DOI:
10.1039/D4DT00286E
(Paper)
Dalton Trans., 2024,
53, 8555-8562
Effective photosensitized emission of a Tb(III) complex using a β-diketonate photosensitizer and an oxygen barrier system in a thermally populated triplet state†
Received
31st January 2024
, Accepted 20th March 2024
First published on 3rd April 2024
Abstract
Photosensitizer design of luminescent terbium (Tb(III)) complexes with narrow bandwidths is important for advancing luminescent materials. In this study, we report an effective photosensitizer model in a thermally populated lowest excited triplet (T1) state during Tb(III) emission. The Tb(III) complex comprises a Tb(III) ion (serving as an emission center), hexafluoroacetylacetonates (acting as photosensitizer ligands), and bulky cyclohexyl group-attached phosphine-oxide-type ligands (functioning as an oxygen barrier system). Emission properties including emission and excitation spectra, ligand-excited emission quantum yields, and emission lifetimes were evaluated in the absence and presence of oxygen. Coordination geometry structures were determined through analysing single-crystal structures. The electronic structure based on 4f-orbitals was estimated from radiative rate constants and quantum chemical calculations. The bulky phosphine oxide ligand not only provides an oxygen barrier system but also induces an electronic structural modulation based on 4f-orbitals, allowing for effective photosensitized Tb(III) emission in a thermally populated ligand T1 state in air.
Introduction
Trivalent terbium complexes (Tb(III)) have received considerable attention as green luminescent materials owing to their narrow luminescence bandwidths.1,2 Their enhanced luminescence properties result from a photosensitization effect based on energy transfer (EnT) from organic ligands to Tb(III).3 Designing ligands strategically for achieving effective photosensitized emission from Tb(III) complexes is crucial in advancing applications for luminescent materials, including light-emitting diodes,4–7 sensors,1,8–13 and security inks.14–16
In the photosensitized emission process, the organic ligands undergo intersystem crossing from the lowest excited singlet (S1) state to the lowest excited triplet (T1) state upon excitation. Subsequently, the ligands transfer their electronic energy to the Tb(III) emission level (5D4).2 Intrinsically long-lived 5D4 states (>0.5 ms) often cause emission quenching through back energy transfer (BEnT) from 5D4 to T1 states.2 Latva et al. performed photophysical analyses of Tb(III) complexes with amino-carboxylate-type ligands.17 They empirically determined that a sufficiently higher T1 level compared to the Tb(III) emission level (ΔET1-5D4 > 1850 cm−1) is essential for achieving efficient photosensitized emission. Based on these findings, energy-gap (ΔET1-5D4) control has been reported for highly luminescent Tb(III) complexes using benzoate-,18 β-diketonate-,19 and bipyridine20-type ligands. Recently, the importance of the first excited-state (7F5 level: 2050 cm−1) population of Tb(III) for energy transfer has been reported.21,22
On the other hand, further studies revealed the critical importance of the long-lived T1 state for overcoming emission quenching by BEnT and achieving efficient photosensitized Tb(III) emission, in situations characterized by a small energy gap (Fig. 1a). The long T1 lifetime allows for efficient Ln(III) emission in the thermally populated T1 state.23 Recently, we demonstrated an effective photosensitization model in the thermally populated T1 state during Tb(III) emission using a typical β-diketonate (hfa: hexafluoroacetylacetonate, T1 ∼ 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1, ΔET1-5D4 ∼ 1500 cm−1), with long-lived T1 state controlled by ancillary phosphine oxide ligands.24 The photosensitization model with a low T1 level is beneficial for enhancing the brightness of Tb(III) emission (ESI in ref. 25).25 However, the emission intensity of the Tb(III) luminophores is diminished by triplet oxygen quenching when using long-lived T1 photosensitizers.10,26–31
000 cm−1, ΔET1-5D4 ∼ 1500 cm−1), with long-lived T1 state controlled by ancillary phosphine oxide ligands.24 The photosensitization model with a low T1 level is beneficial for enhancing the brightness of Tb(III) emission (ESI in ref. 25).25 However, the emission intensity of the Tb(III) luminophores is diminished by triplet oxygen quenching when using long-lived T1 photosensitizers.10,26–31
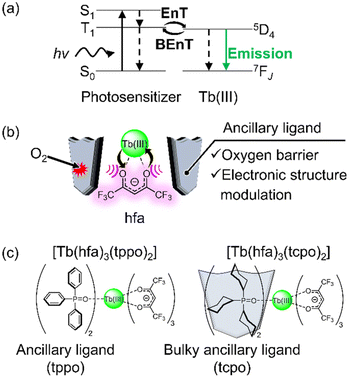 |
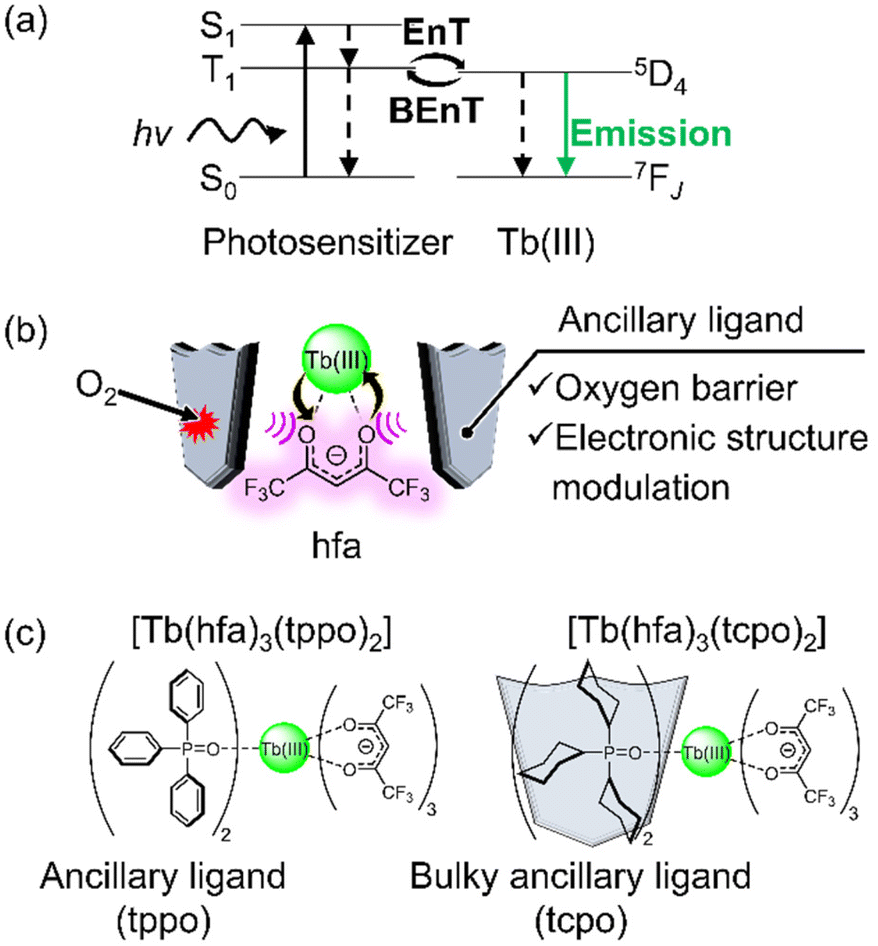
| | Fig. 1 (a) Excited-state dynamics of luminescent Tb(III) complexes with a small energy gap between T1 and 5D4 levels (EnT: energy transfer, BEnT: back energy transfer). (b) Illustration of the strategy for highly luminescent Tb(III) complexes under air conditions. (c) Chemical structures of Tb(III) complexes used in this study. | |
In this study, we report a luminescent Tb(III) complex model using hfa photosensitizer and ancillary ligand with oxygen barrier properties in the solid state (Fig. 1b). A mononuclear Tb(III) complex, [Tb(hfa)3(tcpo)2], was prepared and its photophysical properties were compared to those of [Tb(hfa)3(tppo)2] (Fig. 1c, tppo: triphenylphosphine oxide;32 tcpo: tricyclohexylphosphine oxide). It was found that the bulky cyclohexyl group not only shields the hfa ligand from oxygen but also provides effective modulation of the 4f-orbital-based electronic structure. Gd(III) complexes were also prepared to estimate the dependence of the T1 lifetimes on the oxygen concentration. This system provides new insights into the photosensitizer model with a long-lived excited state for highly luminescent Tb(III) complexes in a thermally populated ligand T1 state in air.
Experimental
Materials
Gadolinium(III) acetate hydrate (99.9%) was purchased from Sigma Aldrich Co., LLC. Terbium(III) acetate tetrahydrate (99.9%) was purchased from FUJIFILM Wako Pure Chemical Corporation. Hexafluoroacetylacetone (>95.0%) and triphenylphosphine oxide (>98.0%) were purchased from Tokyo Chemical Industry Co., Ltd. Tricyclohexylphosphine oxide (>98.0%) was purchased from Kanto Chemical Co., Inc. All other chemicals were reagent grade and used without further purification.
General methods
Electrospray ionization-mass spectrometry (ESI-MS) was performed using a JEOL JMS-T100LP instrument. Elemental analyses were performed using an Exeter Analytical CE440 instrument. Thermogravimetric analyses were performed on a Rigaku Thermo plus EVO2 TG-DTA8122 with Al2O3 as a reference. Infrared spectra were measured using a JASCO FT/IR-4600 instrument.
Preparation of [Ln(hfa)3(tppo)2] (Ln = Gd, Tb)
[Ln(hfa)3(tppo)2] was synthesized according to a previously reported method.32 The mixture of [Ln(hfa)3(H2O)2] (0.90 mmol) and triphenylphosphine oxide (tppo) (0.5 g, 1.80 mmol) was refluxed at 70 °C in methanol (10 mL) for 3 h. The reaction solution was cooled to approximately 20 °C, and the solvent was distilled using a rotary evaporator. The residue was recrystallized from methanol to produce colorless crystals of the target compounds.
[Gd(hfa)3(tppo)2]. Yield: 0.44 g (37%). MS (ESI) m/z: [M − hfa]+ calcd for C46H32F12GdO6P2 1128.07; found 1128.06. Anal.: calcd for C51H33F18GdO8P2: C, 45.89; H, 2.49; found: C, 45.69; H, 2.33%.
[Tb(hfa)3(tppo)2]. Yield: 0.53 g (44%). MS (ESI) m/z: [M − hfa]+ calcd for C46H32F12O6P2Tb 1129.07; found 1129.06. Anal.: calcd for C51H33F18O8P2Tb: C, 45.83; H, 2.49; found: C, 45.75; H, 2.35%.
Preparation of [Ln(hfa)3(tcpo)2] (Ln = Gd, Tb)
A mixture of [Ln(hfa)3(H2O)2] (0.84 mmol) and tricyclohexylphosphine oxide (tcpo) (0.5 g, 1.69 mmol) was refluxed at 70 °C in methanol (10 mL) for 3 h to form a white precipitate. The reaction solution was cooled to approximately 20 °C, and the solvent was distilled using a rotary evaporator. The residue was recrystallized from methanol to produce colorless crystals of the target compounds.
[Gd(hfa)3(tcpo)2]. Yield: 0.80 g (69%). MS (ESI) m/z: [M + Na]+ calcd for C51H69F18GdO8P2Na+ 1394.33; found 1394.34. Anal.: calcd for C51H69F18GdO8P2: C, 44.67; H, 5.07; found C, 44.59; H, 4.98%.
[Tb(hfa)3(tcpo)2]. Yield: 0.68 g (58%). MS (ESI) m/z: [M + Na]+ calcd for C51H69F18O8P2TbNa+ 1395.33; found 1395.33. Anal.: calcd for C51H69F18O8P2Tb: C, 44.62; H, 5.07; found: C, 44.59; H, 5.02%.
Crystallography
Rigaku MicroMax-007HF with Rigaku HyPix-6000HE and VariMax DW (Mo-Kα radiation (λ = 0.71073 nm)) was used for collecting crystallographic data for Tb(III) and Gd(III) complexes. All calculations were performed using Olex2 except for refinement.33 Non-hydrogen atoms were refined anisotropically using the SHELX system.34 Hydrogen atoms were refined using the riding model. CIF files are available in CCDC 2323896 (for Tb(hfa)3(tppo)2), CCDC 2324233 (for Tb(hfa)3(tcpo)2), CCDC 2324192 (for Gd(hfa)3(tppo)2), and CCDC 2324895 (for Gd(hfa)3(tcpo)2).†
Optical measurements
All optical measurements were carried out in the solid state. Emission and excitation spectra and emission decay profiles of Tb(III) complexes were recorded on a HORIBA Fluorolog-3 spectrofluorometer with a cryostat (Thermal Block Company, SA-SB1905HA, vacuum ultimate pressure of the vacuum pump: 6.7 × 10−2 Pa). For emission decay profiles of Tb(III) complexes, a SpectraLED-355 (λex = 356 nm, full width at half maximum (FWHM) = 17 nm) was used as an excitation source. Emission decay profiles of Gd(III) complexes were measured using third-harmonics of a Q-switched Nd:YAG laser (SpectraPhysics, INDI-50, FWHM = 5 ns, λ = 1064 nm) and a photomultiplier (Hamamatsu Photonics, R5108, response time < 1.1 ns). The Nd:YAG laser response was monitored with a digital oscilloscope (Sony Tektronix, TDS3052). Phosphorescence spectra of Gd(III) complexes and emission quantum yields of Tb(III) complexes were measured on a JASCO FP-6300 instrument. A cryostat (Thermal Block Company, SA-SB1905HA) was used for the phosphorescence spectra of Gd(III) complexes. A JASCO ILF-533 integrating sphere unit was used for emission quantum yields of Tb(III) complexes.
Computational details
All quantum chemical calculations were performed using density functional theory (DFT) using the Gaussian 16 package.35 Energy calculations of Tb(III) complexes were evaluated with the long-range corrected BLYP functional (basis set: Stuttgart RSC 1997 for Tb atoms and cc-pVDZ for C, H, O, F, and P atoms).36–38
Results and discussion
Structures of [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2]
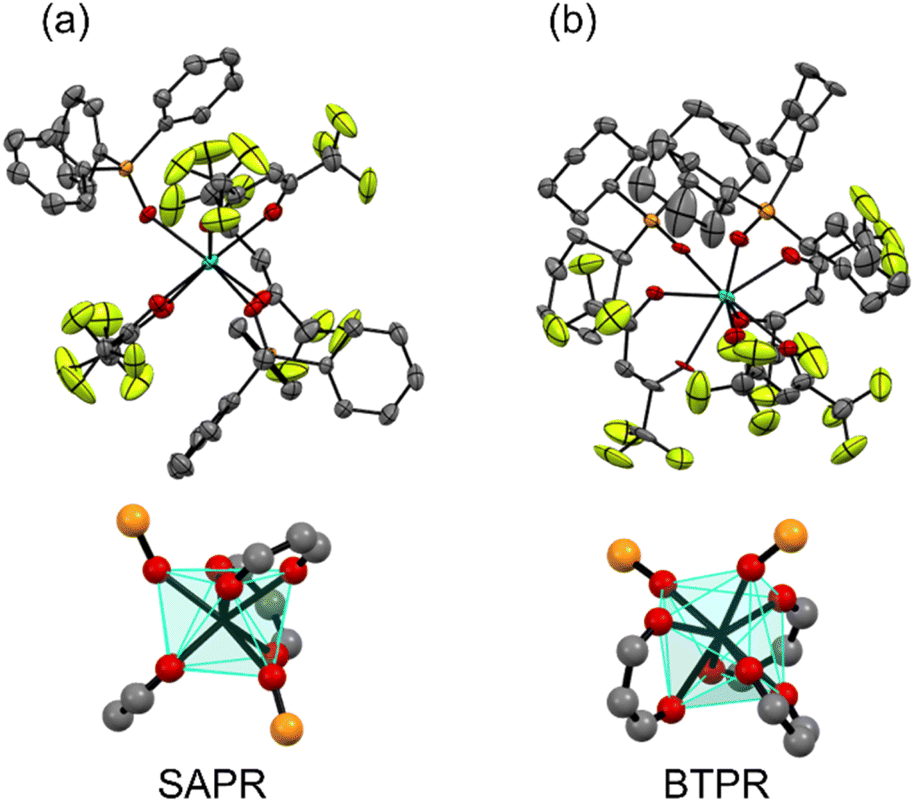
[Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2] were synthesized through refluxing the precursor complex [Tb(hfa)3(H2O)2] with the respective phosphine oxide ligand in methanol. Single crystals of Tb(III) complexes were obtained through recrystallization from methanol. The crystal structures of the complexes are shown in Fig. 2 and their corresponding crystal data are presented in Table 1. Both Tb(III) complexes exhibited eight-coordination structures with three hfa ligands and two phosphine oxide ligands. In both [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2], multiple intra- and intermolecular CH/F interactions were observed. The rigid structures are linked to high thermostability (decomposition point >260 °C, Fig. S1†). In [Tb(hfa)3(tcpo)2], two phosphine oxide ligands were located on the same side of the mononuclear complex as bidentate ligands (Fig. 2b),39 which is distinct from the crystal structure of [Tb(hfa)3(tppo)2] (Fig. 2a). We performed continuous shape measure (CShM) analysis to elucidate the coordination geometry of the unit.40–42 The CShM factor S was calculated to quantitatively estimate the coordination geometry. Based on CShM analysis, the polyhedral structures of [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2] were classified as square antiprism (SAPR) and bicapped trigonal prism (BTPR) structures, respectively (Fig. 2, Table S1†). Additionally, Gd(III) complexes were prepared using the same procedure as that for the Tb(III) complexes. The crystal structures of [Gd(hfa)3(tppo)2] and [Gd(hfa)3(tcpo)2] closely resembled those of [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2], respectively (Fig. S2, Table S2†)
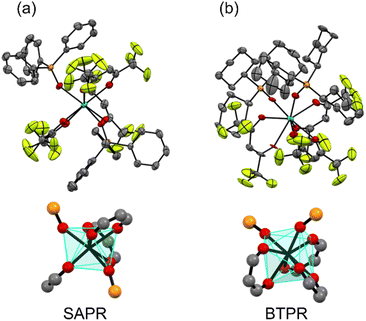 |
| | Fig. 2 ORTEP drawings (top) and coordination geometries (bottom) of the Tb(III) complexes: (a) [Tb(hfa)3(tppo)2] and (b) [Tb(hfa)3(tcpo)2]. Ellipsoid probability was set at 50%. Hydrogen atoms are omitted for clarity. | |
Table 1 Crystal data of [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2]
| |
[Tb(hfa)3(tppo)2] |
[Tb(hfa)3(tcpo)2] |
| Chemical formula |
C51H33F18O8P2Tb |
C51H69F18O8P2Tb |
| Molecular weight |
1336.63 |
1372.92 |
| Crystal system |
Monoclinic |
Orthorhombic |
| Space group |
P21/n |
Pca21 |
|
a/Å |
17.0264(3) |
17.9100(6) |
|
b/Å |
15.4243(3) |
18.7471(6) |
|
c/Å |
20.4521(4) |
17.3371(5) |
|
α/° |
90 |
90 |
|
β/° |
93.879(2) |
90 |
|
γ/° |
90 |
90 |
| Volume/Å3 |
5358.83(18) |
5821.1(3) |
|
Z
|
4 |
4 |
| Density/g cm−3 |
1.657 |
1.567 |
| Temperature/°C |
−150 |
−150 |
|
R
1
|
0.0400 |
0.0804 |
| wR2 |
0.0937 |
0.2366 |
General photophysical properties
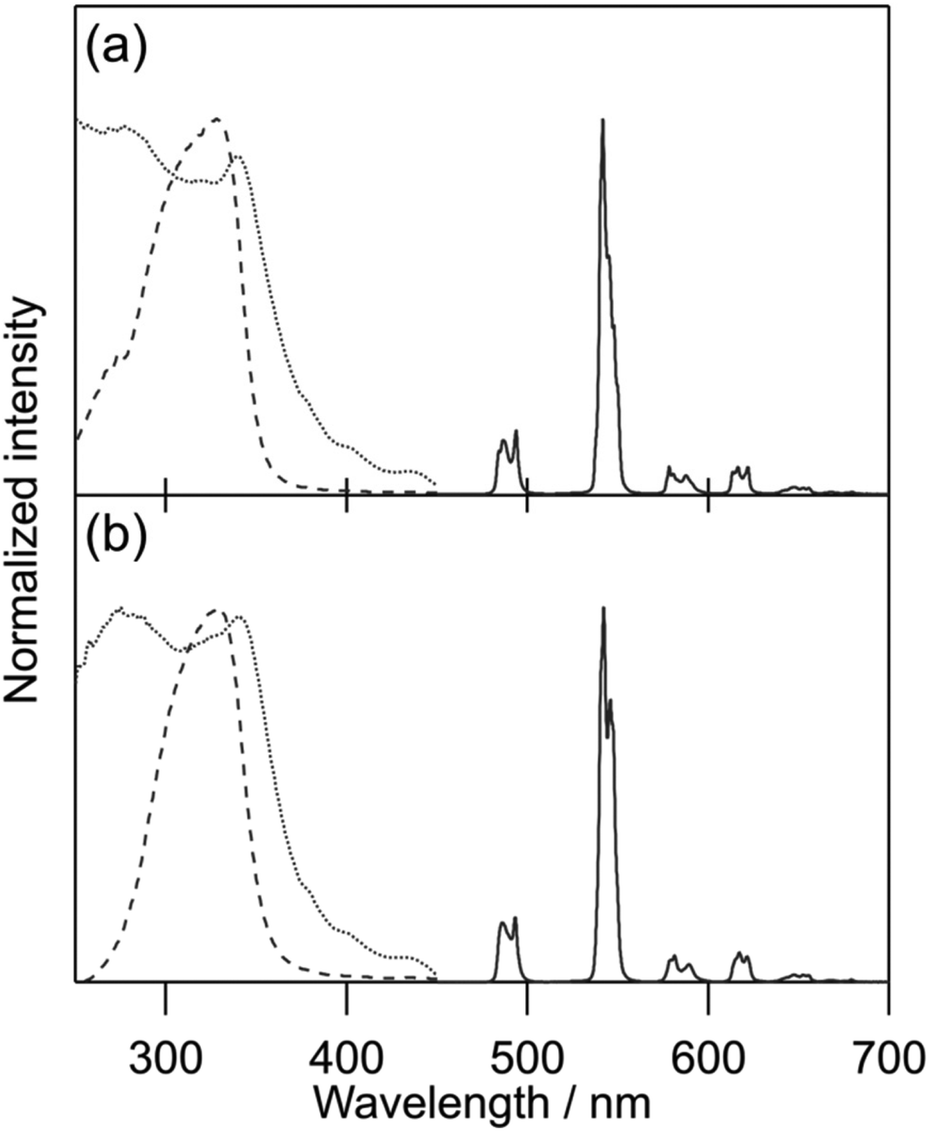
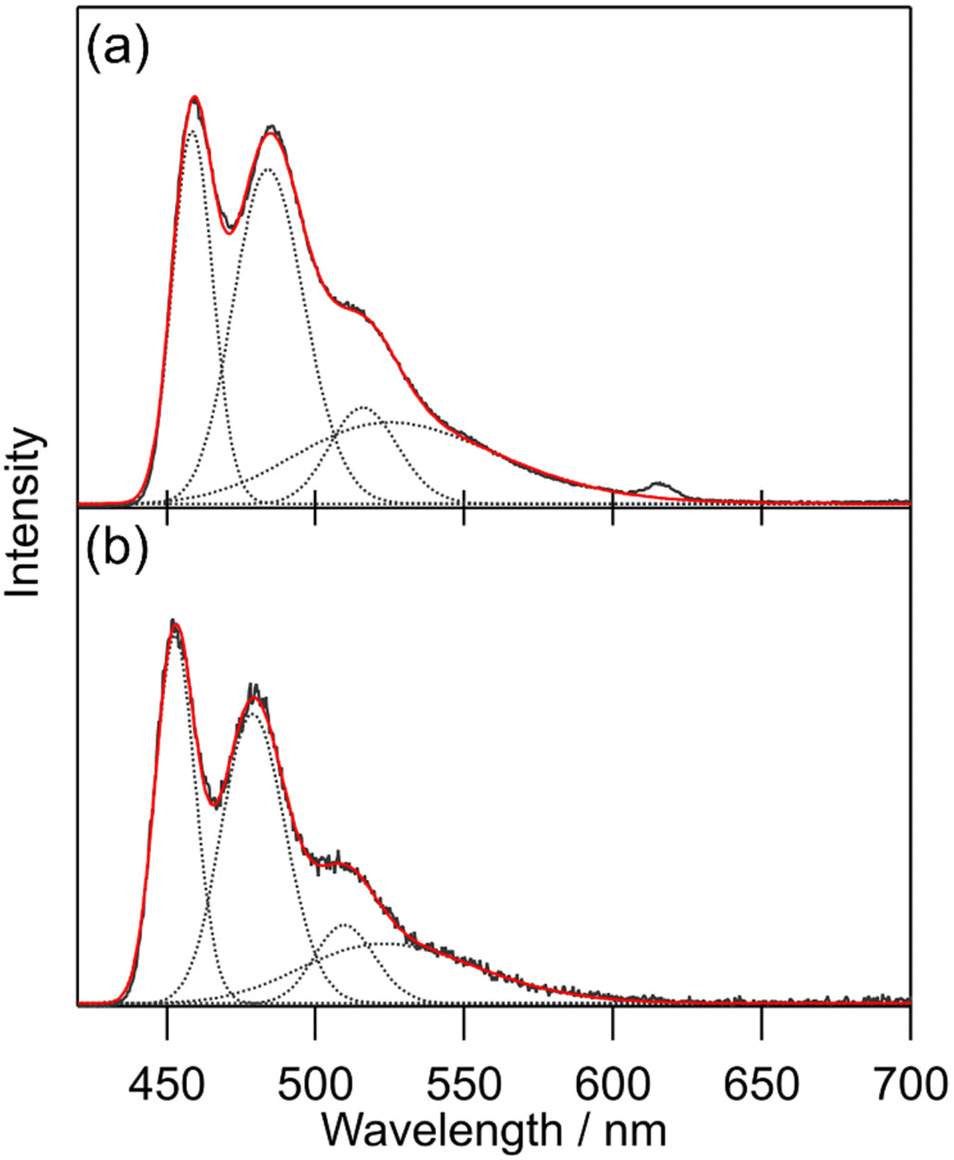
General photophysical properties of [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2] (solid state) were measured in air. Emission, excitation, and diffuse reflectance spectra of the Tb(III) complexes are shown in Fig. 3. Sharp emission bands were observed at around 490, 550, 580, 620, and 650 nm, which are attributed to the 5D4 → 7FJ (J = 6, 5, 4, 3, 2) transition of Tb(III), respectively.1 In the excitation spectra, broad excitation bands attributable to hfa ligands were observed in the ultraviolet region. Emission lifetime (τavg) and ligand-excited emission quantum yield (Φtot) were evaluated in air (Table 2). To estimate the hfa ligand T1 levels, we measured the phosphorescence spectra of [Gd(hfa)3(tppo)2] and [Gd(hfa)3(tcpo)2] (Fig. 4). The phosphorescence spectra were deconvoluted into four vibronic bands and the deconvolution results for the four vibronic bands were designated as 0–0, 0–1, 0–2, and 0–3. The hfa ligand T1 levels (energy gap ΔET1-5D4) were determined based on 0–0 vibronic bands using a wavenumber scale of 21800 cm−1 (1300 cm−1) for [Gd(hfa)3(tppo)2] and 22100 cm−1 (1600 cm−1) for [Gd(hfa)3(tcpo)2].
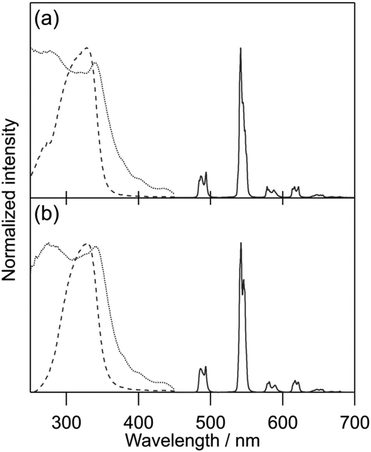 |
| | Fig. 3 Emission (solid line, λex = 360 nm), excitation (dotted line, λem = 542 nm), and diffuse reflectance (dashed line) spectra of Tb(III) complexes in air: (a) [Tb(hfa)3(tppo)2] and (b) [Tb(hfa)3(tcpo)2]. | |
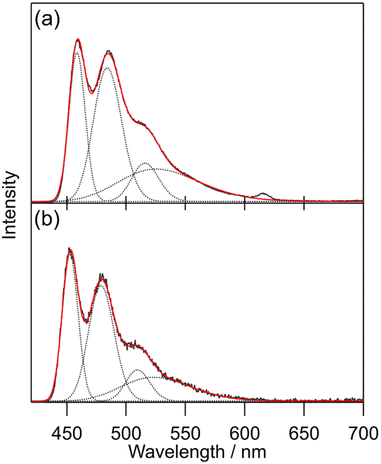 |
| | Fig. 4 Phosphorescence spectra (λex = 350 nm, 100 K, under vacuum, delay = 20 ms) of the Gd(III) complexes (black lines): (a) [Gd(hfa)3(tppo)2] and (b) [Gd(hfa)3(tcpo)2] with peak deconvolution using the Gaussian function (dotted lines). The red lines are the summations of Gaussian functions. | |
Table 2 Photophysical parameters of [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2]
| Entry |
k
ra/s−1 |
k
nrb/s−1 |
k
qc/s−1 |
τ
avgd/μs |
Φ
tote/% |
| Vacuum |
Vacuum |
— |
Air |
Vacuum |
O2 |
Air |
Ar |
|
k
r = 1/τ90 K (vacuum).18,19,43τ90 K: λex = 356 nm, λem = 542 nm, 90 K.
k
nr = 1/τ (vacuum, λex = 495 nm) − kr.
k
q = 1/τ (air) − 1/τ (vacuum).
λ
ex = 356 nm, λem = 542 nm, 293 K. It was estimated by fitting with a double-exponential function. The standard deviations were determined by five measurements.
λ
ex = 370 nm. The standard deviations were determined by measuring five times.
|
| [Tb(hfa)3(tppo)2] |
1.3 × 103 |
8.8 × 103 |
3.8 × 102 |
101 ± 1 |
105 ± 0.4 |
82 ± 0.2 |
9.3 ± 0.3 |
11.2 ± 0.1 |
| [Tb(hfa)3(tcpo)2] |
1.1 × 103 |
1.1 × 103 |
6.2 × 10 |
453 ± 1 |
466 ± 1 |
398 ± 1 |
31.9 ± 0.1 |
33.3 ± 0.3 |
Back energy transfer (BEnT) under vacuum
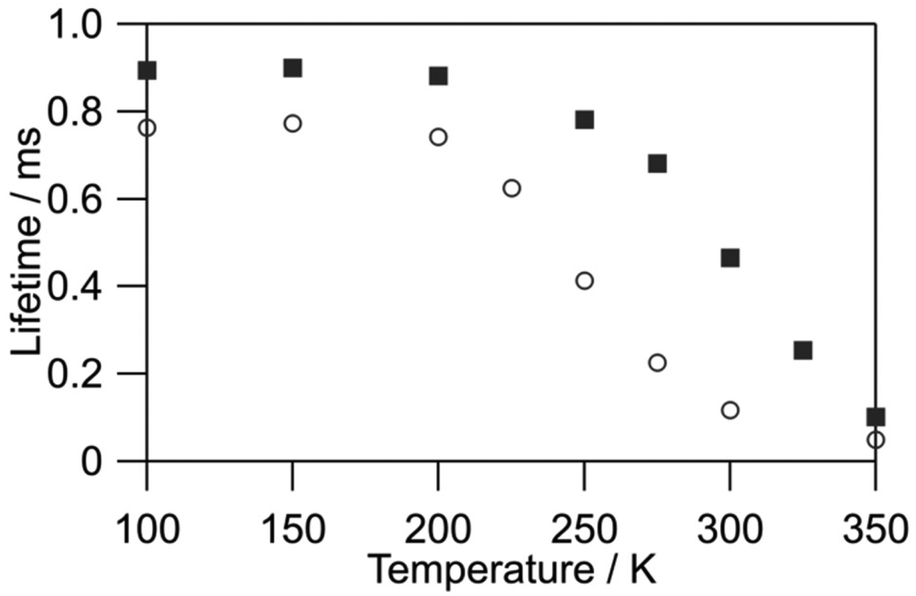
We evaluated the value of kr for [Tb(hfa)3(tppo)2] (kr = 1.3 × 103 s−1) and [Tb(hfa)3(tcpo)2] (kr = 1.1 × 103 s−1) using the emission lifetime at 90 K using a previously reported approximate method.18,19,43 The variation between kr values is attributed to the electronic structure differences between [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2]. The non-radiative rate constants knr were estimated based on kr values and emission decay analyses under vacuum (Fig. S5†). The electronic structure changes also induced a significant difference in knr values of [Tb(hfa)3(tppo)2] (knr = 8.8 × 103 s−1) and [Tb(hfa)3(tcpo)2] (knr = 1.0 × 103 s−1), indicating that BEnT is inefficient in [Tb(hfa)3(tcpo)2] compared with that in [Tb(hfa)3(tppo)2]. This interpretation is consistent with temperature-dependent emission lifetime measurements under vacuum (Fig. 5). The emission lifetime of [Tb(hfa)3(tppo)2] changed significantly in the lower temperature region, suggesting that BEnT from the 5D4 emitting state is more pronounced compared with that of [Tb(hfa)3(tcpo)2]. Under oxygen-free conditions, the ligand-excited emission quantum yield of [Tb(hfa)3(tcpo)2] (Φtot = 33.3%) was significantly higher than that of [Tb(hfa)3(tppo)2] (Φtot = 11.2%). This is also ascribed to the suppression of BEnT in [Tb(hfa)3(tcpo)2].
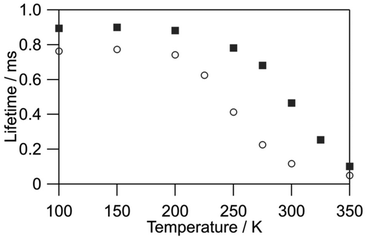 |
| | Fig. 5 Temperature-dependent emission lifetimes of [Tb(hfa)3(tppo)2] (circles) and [Tb(hfa)3(tcpo)2] (squares) (λex = 356 nm, λem = 542 nm, under vacuum). | |
To investigate the BEnT mechanism, we evaluated the emission lifetimes of the Gd(III) complexes. The emission lifetimes under vacuum for [Gd(hfa)3(tppo)2] and [Gd(hfa)3(tcpo)2] were estimated to be 237 and 52 μs, respectively, using emission decay analysis (Fig. S8†). The phosphorescence properties imply that, ideally, [Tb(hfa)3(tppo)2] should show an effective photosensitized Tb(III) emission due to a long-lived T1 state. However, [Tb(hfa)3(tcpo)2] was found to demonstrate effective photosensitized Tb(III) emission with a larger Φtot. To further clarify the origins, we evaluated the photophysical properties of the Tb(III) and Gd(III) complexes in the presence of oxygen.
BEnT mechanism in a thermally populated T1 state during Tb(III) emission
The energy transfer from the T1 state of an organic ligand to Tb(III) is reversible when the T1 state is thermally re-populated from the Tb(III) 5D4 state. This re-population of the T1 state competes with Tb(III) luminescence.26 When the EnT rate between T1 and 5D4 states is significantly higher than the deactivation rates of T1 → S0 and 5D4 → 7FJ transitions, investigating the oxygen concentration dependence of Tb(III) emission lifetimes is useful for examining the population of the T1 level during Tb(III) emission.10,26–31,44
This measurement relies on the distinct sensitivity property towards O2, that is the ligand T1 state exhibits sensitivity to O2, while the Tb(III) 5D4 state is insensitive to O2. The decrease of emission lifetimes with the increase of oxygen concentration was observed both in [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2] (Table 2, Fig. S5†), revealing the T1 state population during Tb(III) emission. As indicated by the photophysical analysis of [Gd(hfa)3(tcpo)2] (τavg = 52 μs), [Tb(hfa)3(tcpo)2] exhibits a shorter T1 lifetime. Thus, the crucial factor contributing to the emission quantum yield of [Tb(hfa)3(tcpo)2] being higher than that of [Tb(hfa)3(tppo)2] is the difference between EnT and BEnT rates between the two Tb(III) complexes. These energy transfer rate differences between the Tb(III) complexes are attributed to the higher T1 level of [Tb(hfa)3(tcpo)2] (T1 = 22100 cm−1) compared to that of [Tb(hfa)3(tppo)2] (T1 = 21800 cm−1). Based on the different 4f–4f radiative rate constant values, the 4f-orbital-based electronic structures of [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2] are also different (Table 2). We consider that the 4f-orbital-based electronic structural differences also affect the energy transfer rates. To investigate the effect of the 4f-orbital-based electronic structure on energy transfer rates, the activation energy Ea and frequency factor A in the energy transfer were estimated from an Arrhenius plot using the temperature-dependent emission lifetime (Fig. S7, Table S10†).45 In particular, the A value, which incorporates electronic frequency, is associated with the electronic coupling of 5D4 with the T1 state.46,47 As a consequence, the Ea values were calculated to be identical (2000 cm−1) in Tb(III) complexes, whereas the A values were calculated to be 1.2 × 108 s−1 for [Tb(hfa)3(tppo)2] and 1.9 × 107 s−1 for [Tb(hfa)3(tcpo)2] (Table S10†). The consistent Ea values stem from a balance between different ΔET1-5D4 ([Tb(hfa)3(tppo)2] < [Tb(hfa)3(tcpo)2]) and contrasting T1 lifetimes ([Tb(hfa)3(tppo)2] > [Tb(hfa)3(tcpo)2]). The smaller A value in [Tb(hfa)3(tcpo)2] indicates less electronic coupling. As 5D4 weakly interacts with the T1 state in [Tb(hfa)3(tcpo)2], BEnT is passivated, therefore, effective photosensitized Tb(III) emission is observed.
Evaluation of the oxygen barrier ability
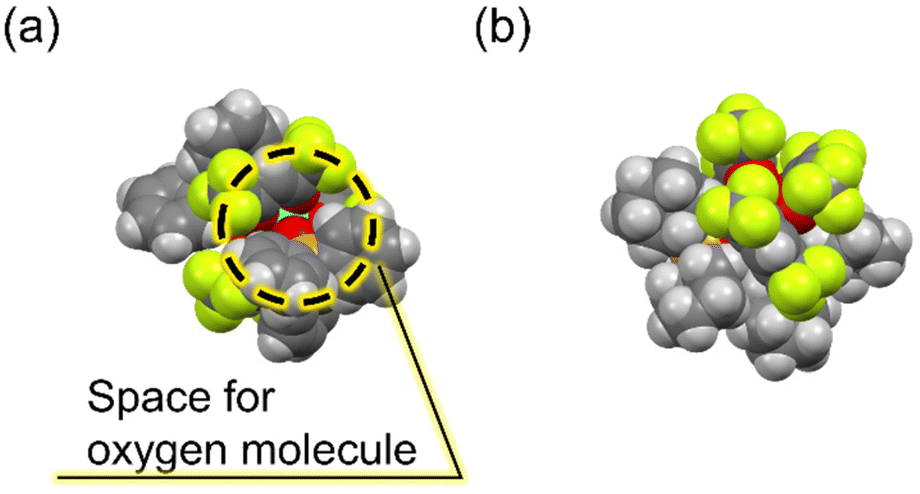
Notably, despite the emission quenched by oxygen in the populated T1 state during Tb(III) emission, [Tb(hfa)3(tcpo)2] exhibits excellent emission properties in air and under oxygen. To elucidate the oxygen barrier ability, the indirect emission quenching constant of oxygen (kq) was evaluated using the Tb(III) emission lifetimes in air and under vacuum (kq = 1/τ (air) − 1/τ (vacuum)). The kq value of [Tb(hfa)3(tcpo)2] was calculated to be 6.2 × 10 s−1, significantly smaller than that of [Tb(hfa)3(tppo)2] (3.8 × 102 s−1) (Table 2). The kq is substantially reduced by the presence of bulky tcpo ligands, which is consistent with the calculated kq in [Gd(hfa)3(tcpo)2] (Table 3). The energy transfer efficiency is expected to be dependent on the oxygen concentration, although the energy transfer efficiency cannot be estimated precisely at present.46 It has been reported that quenching of the ligand T1 state by oxygen originates from orbital overlap between the π-conjugated orbitals of hfa and molecular oxygen.10 The bulky tcpo ligands inhibit orbital overlapping, providing an oxygen barrier ability (Fig. 6 and S9†). Consequently, we have successfully demonstrated a luminescent Tb(III) complex model in a thermally populated T1 state with an oxygen barrier system based on ancillary ligands.
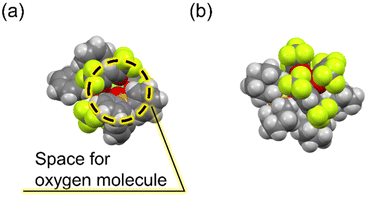 |
| | Fig. 6 Space-fill drawing models of (a) [Tb(hfa)3(tppo)2] and (b) [Tb(hfa)3(tcpo)2]. | |
Table 3 Photophysical parameters of [Gd(hfa)3(tppo)2] and [Gd(hfa)3(tcpo)2]
| Entry |
τ
avga,b/μs |
k
q/s−1 |
T1 levelc/cm−1 |
| Air |
Vacuum |
O2 |
|
λ
ex = 355 nm, λem = 500 nm (using a 510 nm short-pass filter with a 490 nm long-pass filter), 293 K.
It was estimated by fitting with a double-exponential function. The standard deviations were determined by ten measurements.
Estimated from phosphorescence spectra.
|
| [Gd(hfa)3(tppo)2] |
64 ± 1 |
237 ± 1 |
16 ± 0.2 |
1.1 × 104 |
21800 |
| [Gd(hfa)3(tcpo)2] |
50 ± 0.3 |
52 ± 0.5 |
29 ± 0.2 |
7.3 × 102 |
22100 |
Quantum chemical calculations
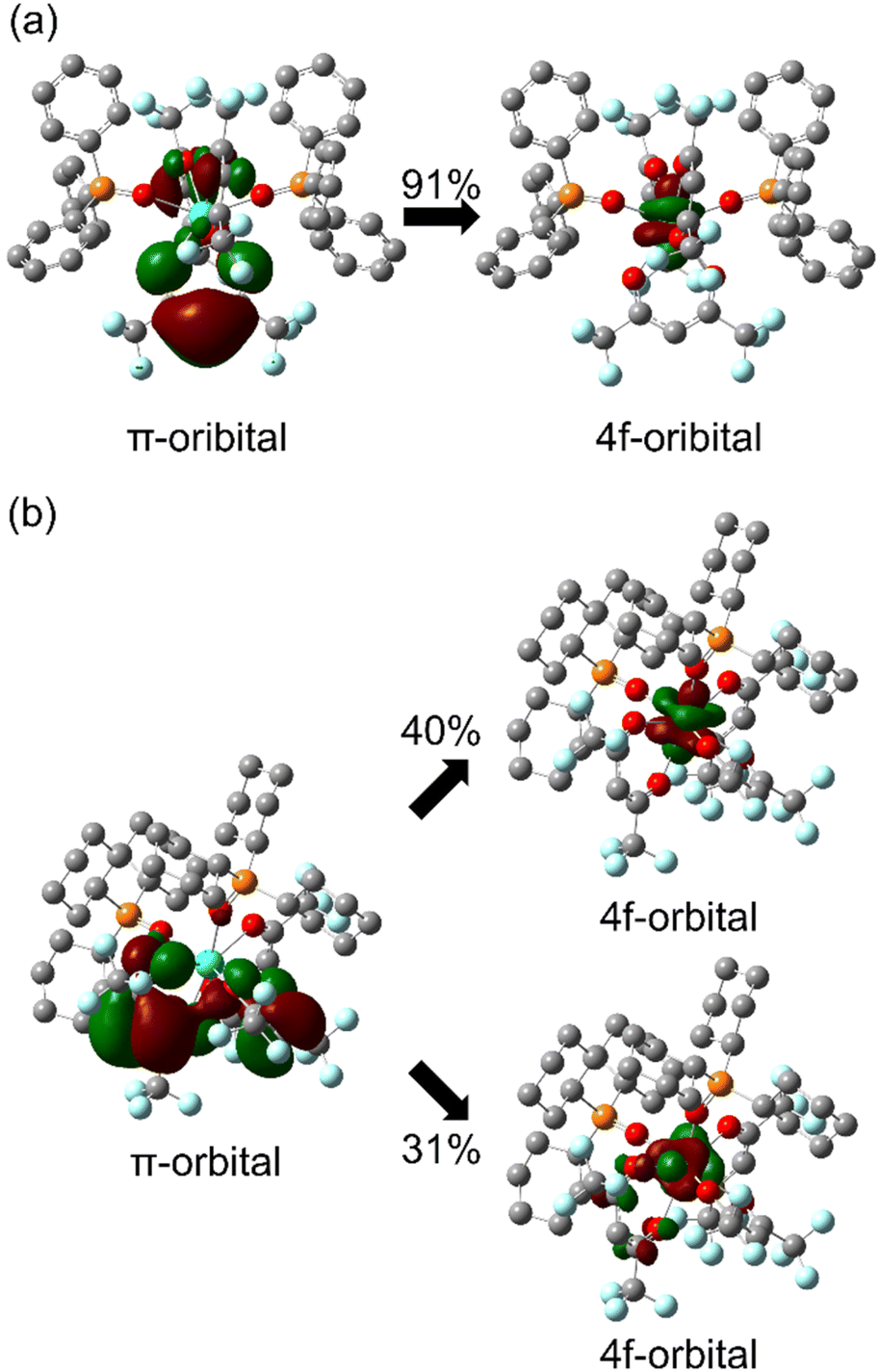
In photophysical analyses of the Tb(III) complexes, different kr values were considered to result from the 4f-orbital-based electronic structure differences between [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2]. The 4f–4f transition characteristics are affected by ligand polarization48–53 and/or the charge transfer excited state between the π-orbital and the 4f-orbital.54,55 According to the dynamic coupling model, the 4f–5d excited states are mixed with the 4f–4f excited state by the interaction between the induced electronic dipole moment of the ligands and the transition electric quadrupole moment of the lanthanide. Thus, the 4f–4f oscillator strength f depends on the ligand polarizability, and can be approximately described as follows:56where α and R are the ligand polarizability and the distance between the coordination atom and lanthanide ion, respectively. To further clarify the Tb(III) 4f-orbital-based electronic structural differences, the quantum chemical calculations were performed for [Tb(hfa)3(tppo)2] and [Tb(hfa)3(tcpo)2] using crystal structures (ULC-BLYP/Stuttgart RSC 1997 for Tb atoms, cc-pVDZ for C, H, O, F, and P atoms).35–38 The summation of the α2R−8 value of [Tb(hfa)3(tppo)2] did not differ from that of [Tb(hfa)3(tcpo)2] (Table S13†).
In contrast, Henrie reported an equation for the 4f–4f oscillator strength fpt borrowed from the charge-transfer (CT) transition between the π and 4f orbitals as follows:54
where
ECT and
fCT are the excitation energy of the CT state and the oscillator strength of the CT transition, respectively. The lowest energy level in the CT transition originates from the ligand (π-orbital) to metal (4f-orbital) charge transfer (LMCT) transition (
Fig. 7, Tables S14 and S15
†). The 4f-orbital shapes depend on the coordination geometry. The average of calculated parameter (
ECT−3fCT) of [Tb(hfa)
3(tppo)
2] (1.05 × 10
−16 cm
3) is significantly larger than that of [Tb(hfa)
3(tcpo)
2] (1.11 × 10
−17 cm
3) (
Table 4). The differences in the
kr values and EnT rate of the Tb(
III) complexes might be attributed to the electronic structure difference in the perturbations of the LMCT states to the 4f–4f states, in addition to the difference in the coordination geometry.
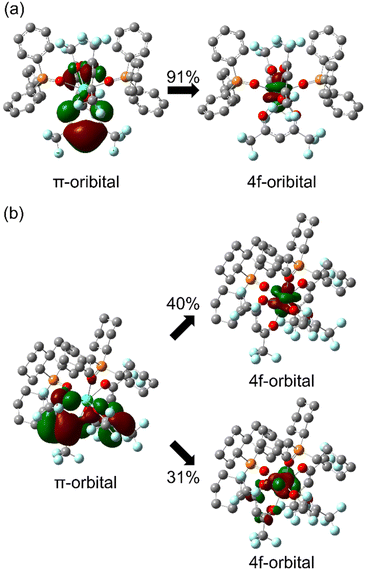 |
| | Fig. 7 Molecular orbital characteristics of the lowest LMCT transition of Tb(III) complexes: (a) [Tb(hfa)3(tppo)2] and (b) [Tb(hfa)3(tcpo)2]. | |
Table 4 Parameters expressing the contribution of the LMCT state with respect to the 4f–4f states of the Tb(III) complexes for oscillator strength of the 4f–4f transition
| Entry |
E
CT
−3
f
CTa/cm3 |
|
Average of the approximate parameters calculated from excitation energies and oscillator strengths for the bottom 20 LMCT levels (Tables S14 and S15†).
|
| [Tb(hfa)3(tppo)2] |
1.05 × 10−16 |
| [Tb(hfa)3(tcpo)2] |
1.11 × 10−17 |
Conclusion
In this study, an effective photosensitized emission was demonstrated by fabricating a Tb(III) complex with a typical hexafluoroacetylacetonate photosensitizer in the thermally populated T1 state. The bulky ancillary ligand structure acts as a modulator of the 4f-orbital-based electronic structure and an oxygen barrier to suppress orbital overlap between the β-diketonate photosensitizer and molecular oxygen. These results provide new insights into the photosensitizer model based on thermally populated T1 states during Tb(III) emission for the improvement of brightness in luminescent Tb(III) complexes in the presence of oxygen, such as in air. Studies on oxygen barrier control methods using polyaromatic photosensitizers for bright lanthanide emission25 are currently in progress.
Author contributions
Yuichi Kitagawa designed this research. Kota Inage performed all the syntheses, optical measurements, and structural analyses and wrote the paper. Kota Inage, Mengfei Wang, Yasuchika Hasegawa, and Yuichi Kitagawa discussed this work. All the authors have reviewed the final version of the paper.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was partially supported by a grant-in-aid with JSPS KAKENHI Grant Numbers JP20H02748, JP20H04653, JP20H05197, JP20K21201, JP21K18969, JP22H02152, JP22H04516, and JP23K17925. This work is supported by the Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from the Japan Science and Technology Agency (JST), Japan Grant Number JPMJTR23T5. This work was also supported by the Institute for Chemical Reaction Design and Discovery (ICReDD), established by the World Premier International Research Center Initiative (WPI) of the MEXT, Japan.
References
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- A. B. Dias, Dalton Trans., 2007, 2229–2241 RSC.
- L. Wang, Z. Zhao, C. Wei, H. Wei, Z. Liu, Z. Bian and C. Huang, Adv. Opt. Mater., 2019, 7, 1801256 CrossRef.
- N. Sun, L. Li, Y. Yang, A. Zhang, H. Jia, X. Liu and B. Xu, Opt. Mater., 2015, 49, 39–45 CrossRef CAS.
- S. F. H. Correia, R. L. Fernandes, L. Fu, M. M. Nolasco, L. D. Carlos and R. A. S. Ferreira, Eur. J. Inorg. Chem., 2020, 2020, 1736–1742 CrossRef CAS.
- C. D. S. Brites, S. Balabhadra and L. D. Carlos, Adv. Opt. Mater., 2019, 7, 1801239 CrossRef.
- K. Miyata, Y. Konno, T. Nakanishi, A. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed., 2013, 52, 6413–6413 CrossRef CAS.
- Y. Kitagawa, T. Nakai, S. Hosoya, S. Shoji and Y. Hasegawa, ChemPlusChem, 2023, 88, e202200445 CrossRef CAS.
- K. Iman and M. Shahid, New J. Chem., 2019, 43, 1094–1116 RSC.
- D. Parker, J. D. Fradgley and K. L. Wong, Chem. Soc. Rev., 2021, 50, 8193–8213 RSC.
- D. Parker, Coord. Chem. Rev., 2000, 205, 109–130 CrossRef CAS.
- Z. Gao, B. Xu, T. Zhang, Z. Liu, W. Zhang, X. Sun, Y. Liu, X. Wang, Z. Wang, Y. Yan, F. Hu, X. Meng and Y. S. Zhao, Angew. Chem., Int. Ed., 2020, 59, 19060–19064 CrossRef CAS PubMed.
- Z. Gao, S. Yang, B. Xu, T. Zhang, S. Chen, W. Zhang, X. Sun, Z. Wang, X. Wang, X. Meng and Y. S. Zhao, Angew. Chem., Int. Ed., 2021, 60, 24519–24525 CrossRef CAS.
- J. Andres, R. D. Hersch, J. E. Moser and A. S. Chauvin, Adv. Funct. Mater., 2014, 24, 5029–5036 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- A. R. Ramya, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, Inorg. Chem., 2010, 49, 2407–2415 CrossRef CAS.
- K. Yanagisawa, T. Nakanishi, Y. Kitagawa, T. Seki, T. Akama, M. Kobayashi, T. Taketsugu, H. Ito, K. Fushimi and Y. Hasegawa, Eur. J. Inorg. Chem., 2015, 2015, 4769–4774 CrossRef CAS.
- P. R. Matthes, J. Nitsch, A. Kuzmanoski, C. Feldmann, A. Steffen, T. B. Marder and K. Müller-Buschbaum, Chem. – Eur. J., 2013, 19, 17369–17378 CrossRef CAS.
- A. S. Souza, L. A. Nunes, M. C. F. C. Felinto, H. F. Brito and O. L. Malta, J. Lumin., 2015, 167, 167–171 CrossRef CAS.
- A. N. C. Neto, E. Kasprzycka, A. S. Souza, P. Gawryszewska, M. Suta, L. D. Carlos and O. L. Malta, J. Lumin., 2022, 248, 118933 CrossRef.
- Y. Kitagawa, F. Suzue, T. Nakanishi, K. Fushimi, T. Seki, H. Ito and Y. Hasegawa, Commun. Chem., 2020, 3, 3 CrossRef CAS.
- Y. Kitagawa, R. Moriake, T. Akama, K. Saito, K. Aikawa, S. Shoji, K. Fushimi, M. Kobayashi, T. Taketsugu and Y. Hasegawa, ChemPlusChem, 2022, 87, e202200151 CrossRef CAS.
- Y. Kitagawa, K. Shima, T. Nakai, M. Kumagai, S. Omagari, P. P. Ferreira da Rosa, S. Shoji, K. Fushimi and Y. Hasegawa, Commun. Chem., 2023, 6, 122 CrossRef CAS.
- N. Sabbatini, M. Guardigli, I. Manet, F. Bolletta and R. Ziessel, Inorg. Chem., 1994, 33, 955–959 CrossRef CAS.
- A. Beeby, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1999, 2, 493–504 RSC.
- S. Blair, R. Kataky and D. Parker, New J. Chem., 2002, 26, 530–535 RSC.
- G. L. Law, R. Pal, L. O. Palsson, D. Parker and K. L. Wong, Chem. Commun., 2009, 7321–7323 RSC.
- J. Lehr, M. Tropiano, P. D. Beer, S. Faulkner and J. J. Davis, Chem. Commun., 2015, 51, 15944–15947 RSC.
- T. J. Sørensen, A. M. Kenwright and S. Faulkner, Chem. Sci., 2015, 6, 2054–2059 RSC.
- S. Katagiri, Y. Tsukahara, Y. Hasegawa and Y. Wada, Bull. Chem. Soc. Jpn., 2007, 80, 1492–1503 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Cryst. A, 2008, 64, 112–122 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallin, 2016 Search PubMed.
- M. Dolg, H. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- K. Miyata, T. Nakagawa, R. Kawakami, Y. Kita, K. Sugimoto, T. Nakashima, T. Harada, T. Kawai and Y. Hasegawa, Chem. – Eur. J., 2011, 17, 521–528 CrossRef CAS.
-
SHAPE, version 2.1. Continuous shape measures calculations. Electronic Structure Group, Universitat de Barcelona, Spain, 2010 Search PubMed.
- D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. – Eur. J., 2005, 11, 1479–1494 CrossRef CAS PubMed.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- R. Pavithran, N. S. S. Kumar, S. Biju, M. L. P. Reddy, S. A. Junior and R. O. Freire, Inorg. Chem., 2006, 45, 2184–2192 CrossRef CAS.
- R. Hueting, M. Tropiano and S. Faulkner, RSC Adv., 2014, 4, 44162–44165 RSC.
- K. Yanagisawa, Y. Kitagawa, T. Nakanishi, T. Akama, M. Kobayashi, T. Seki, K. Fushimi, H. Ito, T. Taketsugu and Y. Hasegawa, Eur. J. Inorg. Chem., 2017, 2017, 3843–3848 CrossRef CAS.
- S. Omagari and M. Vacha, Phys. Chem. Chem. Phys., 2020, 22, 3683–3690 RSC.
- V. M. Korshunov, M. A. Kiskin and I. V. Taydakov, J. Lumin., 2022, 251, 119235 CrossRef CAS.
- R. D. Peacock, J. Mol. Struct., 1978, 46, 203–207 CrossRef CAS.
- S. F. Mason, R. D. Peacock and B. Stewart, Chem. Phys. Lett., 1974, 29, 149–153 CrossRef CAS.
- M. F. Reid and F. Richardson, J. Phys. Chem., 1984, 88, 3579–3586 CrossRef CAS.
- M. F. Reid and F. S. Richardson, J. Chem. Phys., 1983, 79, 5735–5742 CrossRef CAS.
- L. Hu, M. F. Reid, C.-K. Duan, D. J. Newman and G. Balasubramanian, J. Phys. C: Solid State Phys., 1975, 8, 37–44 CrossRef.
- M. F. Reid and F. S. Richardson, Chem. Phys. Lett., 1983, 95, 113–118 CrossRef.
- D.-E. Henrie, R.-L. Fellows and G. R. Choppin, Coord. Chem. Rev., 1976, 18, 199–224 CrossRef CAS.
- Y. Kitagawa, P. P. Ferreira Da Rosa and Y. Hasegawa, Dalton Trans., 2021, 50, 14978–14984 RSC.
- M. Hatanaka and S. Yabushita, Chem. Phys. Lett., 2011, 504, 193–198 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc and
Yuichi
Kitagawa
*bc and
Yuichi
Kitagawa