
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium(III)-based minor groove binding complexes as DNA photocleavage agents†
Stephen O.
Aderinto
a,
Torsten
John‡
 b,
Abdulmujeeb
Onawole
c,
Raphael Peter
Galleh
d and
Jim A.
Thomas
*a
b,
Abdulmujeeb
Onawole
c,
Raphael Peter
Galleh
d and
Jim A.
Thomas
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: james.thomas@sheffield.ac.uk
bDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cInstitute for Molecular Bioscience, The University of Queensland, St. Lucia, QLD 4072, Australia
dSchool of Clinical Dentistry, University of Sheffield, Sheffield, S10 2TA, UK
First published on 11th March 2024
Abstract
Transition metal complexes containing the qtpy ligand (2′:4,4′′:4′,4′′′-quaterpyridyl) are known to be DNA intercalators or minor groove binders. In this study, new tricationic iridium(III) complexes of qtpy are reported. Both [Ir(bpy)2(qtpy)]3+1 and [Ir(phen)2(qtpy)]3+2 display good water solubility as chloride salts. The complexes possess high-energy excited states, which are quenched in the presence of duplex DNA and even by the mononucleotides guanosine monophosphate and adenosine monophosphate. Further studies reveal that although the complexes bind to quadruplex DNA, they display a preference for duplex structures, which are bound with an order of magnitude higher affinities than their isostructural dicationic RuII-analogues. Detailed molecular dynamics simulations confirm that the complexes are groove binders through the insertion of, predominantly, the qtpy ligand into the minor groove. Photoirradiation of 1 in the presence of plasmid DNA confirms that this class of complexes can function as synthetic photonucleases by cleaving DNA.
Introduction
Following the discovery of the biological activity of cisplatin and its successful development as an anticancer therapy,1,2 the interaction of transition metal complexes with DNA has been much studied. Initially, PtII and PtIV complexes that irreversibly bind DNA formed the basis of research.3–6 However, with the aim of developing new therapeutic actions, systems capable of reversible intercalation into DNA were soon identified.7–9Over the last few decades, the range of metals used in these studies progressively widened. Fuelled by the discovery of the DNA light-switch complex, [Ru(LL)2(dppz)]2+ (dppz = dipyridylphenazine, LL = 2,2′-bipyridine (bpy) or 1,10-phenanthroline (phen)), a metallo-intercalator that displays an “off-on” metal-to-ligand charge-transfer (MLCT)-based luminescence response to DNA-binding,10,11 polypyridyl RuII complexes have attracted a particularly large amount of attention.12–17 Although research on the light-switch complex and other RuII analogues first focused on duplex DNA, studies on non-canonical structures such as single-stranded18 and triplex DNA19,20 soon emerged. In this context, inspired by the growing interest in quadruplex nucleic acid structures and their myriad putative biological functions, the Thomas group made the first report on a “quadruplex light-switch”, identifying a dinuclear complex, [{Ru(phen)2}2(tpphz)]4+ (tpphz = tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine), that threads into quadruplex loops resulting in a “switched-on” state that is blue-shifted and more intense than the emission induced by its non-intercalative duplex binding;21 an effect that could also be used to differentiate between duplex and quadruplex structures.22 Over the following years, a large number of studies on RuII complexes and their interaction with quadruplexes and other related structures have been reported.23–27
More recently, related research into IrIII complexes has developed28,29 and cyclometalated polypyridyl IrIII metallo-intercalating complexes have proven to be particularly attractive as their photoexcited states are potentially tunable.30–33 Yet, while such systems have been investigated as therapeutics and cell probes,34–44 their use is often restricted as – due to their lower cationic charge – they frequently display poor inherent water solubility and lower DNA binding affinities.45,46 To address this issue, we recently developed routes to dicationic [Ir(phen)2(bppz)]2+ (bppz = benzopyridophenazine) and tricationic [Ir(bpy)2(dppz)]3+, IrIII-based isostructural analogues of the original light-switch complex containing N5C and N6 Ir coordination spheres respectively.47 As might be expected, due to their increased charge, these complexes bind to duplex DNA with affinities that are two orders of magnitude higher than previously reported monocationic IrIII(dppz)-based systems and are comparable with RuII(dppz) analogues. Furthermore, the photoexcited states of these two new complexes are very different to their RuII analogue. In more recent work, [Ir(phen)3]3+ derivatives incorporating extended aromatic moieties were found to possess long-lived intraligand-charge-transfer (ILCT) excited states that offer potential as photosensitisers for photodynamic therapy, PDT.48
Although the intercalating/threading ligands employed in all the studies discussed above are planar polyaromatic systems, “non-classical intercalators” consisting of unfused rings are also known.49,50 Indeed, the Thomas group has identified that RuII and cyclometallated IrIII complexes incorporating the qtpy ligand (2,2′:4,4′′:4′,4′′′-quaterpyridyl) can bind to duplex DNA through intercalation or groove binding, depending on the metal centre coordinated to the DNA-interacting qtpy ligand.51 Although these studies initially included cyclometallated IrIII systems, such as [Ir(phenylpyridine)2(qtpy)]+ and [Ir(benzo[h]quinoline)2(qtpy)]+, like many other cyclometalated IrIII complexes containing extended aromatic ligands, even their chloride salts displayed very low water solubility so that DNA-binding studies could not be carried out.51
Given the increased solubility of N6IrIII complexes and their potentially novel excited states, we set out to investigate the photophysical and biophysical properties of such metal centres when coordinated to qtpy. Herein, we report new non-cyclometalated, N6-coordination IrIII complexes and provide an initial investigation into their interaction with a G-quadruplex structure. Our studies reveal that the IrIII complexes presented here are groove binders possessing high-energy excited states capable of photocleaving DNA.
Results and discussion
Synthesis and characterisation
Two new complexes, [Ir(bpy)2(qtpy)]3+1 and [Ir(phen)2(qtpy)]3+2, Fig. 1, were synthesised in multistep syntheses starting from [Ir(bpy)2Cl2]PF6 and [Ir(phen)2Cl2]PF6via [Ir(bpy)2(CF3SO3)2]CF3SO3 and [Ir(bpy)2(CF3SO3)2]CF3SO3, respectively, through adapted reported methods.47,52 Specifically, it was found that the final step – the reaction of [Ir(LL)2(CF3SO3)2]CF3SO3 (where LL = bpy or phen) with qtpy in EtOH – could only be accomplished through microwave heating over 9–12 hours, reflecting the kinetically inert nature of the IrIII oxidation state – see ESI† for details. | ||
| Fig. 1 Newly synthesised complexes 1 and 2. | ||
To study the DNA binding properties of the complexes, the triflate salts of 1 and 2 were converted to chloride salts and their absorption and emission spectra in aqueous buffer were investigated.
Unlike their cyclometalated, monocationic analogues, [Ir(ppy)2(qtpy)]+ and [Ir(bhq)2(qtpy)]+ (ppy = 2-phenylpyridine, bhq = benzo[h]quinoline),511 and 2 display good water solubility. The absorption spectra of the new complexes show characteristic absorption bands29 in the UV region (200–340 nm) corresponding to ligand-based π–π* transitions and, at lower energies (340–400 nm), MLCT transitions tailing out to 450 nm are observed (see ESI Fig. S1 and S2,† data summarised in Table 1).
| λ abs (nm), (ε (M−1 cm−1)) | λ ex (nm) | λ em (nm) | |
|---|---|---|---|
| 1 | 200 (41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 710), 250 (35365), 305 (25055), 380 (2880) 710), 250 (35365), 305 (25055), 380 (2880) |
310 | 458 (sh), 489, 520 (sh) |
| 2 | 203 (48985), 225 (46595), 273 (46205), 357 (3805) |
315 | 466, 496, 535 (sh) |
In contrast to their isostructural RuII analogues, the new IrIII complexes displayed vibronically structured emission when photoexcited into their MLCT bands – see ESI Fig. S3 and S4.† These observations are consistent with previous studies indicating that the excited states of such complexes have a large contribution from π–π* states.
The nature of this luminescence is dependent on the ligand attached to the IrIII(qtpy) core. The emission of 1 displays maxima at 458 nm, 489 nm, and 520 nm, and possesses a low energy shoulder that stretches out beyond 600 nm, while the luminescence of 2 shows peaks at 466 nm and 496 nm, with a shoulder at 535 nm that again stretches out to ∼610 nm.
DFT calculations
To gain a deeper understanding of the excited state properties of 1 and 2, they were investigated using quantum chemistry-based methods. The optimised geometries showed that all the complexes form an expected quasi-octahedral coordination geometry with 1 being the most compact – Fig. 2 (see ESI, Table 1†). This is due to the lower steric hindrance of bpy ligands compared to phen.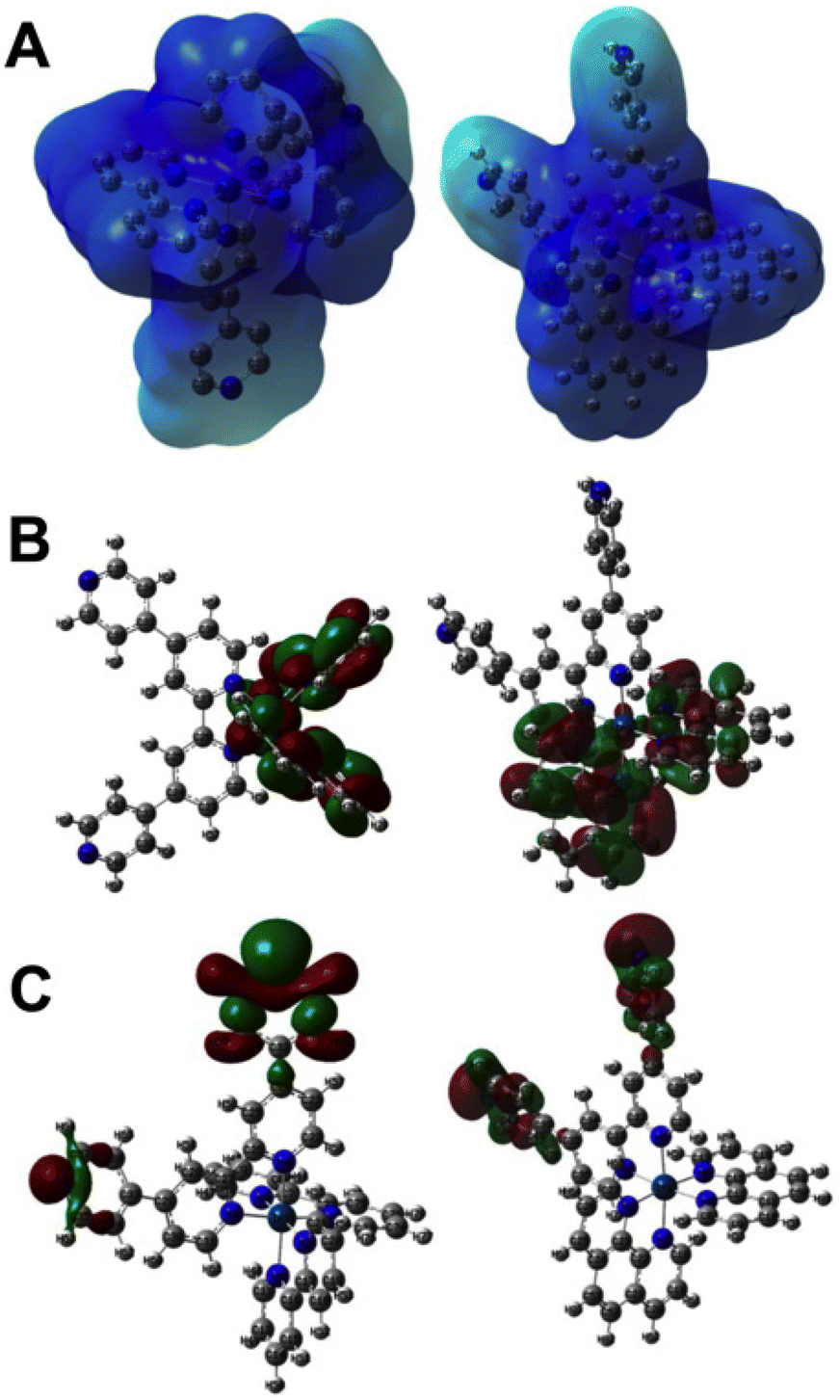 | ||
| Fig. 2 Results of DFT calculations. (A) Electrostatic potential map of complexes 1 and 2. (B) Calculated HOMO−1 of each complex. (C) Calculated LUMO−1 of each complex. | ||
Generated electrostatic potential maps depicting charge distributions are shown in Fig. 2A These indicate that complex 1 has the most localised positive charge and this is expected as the bpy ligands attached to the IrIII centre have less extended aromatic rings to delocalise over compared to phen.
The calculated frontier orbitals, shown in Fig. 2B, clearly indicate that, although the HOMOs extend over the 5d-orbital on the iridium(III) metal centre, the LUMOs for each complex are exclusively distributed over the qtpy ligands, which is consistent with excited states possessing a contribution from IrIII → qtpy MLCT processes.
Interaction with duplex DNA
To parameterise the binding properties of the two complexes with duplex DNA, luminescent titrations were used. Each complex displayed similar DNA-induced behaviour. Addition of DNA to a solution of the complexes resulted in progressive quenching of luminescence, as illustrated for 2 in Fig. 3 – see ESI† for similar data on 1 (Fig. S5†). These observations are in stark contrast to their isostructural RuII analogues [Ru(bpy)2(qpty)]2+ and [Ru(phen)2(qpty)]2+ whose emission is unaffected by addition of DNA.51,53,54 | ||
| Fig. 3 Example of changes in the luminescence of a buffered aqueous 100 μM solution of 2 on addition of calf thymus-DNA, CT-DNA, up to excess. Inset: binding curve constructed from these data, R is the binding ratio. | ||
The changes observed for 1 and 2 were fit to the McGhee-von Hippel model for non-cooperative binding55 to produce the binding parameters presented in Table 2 (see ESI Fig. S6†). The resulting estimated binding affinities for 1 and 2 with duplex DNA are significantly larger than that obtained for its isostructural RuII-dications, with Kb for 1 being more than an order of magnitude higher53 than that for [Ru(bpy)2(qtpy)]2+ – indicating that the increased charge of the IrIII complexes makes an appreciable contribution to this interaction. Interestingly, fits to the McGhee-von Hippel model for both complexes reveal a larger site size of 8 bp than that observed for most metallo-intercalators, which typically display site sizes of ∼3 bp, suggesting that they display some sequence selectivity.
| Complex | K b (M−1) for duplex (site size) | K b (M−1) for HTS |
|---|---|---|
| 1 | 3.7 × 106 (8 bp) | 1.68 × 105 |
| 2 | 1.5 × 106 (8 bp) | 4.5 × 105 |
The decreasing emission of 1 and 2 on addition of DNA is consistent with the kind of photo-induced redox processes frequently observed with metal complexes possessing photo-oxidising excited states.56–58 Indeed, we have previously reported on a dinuclear RuII-complex containing electron deficient tetraazaphenanthrene ligands59,60 as well as organic cationic intercalators61–63 that exhibit similar behaviour. In such systems, quenching occurs on binding to DNA as their high energy excited states oxidise specific base sites within the duplex. Usually this process involves guanine sites, as they have the most accessible one-electron oxidation (1.29 V vs. NHE), but if excited states are sufficiently energetic photo-oxidation of adenine (2.03 V vs. NHE) can also occur.61,64,65 To investigate this question, the effect of increasing concentrations of nucleotide monophosphates on the emission spectrum of 1 and 2 was explored.
Emission quenching by nucleotide phosphates
The addition of either guanosine monophosphate (GMP) or adenosine monophosphate (AMP) led to emission quenching of solutions of 1 and 2, as the examples in Fig. 4 and Fig. S7 (see ESI†) demonstrate. This confirms that the excited-states of these complexes can photo-oxidise both AMP and GMP. Classical linear Stern–Volmer plots are observed for the quenching of 1 by GMP and AMP and 2 with GMP (see Fig. 4B); however, the plots associated with the addition of AMP to 2 display distinctive positive curvatures (Fig. 4D). | ||
| Fig. 4 Examples of the aqueous solution quenching of luminescence of complexes by nucleotides. (A) Change in emission from an aqueous solution of 1 upon successive addition of 5′-GMP. (B) Stern–Volmer plot derived from data shown in A. (C) Change in emission from an aqueous solution of 2 upon successive addition of 5′-AMP (D) Nonlinear Stern–Volmer plot derived from data shown in C. | ||
This phenomenon is indicative of static quenching, likely due to a ground state interaction between the complex and AMP. Certainly, previous studies have shown that complexes containing extended aromatic ligands, including qtpy, can interact with nucleotides through π–π stacking interactions.59,66–69 In this case, it seems the extended aromatic surfaces of the phen ligand are required for this interaction.
Taken together, these experiments and the data from the DNA binding studies indicate that the complexes interact with duplex DNA and can photo-oxidise both A and G sites within a duplex. These studies were then extended to investigate a representative G-quadruplex DNA sequence.
Binding to quadruplex DNA
In recent decades there has been an explosion of interest in the structure and function of G-rich sequences capable of forming quadruplexes.70–73 These studies have revealed putative roles for quadruplex DNA in a wide range of cellular processes including fundamental genetics, genetic disease, and ageing.74–82 A much-studied example is provided by telomeric DNA.Although telomere sequences found at the end of chromosomes83 are single-stranded, they are known to fold into quadruplex structures.84–88 Significantly, telomeres have a role in defining the Hayflick limits for cell division, as each cell division results in shortening of the telomere sequence, until a critical length is reached, and senescence is triggered.82,89–92 It is also established that telomere length maintenance is a key marker of cell immortalisation in carcinogenesis.93–95 For these reasons, methods to selectively bind to telomeric quadruplexes96–99 and/or shorten telomere length in cancer cells have been investigated.27,59,100–103 Given that such studies have included transition metal complexes as binding substrates,25,104 and previous work within the Thomas group has identified RuII complexes that bind to quadruplex-folded human telomere sequence (HTS) with high affinities,21,22,105,106 analogous experiments were carried out with 1 and 2. These were of particular interest as a very recent report from the Keyes group107 has shown that a RuII-complex containing a ligand derived from phen-DC3108 – a high-affinity quadruplex-binding substrate109 that has some structural similarities to qtpy – actually disrupts quadruplex structures. The addition of HTS to solutions of 1 and 2 also resulted in quenching, Fig. 5 and ESI Fig. S8A,† with progressive addition of quadruplex.
 | ||
| Fig. 5 (A) Details of the luminescence response induced through progressive addition of 0.85 μM of quadruplex folded HTS to complex 1. (B) Binding curve for 1 with HTS constructed using emission data – continuous line shows fitting to binding to one-set-of-sites. | ||
Again, the changes in emission were used to estimate the binding affinities of the complexes to HTS. In this case, the data was best fitted to a one-set-of-sites binding model – Fig. 5B, ESI Fig. S8B† and Table 2. In both cases, the estimated Kb values for binding to quadruplex were lower than those for duplex DNA, however this difference was much larger for 1 – where Kb for duplex is over ×20Kb of that of the quadruplex – than for 2, where the difference is only three-fold.
The effect of the complexes on the stability of the folded quadruplex was explored with circular dichroism, CD. Although the racemic mixtures of each complex are not active themselves, the folded quadruplex is chiral and has a characteristic CD signature, with a negative peak at 260 nm and positive peak maximum at 290 nm.110 As illustrated in Fig. 6 and ESI Fig. S9,† it was found that even after addition of excess complex, neither 1 nor 2 produced any real change in the CD of the folded oligonucleotide, indicating that unlike the structures reported by the Keyes group, complexes 1 and 2 do not disrupt the folded structure on interacting with the quadruplex.
 | ||
| Fig. 6 CD spectrum of 5.65 μM HTS (black broken line) and in the presence of 37.36 μM of 2 (red line) in PBS buffer, pH 7.41 at 25 °C. | ||
Molecular dynamics studies
Although we have previously used molecular dynamics (MD) simulations to provide insights into the quadruplex DNA binding properties of dinuclear RuII complexes,106 simulations on the interaction of metal complexes with DNA – particularly noncanonical structures – are still very rare. While our previous studies had shown that qtpy complexes can interact with duplex DNA through intercalation or groove binding, further insights into the interactions of 1 and 2 with the two different DNA structures were sought through detailed MD simulations.Geometry optimised structures of 1 and 2 were first parameterised and then simulated in Amber and Ambertools22111 (see ESI† for details); likewise, the CT-DNA used in experiments was modelled by a DNA duplex (B-form, sequence: AATTGGCCAATTGGCCAATT), and for the G-quadruplex, the unimolecular 22-nt human telomeric DNA, d[AGGG-(TTAGGG)3] structure112 from the Protein Data Bank (PDB entry: 143D) was used (see ESI† for full details). The MD simulations were analysed to identify dominant binding motifs of the complexes to the DNA structures. Clustering of the simulation trajectories was performed over all replicates for each system. In the simulations with the duplex, two types of interaction common to both complexes were observed. The first was an end-stacking interaction onto the blunt ends of the duplex (see ESI, Fig. S10 and S11†). Such interactions have been observed in X-ray structures of polypyridyl RuII systems before. The second common binding motif is more likely to be representative of what occurs experimentally on interaction with CT-DNA as these are much longer and statistically less ends prominent. In this arrangement, illustrated by the interaction of 1 with duplex DNA depicted in Fig. 7A, the qtpy ligand is inserted parallel to the minor groove, with its ring system tracking the curvature of the double helix. Complex 2 is seen to bind into the duplex in a similar manner to that observed for 1 (Fig. 7B) with the qtpy ligand inserted parallel to the groove. Thus, our simulations indicate that both 1 and 2 are minor groove binders. Complex 2 also displayed a second dominant binding geometry within the minor groove in which a phen ligand inserts into the groove (Fig. 7B). This “semi-intercalation” motif has been observed in several crystal structures of [Ru(LL)2(dppz)]2+ (where LL = phenanthroline or 1,4,5,8-tetraazaphenanthrene)113–115 bound to duplexes. Related studies involving HTS resulted in a more flexible and a more diverse set of structures.
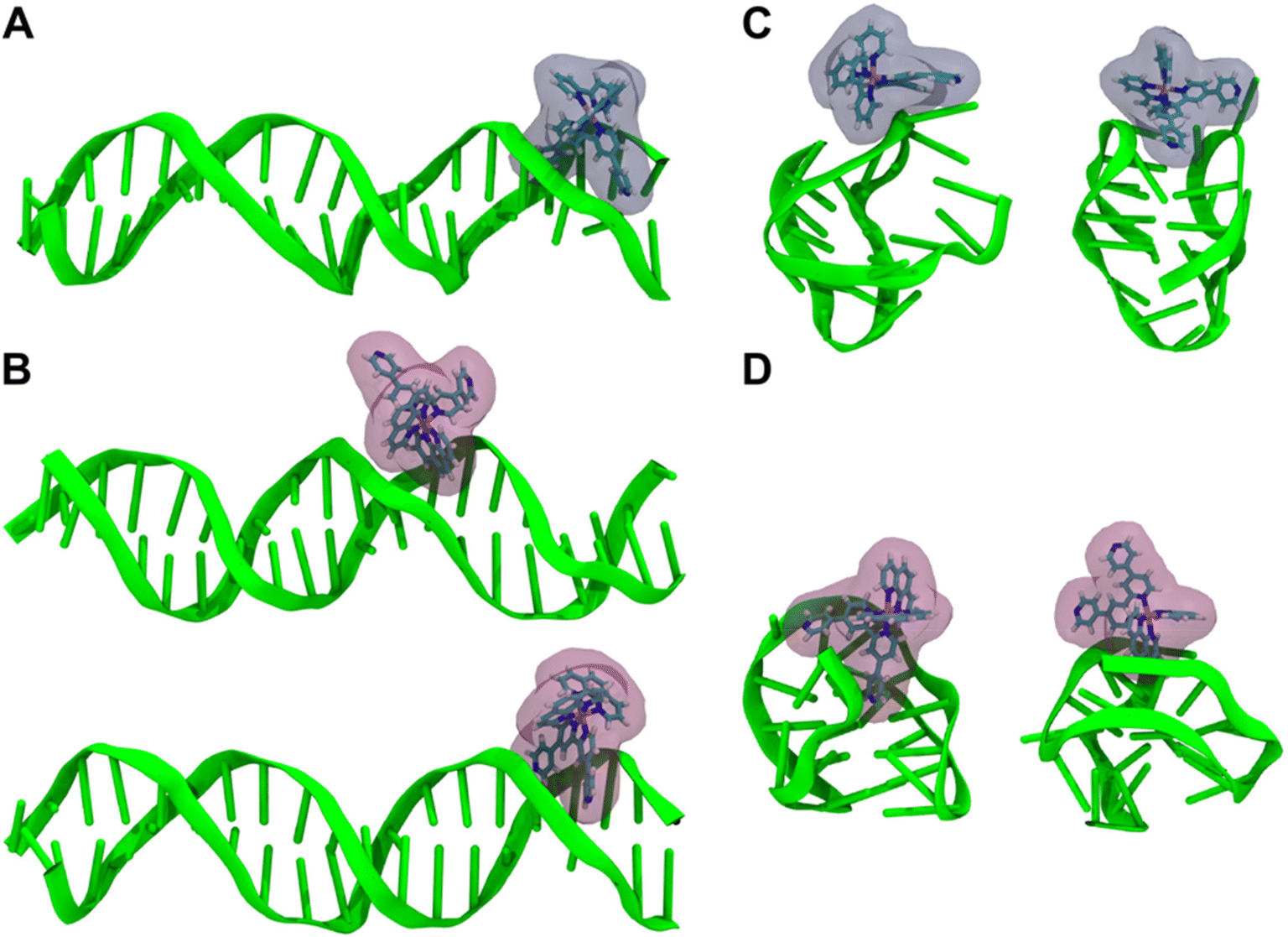 | ||
| Fig. 7 MD simulations for the interaction of complexes 1 and 2 with duplex and quadruplex DNA. (A) Minor groove binding motif of complex 1 to duplex DNA. (B) Dominant binding modes of complex 2 to duplex DNA, showing “minor facial” binding of phen ligands (top) and minor groove binding with qtpy inserted into groove (bottom). (C) Dominant end-pasting motifs for the interaction of 1 with quadruplex-folded HTS. (D) Dominant end-pasting motifs for the interaction of 2 with quadruplex-folded HTS. | ||
For both 1 and 2, the simulations resulted in several binding poses with folded HTS that are represented in Fig. 7C and D. For 1, the metal complex either sits externally to the groove of the quadruplex or in a more intimate interaction with the lateral loop end of the folded structure. In the latter structure, the qpty ligand is orientated with the cleft defined by the two loops and uncoordinated pyridyl rings are parallel with A residues in each of the lateral loop. Binding motifs involving complex 2 also involved external groove binding as well as a contrasting close interaction involving qtpy. In this case, the complex was found at the opposite, diagonal loop end of the folded quadruplex binding into its wide groove in a manner that is much closer to the duplex groove binding motif. Next, to gain quantitative insights into the relative binding strengths with the two different DNA structures, analysis was extended to investigate the solvent accessibility of interacting surfaces and the comparative flexibility of the binding motifs. The solvent accessible surface area (SASA) of the Ir(III)-complexes, obtained using the LCPO algorithm,116 was calculated for the initial 10 ns simulation time and the last 100 ns simulation time and averaged for the respective simulations of complex-duplex and complex-G-quadruplex and changes in SASA were used to qualitatively compare the binding affinity of the different complexes to the DNA structures (Fig. 8A).
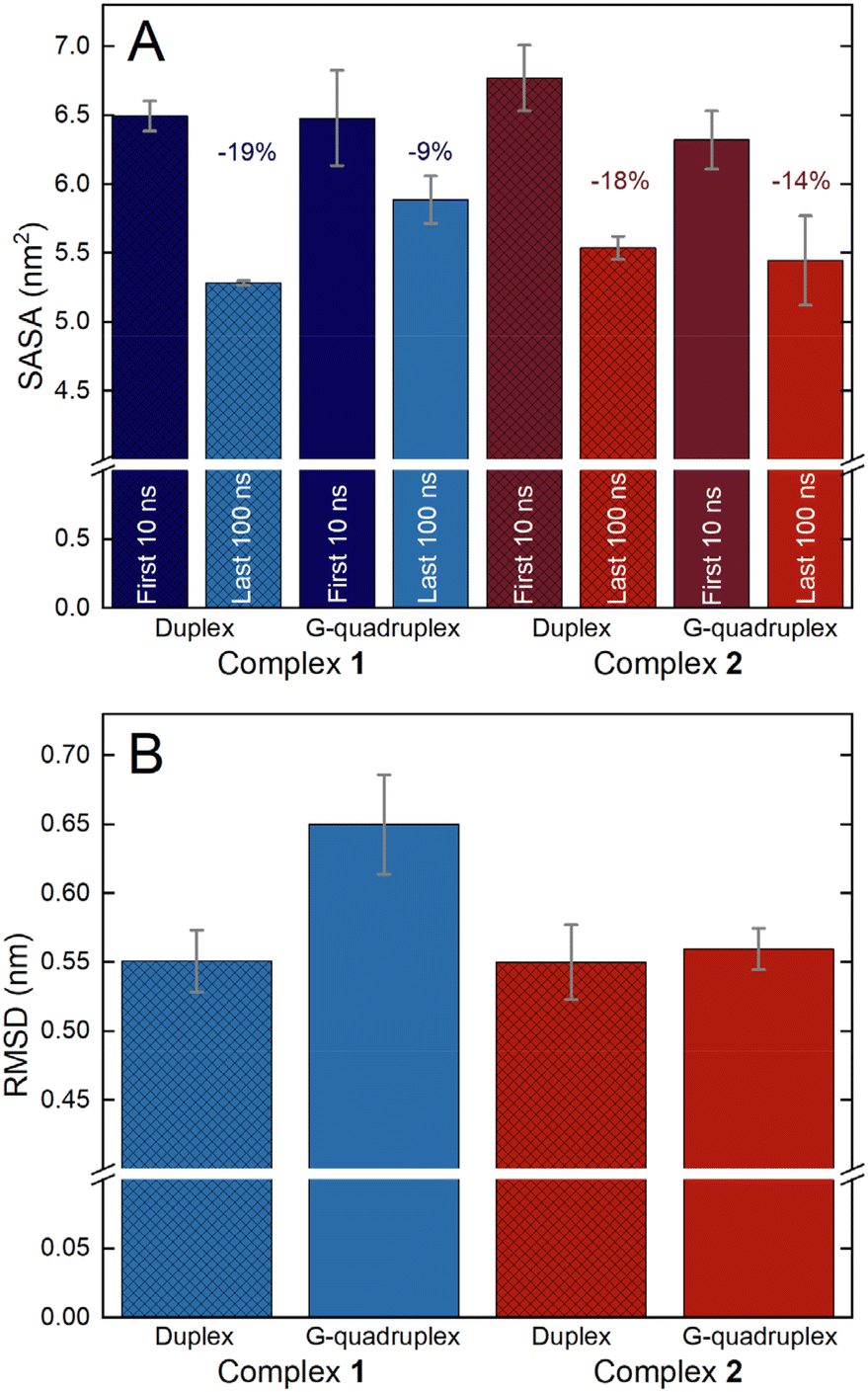 | ||
| Fig. 8 (A) The SASA of the Ir(III)-complexes was calculated using the LCPO algorithm. Error bars indicate standard errors of the mean. SASAs were determined for the first 10 ns simulation time to reflect the initial SASA of the complex in solvent, and for the last 100 ns simulation time to reflect the DNA-bound states. Reported values are averages over all repetitions. The decrease in SASA upon DNA binding is shown. (B) The RMSD of the Ir(III)-complexes was calculated for the last 100 ns simulation time to reflect the DNA-bound states. Error bars indicate standard errors of the mean. Reported values are averages over all replicates. | ||
These figures showed larger changes in SASA for the interactions with duplex over G-quadruplex for both complexes, but this was particularly apparent for complex 1.
To further probe the interaction of the complexes with the two DNA structures, their root mean square displacements (RMSD) were calculated for the last 100 ns simulation time and averaged for the respective simulations of complex-duplex and complex-G-quadruplex binding to quantify the flexibility of the different complexes to the DNA structures (Fig. 8B). These calculations reveal that while complex 2 showed similar flexibility when bound to duplex and G-quadruplex, complex 1 was appreciably more rigidly bound to the duplex than the G-quadruplex structure.
Taken together, the SASA and RMSD calculations indicate that complex 1 displays more pronounced binding selectivity than complex 2, as it appears to bind more tightly to duplex than quadruplex; a conclusion that agrees with the experimental observations.
DNA photocleavage
Given that the excited states of 1 and 2 are quenched on binding to DNA, it seemed likely they were capable of photodamaging nucleotides. Therefore, photocleavage experiments with plasmid DNA were carried out using complex 1 as the representative example.The photonuclease activity of 1 was assessed using the supercoiled pBR322 plasmid under both unilluminated and illuminated conditions. In the dark, no cleavage was observed. However, as illustrated by Fig. 9, subsequent photoirradiation of the supercoiled form leads to the generation of relaxed singly nicked plasmid in a concentration-dependent manner. At higher complex loading, or extended illumination periods further degradation of the DNA through double nicking is evident, confirming that this class of complexes offer promise as photocleavage agents.
 | ||
| Fig. 9 Photocleavage of supercoiled pBR322 DNA by 1 in 5 mM Tris, 25 mM NaCl buffer. (Right to left) lane 1: DNA control; lane 2: DNA + 10 μM complex 1; lane 3: DNA + 20 μM complex 1; lane 4: DNA + 30 μM complex 1. Illumination conditions: λex = 365 nm, 125 mV, 10 minutes. | ||
Conclusions
Although there is a huge amount of work on polypyridyl RuII complexes as metallointercalators, very few studies have investigated the potential of such systems as groove binders, even though minor groove binders often possess higher affinities and selectivity. Furthermore, there has been no work on tricationic N6IrIII complexes in this context. Herein, we demonstrate that these relatively easily accessed class of compounds offer an opportunity to develop systems that can bind to DNA structures with affinities that are considerably higher than their RuII analogues. The photophysical properties of this class of compounds are very different to equivalent RuII and conventional cyclometallated IrIII complexes, as they can display distinctive photonuclease activity as they are photoredox quenched by both G and A-steps.In future reports we will explore the interaction of this class of complexes with other biomolecules. Although these compounds possess higher charge than their RuII analogues, which can often decrease cell-membrane permeance, this is not always the case;105,117,118 therefore, their cell uptake and potential as novel phototherapeutics will also be investigated and assessed.
Author contributions
The study was designed and conceptualised by S. O. A. and J. A. T. Synthesis and experimental characterisation were performed by S. O. A. Biophysical studies were carried out by S. O. A. and R. P. G. Geometry optimisation and DFT to study excited states were performed and analysed by A. O. Force field parameterisation, including DFT, and MD simulations were performed and analysed by T. J. The results were discussed and interpreted by S. O. A., T. J., A. O. and J. A. T. The research was supervised by J. A. T. The first draft of the manuscript was written by J. A. T. and revised by S. O. A. and T. J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. O. is grateful for the award of a University of Sheffield Research Scholarship. T. J. thanks Prof. Dr. Mark Bathe (MIT, Cambridge, MA, USA) for his mentorship and project funding. T. J. was supported by the National Science Foundation (NSF, CCF-1956054) and the Office of Naval Research (ONR, N00014-21-1-4013), and acknowledges financial support from the Alexander von Humboldt Foundation through a Feodor-Lynen Research Fellowship. The authors acknowledge the MIT SuperCloud and Lincoln Laboratory Supercomputing Center as well as the MIT Engaging Cluster for providing HPC resources that have contributed to the research results reported within this work.References
- B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- D. Wang, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS PubMed.
- K. M. Deo, D. L. Ang, B. McGhie, A. Rajamanickam, A. Dhiman, A. Khoury, J. Holland, A. Bjelosevic, B. Pages, C. Gordon and J. R. Aldrich-Wright, Coord. Chem. Rev., 2018, 375, 148–163 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839–3843 CrossRef CAS PubMed.
- R. W.-Y. Sun, A. L.-F. Chow, X.-H. Li, J. J. Yan, S. S.-Y. Chui and C.-M. Che, Chem. Sci., 2011, 2, 728–736 RSC.
- B. J. Pages, F. Li, P. Wormell, D. L. Ang, J. K. Clegg, C. J. Kepert, L. K. Spare, S. Danchaiwijit and J. R. Aldrich-Wright, Dalton Trans., 2014, 43, 15566–15575 RSC.
- A. E. Friedman, J. K. Barton, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- C. Hiort, P. Lincoln and B. Nordén, J. Am. Chem. Soc., 1993, 115, 3448–3454 CrossRef CAS.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 7345, 4565–4579 RSC.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- A. W. McKinley, P. Lincoln and E. M. Tuite, Coord. Chem. Rev., 2011, 255, 2676–2692 CrossRef CAS.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- M. Lin, S. Zou, X. Liao, Y. Chen, D. Luo, L. Ji and H. Chao, Chem. Commun., 2021, 57, 4408–4411 RSC.
- C. G. Coates, J. J. McGarvey, P. L. Callaghan, M. Coletti and J. G. Hamilton, J. Phys. Chem. B, 2001, 105, 730–735 CrossRef CAS.
- S. D. Choi, M. S. Kim, S. K. Kim, P. Lincoln, E. Tuite and B. Nordén, Biochemistry, 1997, 36, 214–223 CrossRef CAS PubMed.
- M. N. Peng, Z. Y. Zhu and L. F. Tan, Inorg. Chem., 2017, 56, 7312–7315 CrossRef CAS PubMed.
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem. – Eur. J., 2006, 12, 4611–4619 CrossRef CAS PubMed.
- T. Wilson, M. P. Williamson and J. A. Thomas, Org. Biomol. Chem., 2010, 8, 2617–2621 RSC.
- K. McQuaid, J. P. Hall, L. Baumgaertner, D. J. Cardin and C. J. Cardin, Chem. Commun., 2019, 55, 9116–9119 RSC.
- K. T. McQuaid, S. Takahashi, L. Baumgaertner, D. J. Cardin, N. G. Paterson, J. P. Hall, N. Sugimoto and C. J. Cardin, J. Am. Chem. Soc., 2022, 144, 5956–5964 CrossRef CAS PubMed.
- E. Palma, J. Carvalho, C. Cruz and A. Paulo, Pharmaceuticals, 2021, 14, 605 CrossRef CAS PubMed.
- J. Jiang, T. Teunens, J. Tisaun, L. Denuit and C. Moucheron, Molecules, 2022, 27, 1541 CrossRef CAS PubMed.
- M. Gillard, G. Piraux, M. Daenen, M. Abraham, L. Troian-Gautier, L. Bar, H. Bonnet, F. Loiseau, H. Jamet, J. Dejeu, E. Defrancq and B. Elias, Chem. – Eur. J., 2022, 28, e20220225 CrossRef PubMed.
- I. M. Dixon, J.-P. Collin, J.-P. Sauvage, L. Flamigni, S. Encinas and F. Barigelletti, Chem. Soc. Rev., 2000, 29, 385–391 RSC.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed.
- S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2013, 2985–3007 CrossRef CAS.
- Ł. Skórka, M. Filapek, L. Zur, J. G. Małecki, W. Pisarski, M. Olejnik, W. Danikiewicz and S. Krompiec, J. Phys. Chem. C, 2016, 120, 7284–7294 CrossRef.
- L. Lu, L. J. Liu, W. C. Chao, H. J. Zhong, M. Wang, X. P. Chen, J. J. Lu, R. N. Li, D. L. Ma and C. H. Leung, Sci. Rep., 2015, 5, 14544–14549 CrossRef CAS PubMed.
- J. J. Cao, C. P. Tan, M. H. Chen, N. Wu, D. Y. Yao, X. G. Liu, L. N. Ji and Z. W. Mao, Chem. Sci., 2016, 8, 631–640 RSC.
- J. Kasparkova, A. Hernández-García, H. Kostrhunova, M. Goicuría, V. Novohradsky, D. Bautista, L. Markova, M. D. Santana, V. Brabec and J. Ruiz, J. Med. Chem., 2024, 67, 691–708 CrossRef CAS PubMed.
- K. K. W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- X. Tian, C. De Pace, L. Ruiz-Perez, B. Chen, R. Su, M. Zhang, R. Zhang, Q. Zhang, Q. Wang, H. Zhou, J. Wu, Z. Zhang, Y. Tian and G. Battaglia, Adv. Mater., 2020, 32, e2003901 CrossRef PubMed.
- C. Caporale, C. A. Bader, A. Sorvina, K. D. M. MaGee, B. W. Skelton, T. A. Gillam, P. J. Wright, P. Raiteri, S. Stagni, J. L. Morrison, S. E. Plush, D. A. Brooks and M. Massi, Chem. – Eur. J., 2017, 23, 15666–15679 CrossRef CAS PubMed.
- A. H. Day, M. H. Übler, H. L. Best, E. Lloyd-Evans, R. J. Mart, I. A. Fallis, R. K. Allemann, E. A. H. H. Al-Wattar, N. I. Keymer, N. J. Buurma and S. J. A. A. Pope, Chem. Sci., 2020, 115, 195–198 Search PubMed.
- P. Y. Ho, C. L. Ho and W. Y. Wong, Coord. Chem. Rev., 2020, 413, 213267 CrossRef CAS.
- H. Yuan, Z. Han, Y. Chen, F. Qi, H. Fang, Z. Guo, S. Zhang and W. He, Angew. Chem., Int. Ed., 2021, 60, 8174–8181 CrossRef CAS PubMed.
- B. Das, S. Gupta, A. Mondal, K. J. Kalita, A. I. Mallick and P. Gupta, J. Med. Chem., 2023, 66, 15550–15563 CrossRef CAS PubMed.
- K. K. W. Lo, C. K. Chung and N. Zhu, Chem. – Eur. J., 2006, 12, 1500–1512 CrossRef CAS PubMed.
- F. Shao, B. Elias, W. Lu and J. K. Barton, Inorg. Chem., 2007, 46, 10187–10199 CrossRef CAS PubMed.
- S. Stimpson, D. R. Jenkinson, A. Sadler, M. Latham, A. Wragg, A. J. H. M. Meijer and J. A. Thomas, Angew. Chem., Int. Ed., 2015, 54, 3000–3003 CrossRef CAS PubMed.
- L. Wang, S. Monro, P. Cui, H. Yin, B. Liu, C. G. Cameron, W. Xu, M. Hetu, A. Fuller, S. Kilina, S. A. McFarland and W. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 3629–3644 CrossRef CAS PubMed.
- L. Strekowski and B. Wilson, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2007, 623, 3–13 CrossRef CAS PubMed.
- R. D. Snyder, D. E. Ewing and L. B. Hendry, Environ. Mol. Mutagen., 2004, 44, 163–173 CrossRef CAS PubMed.
- H. Ahmad, A. Wragg, W. Cullen, C. Wombwell, A. J. H. M. Meijer and J. A. Thomas, Chem. – Eur. J., 2014, 20, 3089–3096 CrossRef CAS PubMed.
- M. Pandrala, F. Li, L. Wallace, P. J. Steel, B. Moore II, J. Autschbach, J. G. Collins and F. R. Keene, Aust. J. Chem., 2013, 66, 1065–1073 CrossRef CAS.
- R. J. Morgan, S. Chatterjee, A. D. Baker and T. C. Strekas, Inorg. Chem., 1991, 30, 2687–2692 CrossRef CAS.
- H. Ahmad, D. Ghosh and J. A. Thomas, Chem. Commun., 2014, 50, 3859–3861 RSC.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS PubMed.
- B. Elias and A. Kirsch-De Mesmaeker, Coord. Chem. Rev., 2006, 250, 1627–1641 CrossRef CAS.
- L. Herman, S. Ghosh, E. Defrancq and A. Kirsch-De Mesmaeker, J. Phys. Org. Chem., 2008, 21, 670–681 CrossRef CAS.
- J. Ghesquiere, S. Le Gac, L. Marcelis, C. Moucheron and A. Kirsch-De Mesmaeker, Curr. Top. Med. Chem., 2012, 12, 185–196 CrossRef CAS PubMed.
- S. A. Archer, A. Raza, F. Dröge, C. Robertson, A. J. Auty, D. Chekulaev, J. A. Weinstein, T. Keane, A. J. H. M. Meijer, J. W. Haycock, S. Macneil and J. A. Thomas, Chem. Sci., 2019, 10, 3502–3513 RSC.
- A. Raza, S. A. Archer, S. D. Fairbanks, K. L. Smitten, S. W. Botchway, J. A. Thomas, S. MacNeil and J. W. Haycock, J. Am. Chem. Soc., 2020, 142, 4639–4647 CrossRef CAS PubMed.
- T. Phillips, I. Haq, A. J. H. M. Meijer, H. Adams, I. Soutar, L. Swanson, M. J. Sykes and J. A. Thomas, Biochemistry, 2004, 43, 13657–13665 CrossRef CAS PubMed.
- T. Phillips, C. Rajput, L. Twyman, I. Haq and J. A. Thomas, Chem. Commun., 2005, 44, 4327–4329 RSC.
- T. Phillips, I. Haq and J. A. Thomas, Org. Biomol. Chem., 2011, 9, 3462–3470 RSC.
- C. A. M. Seidel, A. Schulz and M. H. M. Sauer, J. Phys. Chem., 1996, 100, 5541–5553 CrossRef CAS.
- F. A. Baptista, D. Krizsan, M. Stitch, I. V. Sazanovich, I. P. Clark, M. Towrie, C. Long, L. Martinez-Fernandez, R. Improta, N. A. P. Kane-Maguire, J. M. Kelly and S. J. Quinn, J. Am. Chem. Soc., 2021, 143, 14766–14779 CrossRef CAS PubMed.
- H. Ahmad, B. W. Hazel, A. J. H. M. Meijer, J. A. Thomas and K. A. Wilkinson, Chem. – Eur. J., 2013, 19, 5081–5087 CrossRef CAS PubMed.
- A. M. Agafontsev, A. Ravi, T. A. Shumilova, A. S. Oshchepkov and E. A. Kataev, Chem. – Eur. J., 2019, 25, 2684–2694 CrossRef CAS PubMed.
- A. M. Agafontsev, T. A. Shumilova, A. S. Oshchepkov, F. Hampel and E. A. Kataev, Chem. – Eur. J., 2020, 26, 9991–9997 CrossRef CAS PubMed.
- L. Catti, R. Sumida and M. Yoshizawa, Coord. Chem. Rev., 2022, 460, 214460 CrossRef CAS.
- P. Baumann, Nat. Struct. Mol. Biol., 2005, 12, 832–833 CrossRef CAS PubMed.
- G. N. Parkinson, in Quadruplex Nucleic Acids, ed. S. Neidle and S. Balasubramanian, 2007, pp. 1–30 Search PubMed.
- J. L. Huppert, Chem. Soc. Rev., 2008, 37, 1375–1384 RSC.
- E. Y. N. Lam, D. Beraldi, D. Tannahill and S. Balasubramanian, Nat. Commun., 2013, 4, 1796 CrossRef PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed.
- T. Tian, Y. Q. Chen, S. R. Wang and X. Zhou, Chem, 2018, 4, 1314–1344 CAS.
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, Nat. Genet., 2016, 48, 1267–1272 CrossRef PubMed.
- Y. Wang, J. Yang, A. T. Wild, W. H. Wu, R. Shah, C. Danussi, G. J. Riggins, K. Kannan, E. P. Sulman, T. A. Chan and J. T. Huse, Nat. Commun., 2019, 10, 1–14 CrossRef PubMed.
- R. Simone, P. Fratta, S. Neidle, G. N. Parkinson and A. M. Isaacs, FEBS Lett., 2015, 589, 1653–1668 CrossRef CAS PubMed.
- A. Malousi, A. Z. Andreou, E. Georgiou, G. Tzimagiorgis, L. Kovatsi and S. Kouidou, Epigenetics, 2018, 13, 808–821 CrossRef PubMed.
- D. Liano, S. Chowdhury and M. Di Antonio, J. Am. Chem. Soc., 2021, 143, 20988–21002 CrossRef CAS PubMed.
- M. A. Blasco, Nat. Chem. Biol., 2007, 3, 640–649 CrossRef CAS PubMed.
- E. H. Blackburn, Nature, 2000, 408, 53–56 CrossRef CAS PubMed.
- K. Paeschke, T. Simonsson, J. Postberg, D. Rhodes and H. J. Lipps, Nat. Struct. Mol. Biol., 2005, 12, 847–854 CrossRef CAS PubMed.
- K. Paeschke, S. Juranek, T. Simonsson, A. Hempel, D. Rhodes and H. J. Lipps, Nat. Struct. Mol. Biol., 2008, 15, 598–604 CrossRef CAS PubMed.
- A. L. Moye, K. C. Porter, S. B. Cohen, T. Phan, K. G. Zyner, N. Sasaki, G. O. Lovrecz, J. L. Beck and T. M. Bryan, Nat. Commun., 2015, 6, 7643 CrossRef PubMed.
- J. S. Smith, Q. Chen, L. A. Yatsunyk, J. M. Nicoludis, M. S. Garcia, R. Kranaster, S. Balasubramanian, D. Monchaud, M. P. Teulade-Fichou, L. Abramowitz, D. C. Schultz and F. B. Johnson, Nat. Struct. Mol. Biol., 2011, 18, 478–486 CrossRef CAS PubMed.
- S. Neidle, FEBS J., 2010, 277, 1118–1125 CrossRef CAS PubMed.
- C. B. Harley, A. B. Futcher and C. W. Greider, Nature, 1990, 345, 458–460 CrossRef CAS PubMed.
- M. Z. Levy, R. C. Allsopp, A. B. Futcher, C. W. Greider and C. B. Harley, J. Mol. Biol., 1992, 225, 951–960 CrossRef CAS PubMed.
- F. Sampedro Camarena, G. Cano Serral and F. Sampedro Santaló, Clin. Transl. Oncol., 2007, 9, 145–154 CrossRef CAS PubMed.
- J. W. Shay, Curr. Opin. Cell Biol., 2018, 52, 1–7 CrossRef CAS PubMed.
- J. D. Henson, A. A. Neumann, T. R. Yeager and R. R. Reddel, Oncogene, 2002, 21, 598–610 CrossRef CAS PubMed.
- S. E. Artandi and R. A. DePinho, Carcinogenesis, 2009, 31, 9–18 CrossRef PubMed.
- A. J. Cesare and R. R. Reddel, Nat. Rev. Genet., 2010, 11, 319–330 CrossRef CAS PubMed.
- S. Neidle, Curr. Opin. Struct. Biol., 2009, 19, 239–250 CrossRef CAS PubMed.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- P. J. Campbell, Cell, 2012, 148, 633–635 CrossRef CAS PubMed.
- Y. Xu, Chem. Soc. Rev., 2011, 40, 2719–2740 RSC.
- W. Chen, Y. Zhang, H.-B. Yi, F. Wang, X. Chu and J.-H. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202300162 CrossRef CAS PubMed.
- J. Weynand, A. Diman, M. Abraham, L. Marcélis, H. Jamet, A. Decottignies, J. Dejeu, E. Defrancq and B. Elias, Chem. – Eur. J., 2018, 24, 19216–19227 CrossRef CAS PubMed.
- K. Kawauchi, R. Urano, N. Kinoshita, S. Kuwamoto, T. Torii, Y. Hashimoto, S. Taniguchi, M. Tsuruta and D. Miyoshi, Genes, 2020, 11, 1340 CrossRef CAS PubMed.
- E. Cadoni, L. De Paepe, G. Colpaert, R. Tack, D. Waegeman, A. Manicardi and A. Madder, Nucleic Acids Res., 2023, 51, 4112–4125 CrossRef CAS PubMed.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- T. Wilson, P. J. Costa, V. V. Félix, M. P. Williamson and J. A. Thomas, J. Med. Chem., 2013, 56, 8674–8683 CrossRef CAS PubMed.
- L. Holden, K. S. Gkika, C. S. Burke, C. Long and T. E. Keyes, Inorg. Chem., 2022, 62, 2213–2227 CrossRef PubMed.
- A. De Cian, E. DeLemos, J. L. Mergny, M. P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856–1857 CrossRef CAS PubMed.
- W. J. Chung, B. Heddi, F. Hamon, M. P. Teulade-Fichou and A. T. Phan, Angew. Chem., Int. Ed., 2014, 53, 999–1002 CrossRef CAS PubMed.
- M. Vorlíčková, I. Kejnovská, K. Bednářová, D. Renčiuk and J. Kypr, Chirality, 2012, 24, 691–698 CrossRef PubMed.
- D. A. Case, H. M. Aktugla, K. Belfon, I. Y. Ben-Shalom, J. T. Berryman, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, N. Forouzesh, G. Giambaşu, T. Giese, M. K. Gilson, H. Gohlke, A. W. Goetz, J. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, A. Shajan, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, J. Wang, H. Wei, X. Wu, Y. Wu, Y. Xiong, Y. Xue, D. M. York, S. Zhao, Q. Zhu and P. A. Kollman, Amber22 and AmberTools22, University of California, San Francisco, 2022 Search PubMed.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef CAS PubMed.
- J. P. Hall, K. O'Sullivan, A. Naseer, J. A. Smith, J. M. Kelly and C. J. Cardin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17610–17614 CrossRef CAS PubMed.
- H. Niyazi, J. P. Hall, K. O'Sullivan, G. Winter, T. Sorensen, J. M. Kelly and C. J. Cardin, Nat. Chem., 2012, 4, 621–628 CrossRef CAS PubMed.
- J. P. Hall, P. M. Keane, H. Beer, K. Buchner, G. Winter, T. L. Sorensen, D. J. Cardin, J. A. Brazier and C. J. Cardin, Nucleic Acids Res., 2016, 44, 9472–9482 CrossRef CAS PubMed.
- J. Weiser, P. S. Shenkin and W. C. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS.
- M. G. Walker, P. J. Jarman, M. R. Gill, X. Tian, H. Ahmad, P. A. N. Reddy, L. McKenzie, J. A. Weinstein, A. J. H. M. Meijer, G. Battaglia, C. G. W. Smythe and J. A. Thomas, Chem. – Eur. J., 2016, 22, 5996–6000 CrossRef CAS PubMed.
- Z. Zhou, J. Liu, T. W. Rees, H. Wang, X. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5664–5669 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00171k |
| ‡ Current address: Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |