 | 181.62(18) | 182.33(18) | 181.91(13) |
| C1–P1–C10 | 108.07(8) | 107.68(8) | 107.65(6) |
| E1–P1–C1 | 113.94(8) | 113.46(6) | 122.17(4) |
| E1–P1–C10 | 116.64(8) | 121.14(6) | 113.42(5) |
The bond lengths of Mes2P(E)H are very similar to those found for Ph2P(E)H (E = O,13 S,8 Se14), a slight but systematic elongation of the P–C bonds is caused by bulkier mesityl substituents. The distortion of the tetrahedral environment of P1 is more striking with an increasing difference Δ between the proximal and distal E1–P1–C1/C10 bond angles for the heavier chalcogenides (Δ = 2.7° for O, 7.7° for S, 8.8° for Se) due to steric repulsion between an ortho-methyl substituent and the chalcogen atom.
Alkali metal diarylphosphinites crystallize preferably with tetranuclear structures with central A4O4 heterocubane cages as observed for lithium6 and potassium.8 Bidentate Lewis bases (like 1,2-dimethoxyethane and tmeda) stabilize dinuclear [(dme)Li-OPPh2]27 and [(tmeda)Na-OPMes2]223 with A2O2 rings. Crown ethers such as 18-crown-6 deaggregate the tetranuclear potassium cage compound and even mononuclear [(18C6)K-OPAr2] (Ar = Ph, Mes) crystallizes.16 Substitution of the oxygen atom by sulfur in [(thp)K-SPPh2]∞ changes the structure in the solid phase drastically and a wavy strand structure forms8 whereas in [(tmeda)Li-SPPh2]2 the expected four-membered Li2S2 ring is observed.24 However, the homologous dinuclear selenium congener [(tmeda)Li-SePPh2]2 shows a unique molecular structure and this derivative crystallizes with a five-membered Li2Se2P ring.25 Based on this diverse coordination chemistry of diaryl-chalcogenophosphinites, we became interested in the dimesityl derivatives with bulkier aryl substituents to elucidate structural and NMR spectroscopic trends in dependency of the chalcogen and alkali metal atoms as well as of the neutral co-ligand.
Molecular structures and atom labelling schemes of [(thf)2K-SPMes2]2 (2a) and of [(thp)2K-SePMes2]2 (3b) are depicted in Fig. 2 and 3, molecular structures of [(thp)2K-SPMes2]2 (3a) and of [(thf)2K-SePMes2]2 (2b) are shown in the ESI.† THF is a stronger Lewis base than THP but very similar aggregation degrees and discrete dinuclear molecules are observed.
 | ||
| Fig. 2 Solid state molecular structure and atom labelling scheme of centrosymmetric [(thf)2K-SPMes2]2 (2a). The ellipsoids represent a probability of 30%, H atoms are omitted for clarity reasons. Symmetry-equivalent atoms are marked with the letter “A”. Selected bond lengths (pm): K1–S1 323.71(7), K1–S1A 314.77(7), P1–S1 207.19(7), P1–C1 186.43(18), P1–C10 186.95(18), K1–O1 269.47(19), K1–O2 278.8(2); angles (°): C1–P1–C10 100.38(7), S1–P1–C1 99.98(6), S1–P1–C10 114.52(6), K1–S1–K1A 91.475(18), K1–S1–P1 95.03(2), K1A–S1–P1 86.42(2), S1–K1–S1A 88.525(18), S1–K1–O1 174.92(5), S1–K1–O2 97.54(4), S1A–K1–O1 94.53(4), S1A–K1–O2 139.51(5), O1–K1–O2 82.77(5). | ||
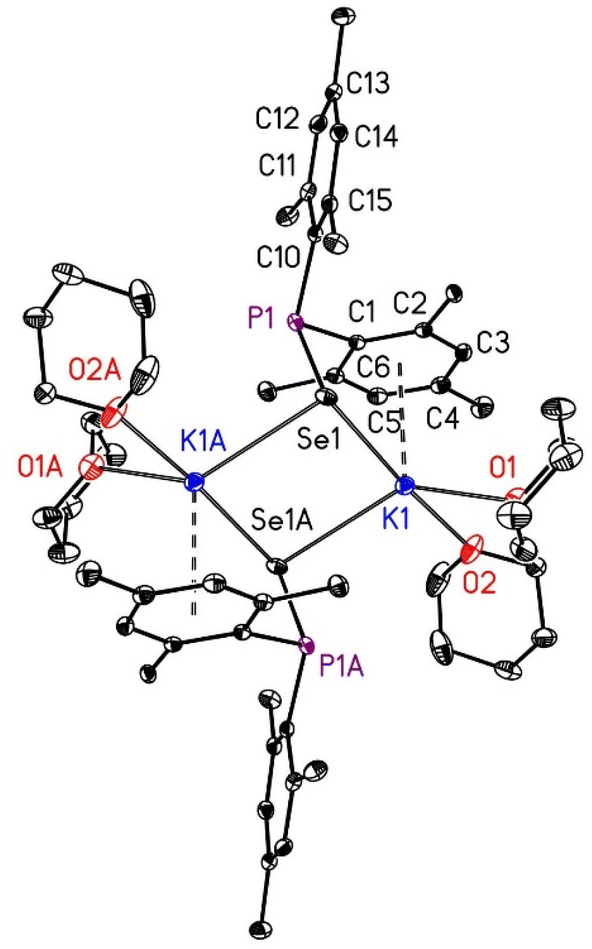 | ||
| Fig. 3 Solid state molecular structure and atom labelling scheme of centrosymmetric [(thp)2K-SePMes2]2 (3b). The ellipsoids represent a probability of 30%, H atoms are neglected for the sake of clarity. Symmetry-related atoms are marked with the letter “A”. Selected bond lengths (pm): K1–Se1 328.61(9), K1–Se1A 322.63(8), P1–Se1 223.88(6), P1–C1 186.57(15), P1–C10 186.82(15), K1–O1 272.35(15), K1–O2 264.59(15); angles (°): C1–P1–C10 102.42(7), Se1–P1–C1 98.68(5), Se1–P1–C10 114.85(5), K1–Se1–K1A 88.530(10), K1–Se1–P1 93.571(13), K1A–Se1–P1 89.51(2), Se1–K1–S1eA 91.471(10), Se1–K1–O1 93.80(3), Se1–K1–O2 175.80(4), Se1A–K1–O1 136.93(4), Se1A–K1–O2 92.57(4), O1–K1–O2 82.45(4). | ||
Potassium dimesityl-chalcogenophosphinites have central K2E2 rings in common and two ether bases bind to the potassium ions. In addition, π-interactions to side-on bound aryl groups saturate the coordination spheres of the alkali metals, thus shielding one side of the Lewis-acidic cation and preventing aggregation to larger molecules or to coordination polymers. In addition, this coordination mode enforces significantly different proximal and distal E1–P1–C1/C10 bond angles. The smaller size of the K2S2 ring compared to the K2Se2 cycle enhances steric repulsion between the mesityl substituent and ligated ether molecules leading to larger K1–O1 and K1–O2 distances. In all potassium chalcogenophosphinites, two strongly different K1–O1 and K1–O2 bond lengths are observed due to repulsive forces between an ortho-methyl group and one of the ether bases.
In Table 3 selected bonding parameters of the dinuclear potassium dimesityl-chalcogenophosphinites of the type [(L)2K-EPMes2]2 are compared. All these derivatives have nearly square K2E2 rings with slightly different K–E bond lengths. The coordination spheres are best described as square pyramidal environments, taking ligated π-bound aryl groups as single ligands. The ortho-methyl group of the side-on bound mesityl group pushes one ether base slightly out of the square O2E2 plane leading to an enhancement of the respective K1–O distance. The larger P–Se and K–Se bond lengths offer more flexibility for the adjustment of the π-interaction between K1 and the aryl group, leading to smaller differences between K1–Cmax and K1–Cmin which is a measure for tilting of the mesityl group with respect to the K1 atom (or the dislocation of the potassium atom K1 from the center of the aromatic ring). It is obvious that the stronger thf ligand enhances the dislocation in comparison to the weaker thp bases.
| 2a | 2b | 3a | 3b | |
|---|---|---|---|---|
| a Shortest potassium–carbon bond to the π-bound mesityl substituent. b Largest K–C distance to the π-bound mesityl substituent. c Difference between largest and smallest K–C bond lengths. d Angle sums of the chalcogen atom E1 and of the phosphorus atom P1. | ||||
| E | S | Se | S | Se |
| L | thf | thf | thp | thp |
| K1–O1 | 269.5(2) | 275.1(2) | 284.4(2) | 272.4(2) |
| K1–O2 | 278.8(2) | 270.2(2) | 275.2(2) | 264.6(2) |
| K1–E1 | 323.71(7) | 334.96(7) | 312.30(11) | 328.61(9) |
| K1–E1A | 314.77(7) | 326.37(7) | 326.77(10) | 322.63(8) |
| K1–Cmina |
313.4(2) | 314.5(3) | 315.6(3) | 319.2(2) |
| K1–Cmaxb |
(358.02) | 351.1(3) | 348.1(3) | 346.1(2) |
| Δ(K–C)c | 44.5 | 36.6 | 32.5 | 26.9 |
| ∑(E1)d | 272.9 | 273.1 | 270.1 | 271.6 |
| ∑(P1)d | 314.9 | 314.2 | 317.3 | 316.0 |
| E1–K1–E1A | 88.52(2) | 88.41(2) | 85.18(3) | 91.47(1) |
| K1–E1–K1A | 91.48(2) | 91.59(2) | 94.45(3) | 88.53(1) |
| O1–K1–O2 | 82.77(5) | 84.30(7) | 86.15(7) | 82.45(4) |
The molecular structures and atom labelling schemes of centrosymmetric lithium (4) and sodium dimesityl-thiophosphinites (5) are depicted in Fig. 4 and 5. The structure of 4 is comparable to the O congener [(dme)Li-OPPh2]27 with a central Li2E2 ring without any π-interactions to the aromatic aryl substituents. The lithium atoms are in distorted tetrahedral coordination environments with large S–Li1–O and smaller O1–Li1–O1 and S1–Li1–S1A bond angles. The Li1–O1 and Li1–O2 bond lengths have rather similar values but steric strain is minimized by tilting of the bridging thiophosphinite ligand leading to different S1–P1–C1 and S1–P1–C10 bond angles.
 | ||
| Fig. 4 Solid state molecular structure and atom labelling scheme of [(thf)2Li-SPMes2]2 (4). The ellipsoids represent a probability of 30%, H atoms are omitted for clarity reasons. Symmetry-related atoms are marked with the letter “A”. Selected bond lengths (pm): Li1–S1 245.8(2), Li1–S1A 248.8(2), P1–S1 207.51(5), P1–C1 186.46(12), P1–C10 186.24(12), Li1–O1 192.6(2), Li1–O2 194.0(2); angles (°): C1–P1–C10 102.08(5), S1–P1–C1 113.32(4), S1–P1–C10 102.60(4), Li1–S1–Li1A 79.49(8), Li1–S1–P1 111.53(6), Li1A–S1–P1 116.88(6), S1–Li1–S1A 100.52(8), S1–Li1–O1 108.47(10), S1–Li1–O2 121.77(11), S1A–Li1–O1 123.27(11), S1A–Li1–O2 101.18(10), O1–Li1–O2 103.05(11). | ||
 | ||
| Fig. 5 Solid state molecular structure and atom labelling scheme of [(thf)2Na-SPMes2]2 (5). The ellipsoids represent a probability of 30%, H atoms are omitted for clarity reasons. Disordering of O2 is not shown. Symmetry-equivalent atoms are marked with the letter “A”. Selected bond lengths (pm): Na1–S1 281.95(13), Na1–S1A 284.11(13), Na1–P1A 314.15(13), P1–S1 207.32(9), P1–C1 185.4(2), P1–C10 186.6(2), Na1–O1 233.9(2), Na1–O2A 231.9(8), Na1–O2B 237.2(18); angles (°): C1–P1–C10 101.65(10), S1–P1–C1 113.52(8), S1–P1–C10 114.26(8), Na1–S1–Na1A 83.73(4), Na1–S1–P1 102.67(4), Na1A–S1–P1 77.74(4), S1–Na1–S1A 96.27(4), S1–Na1–O1 89.98(7), S1–Na1–O2A 149.1(2), S1A–Na1–O1 151.38(8), S1A–Na1–O2 101.61(18), O1–Na1–O2 86.24(18). | ||
The molecular structure and atom labelling scheme of homologous [(thf)2Na-SPMes2]2 (5) are depicted in Fig. 5. The thf ligand of O2 is disordered on two sites verifying reduced intramolecular steric pressure. Due to the larger radius of sodium ions (r(Li+) 73, r(Na+) 113, r(K+) 151 pm),15 steric strain is released and very similar S1–P1–C1 and S1–P1–C10 bond angles exist. The smaller radii of lithium and sodium ions make these alkali metals rather hard Lewis acids preferring hard ether bases whereas for larger and softer potassium ions, the ether bases compete with the soft π-systems of the aryl groups.
Selected structural parameters of [(thf)2A-SPMes2]2 with A = Li (4), Na (5) and K (2a) are compared in Table 4. The small radius of the lithium ion induces intramolecular steric repulsion leading to a large angle sum of S1. The larger Na–S distances release steric pressure, and a smaller ∑(S1) value can be realized and very similar S1–P1–C1 and S1–P1–C10 bond angles are found. Contrary to this finding, steric pressure is slightly enhanced in potassium derivative 2a due to the enlarged coordination number by additional π-interactions to the mesityl rings, again giving an enlarged angle sum at S1 and quite different S1–P1–C1/C10 angles. The small radius of Li+ also affects the O1–Li1–O2 bond angle due to steric repulsion between the thf ligands, whereas significantly smaller O1–A1–O1 values are observed for sodium (5) and potassium (2a).
| A | Li (4) | Na (5) | K (2a) | K (6) |
|---|---|---|---|---|
| a Angle sum of P1. b Angle sum of S1. c There are three groups of O–K–O bond angles: bite angles O(n)–K1–O(n + 1) vary around 60°, O(n)–K1–O(n + 2) bond angles around 120° and O(n)–K1–O(n + 3) values around 160°. | ||||
| P1–S1 | 207.51(5) | 207.32(9) | 207.19(7) | 205.11(7) |
| A1–S1 | 245.8(2) | 282.0(1) | 323.71(7) | 306.85(8) |
| A1–S1A | 248.8(2) | 284.1(1) | 314.77(7) | — |
| ∑(P1)a | 318.00 | 329.43 | 314.88 | 315.75 |
| S1–P1–C1 | 113.32(4) | 113.52(8) | 99.98(6) | 114.53(6) |
| S1–P1–C10 | 102.60(4) | 114.26(8) | 114.52(6) | 99.72(6) |
| C1–P1–C10 | 102.08(5) | 101.7(1) | 100.38(7) | 101.50(8) |
| ∑(S1)b | 307.90 | 264.14 | 272.93 | — |
| A1–S1–A1A | 79.49(8) | 83.73(4) | 91.48(2) | — |
| P1–S1–A1 | 111.53(6) | 102.67(4) | 95.03(2) | 101.91(3) |
| P1–S1–A1A | 116.88(6) | 77.74(4) | 86.42(2) | — |
| O1–A1–O2 | 103.1(1) | 86.2(2) | 82.77(5) | |
The molecular structure and atom labelling scheme of [(18C6)K-SPMes2] (6) are depicted in Fig. 6. The crown ether hinders aggregation and hence a mononuclear complex is stabilized. In comparison to dinuclear [(thf)2K-SPMes2]2 (2a), smaller K1–S1 and P1–S1 bond lengths are observed (Table 4).
 | ||
| Fig. 6 Solid state molecular structure and atom labelling scheme of [(18C6)K-SPMes2] (6). The ellipsoids represent a probability of 30%, H atoms are omitted for clarity reasons. The disordering of the atoms O5A/B and O6A/B is not shown. Selected bond lengths (pm): K1–S1 306.85(8), P1–S1 205.11(7), P1–C1 186.7(2), P1–C10 186.2(2), K1–O1 293.3(1), K1–O2 276.9(1), K1–O3 284.4(2), K1–O4 280.7(2), K1–O5A 286(1), K1–O5B 289(2), K1–O6A 276.2(7), K1–O6B 273(2); bond angles (°): C1–P1–C10 101.50(8), S1–P1–C1 114.53(6), S1–P1–C10 99.72(6), K1–S1–P1 101.91(3), S1–K1–O1 114.14(4), S1–K1–O2 85.54(3), S1–K1–O3 80.92(3), S1–K1–O4 87.54(4), S1–K1–O5A 112.1(2), S1–K1–O5B 115.0(4), S1–K1–O6A 111.3(2), S1–K1–O6B 120.3(3). | ||
In Table 4 selected structural parameters of the alkali metal dimesitylthiophosphinites of lithium, sodium, and potassium are compared. The phosphinite anion shows characteristic structural parameters with C1–P1–C10 bond angles slightly larger than 100° regardless of the alkali metal and of the bridging or terminal position but smaller than observed in Mes2P(S)H (1a, Table 2). Remarkably, the S1–P1–C1/C10 bond angles differ significantly regardless of the bridging or terminal binding mode as shown for the potassium derivatives 2a and 6. The exception is the sodium complex 5 with two rather large S1–P1–C1/C10 angles leading to an enhanced angle sum at P1 although the compounds 4 (A = Li), 5 (A = Na), and 2a (A = K) crystallize as dimeric centrosymmetric molecules with tetracoordinate alkali metal atoms. However, in derivative 2a one mesityl group shows interactions between the potassium atom and the π-system of one mesityl group. This side-on coordination mode of the aryl group leads to distortion of the four-membered K2S2 ring and different K1–S1 and K1–S1A bond lengths. In the sodium derivative 5 the metal-aryl interaction tilts the phosphinite anion and significantly different P1–S1–Na1/Na1A bond angles are observed. In addition, the angles within the phosphinite anion deviate from those of the other congeners and two quite similar S1–P1–C1/C10 bond angles are realized.
Concluding investigations concern the ability of the potassium dimesityl-chalcogenophosphinites to enable catalytic formation of dimesityl-alkenylphosphane oxides via addition of Mes2P(E)H onto alkynes. The potassium-mediated addition of dimesitylphosphane oxide across alkynes (Pudovik reaction) succeeds smoothly with good yields whereas lithium and sodium are unsuitable alkali metals to promote these addition reactions.2 Contrary to this finding, substitution of E = O by the heavier chalcogens sulfur and selenium impedes this catalytic conversion. Several reasons may contribute to this finding:
(i) The heteropolar P–O bond leads to polarization and negatively charged oxygen sites on the one hand and positively charged P atoms on the other whereas the P-E bonds (E = S, Se) are mainly of covalent nature. The positively charged P atoms in K-OPAr2 ease the attack at electron-rich unsaturated compounds such as alkynes and heterocumulenes.
(ii) The enhanced negative charge on the oxygen base enables strong electrostatic bonds to cations whereas the soft and large chalcogen bases can compete with the soft aromatic π-systems. The potassium π-interactions strongly shield the reactive sites of the complexes K-EPAr2 with E = S, Se.
(iii) The dismutation rate of K-E-PMes2 (E = S, Se) into the corresponding phosphinates KE2PMes2 and phosphanides KPMes2 is accelerated in the presence of weak proton sources (Brønsted acids) such as R2P(E)H leading to a decline of the catalyst.
Consequently, alternative routes must be chosen to prepare diaryl-alkenylphosphane sulfides and selenides. A feasible strategy involves several steps, including the Pudovik reaction and synthesis of diaryl-alkenylphosphane oxides, subsequent reduction of these compounds to phosphanes and finally, sulfurization or selenation with the heavy elemental chalcogens.
Experimental
General information
All manipulations were carried out under an inert nitrogen atmosphere using standard Schlenk techniques. The solvents were dried over KOH and subsequently distilled over sodium/benzophenone under a nitrogen atmosphere prior to use. All substrates were purchased from Alfa Aesar, abcr, Sigma Aldrich or TCI and used without further purification. Mes2PH was prepared by an adjusted literature procedure.26 The yields given are not optimized. Purity of the compounds was verified by NMR spectroscopy. Deuterated solvents were dried over sodium, distilled, degassed, and stored under nitrogen over sodium. 1H, 13C{1H}, and 31P NMR spectra were recorded on Bruker Avance I 250 (BBO) Bruker Avance III 400 (BBO, BBFO probes), Avance neo 500 (BBFO Prodigy probe), and Avance III HD 600 (TCI cryoprobe) spectrometers. Chemical shifts (1H, 13C) are reported in parts per million relatively to SiMe4 as an external standard referenced to the solvents residual proton signal using xiref AU program for 13C NMR spectra. The assignment of the NMR signals is depicted in Scheme 6. DOSY-NMR spectra were measured using the convection compensated dstebpgp3s standard pulse sequence. ASAP-HSQC spectra and ASAP-HSQC-DEPT spectra were recorded using the published pulse sequences.27 Elemental analyses of the alkali metal compounds gave no reproducible and reliable results due to loss of ligated ether bases and formation of carbonates during handling and combustion. During heating and combustion analysis, small amounts of 1a and 1b degraded and sublimed; this led to generally too small values and has been taken into account by adjusting the elemental analysis according to the H values. | ||
| Scheme 6 Atom labelling scheme for the assignment of the NMR signals. | ||
Synthesis of Mes2P(S)H (1a)
The suspension of dimesitylphosphane oxide (302 mg, 1.1 mmol, 1.0 equiv.) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide (Lawessons reagent, 191 mg, 0.47 mmol, 0.4 equiv.) in 10 mL of THF was heated under reflux for 2 h. During this time a slightly yellow clear solution formed. Then the solvent was removed in vacuo and the residue recrystallized from acetonitrile (6 mL), yielding dimesitylphosphane sulfide (1a) as a colorless solid (119 mg, 40%), mp: 152–156 °C (dec.). 1H NMR (400.13 MHz, CDCl3, 298 K): δ = 8.52 (d, 1H, 1JPH = 471.8 Hz, H–P), 6.86 (d, 4H, 4JPH = 4.4 Hz, H3), 2.49 (s, 12H, H5), 2.27 (s, 6H, H6). 31P NMR (CDCl3, 161.98 MHz, 298 K): δ = −1.45 (d, 1JPH = 468.6 Hz). 13C{1H} NMR (CDCl3, 100.61 MHz, 298 K): δ = 141.7 (d, 5JPC = 3.3 Hz, C4), 141.4 (d, 2JPC = 10.5 Hz, C2), 131.1 (d, 3JPC = 10.9 Hz, C3), 125.6 (d, 1JPC = 81.1 Hz, C1), 21.3 (d, 3JCP = 8.8 Hz, C5), 21.2 (s, C6). MS (m/z): 325 [M + Na]+, 303 [M + H]+. Elemental analysis: found: C 71.07, H 7.67, S 9.80; calc. for C18H23PS: C 71.49, H 7.67, S 10.60; IR (ATR, cm−1): 2913 (m), 2378 (m), 1601 (s), 1447 (s), 673 (vs), 630 (s).Synthesis of Mes2P(Se)H (1b)
Dimesitylphosphane (2.72 g, 10 mmol, 1.0 equiv.) and grey selenium granules (797 mg, 10 mmol, 1.0 equiv.) were heated under reflux in 15 mL of toluene for 4 h. Unreacted selenium was removed by filtration and thereafter the solvent was removed by distillation in vacuo. The residue was heated under reflux in n-hexane for 2 h. Then the product dimesitylphosphane selenide (1b, 1.26 g, 36%) was collected by filtration. This compound was a colorless solid which quickly degraded under aerial conditions. 1H NMR (400.13 MHz, C6D6, 298 K): δ = 8.09 (d, 1H, 1JPH = 463 Hz, P–H), 6.53 (d, 4H, 3JPH = 4 Hz, H3), 2.44 (s, 12H, H5), 1.97 (s, 6H, H6). 31P NMR (161.98 MHz, C6D6, 298 K): δ = −27.7 (d, 1JPH = 461 Hz, satellites 1JPSe = 726 Hz). 13C{1H} NMR (100.61 MHz, C6D6, 298 K): δ = 141.7 (d, 2JPC = 10.1 Hz, C2), 141.2 (d, 4JPC = 3.4 Hz, C4), 131.2 (d, 3JPC = 10.3 Hz, C3), 124.6 (d, 1JPC = 71.6 Hz, C1), 21.4 (d, 3JPC = 8.5 Hz, C5), 20.9 (s, C6). 77Se NMR (76.31 MHz, C6D6, 298 K): δ = −274.12 (d, 1JPSe = 726 Hz). MS (m/z): 373 [M + Na]+, 351 [M + H]+, 349 [M − H]+. Elemental analysis: calcd: C 61.89, H 6.64; found: C 61.77, H 6.64. IR (ATR, cm−1): 2912 (w), 2387 (w), 1601 (m), 1442 (s), 843 (s), 622 (vs), 553 (vs), 492 (vs). Dec. above 130 °C (without melting).Synthesis of [(thf)2K-SPMes2]2 (2a)
Potassium hydride (300 mg, 7.5 mmol, 4.3 equiv.) and Mes2P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)H (527 mg, 1.74 mmol, 1.0 equiv.) were combined in a Schlenk flask and cooled to −20 °C. Then 20 mL of THF were added and the suspension stirred for 2.5 h whereat a yellow solution formed. Then the volume of the solution was reduced to a third of the original volume and stored at −20 °C yielding yellow crystalline 2a (yield: 444 mg, 52%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ = 6.52 (s, 8H, H3), 2.49 (s, 24H, H5), 2.09 (s, 12H, H6). 31P NMR (161.98 MHz, THF-d8, 253 K): δ = 21.0 (s). 13C{1H} NMR (100.61 MHz, THF-d8, 253 K): δ = 143.8 (d, 1JCP = 50.2 Hz, C1), 141.5 (d, 2JCP = 15.0 Hz, C2), 134.7 (s, C4), 129.8 (s, C3), 22.6 (d, 3JCP = 16.4 Hz, C5), 20.8 (s, C6). IR (ATR, cm−1): 2919 (m), 1601 (m), 1445 (s), 1055 (s), 1025 (s), 846 (s), 632 (vs). Dec. above 245 °C (without melting).
S)H (527 mg, 1.74 mmol, 1.0 equiv.) were combined in a Schlenk flask and cooled to −20 °C. Then 20 mL of THF were added and the suspension stirred for 2.5 h whereat a yellow solution formed. Then the volume of the solution was reduced to a third of the original volume and stored at −20 °C yielding yellow crystalline 2a (yield: 444 mg, 52%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ = 6.52 (s, 8H, H3), 2.49 (s, 24H, H5), 2.09 (s, 12H, H6). 31P NMR (161.98 MHz, THF-d8, 253 K): δ = 21.0 (s). 13C{1H} NMR (100.61 MHz, THF-d8, 253 K): δ = 143.8 (d, 1JCP = 50.2 Hz, C1), 141.5 (d, 2JCP = 15.0 Hz, C2), 134.7 (s, C4), 129.8 (s, C3), 22.6 (d, 3JCP = 16.4 Hz, C5), 20.8 (s, C6). IR (ATR, cm−1): 2919 (m), 1601 (m), 1445 (s), 1055 (s), 1025 (s), 846 (s), 632 (vs). Dec. above 245 °C (without melting).
Synthesis of [(thf)2K-SePMes2]2 (2b)
A solution of Mes2P(Se)H (171 mg, 0.5 mmol, 1.0 equiv.) in 10 mL of THF was added at −40 °C to a suspension of potassium hydride (33 mg, 0.8 mmol, 1.6 equiv.) in THF. Hydrogen gas was liberated, and the suspension turned yellow. After four hours of stirring the volume of the solution was reduced to half of the original volume. During storage at −20 °C a few yellow crystals of 2b precipitated (yield was approx. 5%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ = 6.58 (s, 8H, H3), 2.54 (s, 24H, H5), 2.16 (s, 12H, H6). 31P NMR (161.98 MHz, THF-d8, 253 K): δ = −8.9 (s, satellites 1JPSe = 350.8 Hz). 77Se NMR (76.31 MHz, THF-d8, 253 K): δ = −167.4 (d, 1JPSe = 354.3 Hz). IR (ATR, cm−1): 2919 (m), 1601 (m), 1445 (s), 1055 (s), 847 (s). Dec. above 120 °C (without melting).Synthesis of [(thp)2K-SPMes2]2 (3a)
Potassium hydride (364 mg, 9.1 mmol, 3.5 equiv.) was suspended in 5 mL of THP and cooled to −30 °C. Then dimesitylphosphane sulfide (791 mg, 2.6 mmol, 1.0 equiv.), dissolved in 20 mL of THP, was added dropwise within 15 min. The solution turned yellow and hydrogen gas evolved. The reaction mixture was stirred for 3 h and then all solids were removed by filtration. The volume of the solution was reduced and at −20 °C yellow [K(SPMes2)(thp)2]2 (3a) crystallized (yield: 234 mg, 24%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ = 6.60 (s, 8H, H3), 3.57 (s, 5H, H7), 2.52 (s, 24H, H5), 2.12 (s, 12H, H6), 1.60 (d, 8H, H9), 1.49 (m, 16H, H8). 31P NMR (161.98 MHz, THF-d8, 253 K): δ = 19.4 (s). 13C{1H} NMR (100.61 MHz, THF-d8, 253 K): δ = 143.5 (d, 1JCP = 50.3 Hz, C1), 141.5 (d, 2JCP = 15.6 Hz, C2), 134.8 (s, C4), 129.9 (s, C3), 68.8 (s, C7), 27.4 (s, C8), 24.3 (s, C9), 22.6 (d, 3JCP = 17.3 Hz, C5), 20.8 (s, C6). IR (ATR, cm−1): 2919 (s), 1600 (m), 1441 (s), 1087 (s), 844 (s), Dec. above 287 °C (without melting).Synthesis of [(thp)2K-SePMes2]2 (3b)
Potassium hydride (100 mg, 2.5 mmol, 1.9 equiv.) was placed in a flask at −30 °C and dimesitylphosphane selenide (470 mg, 1.3 mmol, 1.0 equiv.), dissolved in THP (21.5 mL), was added within 5 min. Evolution of hydrogen gas and formation of a yellow solution was observed. The reaction mixture was stirred for 2 h and then half of the solvent was removed in vacuo. At −20 °C yellow crystalline [K(SePMes2)(thp)2]2 crystallized (yield: 273 mg, 30%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ (ppm) = 6.54 (s, 8H, H3), 3.53 (s, 6H, H7), 2.49 (s, 24H, H5), 2.12 (s, 12H, H6), 1.60 (d, 3H, H9), 1.49 (m, 6H, H8). 31P NMR (161.98 MHz, THF-d8, 253 K): δ (ppm) = −10.5 (s, satellites 1JPSe = 353.1 Hz). 13C NMR (100.61 MHz, THF-d8, 253 K): δ (ppm) = 141.7 (d, 1JCP = 52.9 Hz, C1), 141.7 (d, 2JCP = 14.7 Hz, C2), 134.6 (s, C4), 129.7 (s, C3), 68.8 (s, C7), 27.4 (s, C8), 24.3 (s, C9), 23.0 (d, 3JCP = 16.5 Hz, C5), 20.8 (s, C6). 77Se NMR (76.31 MHz, THF-d8, 253 K): δ (ppm) = −166.4 (d, 1JPSe = 353.1 Hz). IR (ATR, cm−1): 2918 (m), 1603 (m), 1402 (m), 1172 (m), 1086 (s), 645 (s).Synthesis of [(thf)2Li-SPMes2]2 (4)
Dimesitylphosphane sulfide (496 mg, 1.6 mmol, 1.0 equiv.) was dissolved in THF (5 mL) and a solution of nBuLi (1.6 M, 1.1 ml, 1.1 equiv.) in n-heptane was added dropwise. The yellow solution was cooled to −20 °C, stirred for 2 h and then the volume of the solution was reduced to half of the original volume. Yellow crystalline [Li(SPMes2)(thf)2]2 (4) precipitated during storage at −20 °C (yield: 152 mg, 21%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ = 6.49 (s, 8H, H3), 2.47 (s, 24H, H5), 2.08 (s, 12H, H6). 31P NMR (161.98 MHz, THF-d8, 253 K): δ = 16.4 (s). 13C{1H} NMR (100.61 MHz, THF-d8, 253 K): δ = 143.2 (d, 1JCP = 50.3 Hz, C1), 141.7 (d, 2JCP = 15.6 Hz, C2), 134.5 (s, C4), 129.7 (s, C3), 22.6 (d, 3JCP = 16.5, C5), 20.8 (s, C6). 7Li NMR (155.51 MHz, THF-d8, 253 K): δ = 0.36. IR (ATR, cm−1): 2918 (m), 1602 (m), 1444 (s), 1053 (s), 622 (vs). Dec. above 190 °C (without melting).Synthesis of [(thf)2Na-SPMes2]2 (5)
Sodium hydride (62 mg, 2.6 mmol, 1.1 equiv.) was placed in a Schlenk flask and dimesitylphosphane sulfide (706 mg, 2.3 mmol, 1.0 equiv.), dissolved in 7 mL of THF, was added. After gas evolution ceased, the solution was concentrated and cooled to −20 °C. At −20 °C yellow crystalline [Na(SPMes2)(thf)2]2 (5) precipitated (yield: 436 mg, 40%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ (ppm) = 6.54 (s, 8H, H3), 2.51 (s, 24H, H5), 2.12 (s, 12H, H6). 31P NMR (161.98 MHz, THF-d8, 253 K): δ (ppm) = 16.9 (s). 13C NMR (100.61 MHz, THF-d8, 253 K): δ (ppm) = 143.0 (d, 1JCP = 49.4 Hz, C1), 141.6 (d, 2JCP = 14.7 Hz, C2), 134.7 (s, C4), 129.8 (s, C3), 22.6 (d, 3JCP = 17.3 Hz, C5), 20.8 (s, C6). IR (ATR, cm−1): 2918 (m), 1602 (m), 1444 (s), 1053 (s), 847 (s), 636 (vs). Dec. above 170 °C (without melting).Synthesis of [(18C6)K-SPMes2] (6)
Dimesitylphosphane sulfide (1a, 486 mg, 1.6 mmol, 1.0 equiv.) in 10 mL of toluene was added dropwise at room temperature to a suspension of KH (99 mg, 2.5 mmol, 1.6 equiv.) and [18]crown-6 (429 mg, 1.6 mmol, 1.0 equiv.) in 10 mL of toluene. After one hour all solids were removed by filtration. The filtrate was cooled to −20 °C yielding 350 mg of yellow crystals of 6 (0.6 mmol, 38%). 1H NMR (400.13 MHz, THF-d8, 253 K): δ (ppm) = 6.49 (s, 4H, H3), 3.55 (s, 24H, H7), 2.54 (s, 12H, H5), 2.10 (s, 6H, H6). 31P NMR (161.98 MHz, THF-d8, 253 K): δ (ppm) = 24.3 (s). 13C{1H} NMR (100.61 MHz, THF-d8, 253 K): δ (ppm) = 145.6 (d, 1JCP = 53.8 Hz, C1), 141.8 (d, 1JCP = 14,7 Hz, C2), 133.8 (s, C4), 129.6 (s, C3), 71.0 (s, C7), 22.8 (d, 1JCP = 16.5 Hz, C5), 21.0 (s, C6). IR (ATR, cm−1): 2886 (m), 1468 (m), 1451 (m), 1349 (s) 1102 (s), 960 (s), 835 (s), 641 (s). Dec. above 230 °C (without melting).Crystal structure determinations
The intensity data for the compounds were collected on a Nonius KappaCCD diffractometer equipped with a Mo-Kα IμS microfocus source and an Apex2 detector. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple-scans.28–31 The structures were solved by intrinsic phasing methods (SHELXT-2018)32 and refined by full-matrix least squares techniques against Fo2 (SHELXL-2018).33 The hydrogen atoms bonded to the phosphorus atom P1 of 1a and 1b were located by difference Fourier synthesis and refined isotropically. All other hydrogen atoms were included at calculated positions with fixed thermal parameters. All non-hydrogen atoms were refined anisotropically.33 Crystallographic data as well as structure solution and refinement details are summarized in Table S1.† XP34 and POV-Ray35 were used for structure representations.Conclusions
Metalation of dimesitylphosphane sulfide (1a) and homologous selenide (1b) with n-butyllithium, NaH, and KH yields the following dinuclear complexes of the type [(L)2A-S-PMes2]2 depending on the solvent: A = Li: L = thf (4); A = Na: L = thf (5); A = K: L = thf (2a), thp (3a) as well as [(L)2K-Se-PMes2]2 with L = thf (2b), thp (3b). Deaggregation occurs in the presence of 18-crown-6 ether and mononuclear [(18C6)K-S-PMes2] forms. The preferential formation of dinuclear complexes with monodentate Lewis bases is in contrast to the tetranuclear potassium dimesitylphosphinites [(L)K-O-PMes2]4 with a central K4O4 heterocubane-type cage. These dimesityl-thiophosphinites and -selenophosphinites degrade into the corresponding phosphinates AE2PMes2 and phosphanides APMes2; this degradation is accelerated in the presence of weak Brønsted acids. This behavior hampers the application of these compounds as catalysts in the Pudovik reaction, i.e. the addition of Mes2P(E)H onto alkynes.These properties which are strikingly deviating from those of the lighter homologous phosphinites A-O-PMes2 have several reasons. The phosphinites A-O-PMes2 contain the hard and negatively charged oxygen base which can easily displace ether bases. Contrary, the soft and significantly less electronegative sulfur and selenium bases are significantly less basic and especially for the hard lithium cation coordination of neutral oxygen or nitrogen coligands is beneficial. For the softer potassium cation, coordination of the soft π-system of the mesityl groups can compete with the heavier chalcogen bases of the thio- and selenophosphinites.
Author contributions
R. C. C. D.: Conceptualization, data analysis and methodology, data acquisition, writing original draft and editing; P. L. and H. G.: X-ray structure determinations at single crystals and editing of manuscript; M. W.: conceptualization and supervision, writing original draft and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the valuable support of the NMR and MS service platforms (https://www.nmr.uni-jena.de; https://www.ms.uni-jena.de) of the Faculty of Chemistry and Earth Sciences of the Friedrich Schiller University Jena, Germany.References
- Z.-Y. Wang, Q. Guo, S. Xu and K.-K. Wang, Synthesis, 2021, 3683–3698 CrossRef CAS.
- B. E. Fener, P. Schüler, N. Ueberschaar, P. Bellstedt, H. Görls, S. Krieck and M. Westerhausen, Chem. – Eur. J., 2020, 26, 7235–7243 CrossRef CAS PubMed.
- A. W. J. Platten, A. M. Borys and E. Hevia, ChemCatChem, 2022, 14, e202101853 CrossRef CAS.
- M. A. Beswick, N. L. Cromhout, C. N. Harmer, J. S. Palmer, P. R. Raithby, A. Steiner, K. L. Verhorevoort and D. S. Wright, Chem. Commun., 1997, 583–584 RSC.
- D. Martin, D. Moraleda, T. Achard, L. Giordano and G. Buono, Chem. – Eur. J., 2011, 17, 12729–12740 CrossRef CAS PubMed.
- (a) A. J. Hoskin and D. W. Stephan, Organometallics, 1999, 18, 2479–2483 CrossRef CAS; (b) C. Berthold, L. R. Thomas-Hargreaves, S. I. Ivlev and M. R. Buchner, Z. Naturforsch., 2021, 76b, 651–658 CrossRef.
- M. Westerhausen, Monolithiumphosphid·DME und -arsenid·DME – wertvolle Edukte für die Synthese von Acylphosphanen und -arsanen und verwandten Verbindungen, Ph.D. thesis, Marburg/Lahn, Germany, 1987 Search PubMed.
- S. M. Härling, H. Görls, S. Krieck and M. Westerhausen, Inorg. Chem., 2016, 55, 10741–10750 CrossRef PubMed.
- L. Horner, P. Beck and V. G. Toscano, Chem. Ber., 1961, 94, 1317–1322 CrossRef CAS.
- K. Issleib, B. Walther and E. Fluck, Z. Chem., 1968, 8, 67 CrossRef CAS.
- K. Goda, H. Gomi, M. Yoshifuji and N. Inamoto, Bull. Chem. Soc. Jpn., 1977, 50, 545–546 CrossRef CAS.
- B. Bildstein and F. Sladky, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 341–347 CrossRef CAS.
- S. Härling, J. Greiser, T. M. A. Al-Shboul, H. Görls, S. Krieck and M. Westerhausen, Aust. J. Chem., 2013, 66, 1264–1273 CrossRef.
- F. Dornhaus, H.-W. Lerner and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o4917 CrossRef CAS.
- A. F. Holleman, E. Wiberg and N. Wiberg, Inorganic Chemistry, Academic Press, San Diego, 1995 Search PubMed.
- S. M. Härling, S. Krieck, H. Görls and M. Westerhausen, Inorg. Chem., 2017, 56, 9255–9263 CrossRef PubMed.
- T. L. Gianetti, R. E. Rodriguez-Lugo, J. R. Harmer, M. Trincado, M. Vogt, G. Santiso-Quinones and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 15323–15328 CrossRef CAS PubMed.
- D. Semenzin, G. Etemad-Moghadam, D. Albouy, O. Diallo and M. Koenig, J. Org. Chem., 1997, 62, 2414–2422 CrossRef CAS PubMed.
- M. McFarlane and D. S. Rycroft, J. Chem. Soc., Chem. Commun., 1972, 902–903 RSC.
- G. Koshiro, G. Hideyuki, Y. Masaaki and I. Naoki, Bull. Chem. Soc. Jpn., 1977, 50, 545–546 CrossRef.
- R. P. Davies and M. G. Martinelli, Inorg. Chem., 2002, 41, 348–352 CrossRef CAS PubMed.
- A. J. Veinot, K. Ramgoolam, N. A. Giffin and J. D. Masuda, Molbank, 2017, 2017, M957 CrossRef.
- S. M. Härling, s-Block-Metall-vermittelte Hydrophosphoranylierung von Heterokumulenen und Alkinen, Ph.D. thesis, Jena/Germany, 2018 Search PubMed. Structural data of centrosymmetric [(tmeda)Na-OPMes2]2: P–O 156.39(15), P–C 186.88(19) and 187.2(2), av. Na–O 224.2 pm, C–P–C 100.32(8)°; molecular structure and atom labelling scheme are depicted in the ESI.† Crystallographic and refinement data have been deposited at the Cambridge Crystallographic Data Centre under CCDC 2184163.†.
- N. E. Mansfield, M. P. Coles and P. B. Hitchcock, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 2685–2702 CrossRef CAS.
- R. P. Davies and M. G. Martinelli, Inorg. Chem., 2002, 41, 348–352 CrossRef CAS PubMed.
- C. Bianchini, G. Lenoble, W. Oberhauser, S. Parisel and F. Zanobini, Eur. J. Inorg. Chem., 2005, 23, 4794–4800 CrossRef.
- (a) D. Schulze-Sünninghausen, J. Becker, M. R. M. Koos and B. Luy, J. Magn. Reson., 2017, 281, 151–161 CrossRef PubMed; (b) D. Schulze-Sünninghausen, J. Becker and B. Luy, J. Am. Chem. Soc., 2014, 136, 1242–1245 CrossRef PubMed.
- R. Hooft, COLLECT, Data Collection Software, Nonius B.V., Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, Processing of X-Ray Diffraction Data Collected in Oscillation Mode, in Methods in Enzymology, ed. C. W. Carter and R. M. Sweet, Macromolecular Crystallography, Part A, Academic Press, San Diego, USA, 1997, vol. 276, pp. 307–326 Search PubMed.
- (a) SADABS 2.10, Bruker-AXS Inc., Madison, WI, USA, 2002 Search PubMed; (b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, SADABS 2016/2, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- Bruker APEX3 Bruker AXS LLC, Madison, WI, USA, 2020 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- XP, Siemens Analytical X-ray Instruments Inc., Karlsruhe, Germany, 1990; Madison, WI, USA, 1994.
- POV-Ray, Persistence of Vision Raytracer, Victoria, Australia, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and IR spectra as well as the molecular structure of 1b. CCDC 2184155 for 1a, 2184156 for 1b, 2184157 for 2a, 2184158 for 2b, 2184159 for 3a, 2184160 for 3b, 2184161 for 4, 2184162 for 5, 2184163 for (tmeda)NaOPMes2, and 2254279 for 6. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00264d |
| This journal is © The Royal Society of Chemistry 2024 |