
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi(2,2′:6′,2′′-terpyridine)2: a high-spin octahedral formal Ni(0) complex†
Natalia
Cabrera-Lobera
a,
Estefanía
del Horno
a,
M. Teresa
Quirós
 b,
Elena
Buñuel
a,
Magali
Gimeno
cd,
William W.
Brennessel
c,
Michael L.
Neidig
d,
José Luis
Priego
e and
Diego J.
Cárdenas
*a
b,
Elena
Buñuel
a,
Magali
Gimeno
cd,
William W.
Brennessel
c,
Michael L.
Neidig
d,
José Luis
Priego
e and
Diego J.
Cárdenas
*a
aDepartment of Organic Chemistry, Facultad de Ciencias, Universidad Autónoma de Madrid, Institute for Advanced Research in Chemical Sciences (IAdChem), Red ORFEO-CINQA, Av. Francisco Tomás y Valiente 7, Campus de Cantoblanco, 28049, Madrid, Spain. E-mail: diego.cardenas@uam.es
bDepartment of Organic Chemistry and Inorganic Chemistry, Facultad de Farmacia, Universidad de Alcalá de Henares, Campus Universitario, 28871, Madrid, Spain
cDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA
dInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QR, UK
eDepartment of Inorganic Chemistry, Universidad Complutense de Madrid, Ciudad Universitaria, 28040, Madrid, Spain
First published on 1st May 2024
Abstract
We have synthesised and characterised the complex Ni(tpy)2 (tpy = 2,2′:6′,2′′-terpyridine). This formally Ni(0) complex is paramagnetic both in the solid state and in solution (S = 2). The crystal structure shows an octahedral geometry, with molecules arranged in independent dimers involving π-stacking between pairs of complexes. Magnetic measurementes and DFT calculations suggest the existence of temperature-dependent intermolecular antiferromagnetic coupling in the solid state.
The importance of Ni complexes as catalysts for synthetically useful reactions has experienced a renaissance in recent years.1 The accessibility of a wide variety of oxidation states has allowed the discovery of novel activation pathways of simple substrates and the development of multicomponent reactions for the ready preparation of complex organic substrates.2 Especially relevant are Ni(I) derivatives, which have been proposed as intermediates in C–C bond formation reactions such as cross-couplings of alkyl and aryl halides with organomagnesium and organozinc reagents,3 and haloalkane carboxylation reactions.4 The occurrence of several oxidation states along the catalytic cycles allows complex cascade processes leading to the formation of several C–C and C–B bonds, such as cyclization-couplings,5 difunctionalization of alkenes with alkyl and aryl halides,6 and diborylative cyclization of enynes.7 Part of the success of these reactions relies on the use of redox-active ligands capable of stabilizing different oxidation states.8 A general feature of such ligands in Ni-based catalytic systems is their capability to tune their reactivity by delocalizing electrons from the metal. Thus, the spin density in an open shell complex will not be localized on the metal centre but it will be partially delocalized on the ligands. This effect may confer special stability and modulate the reactivity of radical metal complexes. 2,2′:6′,2′′-Terpyridine (tpy) is an important ligand in synthetically useful Ni-catalyzed reactions,9 and it may stabilize low-valent Ni complexes. In fact, it participates in the first isolated Ni(I) organometallic derivative (CH3)Ni(tpy), a 17-electron square-planar complex.10 Calculations show the unpaired electron lying mainly on the π* orbitals of tpy, rendering an anionic ligand coordinated to a Ni(II) center. However, the actual behavior depends on the rest of the ligands on the coordination sphere. Thus, the analogous Br derivative shows a metal centered SOMO instead (Fig. 1).11
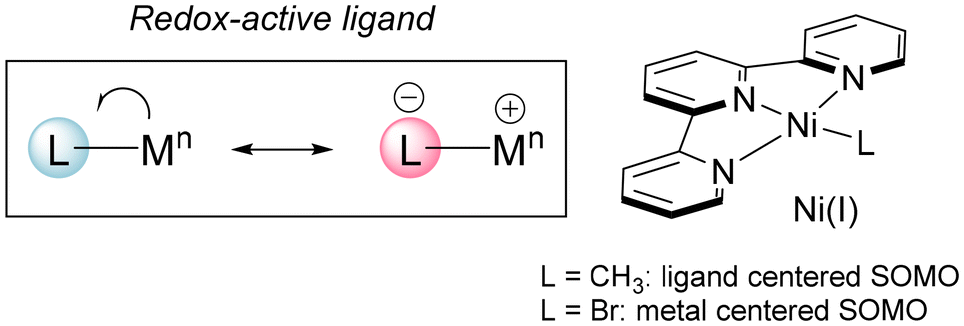 | ||
| Fig. 1 Terpyridine may act as a redox-active ligand in Ni(I) complexes. | ||
Polydentate pyridine-based ligands have been extensively used in Ni-catalyzed synthetically useful reactions involving organometallic intermediates.12 The accessibility of Ni(I) is especially relevant in this context, and the formation and the electronic structure of these kinds of complexes have been studied both experimentally and computationally.4c,10,13 Some years ago, preparation of a complex with Ni(tpy)2 formula was published, and the electronic structure of different putative geometries for this molecular formula were studied by DFT methods.14 Nevertheless, no spectroscopic or crystal structure data were reported. On the basis of its diamagnetic character in the solid state and calculations, it was proposed to have the tetrahedral arrangement that could be expected for a common Ni(0) complex. For an alternative octahedral arrangement, calculations suggested electron delocalization on both terpyridine ligands, as it could be expected from previous studies on formally Ni(I) organometallic complexes with this and related ligands.
We became interested in the actual structure of Ni(0)-tpy complexes, since they may presumably be involved as precatalysts in the Ni-catalyzed diborylative cyclization of enynes we have previously reported.7 In addition, determination of crystal structures may help to shed light on their electronic structure. Thus, we tried to isolate and characterize species formed when Ni(0) precatalysts react with tpy in the absence of other reagents. In contrast to previous report,14 the reaction of Ni(cod)2 with tpy at room temperature in toluene led to a dark blue solution from which crystals of Ni(tpy)2 (1) were obtained (Fig. 2).15 It is important to mention that these conditions differ from those previously reported for the preparation of the diamagnetic complex with the same empirical formula tetrahedral isomer (cyclohexane, 12 h, 80 °C). This emphasizes that solvent and reaction conditions are critical in isolating the octahedral compound.
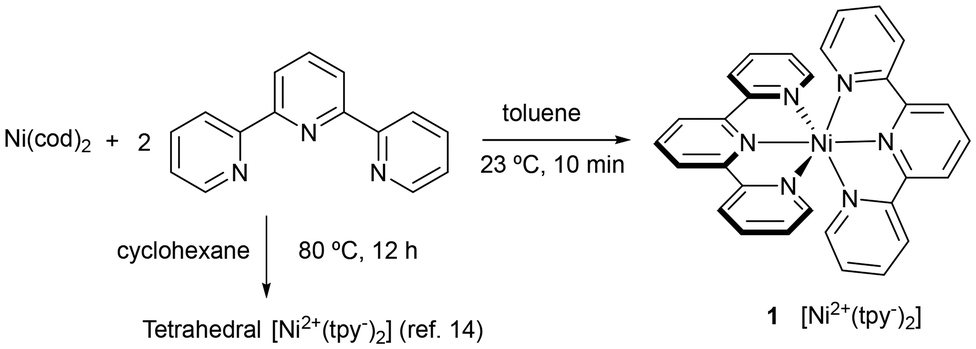 | ||
| Fig. 2 Preparation of Ni(tpy)2 complexes. | ||
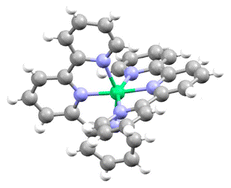
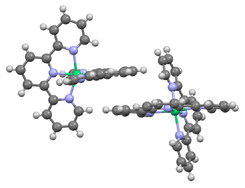
After extensive experimentation, we found that slow vapor diffusion of pentane at −25 °C (24 h) over a solution of a mixture of Ni(cod)2 and tpy afforded dark blue crystals suitable for X-ray diffraction. Crystal structure determination showed an unexpected octahedral Ni(tpy)2 complex with the asymmetric unit containing two [Ni(tpy)2]0 molecules in general positions (Fig. 3).
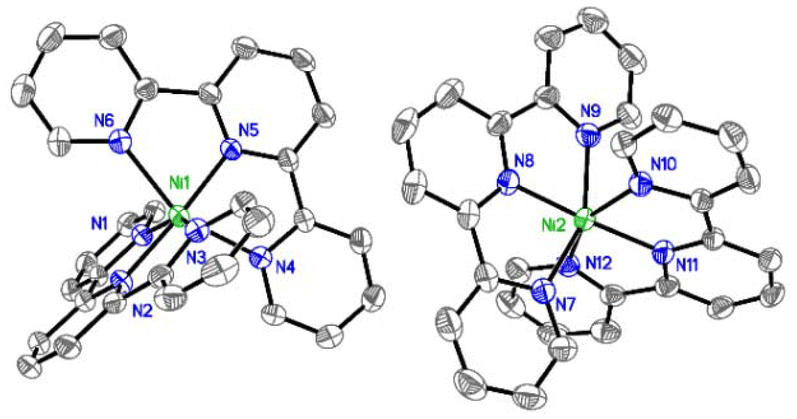 | ||
| Fig. 3 Anisotropic displacement ellipsoid plot at the 50% probability level with hydrogen atoms omitted. | ||
The average Ni–N bond lengths of 2.127(7) [outer] and 1.985(4) [inner] Å slightly differ from the ones found for NiII(tpy)2Cl2 (2.02–2.14) Å,16 and are similar those found for dicationic [Ni(ftpy)2]2+ (ftpy = 4′-(furan-2-yl)-tpy) of 2.107(8) [outer] and 1.991(5) [inner] Å.17 More telling, however, are the average C–C bond lengths between pyridine rings, which is 1.456(12) Å for our structure and 1.493(12) Å for [Ni(ftpy)2]2+,17 the latter which is consistent with that of free tpy at 100 K.18 This trend is seen in the Fe system in which the average C–C bond lengths between pyridine rings shorten in going from [Fe(tpy)2]2+ to [Fe(tpy)2]0, 1.467(4) to 1.452(4) Å, which Wieghardt attributes to reduction of the ligands.19 Moreover, Ni0(bpy)2 shows shorter Ni–N distances, around 1.92 Å,14 and NiI(tpy)R (R = Br, Me) range between 1.92–2.04.10,11 Thus, our complex is best described as [Ni2+(tpy−˙)2]0, a nickel(II) center with two radical anionic tpy ligands.
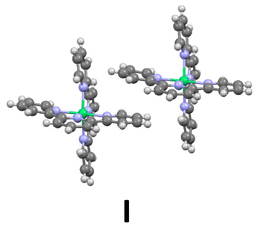
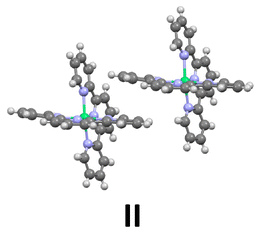
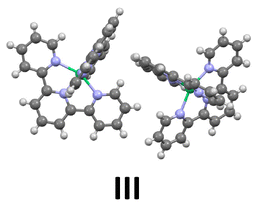
Inspection of the crystal packing shows that molecules of the complex interact by stacking of tpy ligands with some of their neighbours, whereas other close molecules show just edge-to-face interaction between the ligands. Thus, three kinds of dimeric units can be considered in this structure (Fig. 4).
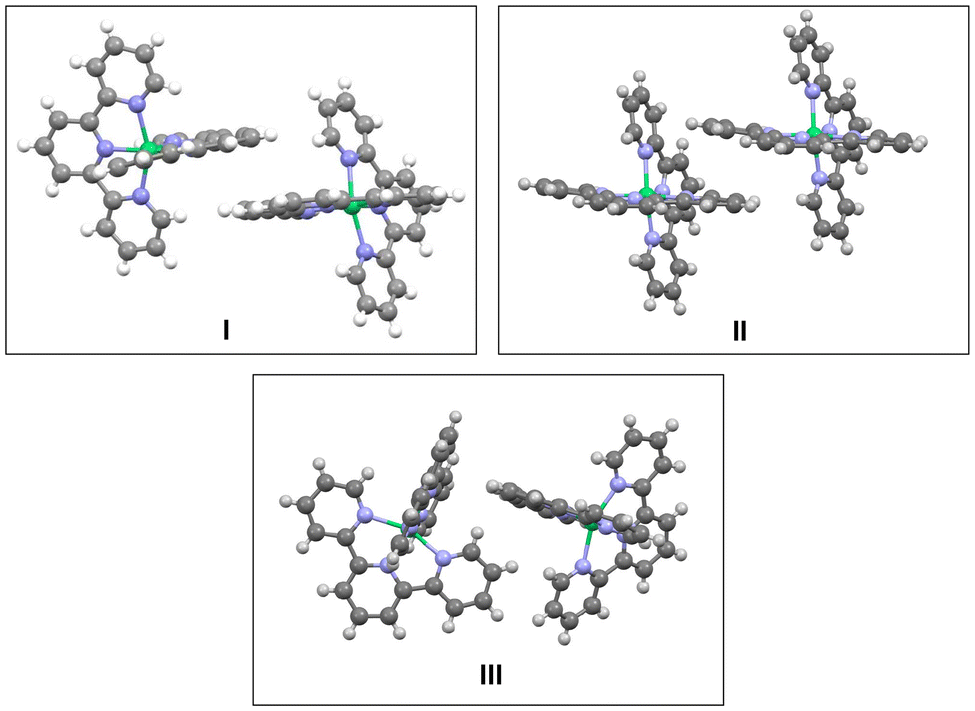 | ||
| Fig. 4 Three different arrangements for neighbouring molecules of Ni(tpy)2. I and II show stacking pyridine ligands and different edge-to-face interactions; III presents just edge-to-face interactions, without stacking rings. | ||
The 1H-NMR in spectrum of the isolated crystals dissolved in d8-THF shows the paramagnetic character of this compound, with signals expanding between 160 and −140 ppm (see ESI).20 On the other hand, quantitative EPR shows a signal corresponding to a complex with S = 1/2. However, this species only represents 1% of the total sample which suggests that the major species has S = 1 or 2. Following the Evans method, which uses difference in the NMR chemical shift in a solvent caused by the presence of a paramagnetic species,17 we measured the effective magnetic moment (μeff) in the solution of complex 1, obtaining a value of 4.9(2)μB in THF and 5.2(2)μB in toluene. These results are consistent with a S = 2 ground-state for 1 in solution.
This complex is also paramagnetic in the solid state, in contrast to previously reported species which is diamagnetic.14 Its magnetic moment decreases from 3.8μB at 298 K to 1.4 at 2 K (Fig. 5). In contrast, the analogous M0(tpy)2 complexes of Ti,21 Cr,22 Mo,22 and W,22 are diamagnetic (S = 0) despite they show very similar crystal structures. On the other hand, Fe and Ru derivatives show S = 1,19 showing same unit cell and packing as 1, as well.
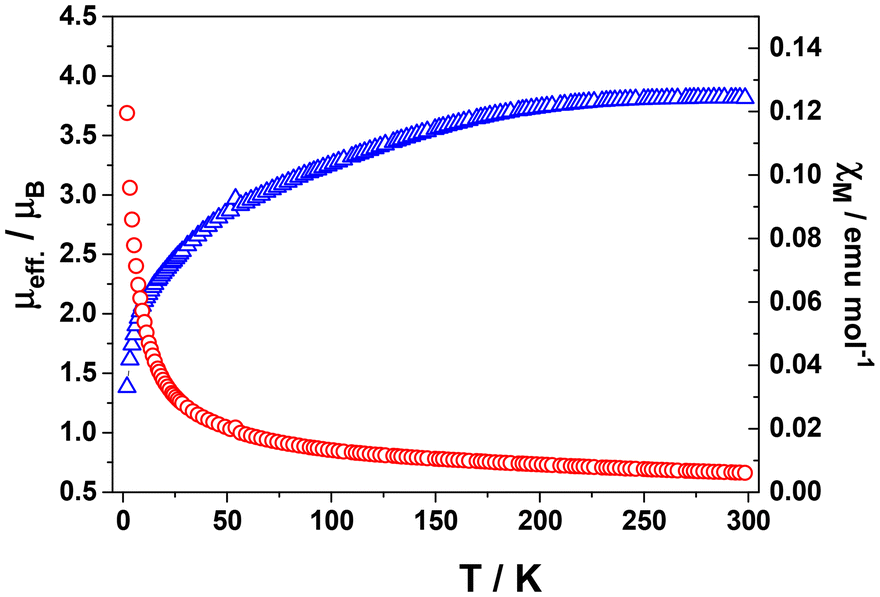 | ||
| Fig. 5 Temperature dependence of the molar susceptibility χM (red circles) and μeff. (blue triangles) for solid Ni(tpy)2 at 0.5 T. | ||
We studied the electronic structure of the octahedral complex by DFT calculations at M06-2x/6-31G(d) (C,H,N), Ni(LANL2DZ) level, functional for different spin multiplicities.23
For isolated Ni(tpy)2 molecules of complex 1, which are the expected to be in solution, the quintet ground state is far more stable compared with the singlet and triplet states (Table 1).24
| Ni(tpy)2 optimized structures | |||
|---|---|---|---|
| a Using SCF = QC as option in Gaussian optimization. See ref. 24. | |||
|
S = 0, m = 1 (singlet) | S = 1, m = 3 (triplet) | S = 2, m = 5 (quintet) |
| Relative E | 0 | −28.9 | −44.2 |
| Relative G (298 K) | 0 | −31.8 | −46.8 |
| Relative energy for different [Ni(tpy) 2 ] 2 Single-point calculation on crystal structure fragments. | |||
| S = 4, m = 9 (nonaplet) | S = 0 (two triplets) | S = 0 (two quintets) | |
|
0 | 28.7 | 4.0 |
|
2.2 | 43.1 | 4.2 |
|
2.0 | 30.5 | 39.4 |
| [Ni(tpy) 2 ] 2 optimized structures | |||
|
S = 4, m = 9 (nonaplet) | S = 0 (two triplets) | S = 0 (two quintets) |
| Relative E | 0 | 5.7 | +1.7 (−0.6)a |
| Relative G (298 K) | 0 | 10.9 | 2.3 |
Moreover, the quintuplet structure shows an octahedral arrangement, whereas the calculated triplet has a distorted tetrahedral structure, with two non-coordinated N atoms (one of each ligand) and the singlet being highly distorted. The high stability of the quintet is in accord with the Evans measurement in solution, which suggested S = 2 for complex 1. On the other hand, the singly-occupied molecular orbitals for the four unpaired electrons are all ligand-centered (Fig. 6) which further supports a Ni(II) metal center coordinated to two anionic tpy ligands, as suggested by the crystal structure.
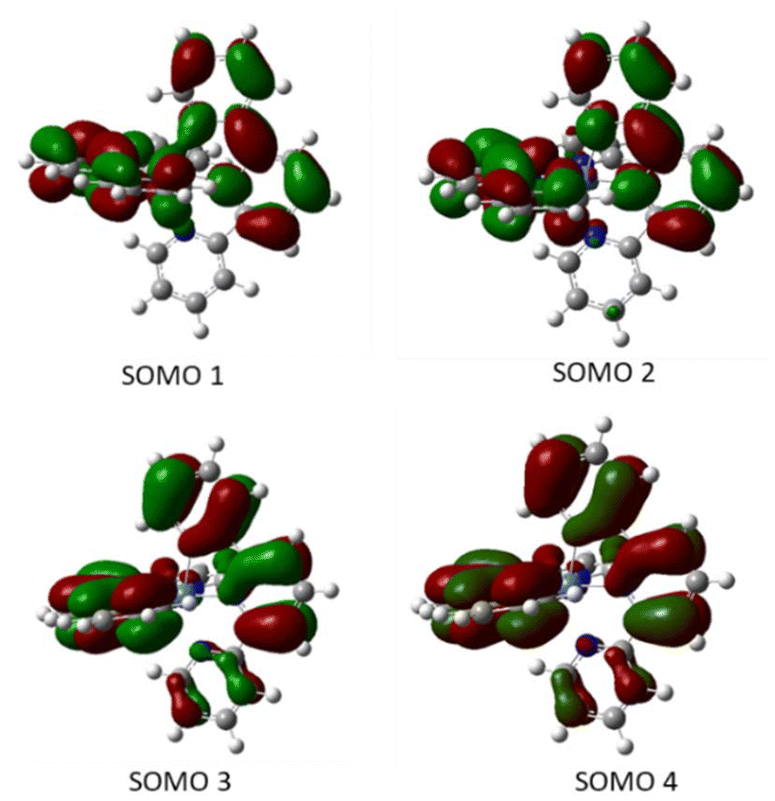 | ||
| Fig. 6 Calculated singly-occupied molecular orbitals for Ni(tpy)2 in the quintuplet state in decreasing order of energy from SOMO1 to SOMO 4. | ||
Then we extended our study to pairs of molecules, and considered the three kinds of arrangements, which can be distinguished in the crystal structure (Fig. 4), as models.
We computed the energy of these dimers for different spin states by broken-symmetry (BS) DFT calculations,25 which allow to assign different spin multiplicities to the Ni complexes in the dimer to determine the energy for complexes with possible antiferromagnetic coupling. We took the atomic coordinates from the crystal structure and calculated the energy for the three models. As it can be seen in Table 1 (center), the most stable arrangement corresponds to the nonaplet dimer I showing π-stacking interaction between pyridines of two different complexes. The alternative nonaplet species are slightly less stable (2.2 and 2.0 kcal mol−1 for II and III, respectively). These results indicate that π-stacking does not stabilize nonaplet spin states significantly. On the other hand, singlet states for dimer III, which lacks stacking interactions, show much higher energies, regardless they are formed by either two triplet or quintet complexes (30.5 and 39.4 kcal mol−1, respectively). On the other hand, regarding the dimers with π-stacked pyridines (I and II), the singlet (two-triplet) states are also considerably unstable compared to the nonaplets (28.7 and 43.1 kcal mol−1, respectively). The results are in accord with the high magnetic moment measured in the solid state at room temperature. In contrast and noticeably, the corresponding singlet dimers composed of two quintets for both I and II lye only 4.0 and 4.2 kcalmol−1 above the nonaplet, in sharp contrast with the energy obtained for the edge-to-face dimer without stacking interactions (III) which is much more eunstable (ΔE = 39.4 kcal·mol−1). These results suggest that antiferromagnetic coupling is feasible for the stacked molecules, considering the error inherent to the computational approach. The relative energy values are significant in this case to derive these conclusions.
Then, we performed structure optimizations for the π-stacked dimer in the three different spin states. This leads to structures different from those found in the crystal but may provide further structural information. As shown in Table 1 (bottom), the most stable structure at room temperature corresponds to the nonaplet dimer (in terms of G). However, the energy of the antiferromagnetic singlet dimer in which both Ni(tpy)2 units are quintets might be more stable at low temperature, according to the calculated electronic energy.24 In any case, the energy difference compared to the nonaplet is very small and within the error of the calculations. Population of the high-spin state accounts for the overall magnetic moment observed at room temperature. Inspection of the molecular orbitals of the dimeric units for the singlet dimers containing two quintet units (HOMO to HOMO-5), show that external electrons are located on the ligands π* orbitals and not on the metal centers (Fig. 7).
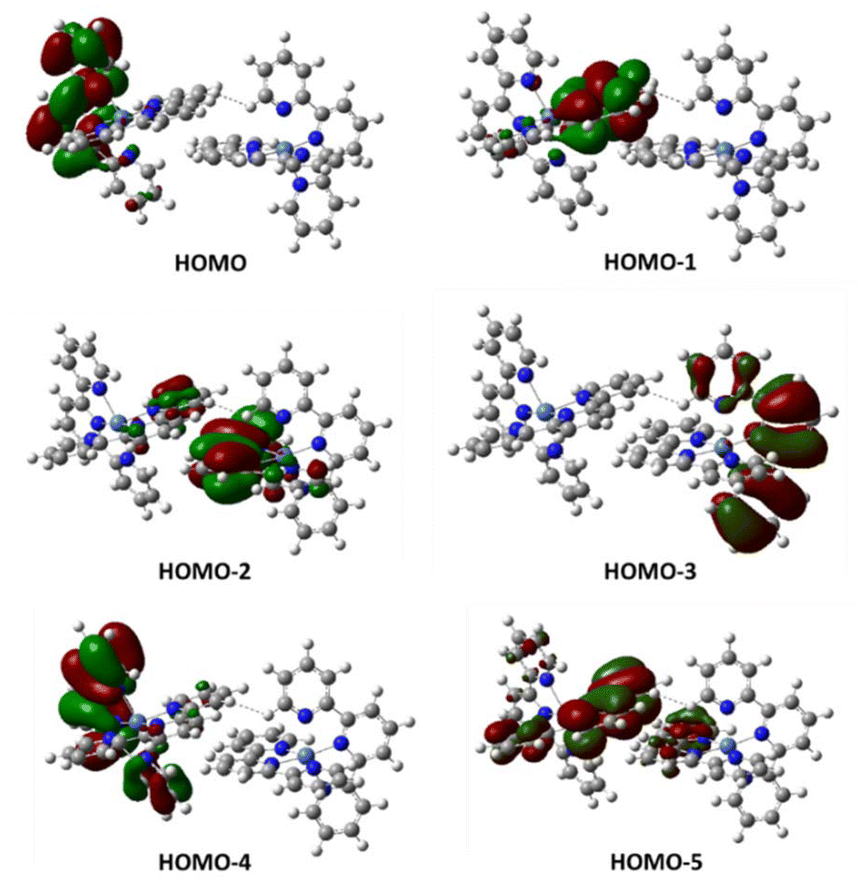 | ||
| Fig. 7 Calculated two-electron occupied molecular orbitals for singlet dimeric units containing two quintuplet complexes 1. HOMO-2 and HOMO-5 are responsible for the interaction between subunits. | ||
Comparison of the distances between centroids of pyridine rings in the crystal structure between 1 and the Fe and Ru analogues suggests that this kind of interaction could also operate in these derivatives (see ESI†).
In conclusion, coordination of two terpyridine ligands to Ni(0) induces the electron transfer to the π* orbitals of the ligands, affording an octahedral quintet complex which should be considered as a Ni(II) species with two anionic tridentate ligands. In the solid state, close molecules may interact through ligand stacking and may couple antiferromagnetically, being the global magnetic moment dependent on the temperature. The high stability of the singlet dimers composed of quintets compared to other spin states, along with the difference of energy calculated on models for the different arrangements of molecules found in the crystal structure supports this proposal. This could be also the case for complexes of other transition metals. We can envision that incorporation of functional groups in the terpyridine ligands, or the use of other polydentate ligands may lead to new structures with extended electron delocalization. that may lead to new interesting solid-state structures or novel Ni precatalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the support by MICIU (PID2019-109088GB-I00 to D. J. C. and a Juan de la Cierva fellowship to M. T. Q) and INVESTIGO-CAM program (for a fellowship to E. d. H). We acknowledge the National Institutes of Health (R01GM111480 to M. L. N.). We also thank the Centro de Computación Científica-UAM for computation time.References
- (a) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542 CrossRef CAS PubMed; (b) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237 CrossRef CAS PubMed; (c) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- (a) C.-Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347 RSC; (b) X. Hu, Chem. Sci., 2011, 2, 1867 RSC; (c) V. B. Phapale and D. J. Cárdenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC.
- (a) G. M. Schwarzwalder, C. D. Matier and G. C. Fu, Angew. Chem., Int. Ed., 2019, 58, 3571 CrossRef CAS PubMed; (b) Z.-C. Cao, S.-J. Xie, H. Fang and Z.-J. Shi, J. Am. Chem. Soc., 2018, 140, 13575 CrossRef CAS PubMed; (c) R. Soler-Yanes, I. Arribas-Álvarez, M. Guisán-Ceinos, E. Buñuel and D. J. Cárdenas, Chem. – Eur. J., 2017, 23, 1584 CrossRef CAS PubMed; (d) R. Soler-Yanes, M. Guisán-Ceinos, E. Buñuel and D. J. Cárdenas, Eur. J. Org. Chem., 2014, 6625 CrossRef CAS; (e) P. M. P. Garcia, T. Di Franco, A. Epenoy, R. Scopelliti and X. Hu, ACS Catal., 2016, 6, 258 CrossRef; (f) O. Vechorkin, A. Godinat, R. Scopelliti and X. Hu, Angew. Chem., Int. Ed., 2011, 50, 11777 CrossRef CAS PubMed; (g) O. Vechorkin and X. Hu, Angew. Chem., Int. Ed., 2009, 48, 2937 CrossRef CAS PubMed; (h) A. J. Oelke, J. Sun and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 2966 CrossRef CAS PubMed; (i) S. W. Smith and G. C. Fu, Angew. Chem. Int. Ed., 2008, 47, 9334 CrossRef CAS PubMed; (j) S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 12645 CrossRef CAS PubMed; (k) V. B. Phapale, M. Guisán-Ceinos, E. Buñuel and D. J. Cárdenas, Chem. – Eur. J., 2009, 15, 12681 CrossRef CAS PubMed.
- (a) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (b) T. Yanagi, R. J. Somerville, K. Nogi, R. Martin and H. Yorimitsu, ACS Catal., 2020, 10, 2117 CrossRef CAS; (c) R. Somerville, C. Odena, M. Obst, N. Hazari, K. Hopmann and R. Martin, J. Am. Chem. Soc., 2020, 142, 10936 CrossRef CAS PubMed.
- (a) M. Guisán-Ceinos, R. Soler-Yanes, D. Collado-Sanz, V. B. Phapale, E. Buñuel and D. J. Cárdenas, Chem. – Eur. J., 2013, 19, 8405 CrossRef PubMed; (b) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790 CrossRef CAS PubMed; (c) J. C. Nieto-Carmona, R. San Román, E. Buñuel and D. J. Cárdenas, Eur. J. Org. Chem., 2022, e202200992 CrossRef CAS.
- (a) X. Wei, W. Shu, A. García-Domínguez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2020, 142, 13515 CrossRef CAS PubMed; (b) Y. Jin, H. Yang and C. Wang, Org. Lett., 2020, 22, 2724 CrossRef CAS PubMed; (c) W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812 CrossRef CAS PubMed.
- N. Cabrera-Lobera, M. T. Quirós, W. W. Brennessel, M. L. Neidig, E. Buñuel and D. J. Cárdenas, Org. Lett., 2019, 21, 6552 CrossRef CAS PubMed.
- Redox-active ligands in catalysis: O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC.
- Y. H. Budnikova, D. A. Vicic and A. Klein, Inorganics, 2018, 6, 18 CrossRef.
- T. J. Anderson, G. C. Jones and D. A. Vicic, J. Am. Chem. Soc., 2004, 126, 8100–8101 CrossRef CAS PubMed . see also Erratum: T. J. Anderson, G. D. Jones and D. A. Vicic, J. Am. Chem. Soc., 2004, 126, 11113 CrossRef.
- J. T. Ciszewski, D. Y. Mikhaylov, K. V. Holin, M. K. Kadirov and Y. H. Budnikova, Inorg. Chem., 2011, 50, 8630–8635 CrossRef CAS PubMed.
- C. Wei, Y. He, X. Shi and Z. Song, Coord. Chem. Rev., 2019, 385, 1 CrossRef CAS PubMed.
- (a) C. S. Day, A. Rentería-Gómez, S. T. Jon, A. R. Gogoi, O. Gutiérrez and R. Martin, Nat. Catal., 2023, 6, 244 CrossRef CAS; (b) M. T. Quirós, D. Collado-Sanz, E. Buñuel-Magdalena and D. J. Cárdenas, Chem. Commun., 2021, 57, 2424 RSC; (c) M. T. Quirós, D. Collado-Sanz, E. Buñuel-Magdalena and D. J. Cárdenas, J. Phys. Chem. A, 2018, 122, 2250 CrossRef PubMed.
- M. Wang, J. England, T. Weyhermüller and K. Wieghardt, Eur. J. Inorg. Chem., 2015, 1511–1523 CrossRef CAS.
- Experimental procedure for the synthesis of complex 1: In a glove-box, a mixture of Ni(cod)2 (74 mg, 0.27 mmol) and terpyridine (125 mg, 0.57 mmol) were dissolved in toluene (3 mL) at room temperature. The resulting dark blue solution is stirred for 10 min, and pentane (5 mL) is added. The resulting precipitate is filtered to give a dark blue solid which is washed with pentane (76 mg, 54%).
- (a) M. I. Arriortua and T. Rojo, Bull. Soc. Chim. Belg., 1982, 91, 337–338 CrossRef CAS; (b) E. C. Constable, J. Lewis, M. C. Liptrot and P. R. Raithby, Inorg. Chim. Acta, 1990, 178, 47 CrossRef CAS.
- B. Z. Momeni, S. K. Anari, M. Torrei and J. Janczak, Appl. Organomet. Chem., 2021, 35, e6179 CrossRef CAS.
- K. F. Bowes, I. P. Clark, J. M. Cole, M. Gourlay, A. M. E. Griffin, M. F. Mahon, L. Ooi, A. W. Parker, P. R. Raithby, H. A. Sparkes and M. Towrie, CrystEngComm, 2005, 7, 269 RSC.
- J. England, C. C. Scarborough, T. Weyhermüller, S. Sproules and K. Wieghardt, Eur. J. Inorg. Chem., 2012, 4605 CrossRef CAS.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003 RSC; (b) E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS; (c) C. Piguet, J. Chem. Educ., 1997, 74, 815 CrossRef CAS.
- M. Wang, T. Weyhermüller, J. England and K. Wieghardt, Inorg. Chem., 2013, 52, 12763 CrossRef CAS PubMed.
- M. Wang, J. England, T. Weyhermüller, S.-L. Kokatam, C. J. Pollock, S. DeBeer, J. Shen, G. P. A. Yap, K. H. Theopold and K. Wieghardt, Inorg. Chem., 2013, 52, 4472 CrossRef CAS PubMed.
- Calculations with other density functionals based on M06 with different correlation-exchange ratios failed. Density functional wB97XD did not work either. On the other hand, as expected, B3LYP did not afford correct structures for the dimeric complexes.
- Calculated energies (E in Table 1) correspond to potential energy minima. Energy values for the singlet dimer composed of two quintet subunits depend on the method used for self-consistent field optimization (QC or XQC). Free energy (G) values are calculated at 298 K. Calculated G values for dimers other than nonaplet could only be computed with SCF = XQC option of Gaussian 09 package.
- (a) L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS; (b) L. Noo-dleman, J. G. Norman, J. H. Osborne, A. Aizman and D. Case, J. Am. Chem. Soc., 1985, 107, 3418 CrossRef CAS; (c) L. Noodleman and E. R. Davidson, Chem. Phys., 1986, 109, 131 CrossRef; (d) L. Noodleman, D. A. Case and A. J. Aizman, J. Am. Chem. Soc., 1988, 110, 1001 CrossRef CAS; (e) L. Noodleman, C. Y. Peng, D. A. Case and J. M. Monesca, Coord. Chem. Rev., 1995, 144, 199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1891622. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04247b |
| This journal is © The Royal Society of Chemistry 2024 |