
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeraurated ruthenium hydride carbonyl clusters: aurophilicity, isolobal analogy, structural isomerism, and fluxionality†
Cristiana
Cesari
 *a,
Marco
Bortoluzzi
b,
Cristina
Femoni
a,
Francesca
Forti
a,
Maria Carmela
Iapalucci
a and
Stefano
Zacchini
a
*a,
Marco
Bortoluzzi
b,
Cristina
Femoni
a,
Francesca
Forti
a,
Maria Carmela
Iapalucci
a and
Stefano
Zacchini
a
aDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
bDipartimento di Scienze Molecolari e Nanosistemi, Ca’ Foscari University of Venice, Via Torino 155, 30175 Mestre, Ve, Italy
First published on 19th January 2024
Abstract
The stepwise addition of increasing amounts of Au(PPh3)Cl to [HRu4(CO)12]3− (1) results in the sequential formation of [HRu4(CO)12(AuPPh3)]2− (2), [HRu4(CO)12(AuPPh3)2]− (3), and HRu4(CO)12(AuPPh3)3 (4). Alternatively, 4 can be obtained upon addition of HBF4·Et2O (two mole equivalents) to 3. Further addition of acid to 3 (three mole equivalents) results in the formation of the tetra-aurated cluster Ru4(CO)12(AuPPh3)4 (5). Compounds 2–5 have been characterized by IR, 1H and 31P{1H} NMR spectroscopies. Moreover, the molecular structures of 3–5 have been determined by single crystal X-ray diffraction as [NEt4][3]·2CH2Cl2, 4-b·2CH2Cl2, 4-a, 5·0.5CH2Cl2·solv, and 5·solv crystalline solids. Two different isomers of 4, that is 4-a and 4-b, have been crystallographically characterized and their rapid interconversion in solution was studied by variable temperature 1H and 31P{1H} NMR spectroscopies. Weak aurophilic Au⋯Au contacts have been detected in the solid state structures of 3–5. Computational studies have been performed in order to elucidate bonding and isomerism, as well as to predict the possible structure of the elusive species 2.
Introduction
Molecular compounds containing two or more [AuPPh3]+ fragments have considerably contributed to the development of the aurophilicity concept.1–5 Indeed, closed-shell d10 Au(I) ions tend to attract themselves, resulting in weak aurophilic interactions, whose strength is somehow comparable to hydrogen bonding.6,7 It has been shown that aurophilic attractions can have electronic and/or dispersive (London forces) nature, which are both due to pronounced relativistic effects on gold atoms.8–10Attractive aurophilic interactions span a wide range of distances, that is ca. 2.75–3.50 Å.11 For comparison, the closest contact of two Au atoms in fcc gold metal is 2.88 Å, the covalent Au–Au bond in dinuclear Au(II) (d9) complexes is ca. 2.57 Å, and the sum of two van der Waals radii of Au(I) is ca. 3.60 Å.2,4,12
Both inter-molecular and intra-molecular aurophilic contacts are well-represented. Inter-molecular interactions have a considerable relevance in supramolecular chemistry and self-assembly phenomena based on Au(I) compounds, with applications in materials science.11,13 Intra-molecular aurophilic contacts contribute to the overall structures of peraurated complexes, which may also show structural isomerism.14–17 In this respect, several molecular metal carbonyl clusters decorated on the surface by [AuPPh3]+ fragments have been reported.18–22
Protons and [AuPPh3]+ fragments are isolobal, and often metal carbonyl clusters containing a single [AuPPh3]+ fragment are isostructural to related hydride derivatives.23–27 However, when two or more [AuPPh3]+ fragments are present, the structural analogy with hydride derivatives fades, due to the formation of weak Au⋯Au aurophilic contacts.1,2,5,6,14 Indeed, often polyhydride and related peraurated carbonyl clusters differ in the geometrical arrangement of the H and AuPPh3 fragments around the metal carbonyl cage of the cluster. This, of course, is more a structural consideration than a general statement. If one ignores for a moment charges and redox states and just looks at H and Au(PPh3), the presence of multiple Au(PPh3) units in a cluster does not invalidate their similarity to a cluster containing the corresponding number of H atoms. Furthermore, it is known that hydrido clusters can be viewed, at least in a limiting form, as dihydrogen complexes (two H atoms close to each other forming H2, like two Au(PPh3) units close to each other forming P–Au–Au–P).28 Such molecules are also often fluxional. The isolobal analogy was not meant to address metallophilic interactions and it still applies to complexes with multiple d10 centres, even if their detailed structure may not be predicted.
Thus, peraurated metal carbonyl clusters are good platform compounds suitable to experimentally test aurophilicity.29–31 Indeed, they contain stronger core interactions, whereas weaker forces operate on their surface. The overall arrangement of the [AuPPh3]+ fragments on the surface of the cluster is, therefore, often the result of a subtle balance between several different weak intramolecular forces, such as Au⋯Au aurophilic contacts, π–π and π–H interactions due to the presence of aromatic groups, as well as steric effects.32 Packing effects may also be relevant in the solid state. As a consequence, structural isomerism and fluxionality are not rare phenomena in the case of peraurated metal carbonyl clusters. In addition, (polarized) heterometallic M–Au bonds may contribute to enhanced catalytic properties.33–38
A few neutral tetrahedral Ru4 carbonyl clusters containing 1–3 [AuPPh3]+ fragments are known, that is, H3Ru4(CO)12(AuPPh3), H2Ru4(CO)12(AuPPh3)2, and HRu4(CO)12(AuPPh3)3.39–42 These are closely related to the tetra-hydride H4Ru4(CO)12, even though structural differences occur upon replacement of hydrides with [AuPPh3]+ fragments, due to the formation of Au⋯Au aurophilic contacts. Also in the case of Os, only neutral tetrahedral clusters have been reported so far, that is H3Os4(CO)12(AuPPh3), H2Os4(CO)12(AuPPh3)2, H3Os4(CO)11(AuPPh3)3, and H2Os4(CO)11(AuPPh3)4.40–45 Conversely, both neutral and anionic clusters are known for Fe, that is HFe4(CO)12(AuPPh3)3 and [HFe4(CO)12(AuPPh3)2]−, respectively.46 It is worth noting that HFe4(CO)12(AuPPh3)3 represents a unique case of a metal carbonyl cluster containing μ4-H within a tetrahedral metal cage. This should be in contrast to HRu4(CO)12(AuPPh3)3, whose hydride is located on the surface of the cluster.
The recent discovery of a straightforward synthesis of [HRu4(CO)12]3− (1)47 prompted a detailed study of its reactivity toward Au(PPh3)Cl. This resulted in the isolation of the first anionic Ru4 carbonyl clusters containing [AuPPh3]+ fragments, that is [HRu4(CO)12(AuPPh3)]2− (2) and [HRu4(CO)12(AuPPh3)2]− (3), an improved synthesis of HRu4(CO)12(AuPPh3)3 (4), as well as the synthesis of tetra-aurated Ru4(CO)12(AuPPh3)4 (5). Details of their preparation, spectroscopic and structural characterization, structural isomerism, fluxional behaviour and theoretical investigation are herein reported.
Results and discussion
The reaction of [HRu4(CO)12]3− (1) with Au(PPh3)Cl: general features and characterization of [HRu4(CO)12(AuPPh3)]2− (2)
The stepwise addition of increasing amounts of Au(PPh3)Cl to [HRu4(CO)12]3− (1), as [NEt4]+ salt, results in the sequential formation of [HRu4(CO)12(AuPPh3)]2− (2), [HRu4(CO)12(AuPPh3)2]− (3), and HRu4(CO)12(AuPPh3)3 (4), as evidenced by IR analyses (Scheme 1, Table 1 and Fig. 1). Alternatively, 4 can be obtained upon addition of HBF4·Et2O (two mole equivalents) to 3. In this case, H4Ru4(CO)12, [H3Ru4(CO)12]− and [H2Ru4(CO)12]2− are obtained as side-products,48–51 as evidenced by IR and 1H NMR spectroscopy. The formation of these homometallic Ru hydride carbonyls is in agreement with the fact that 4 contains more Au than the precursor 3. Addition of a slight excess of acid to 3 (three mole equivalents) results in the formation of the tetra-aurated cluster Ru4(CO)12(AuPPh3)4 (5). As above, H4Ru4(CO)12 and [H3Ru4(CO)12]− are the main side products observed during the synthesis of 5. It is likely that the formation of 4 and 5 upon addition of acids to 3 proceeds via a general rearrangement, producing 4 and 5, which are richer in Au than 3, as well as Au-free clusters H4Ru4(CO)12, [H3Ru4(CO)12]− and [H2Ru4(CO)12]2−, guaranteeing the mass balance.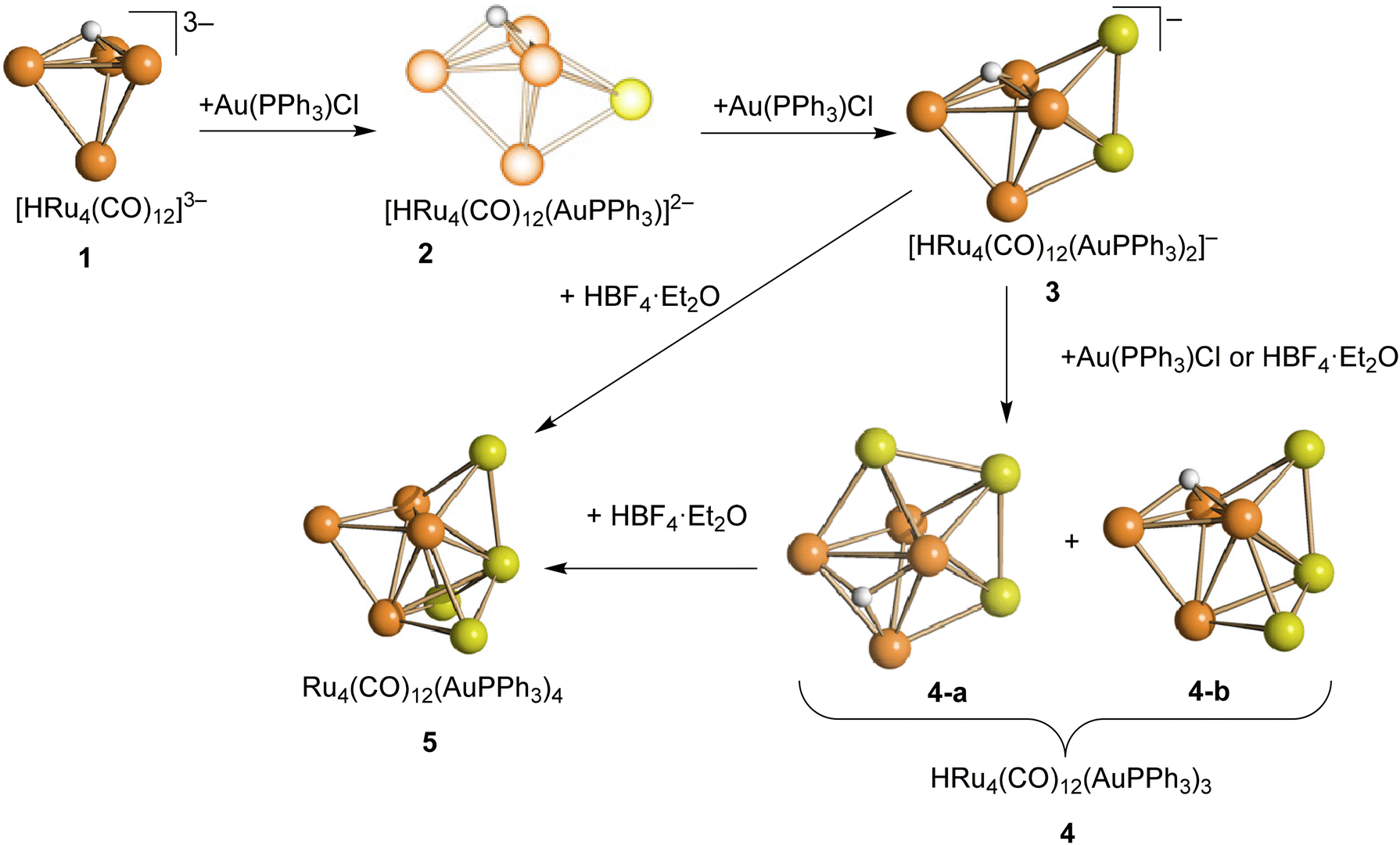 | ||
| Scheme 1 Synthesis of 2–5 from 1 (orange, Ru; yellow, Au; white, H). CO and PPh3 ligands have been omitted for clarity. Two isomers of 4 are obtained, which rapidly interconvert in solution. Compound 2 has been spectroscopically identified and its structure computationally predicted using DFT methods (lighter colors used). All other structures have been determined by SC-XRD (darker colors used). Details may be found in the main text and the Experimental section. | ||
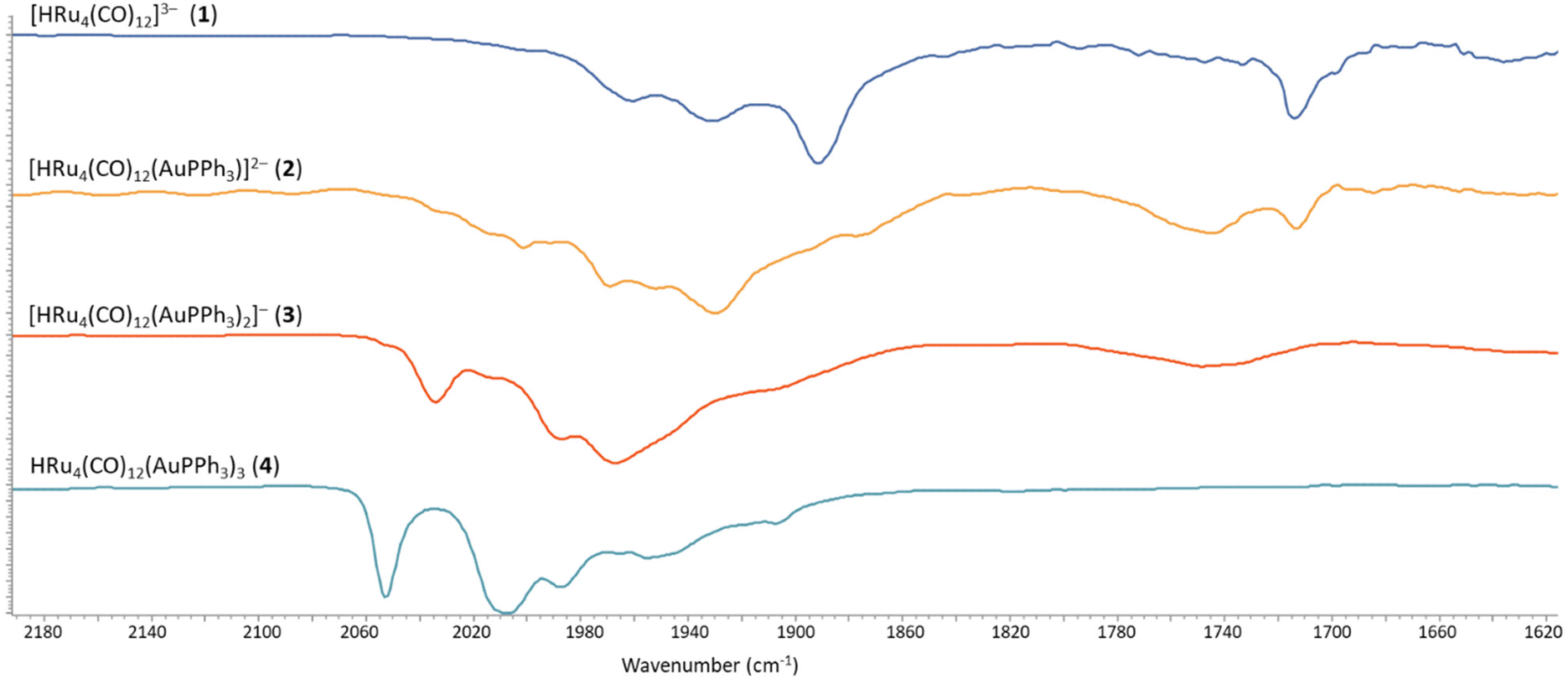 | ||
| Fig. 1 IR spectra in the νCO region of 2–4 obtained by stepwise addition of Au(PPh3)Cl to 1. All spectra have been recorded in CH3CN solution at 298 K. | ||
| IRa (cm−1) | 1H NMR (ppm) | 31P{1H} NMR (ppm) | |
|---|---|---|---|
| a The strongest absorption was used for each νCO value. b From ref. 50 and 51. c From ref. 49 and 50. d From ref. 48. | |||
| 1 | 1928(s), 1898(vs), 1855(m), 1714(m) | −17.65 (br) | — |
[H
2
Ru
4
(CO)
12
]
2−
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
2035(w), 1992(s), 1956(vs), 1750(m) | −19.48 | — |
|
[H
3
Ru
4
(CO)
12
]
−c |
2036(s), 2016(s), 1997(vs), 1975(m) | −17.12 | — |
|
H
4
Ru
4
(CO)
12
d |
2080(s), 2065(vs), 2021(s) | −17.70 | — |
| 2 | 2034(m), 1953(m), 1929(s), 1744(m) | −19.51 (d) | 62.14 |
| 3 | 2033(w), 1986(m), 1967(s), 1749(m) | −13.97 (t) | 63.13 |
| 4 | 2053(s), 2007(vs), 1989(m), 1953 (w) | −13.01 (br) | 60.38 |
| 5 | 2082(w), 2072(m), 2029(s), 1970(m) | — | 68.46 (3P), 67.15 (1P) |
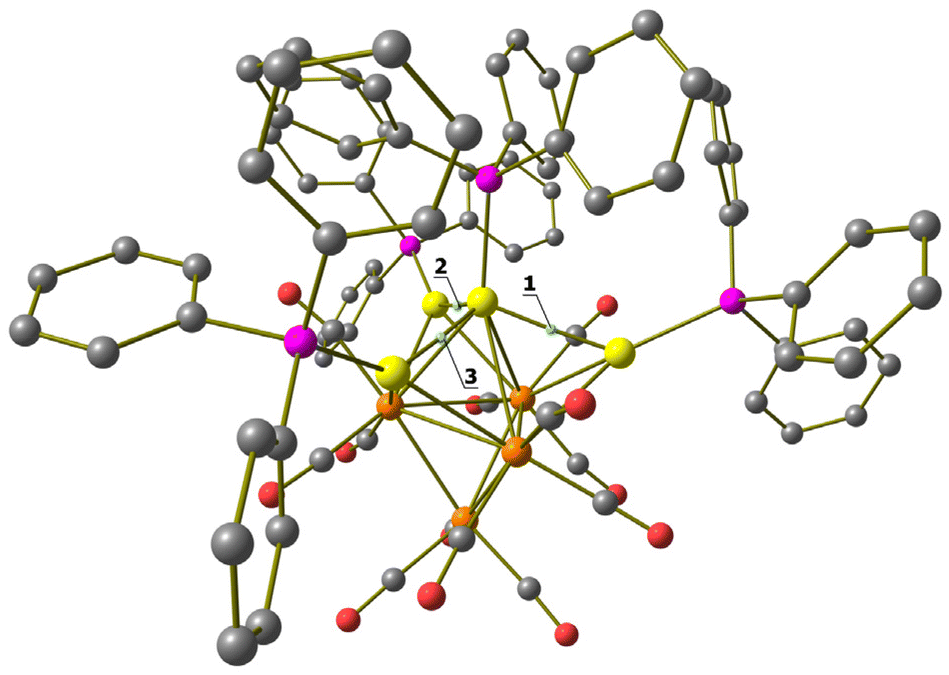
Mono-aurated 2 is very elusive and has been only spectroscopically identified (IR, 1H and 31P{1H} NMR). Conversely, 3–5 have been fully characterized by IR, 1H and 31P{1H} NMR spectroscopies (Fig. S1–S27 in the ESI†), and their molecular structures are determined by SC-XRD as [NEt4][3]·2CH2Cl2, 4-b·2CH2Cl2, 4-a, 5·0.5CH2Cl2·solv (monoclinic, C2/c), and 5·solv (monoclinic, P21/c). The labels 4-a and 4-b refer to the two structurally characterized isomers of 4 (see below for further details).
Two alternative syntheses were previously reported for 4, that is (a) the reaction of H4Ru4(CO)12 or H2Ru4(CO)12(AuPPh3)2 with Au(CH3)(PPh3) and (b) the reaction of [H3Ru4(CO)12]− with [{Au(PPh3)}3O][BF4].41,42 The latter synthesis afforded 4 in combination with H3Ru4(CO)12(AuPPh3) and H2Ru4(CO)12(AuPPh3)2, which were separated by chromatography. It has also been reported that the reaction of [HFe4(CO)12]3− with two mole equivalents of Au(PPh3)Cl affords [HFe4(CO)12(AuPPh3)2]−, which is transformed into HFe4(CO)12(AuPPh3)3 upon addition of HBF4·Et2O.46
The νCO bands are moved 37–40 cm−1 towards higher wavenumbers upon each addition of one [AuPPh3]+ fragment to the 1–4 series of clusters, a result in line with the computational IR simulations (Fig. S28 in the ESI†). This point strongly supports the formation of 2, even if it has not been possible to isolate and structurally characterize it. Moreover, the 1H NMR spectrum of 2 displays a doublet at δH −19.51 ppm with JH–P = 2.0 Hz, in agreement with the presence of one hydride and one AuPPh3 group. This is further supported by the presence of a resonance at δP 62.14 ppm in the 31P{1H} NMR spectrum.
The 1H and 31P{1H} NMR spectra clearly indicate that 2 is always formed in combination with 3 and [H2Ru4(CO)12]2−. This is probably the reason why it was not possible to crystallize 2. Thus, its structure has been predicted and optimized by DFT methods. The ground-state geometry, shown in Fig. 2, is composed of a Ru4 tetrahedron capped on a triangular face by an AuPPh3 fragment, while on another face, a μ3-coordinated hydride is present. The coordination sphere of the Ru centres is completed by nine terminal and three bridging CO ligands. The AIM analysis on the electron density revealed the presence of a (3,−1) bond critical point (BCP) between the Au centre and one of the terminal carbonyl ligands. Selected computed properties at the BCP are provided in the caption of Fig. 2. The negative value of the energy density (E) and the positive value of the Laplacian of electron density (∇2ρ) are in agreement with Bianchi's definition of a dative bond.52 Even if the presence of different attractors does not allow a direct comparison, it is worth noting that the potential energy density (V) at the Au⋯C BCP is about one tenth of the value calculated for the Ru–C BCP involving the same carbonyl ligand, in agreement with a quite weak interaction.
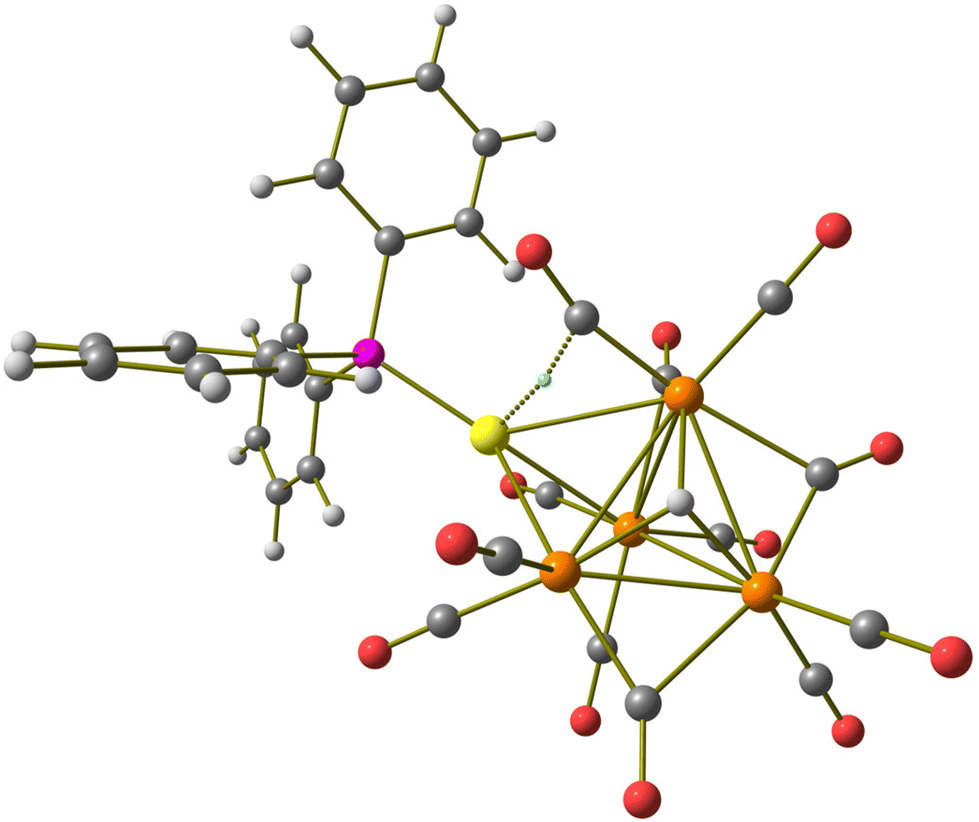 | ||
| Fig. 2 DFT-optimized structure of [HRu4(CO)12(AuPPh3)]2− (2) (orange Ru; yellow Au; purple P; red O; grey C; white H). (3,−1) Au⋯C BCP represented with a small cyan sphere. Selected computed average bond lengths (Å): Ru–Ru 2.918, Ru–H 1.916, and Ru–Au 2.849. Selected properties at the BCP (a.u.): ρ = 0.037, V = −0.026, E = −0.002, and ∇2ρ = 0.086. | ||
Molecular structure, spectroscopic characterization and DFT analysis of [HRu4(CO)12(AuPPh3)2]− (3)
Cluster 3 was obtained in good yield (78%) upon addition of a further mole equivalent of Au(PPh3)Cl to 2. The molecular structure of 3 (Fig. 3 and Table 2) closely resembles that previously reported for [HFe4(CO)12(AuPPh3)2]−.46 It consists of a Ru4 tetrahedron capped on a triangular face by one AuPPh3 fragment adjacent to a Ru3 triangle capped by the μ3-H ligand. The second AuPPh3 fragment is located on the Ru2Au triangle near the hydride ligand. Alternatively, 3 may be viewed as a Ru4Au trigonal bipyramid capped on a Ru3 and a Ru2Au face, sharing a Ru–Ru edge, by μ3-H and μ3-AuPPh3, respectively. Among the 12 CO ligands bonded to the four Ru atoms, 10 are terminal and 2 are edge bridging. The unique hydride ligand has been located in the final Fourier difference map and refined isotropically. The hydride location is the same in 3 and [HFe4(CO)12(AuPPh3)2]−.46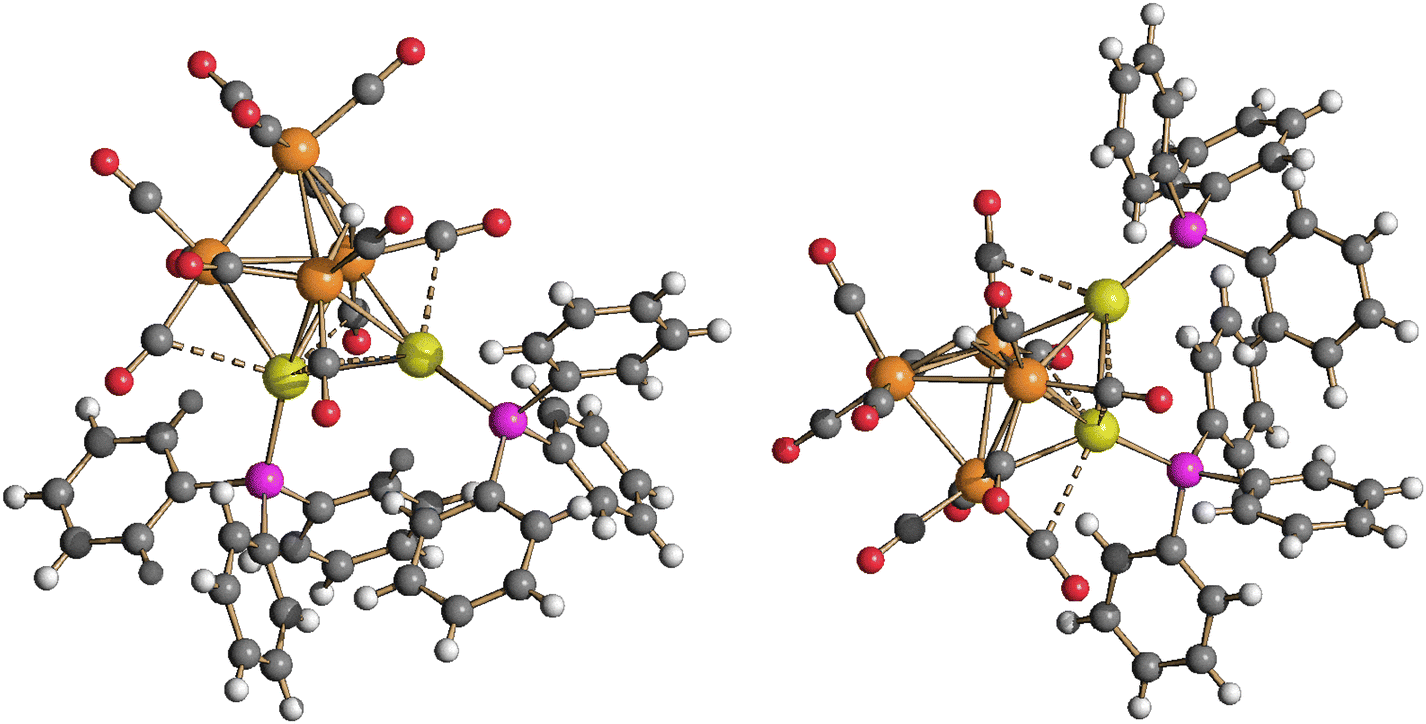 | ||
| Fig. 3 Two views of the molecular structure of [HRu4(CO)12(AuPPh3)2]− (3) (orange Ru; yellow, Au; purple, P; red O; grey C; white H). Au⋯C(O) contacts [2.76–3.06 Å] are represented as fragmented lines. | ||
| Ru–Ru | Ru–H | Ru–Au | Au⋯Au | |
|---|---|---|---|---|
a From ref. 47.
b As [NEt4][3]·2CH2Cl2 (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ).
c As 4-a (triclinic, P) (isomer 4-a).
d As 4-b·2CH2Cl2 (monoclinic, P21/c) (isomer 4-b).
e As 5·0.5CH2Cl2·solv (monoclinic, C2/c).
f As 5·solv (monoclinic, P21/c).
g From ref. 48.
h Isomer C2, from ref. 49 and 50.
i Isomer C3v, from ref. 49 and 50.
j From ref. 49 and 51. ).
c As 4-a (triclinic, P) (isomer 4-a).
d As 4-b·2CH2Cl2 (monoclinic, P21/c) (isomer 4-b).
e As 5·0.5CH2Cl2·solv (monoclinic, C2/c).
f As 5·solv (monoclinic, P21/c).
g From ref. 48.
h Isomer C2, from ref. 49 and 50.
i Isomer C3v, from ref. 49 and 50.
j From ref. 49 and 51.
|
||||
| 1 | 2.8001(11)–2.8113(11), average 2.805(3) | 1.77(4)–1.78(4), average 1.78(7) | — | — |
| 3 | 2.8156(7)–3.0483(6), average 2.9002(15) | 1.73(6)–1.89(6), average 1.82(10) | 2.7808(5)–2.9408(5), average 2.8231(11) | 2.8393(3) |
|
4-ac |
2.770(2)–3.090(2), average 2.902(5) | 1.80(2) | 2.7637(18)–2.9022(18), average 2.826(5) | 2.9247(11)–2.9515(12), average 2.9381(17) |
|
4-bd |
2.7887(6)–2.9888(6), average 2.8865(15) | 1.88(3) | 2.7730(5)–3.0487(5), average 2.8689(13) | 2.8288(3)–2.8623(3), average 2.8456(4) |
| 5 | 2.870(2)–3.0838(16), average 2.977(5) | — | 2.7861(18)–2.9838(14), average 2.860(4) | 2.8180(8)–2.8671(8), average 2.8405(14) |
| 5 | 2.8899(18)–3.213(2), average 3.018(5) | — | 2.8107(16)–3.1396(18), average 2.893(5) | 2.8531(14)–2.8966(14), average 2.880(2) |
|
H
4
Ru
4
(CO)
12
g |
2.7839(8)–2.9565(7), average 2.895(2) | — | — | — |
|
[H
3
Ru
4
(CO)
12
]
−h |
2.7614(5)–2.9423(4), average 2.8504(12) | 1.72(4)–1.80(4), average 1.76(8) | — | — |
|
[H
3
Ru
4
(CO)
12
]
−i |
2.7733(5)–2.9380(5), average 2.8519(12) | 1.72(4)–1.83(4), average 1.76(9) | ||
|
[H
2
Ru
4
(CO)
12
]
2−
j |
2.7526(4)–2.9771(4), average 2.8436(10) | 1.64(4)–1.87(4), average 1.76(6) | — | — |
The Ru–Ru bonds are rather spread [2.8156(7)–3.0483(6) Å, average 2.9002(15) Å] compared to the parent 1 [2.8001(11)–2.8113(11) Å, average 2.805(3) Å] in view of the coordination of two [AuPPh3]+ fragments, which causes swelling of the Ru4 tetrahedron. It must be remarked that this phenomenon is less marked for [H3Ru4(CO)12]− [2.7614(5)–2.9423(4) Å, average 2.8504(12) Å for isomer C2; 2.7733(5)–2.9380(5) Å, average 2.8519(12) Å for isomer C3v], which arises from 1 upon addition of two H+ ligands instead of two [AuPPh3]+ fragments. This supports the fact that the swelling of 3 is mainly due to the presence of bulky AuPPh3 groups, rather than a charge effect. Two isomers of [H3Ru4(CO)12]− are known, and their molecular structures display only edge bridging hydride ligands.49,50 The different stereochemistry observed for the unique hydride and the two [AuPPh3]+ ligands of 3 further supports the evidence that their isolobal analogy fades when two or more Au(I) centers are present, due to the insurgence of aurophilicity.1,2,5,6,14
Cluster 3 displays five Ru–Au bonding contacts [2.7808(5)–2.9408(5) Å, average 2.8231(11) Å] and one aurophilic Au–Au contact [2.8393(3) Å]. Some sub-van der Waals Au⋯CO contacts [2.76–3.06 Å] are also present. As previously reported, the latter Au⋯CO contacts are mainly sterically driven and not attractive bonding interactions.53,54
The 31P{1H} NMR spectrum of 3 recorded in acetone-d6 at 298 K displays a singlet at δP 63.41 ppm, indicating a fluxional behaviour which makes the two AuPPh3 fragments equivalent in the timescale of NMR. This point is further corroborated by the fact that the unique hydride resonates as a triplet at δH −13.74 ppm (JH–P 0.9 Hz) in the 1H NMR spectrum under the same conditions. 1H and 31P{1H} NMR spectra of 3 do not change down to 223 K, indicating a very fast exchange even at a low temperature.
The DFT-optimized structure of 3 is in good agreement with the X-ray data (RMSD = 0.416 Å) and the μ3 coordination mode of the hydride is confirmed by the simulations. As stated before, the HRu4Au core resembles the HFe4Au one in the analogous iron cluster. The best superposition of the metal hydride fragments in 3 and [HFe4(CO)12(AuPPh3)2]−, optimized at the same theoretical level, is shown in Fig. S29 in the ESI.† In contrast to the observation of 2, the AIM analysis of 3 did not localize any (3,−1) BCP related to the Au⋯CO contacts. On the other hand, a (3,−1) BCP between the two Au centres was found, as shown in Fig. 4. The computed properties, summarized in the caption of Fig. 4, are in line with a weak Au⋯Au metal–metal interaction.47 The computed Au⋯Au distance, 2.861 Å, is in excellent agreement with the experimental value, 2.8393(3) Å. Upon comparison, the Au⋯Au interaction in [HFe4(CO)12(AuPPh3)2]−, optimized at the same theoretical level, was found to be slightly stronger (ρ = 0.042 a.u., V = −0.036 a.u.), despite the fact that the computed Au⋯Au distance is the same as in 3, 2.861 Å.
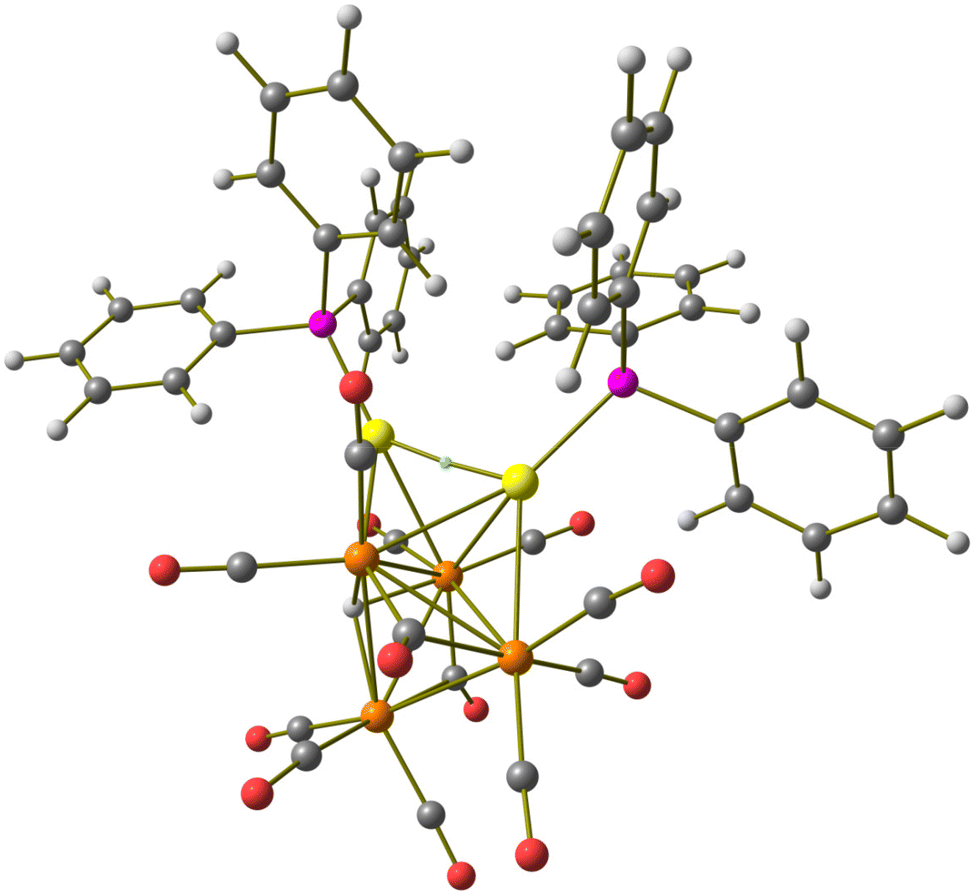 | ||
| Fig. 4 DFT-optimized structure of [HRu4(CO)12(AuPPh3)2]− (3) (orange Ru; yellow Au; purple P; red O; grey C; white H). (3,−1) Au–Au BCP represented with a small cyan sphere. Selected computed average bond lengths (Å): Ru–Ru 2.938, Ru–H 1.895, Ru–Au 2.841, and Au⋯Au 2.861. Selected properties at the BCP (a.u.): ρ = 0.043, V = −0.036, E = −0.007, and ∇2ρ = 0.094. | ||
Attempts to understand the fluxional behaviour of 3 were carried out considering the possible presence of more symmetric structures in solution. In particular, starting geometries with the Cs symmetry of the metal hydride fragment were considered, and quite a symmetric stationary point was achieved (Fig. S30 in the ESI†). The optimized geometry, where the hydride is μ3-coordinated to one triangular face of the Ru4 tetrahedron and the two AuPPh3 fragments are symmetrically capping other two faces, was however meaningfully less stable with respect to the observed isomer of 3, with the Gibbs energy difference around 21.2 kcal mol−1. It is therefore unlikely that such species could have a role in the apparent symmetry observed by means of NMR spectroscopy; thus, the fluxional behaviour appears to be ascribed to the fast exchange of the fragments capping the Ru4 tetrahedron.
Structural, spectroscopic and computational studies of the two isomers of HRu4(CO)12(AuPPh3)3 (4)
Neutral cluster 4 contains one additional [AuPPh3]+ fragment compared to 3. Two different isomers have been characterized in the solid state, that is, isomer 4-a and isomer 4-b (Fig. 5, 6 and Table 1). The latter isomer has been found in the solvate crystals 4-b·2CH2Cl2, whereas isomer 4-a has been found in solid solvent-free 4-a (triclinic, P). The structure of isomer 4-b was previously determined in solid solvent-free crystals (monoclinic, P21/n),41,42 suggesting that crystallization of 4-a or 4-b does not depend on the presence/absence of co-crystallized solvent molecules. The two isomers are rapidly exchanging in solution, as indicated by VT 1H and 31P{1H} NMR experiments (see below). The structures of both 4-a and 4-b differ from that of H4Ru4(CO)1248 due to aurophilic interactions.
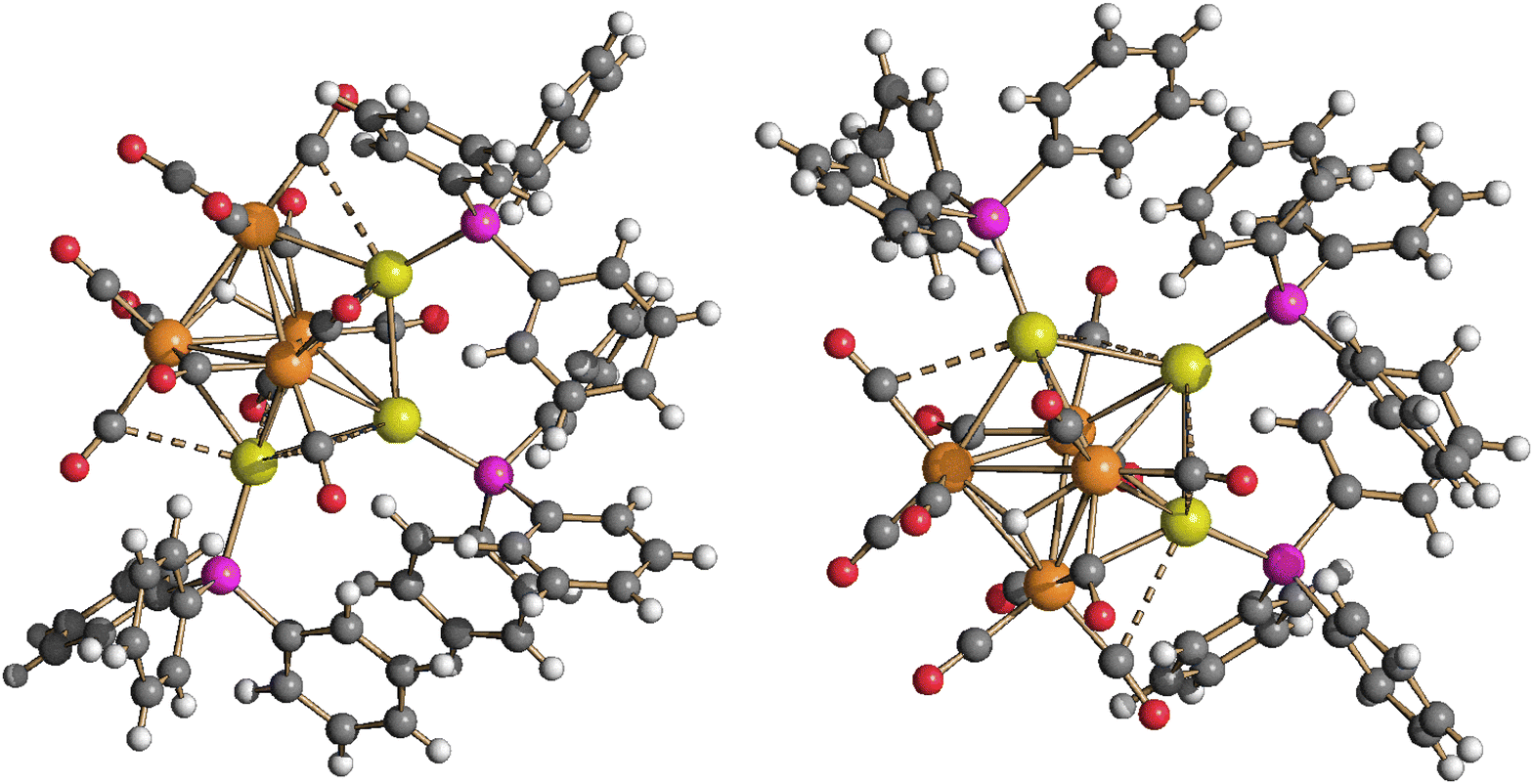 | ||
| Fig. 5 Two views of the molecular structure of HRu4(CO)12(AuPPh3)3, isomer 4-a (orange Ru; yellow, Au; purple, P; red O; grey C; white H). Au⋯C(O) contacts [2.77–3.08 Å] are represented as fragmented lines. | ||
 | ||
| Fig. 6 Two views of the molecular structure of HRu4(CO)12(AuPPh3)3, isomer 4-b (orange Ru; yellow, Au; purple, P; red O; grey C; white H). Au⋯C(O) contacts [2.76–3.26 Å] are represented as fragmented lines. | ||
Isomer 4-a formally arises from the addition of the third [AuPPh3]+ fragment on the triangular face of 3 originally capped by μ3-H, with concomitant migration of the hydride on an adjacent Ru3 face (Scheme 2). In contrast, isomer 4-b originates from the addition of the third [AuPPh3]+ fragment onto a Ru2Au triangular face of 3 without migration of the hydride ligand.
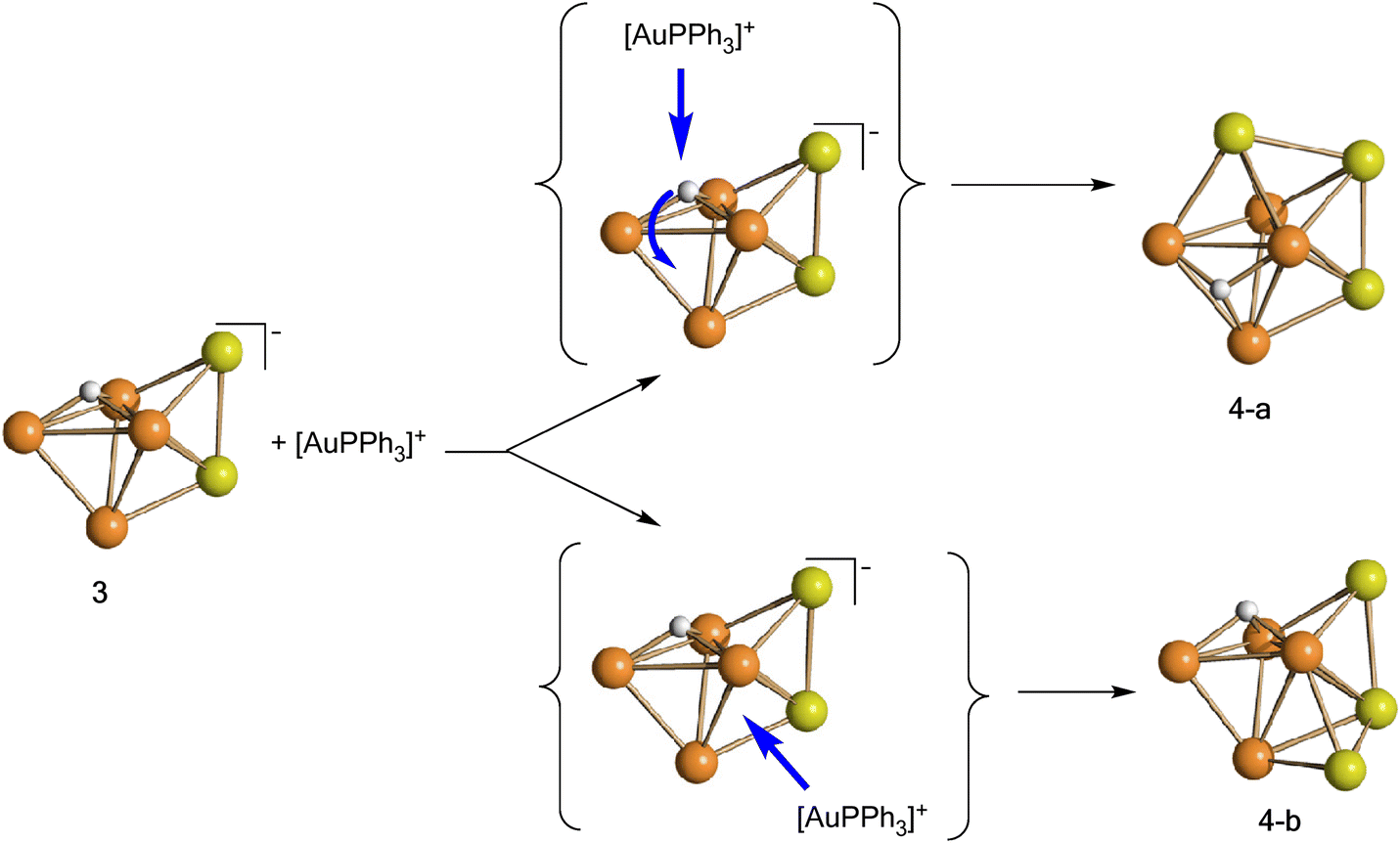 | ||
| Scheme 2 Formal transformation of 3 into 4-a and 4-b, the two isomers of 4 (orange, Ru; yellow, Au; white, H). CO and PPh3 ligands have been omitted for clarity. | ||
The Ru4Au3 metal cage of 4-a may be described as composed of five tetrahedra (Ru4, Ru3Au, Ru2Au2, Ru2Au2, Ru3Au) sharing five triangular faces, resulting in a pentagonal bipyramid. The same metal cage was previously found in HFe4(CO)12(AuPPh3)3,46 even though the unique hydride was located within the tetrahedral Fe4 cage, rather than on a triangular face.
Isomer 4-b is composed of a trigonal bipyramidal Ru4Au core capped on two Ru2Au faces by two further Au atoms. The unique hydride ligand is face capping a triangular Ru3 face.
In both isomers, all the CO ligands are essentially bonded to the four Ru atoms, showing only some weak sub-van der Waals Au⋯C(O) contacts. The nature of the latter contacts is rather debated, as previously discussed.53,54 Disregarding the Au⋯C(O) contacts, all CO ligands are terminally bonded in isomer 4-b, three per Ru atom. Conversely, isomer 4-a contains 10 terminal and two edge bridging CO ligands on two Ru–Ru edges.
The Ru–Ru contacts in isomers 4-a [2.7887(6)–2.9888(6) Å; average 2.8865(15) Å] and 4-b [2.770(2)–3.090(2) Å; average 2.902(5) Å] are rather similar and also comparable to those of 3. This indicates that the addition of a further AuPPh3 fragment does not significantly alter the Ru4 tetrahedron from 3 to 4. Isomer 4-a displays eight Ru–Au bonding contacts [2.7730(5)–3.0487(5) Å; average 2.8689(13) Å], whereas only seven Ru–Au bonds are present in 4-b [2.7637(18)–2.9022(18) Å; average 2.826(5) Å], in view of the different coordination modes of the three AuPPh3 fragments to the Ru4 tetrahedron in the two isomers.
Two aurophilic Au⋯Au contacts are present in both 4-a [2.8288(3)–2.8623(3) Å; average 2.8456(4) Å] and 4-b [2.9247(11)–2.9515(12) Å; average 2.9381(17) Å].
The behavior in solution of 4 has been investigated by VT 1H and 31P{1H} NMR spectroscopy (Fig. 7, 8 and Fig. S17–S26 in the ESI†). Two major differences with the related HFe4(CO)12(AuPPh3)3 cluster are evident. First of all, dissociation of 4 into 3 and [AuPPh3]+ does not occur even in polar solvents such as CH3CN and DMSO, whereas HFe4(CO)12(AuPPh3)3 is stable only in CH2Cl2 and rapidly dissociates into [HFe4(CO)12(AuPPh3)2]− and [AuPPh3]+ in acetone. Moreover, HFe4(CO)12(AuPPh3)3 is fluxional at all temperatures and only one isomer has been detected, whereas the two isomers of 4 rapidly interconvert at room temperature but the process is sensibly slowed down at lower temperatures.
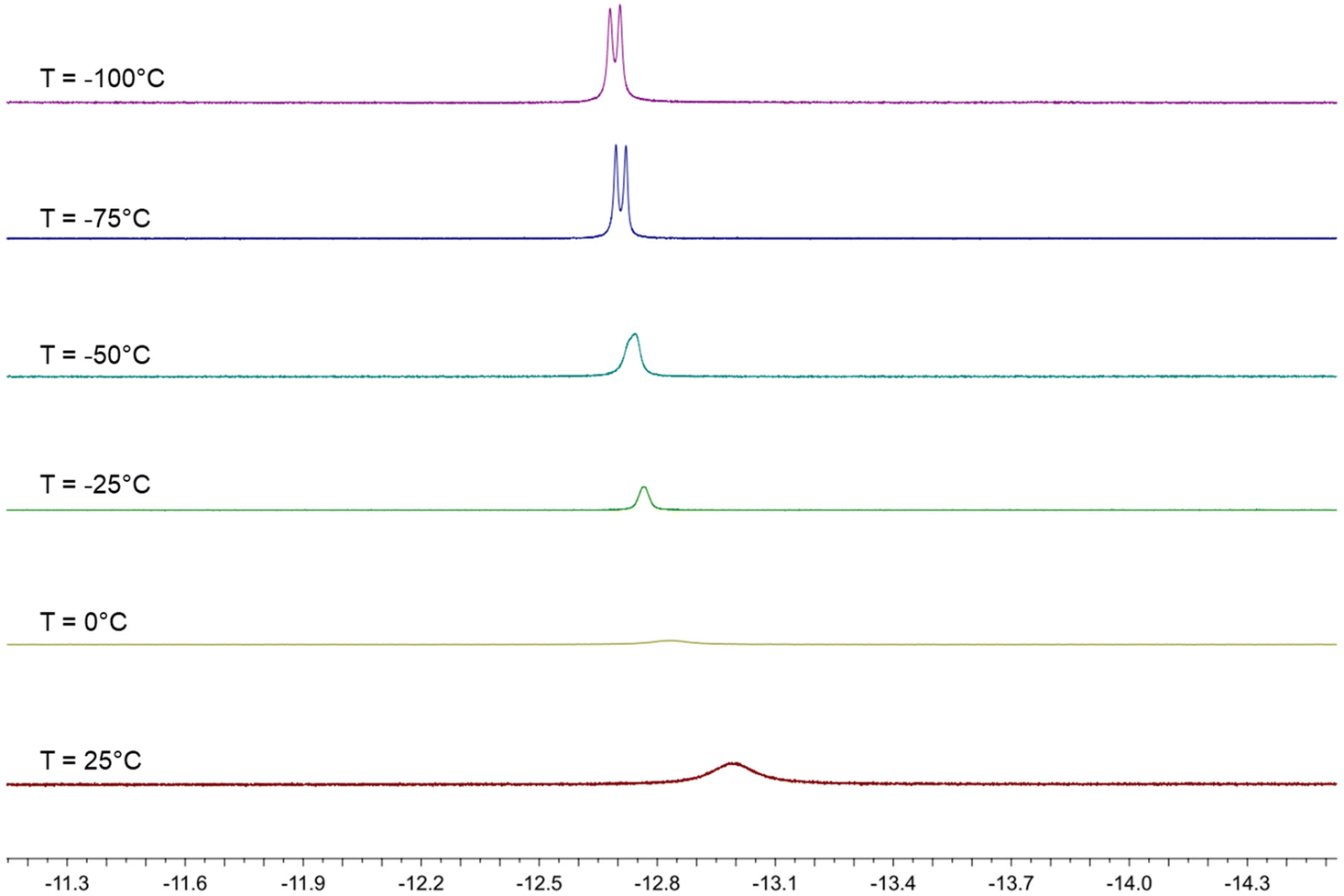 | ||
| Fig. 7 Hydride region of the VT 1H NMR spectra of HRu4(CO)12(AuPPh3)3 (4) in CD2Cl2 (600 MHz). | ||
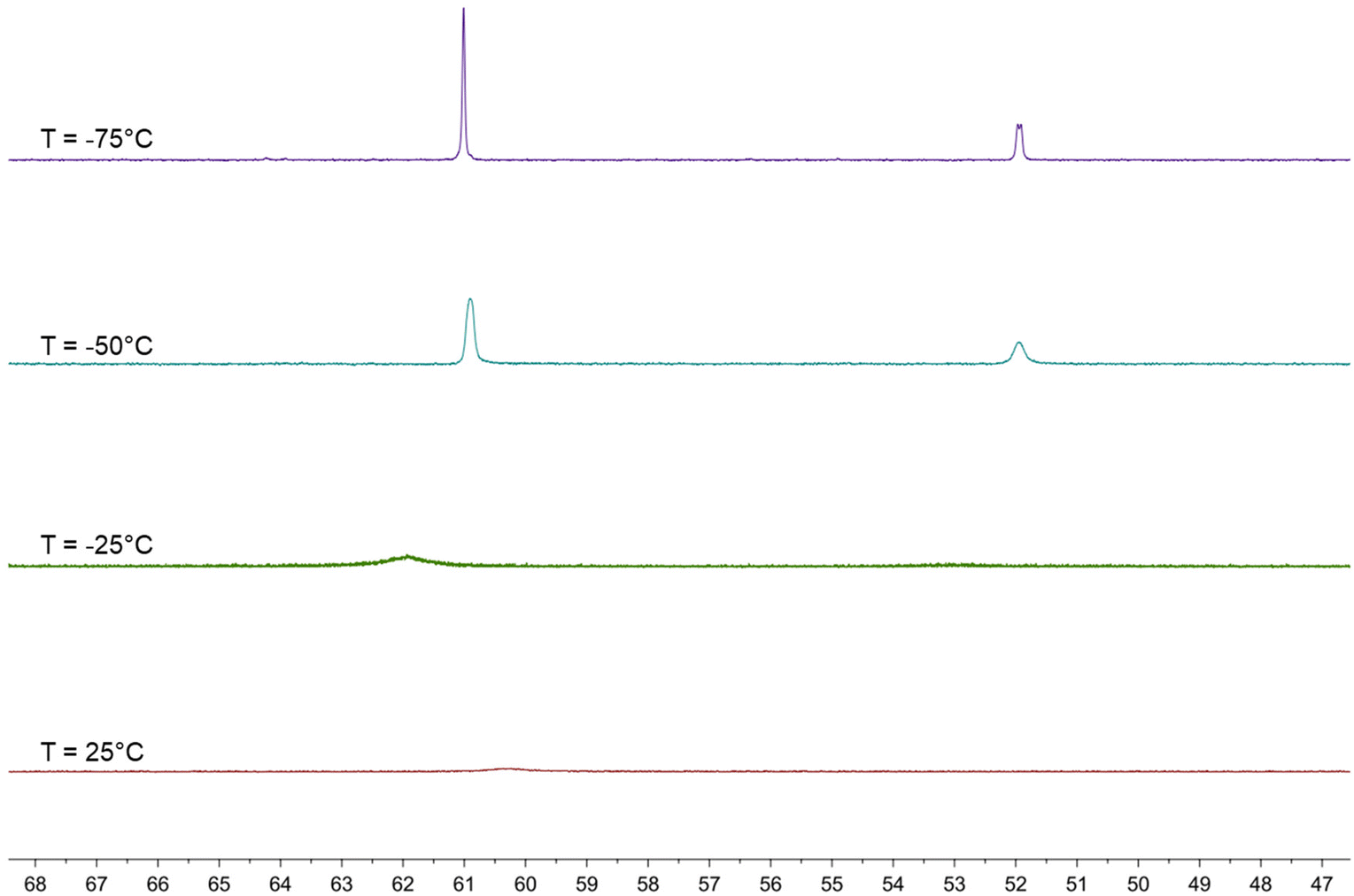 | ||
| Fig. 8 VT 31P{1H} NMR spectra of HRu4(CO)12(AuPPh3)3 (4) in CD2Cl2 (600 MHz). | ||
The 1H NMR spectrum of 4 in CD2Cl2 at 298 K shows a broad resonance at δH −13.01 ppm and almost complete coalescence is observed at 273 K. A quartet at δH −12.80 ppm with JH–P = 5.2 Hz appears, then, at 248 K, indicating that fluxionality makes the three AuPPh3 groups equivalent from the point of view of the unique hydride. This resonance is further broadened at 223 K, and eventually, two equally intense and closely spaced resonances are observed at 198 K and 173 K. These may be interpreted as two singlets due to the presence of a 1:1 mixture of two isomers, or as a doublet where the hydride strongly couples with just one AuPPh3 group. In order to shed more light on this point, VT 31P{1H} NMR experiments have been performed. A broad resonance is observed at 298 K with δP 60.37 ppm and coalescence occurs at 273 K. Two resonances in a 2:1 ratio appear at lower temperatures, which become well resolved at 223 K, with δP 60.91 (2P) and 51.95 (1P) ppm. At 198 K, the lower frequency resonance is further split into two 1:1 resonances at δP 51.96 and 51.92 ppm, whereas that at a higher frequency remains a singlet at δP 61.00 ppm. This observation may be interpreted assuming the presence in solution of two isomers each containing two equivalent AuPPh3 groups and one unique AuPPh3 group, as experimentally found in 4-a and 4-b. For these isomers, the resonances of the unique AuPPh3 group are resolved at 198 K, whereas the resonances of the two equivalent AuPPh3 groups are almost superimposed for the two isomers also at 198 K. This is in agreement with the observation of two isomers in the solid state, which very rapidly exchange in solution.
The DFT-optimized structures of 4-a and 4-b agree with the X-ray data, with RMSD values of 0.928 Å for 4-a and 0.369 Å for 4-b. The localization of the hydrides is confirmed, as observable from the superposition of the HRu4Au3 fragments in Fig. S31 in the ESI.† Gas-phase calculations indicate that 4-b is more stable than 4-a by 4.9 kcal mol−1, and thus, the isolation of both the species is to be ascribed almost in part to different packing forces. Another isomer (4-c) was optimized by changing the position of the hydride in 4-b. The disposition of the metal centres is comparable with respect to 4-b, but the hydride is μ2-coordinated to two Ru centres at the equatorial position of the Ru4Au trigonal bipyramid at the opposite side with respect to the AuPPh3 fragments capping two Ru2Au faces. 4-c was more stable than 4-b by about 4.2 kcal mol−1 in the gas phase. The three isomers are depicted in Fig. 9. It is worth noting that the relative energy values were strongly dependent upon the surrounding medium. The introduction of DMSO as an implicit solvent inverted the stability order of 4-a and 4-b, and the Gibbs energy interval between the less stable isomer (4-b) and the most stable one (4-c) was reduced to only 2.8 kcal mol−1. The position of the hydride is also influenced by the surrounding medium, since the addition of the solvation model caused the change in the coordination mode in 4-a from μ3-H to μ2-H (Fig. S32 in the ESI†). The AIM analyses allowed the localization of all the isomers of (3,−1) BCP related to the Au⋯Au interactions, roughly comparable to that described for 3. Selected data, provided in the caption of Fig. 9, indicate that the Au⋯Au interactions are slightly stronger in 4-b and 4-c, perhaps thanks to the disposition of the Au centres. No (3,−1) BCP was instead localized for the Au⋯C contacts.
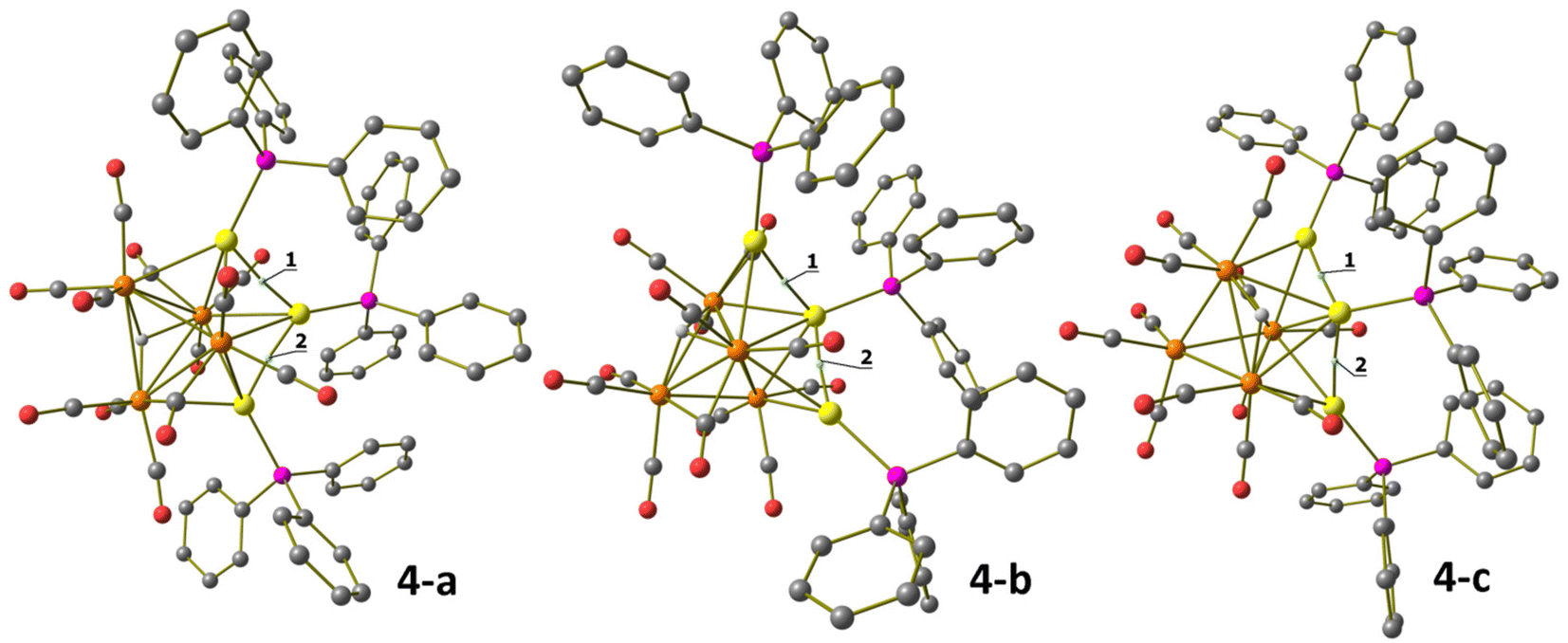 | ||
| Fig. 9 DFT-optimized structures of HRu4(CO)12(AuPPh3)3, isomers 4-a, 4-b and 4-c (orange Ru; yellow Au; purple P; red O; grey C; white H; hydrogen atoms on the phenyl rings are omitted for clarity). (3,−1) Au–Au BCPs represented with small cyan spheres. Selected computed average bond lengths (Å): 4-a, Ru–Ru 3.035, Ru–H 1.840, Ru–Au 2.880, and Au⋯Au 2.948; 4-b, Ru–Ru 2.955, Ru–H 1.865, Ru–Au 2.872, and Au⋯Au 2.887; and 4-c, Ru–Ru 2.914, Ru–H 1.830, Ru–Au 2.911, and Au⋯Au 2.830. Selected properties at the BCP (a.u.): 14−a, ρ = 0.037, V = −0.031, E = −0.005, and ∇2ρ = 0.082; 24−a, ρ = 0.037, V = −0.028, E = −0.004, and ∇2ρ = 0.076; 14−b, ρ = 0.040, V = −0.034, E = −0.006, and ∇2ρ = 0.088; 24−b, ρ = 0.041, V = −0.034, E = −0.006, and ∇2ρ = 0.089; 14−c, ρ = 0.044, V = −0.040, E = −0.007, and ∇2ρ = 0.103; and 24−c, ρ = 0.044, V = −0.039, E = −0.007, and ∇2ρ = 0.100. | ||
The possible formation of isomers with the hydride inside the metal cage was also considered, starting from the X-ray structure of HFe4(CO)12(AuPPh3)3 and replacing the metal centres. On the other hand, an isomer of HFe4(CO)12(AuPPh3)3 with the hydride on the surface of the metal cage was optimized, starting from the geometry of 4-b. The metal hydride cores are compared in Fig. S33 in the ESI.† From a thermodynamic point of view, the migration of the hydride from an M3 triangular face to the M4 cage is more favoured for the Fe cluster with respect to the Ru analogue by about 4.3 kcal mol−1, in agreement with the experimental localizations of the hydride ligands. This is probably due to the steric problems on the surface of the iron cluster. Indeed, Fe being smaller than Ru, it is likely that there is not enough space on the surface of an Fe4 tetrahedron in order to locate 12 CO ligands, three AuPPh3 fragments and a hydride. Thus, the H-atom is forced to migrate inside the Fe4 tetrahedron, whereas there is enough space to remain on the surface of the Ru4 tetrahedron of 4.
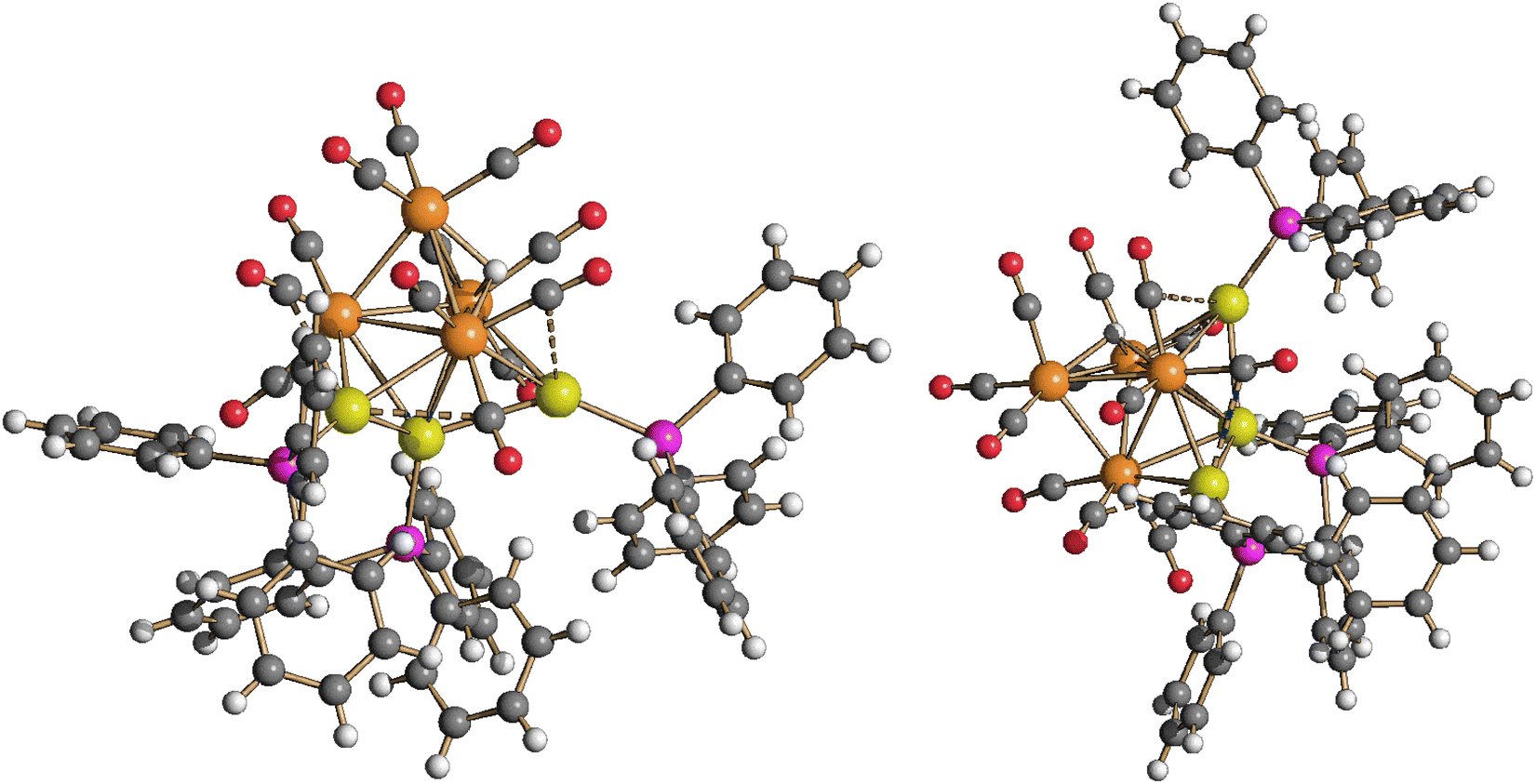
Molecular structure, spectroscopic characterization and DFT analysis of Ru4(CO)12(AuPPh3)4 (5)
The molecular structure of the tetra-aurated cluster 5 has been determined on two solvates, that is, 5·0.5CH2Cl2·solv (monoclinic C2/c) and 5·solv (monoclinic P21/c), displaying almost identical structures and bonding parameters (Fig. 10 and Table 2). Its metal cage may be viewed as a Ru4Au trigonal bipyramid capped on all three Ru2Au faces by Au atoms. The resulting Ru4Au3 core possesses idealized C3v symmetry and is closely related to the metal cage of 4-b. In agreement with this, the 31P{1H} NMR spectrum of 5, recorded in CD2Cl2 at 298 K, displays two resonances at δP 68.46 and 67.15 ppm, with relative intensities at 3:1.
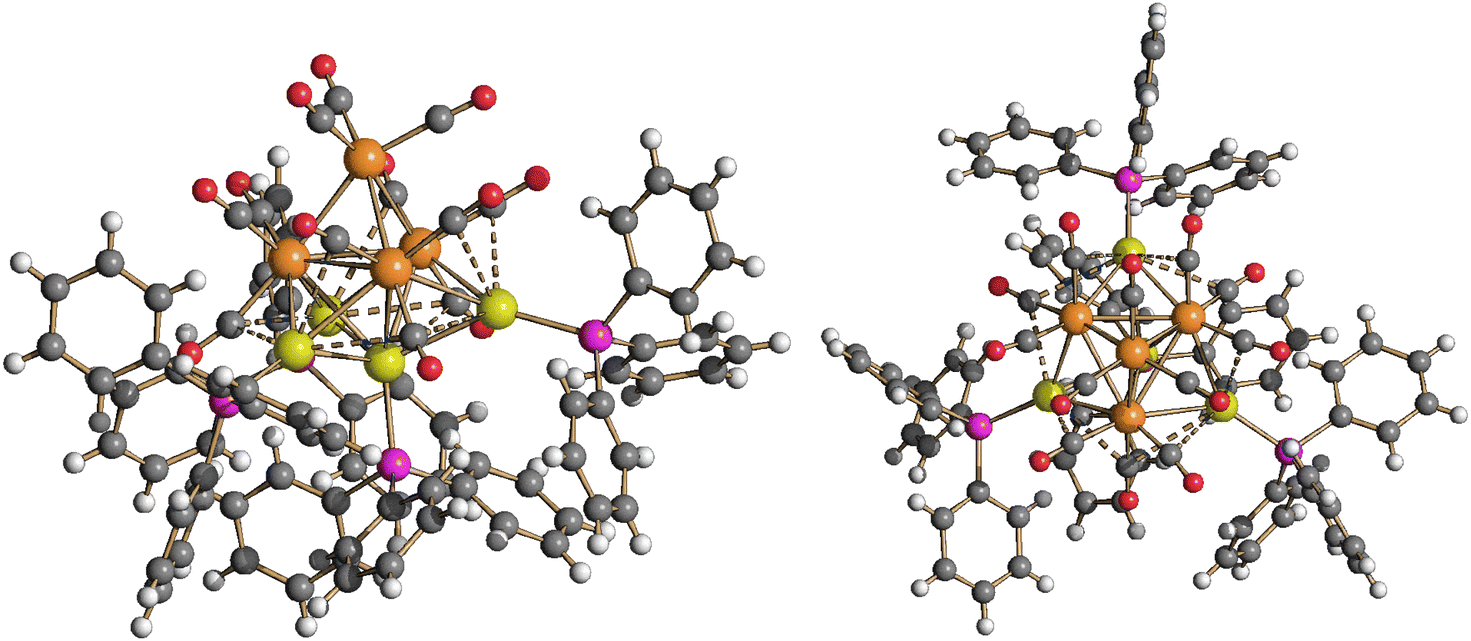 | ||
| Fig. 10 Two views of the molecular structure of Ru4(CO)12(AuPPh3)4 (5) (orange Ru; yellow, Au; purple, P; red O; grey C; white H). Au⋯C(O) contacts [2.70–3.11 Å] are represented as fragmented lines. | ||
Coordination of four AuPPh3 groups to the Ru4 tetrahedron significantly swells the Ru–Ru contacts [2.870(2)–3.0838(16) Å, average 2.977(5) Å for 5·0.5CH2Cl2·solv (monoclinic C2/c); 2.8899(18)–3.213(2) Å, average 3.018(5) Å for 5·solv (monoclinic P21/c)] compared to 3 and 4. Considering only interactions with Ru atoms, the CO ligands are all terminal (three per Ru), even though some sub-van der Waals Au⋯C(O) contacts are present.
Cluster 5 displays nine Ru–Au bonding contacts [2.7861(18)–2.9838(14) Å, average 2.860(4) Å for 5·0.5CH2Cl2·solv (monoclinic C2/c); 2.8107(16)–3.1396(18) Å, average 2.893(5) Å for 5·solv (monoclinic P21/c)] as well as three aurophilic Au⋯Au contacts [2.8180(8)–2.8671(8) Å, average 2.8405(14) Å for 5·0.5CH2Cl2·solv (monoclinic C2/c); 2.8531(14)–2.8966(14) Å, average 2.880(2) Å for 5·solv (monoclinic P21/c)].
The only other species of the general formula M4(CO)12(M'PPh3)4 (M = Fe, Ru, Os; M′ = Cu, Ag, Au) reported prior to this work was Ru4(CO)12(CuPPh3)4 where the four CuPPh3 groups are capping the four triangular faces of the Ru4 tetrahedron without any Cu⋯Cu interaction.54
The DFT-optimized structure of 5 is in good agreement with the X-ray data, the RMSD being 0.588 Å. The Ru4Au4 fragments are superimposed in Fig. S34 in the ESI.† The AIM analysis on 5 revealed the presence of three (3,−1) Au–Au BCPs between the Au centre in the axial position of the Ru4Au trigonal bipyramid and the three AuPPh3 fragments capping the Ru2Au triangular faces (Fig. 11). The properties at the three BCPs are roughly the same (see the caption of Fig. 11), a result in line with the approximate C3v symmetry of the Ru4Au4 fragment (R = 0.077). The values of ρ and V are similar to those obtained for 4-c, suggesting comparable strength of the Au–Au interactions. The presence of bonding overlaps among the Au atoms in 5 can be observed in Fig. 12, where the HOMO is plotted, limited to the contributions of the Au centres.
 | ||
| Fig. 11 DFT-optimized structure of Ru4(CO)12(AuPPh3)4 (5) (orange Ru; yellow Au; purple P; red O; grey C; hydrogen atoms on the phenyl rings are omitted for clarity). (3,−1) Au–Au BCPs represented with small cyan spheres. Selected computed average bond lengths (Å): Ru–Ru 2.929, Ru–Au 2.894, and Au⋯Au 2.818. Selected properties at the BCP (a.u.): 1, ρ = 0.045, V = −0.041, E = −0.007, and ∇2ρ = 0.109; 2, ρ = 0.045, V = −0.040, E = −0.007, and ∇2ρ = 0.104; and 3, ρ = 0.045, V = −0.041, E = −0.007, and ∇2ρ = 0.106. | ||
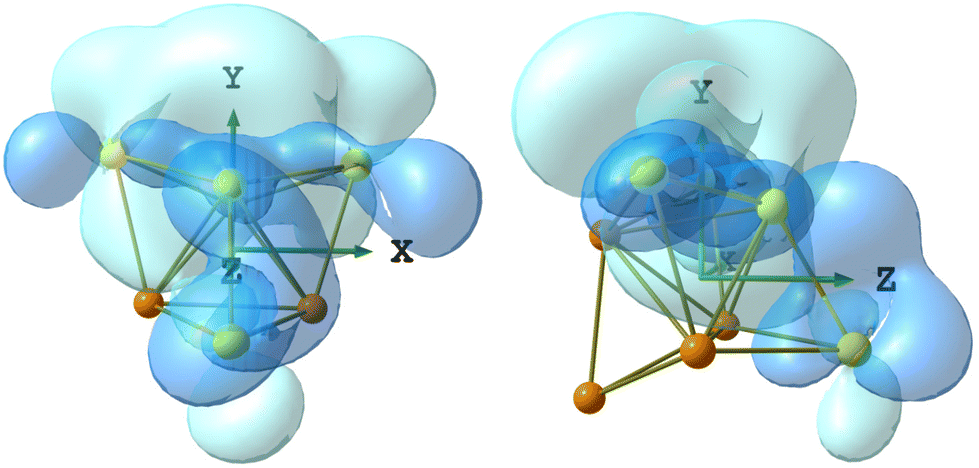 | ||
| Fig. 12 Two views of the Ru4Au4 fragment of 5 (orange Ru; Au yellow) with the HOMO plotted (blue tones), limited to the contributions of the Au centres. Surface isovalue = 0.005 a.u. | ||
Conclusions
The mono-hydride trianionic cluster 1 is a suitable starting material for the addition of one up to four [AuPPh3]+ fragments, resulting in the formation of 2–5. These Ru–Au clusters retain the tetrahedral Ru4 core of 1, which is decorated on the surface by an increasing number of [AuPPh3]+ fragments. Clusters 3–5, which contain two or more Au(I) centres, display aurophilic Au⋯Au contacts, as previously found in other peraurated carbonyl clusters.18–22,29–31 These are soft interactions and thus, rearrangement of the [AuPPh3]+ fragments can be observed. Indeed, two isomers, 4-a and 4-b, have been structurally characterized in the solid state for 4, and their rapid exchange in solution was revealed by VT multinuclear NMR spectroscopy. Interconversion of 4-a and 4-b requires both rearrangement of the three [AuPPh3]+ fragments as well as migration of the unique hydride ligand.The molecular structures of 3–5 differ from the isolobally related hydrides [H3Ru4(CO)12]− and H4Ru4(CO)1248–50 due to the different locations of H and AuPPh3 fragments, because of aurophilicity. Thus, the study of peraurated hydride carbonyl clusters offers the possibility to study at the molecular level the interplay and dynamic behaviour of [AuPPh3]+ and hydride ligands on the surface of the same metal core.
Experimental
General procedures
All reactions and sample manipulations were carried out using standard Schlenk techniques under nitrogen and in dried solvents. All the reagents were commercial products (Aldrich) of the highest purity available and used as received, except for [NEt4]3[1]47 and Au(PPh3)Cl,55 which have been prepared according to the literature. Analyses of C, H and N were performed with a Thermo Quest Flash EA 1112NC instrument. IR spectra were recorded on a PerkinElmer Spectrum One interferometer in CaF2 cells. 1H and 31P{1H} NMR measurements were performed on a Varian Mercury Plus 400 MHz and a Varian Inova 600 MHz instrument. The proton chemical shifts were referenced to the non-deuterated aliquot of the solvent. The phosphorus chemical shifts were referenced to external H3PO4 (85% in D2O). Structure drawings have been performed with SCHAKAL99.56Synthesis of [NEt4]2[HRu4(CO)12(AuPPh3)] ([NEt4]2[2])
Au(PPh3)Cl (0.135 g, 0.274 mmol) was added as a solid in a small portion to a solution of [NEt4]3[1] (0.310 g, 0.274 mmol) in CH3CN (15 mL). The mixture was stirred at room temperature for 1 h and the reaction was monitored by IR spectroscopy. At the end of the reaction, the solvent was removed in vacuo, and the residue was washed with water (2 × 20 mL) and toluene (2 × 10 mL) and extracted with CH2Cl2 (15 mL). IR and NMR analyses on the extraction revealed the presence of [NEt4]2[2] in a mixture with [NEt4][3] (1
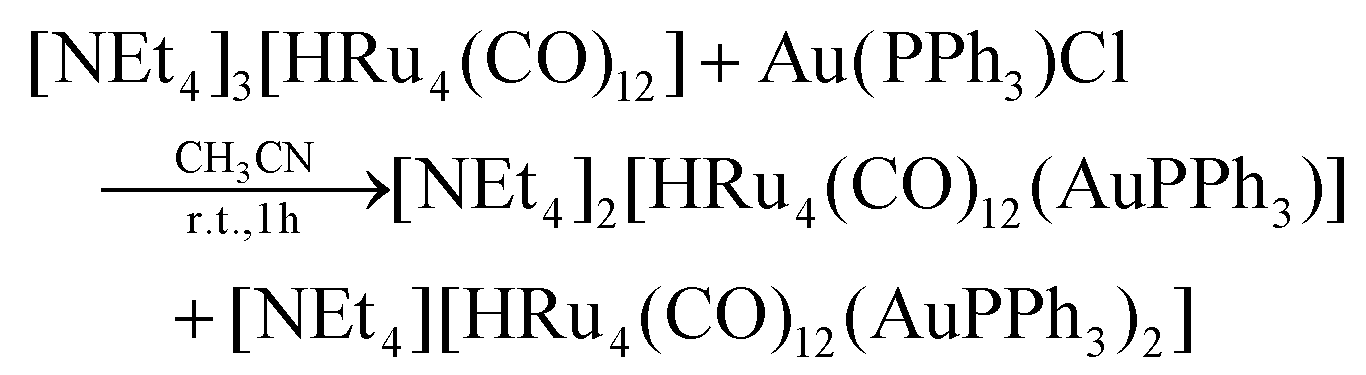 :0.68 ratio by 1H NMR).
:0.68 ratio by 1H NMR).
IR (CH2Cl2, 298 K) νCO: 2034(m), 1953(m), 1929(s), 1744(m) cm−1. 1H NMR (CD2Cl2, 298 K) δ: −19.51 (d, JH–P = 2.0 Hz). 31P{1H} NMR (CD2Cl2, 298 K) δ: 62.15 ppm.
Synthesis of [NEt4][HRu4(CO)12(AuPPh3)2]·2CH2Cl2 ([NEt4][3]·2CH2Cl2)
Au(PPh3)Cl (0.280 g, 0.566 mmol) was added as a solid in a small portion to a solution of [NEt4]3[1] (0.320 g, 0.283 mmol) in CH3CN (15 mL). The mixture was stirred at room temperature for 2 h and the reaction was monitored by IR spectroscopy. At the end of the reaction, the solvent was removed in vacuo, and the residue was washed with water (2 × 20 mL) and toluene (2 × 10 mL) and extracted with CH2Cl2 (15 mL). Crystals of [NEt4][3]·2CH2Cl2 suitable for SC-XRD were obtained by layering n-pentane on the CH2Cl2 solution (yield 78%).
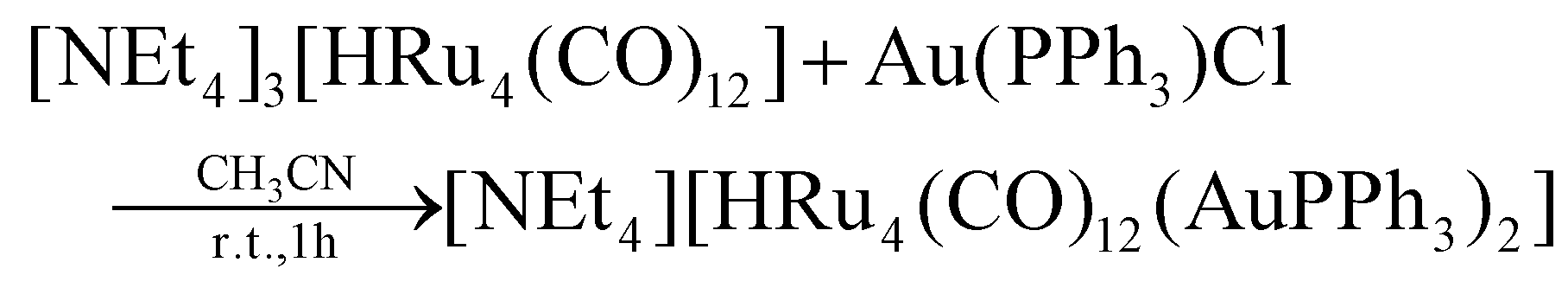
C58H55Au2Cl4NO12P2Ru4 (1959.98): calcd (%): C 35.54, H 2.83, N 0.71; found: C 35.37, H 3.05, N 0.84. IR (CH2Cl2, 298 K) νCO: 2033(w), 1986(m), 1967(s), 1749(m) cm−1. IR (acetone, 298 K) νCO: 2031(w), 1985(m), 1967(s) cm−1. IR (Nujol, 298 K) νCO: 2029(w), 1983(s), 1961(m), 1941(m), 1908(w) cm−1. 1H NMR (Acetone-d6, 298 K) δ: −13.74 ppm (t, JH–P = 0.9 Hz). 31P{1H} NMR (Acetone-d6, 298 K) δ: 63.41 ppm.
Synthesis of HRu4(CO)12(AuPPh3)3 (4)
Au(PPh3)Cl (0.097 g, 0.195 mmol) was added as a solid to a solution of [NEt4][3] (0.350 g, 0.195 mmol) in CH2Cl2 (20 mL), and the mixture was stirred at room temperature for 1 h. At the end of the reaction, the solvent was removed in vacuo, and the residue was washed with water (2 × 20 mL) and extracted with toluene (10 mL). The toluene solution was evaporated to dryness and the green solid obtained was dissolved in CH2Cl2. Crystals of 4-b·2CH2Cl2* suitable for SC-XRD were obtained by layering n-pentane on the dichloromethane solution (yield 68%).
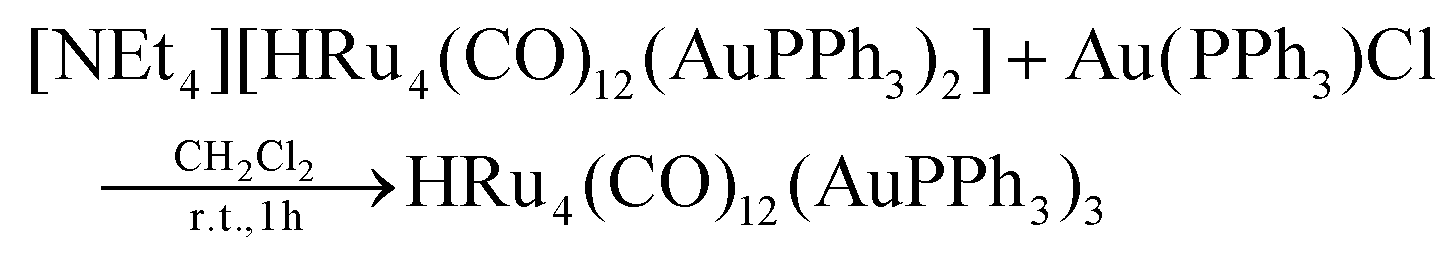
*Sometimes a second isomer crystallized out of the solution as 4-a solvent-free crystals
[NEt4][3] (0.380 g, 0.212 mmol) was dissolved in CH2Cl2 (20 mL), and then HBF4·Et2O (57.8 μL, 0.424 mmol) was added in two portions. The mixture was stirred at room temperature, and the reaction was monitored by IR spectroscopy. At the end of the reaction, the solvent was removed in vacuo, and the residue was washed with water (2 × 20 mL) and extracted with toluene (10 mL). The toluene solution was evaporated to dryness and the solid obtained was dissolved in CH2Cl2. Crystals of 4-b·2CH2Cl2* suitable for SC-XRD were obtained by layering n-pentane on the dichloromethane solution (yield 36%). The IR analysis of the subsequent extractions on the residual solid showed the presence of a series of homometallic ruthenium carbonyl clusters, in particular: H4Ru4(CO)12 in hexane, [H3Ru4(CO)12]− in CH2Cl2 and [H2Ru4(CO)12]2− in acetone.
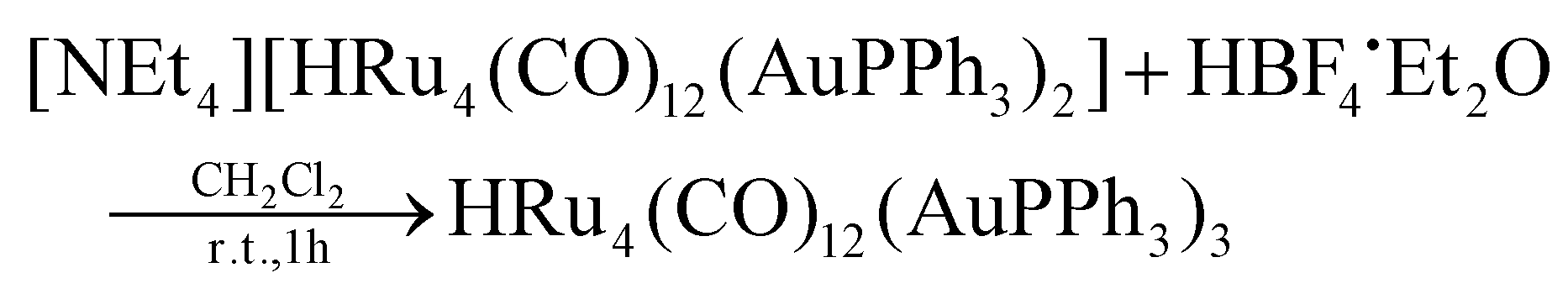
*Sometimes a second isomer crystallized out of the solution as 4-a solvent-free.
C68H50Au3Cl4O12P3Ru4 (2288.97): calcd (%): C 35.68, H 2.20; found: C 35.41, H 2.37. IR (CH2Cl2, 298 K) νCO: 2053(s), 2007(vs), 1989(m), 1953 (w) cm−1. IR (Nujol, 298 K) νCO: 2048(s), 2004(vs), 1981(m), 1960(w), 1924(w) cm−1. 1H NMR (CD2Cl2, 298 K, 400 MHz) δ: −13.01 (br) ppm. 1H NMR (CD2Cl2, 273 K, 400 MHz) δ: −12.84 (br) ppm. 1H NMR (CD2Cl2, 248 K, 400 MHz) δ: −12.80 (q, JH–P = 5.2 Hz) ppm. 1H NMR (CD2Cl2, 223 K, 400 MHz) δ: −12.72 (br), −12.75 (br) ppm. 31P{1H} NMR (CD2Cl2, 298 K, 400 MHz) δ: 60.37 ppm. 31P{1H} NMR (CD2Cl2, 273 K, 400 MHz) δ: 59.80 (br) ppm. 31P{1H} NMR (CD2Cl2, 248 K, 400 MHz) δ: 62.1 (br), 54.1 (br) ppm. 31P{1H} NMR (CD2Cl2, 223 K, 400 MHz) δ: 61.54 (2P), 52.59 (1P) ppm. 1H NMR (CD2Cl2, 298 K, 600 MHz) δ: −13.00 (br) ppm. 1H NMR (CD2Cl2, 273 K, 600 MHz) δ: −12.91 (br) ppm. 1H NMR (CD2Cl2, 248 K, 600 MHz) δ: −12.77 (br) ppm. 1H NMR (CD2Cl2, 223 K, 600 MHz) δ: −12.74 (br) ppm. 1H NMR (CD2Cl2, 198 K, 600 MHz) δ: −12.70 (s), −12.72 (s) or −12.71 (d, J = 15 Hz) ppm. 1H NMR (CD2Cl2, 173 K, 600 MHz) δ: −12.75 (s), −12.77 (s) or −12.76 (d, J = 15 Hz) ppm. 31P{1H} NMR (CD2Cl2, 298 K, 600 MHz) δ: 60.35 (br) ppm. 31P{1H} NMR (CD2Cl2, 248 K, 600 MHz) δ: 61.93 (br, 2P), 53.26 (br, 1P) ppm. 31P{1H} NMR (CD2Cl2, 223 K, 600 MHz) δ: 60.91 (2P), 51.95 (1P) ppm. 31P{1H} NMR (CD2Cl2, 198 K, 600 MHz) δ: 61.00 (2P), 51.96, 51.92 (1P) ppm.
Synthesis of Ru4(CO)12(AuPPh3)4 (5)
HBF4·Et2O (79.6 μL, 0.585 mmol) was added dropwise to a solution of [NEt4][3] (0.350 g, 0.195 mmol) in CH3CN (10 mL). The mixture was stirred at room temperature for 1 h and the reaction was monitored by IR spectroscopy. At the end of the reaction, the solvent was removed in vacuo, and the residue was washed with water (2 × 20 mL) and toluene (2 × 10 mL) and extracted with CH2Cl2 (15 mL). Crystals of 5·0.5CH2Cl2·solv (monoclinic, C2/c)* suitable for X-ray analyses were obtained by layering n-pentane on the CH2Cl2 solution (yield 28%).
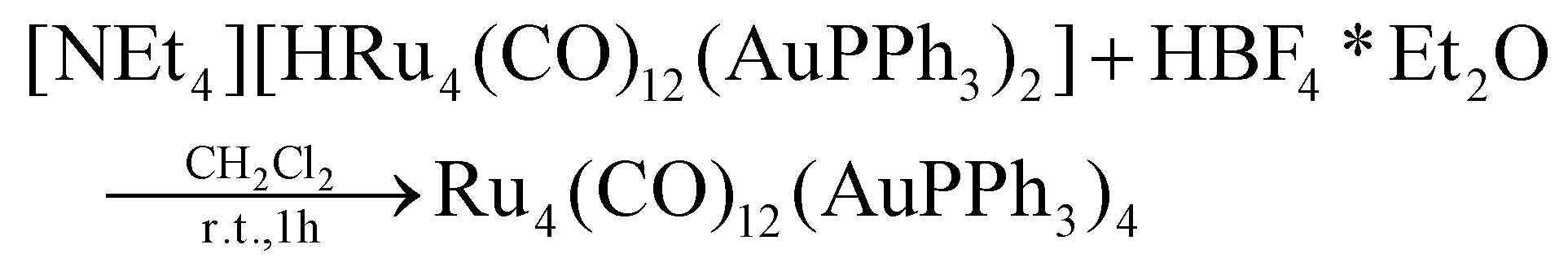
*Sometimes crystals of 5·solv (Monoclinic, P21/c) were obtained instead of 5·0.5CH2Cl2·solv (monoclinic, C2/c).C84.5H61Au4ClO12P4Ru4 (2619.81): calcd (%): C 38.74, H 2.35; found: C 39.03, H 2.61. IR (CH2Cl2, 298 K) νCO: 2082(w), 2072(m), 2029(s), 1970(m) cm−1. IR (Nujol, 298 K) νCO: 2078(m), 2063(m), 2030(s), 2020(vs), 1979(w), 1952(w) cm−1. 31P{1H} NMR (CD2Cl2, 298 K) δ: 68.46 (3P), 67.15 (1P) ppm.
X-ray crystallographic study
Crystal data and collection details for [NEt4][3]·2CH2Cl2, 4-b·2CH2Cl2, 4-a, 5·0.5CH2Cl2·solv, and 5·solv are reported in Table S1 in the ESI.† The diffraction experiments were carried out on a Bruker APEX II diffractometer equipped with a PHOTON2 detector using Mo-Kα radiation. Data were corrected for Lorentz polarization and absorption effects (empirical absorption correction SADABS).57 Structures were solved by direct methods and refined by full-matrix least-squares based on all data using F2.58 Hydrogen atoms were fixed at calculated positions and refined using a riding model, except for hydride ligands which were tentatively located in the Fourier difference map and refined isotropically using the 1.2-fold U value of the parent metal atom. All non-hydrogen atoms were refined with anisotropic displacement parameters, unless otherwise stated. The unit cells of Ru4(CO)12(AuPPh3)4·0.5CH2Cl2·solv and Ru4(CO)12(AuPPh3)4·solv contain additional total potential solvent accessible voids of 1801 and 2115 Å3 (ca. 10 and 22% of the cell volume), respectively, which are likely to be occupied by highly disordered solvent molecules. These voids have been treated using the SQUEEZE routine of PLATON.59,60Computational details
Geometry optimizations were carried out without symmetry constraints using the r2-SCAN-3c method,61 based on the meta-GGA r2SCAN functional62 combined with a tailor-made triple-ζ Gaussian atomic orbital basis set, with ECPs for Ru and Au. The method also includes refitted D4 and geometrical counter-poise corrections for London-dispersion and basis set superposition error.63–65 The C-PCM implicit solvation model was added considering DMSO as a continuous medium.66,67 Calculations were carried out using ORCA 5.0.3,68,69 and the output files were analysed with Multiwfn, version 3.8.70 Cartesian coordinates of the DFT-optimized structures are provided as ESI .xyz file.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the University of Bologna is gratefully acknowledged. CINECA (Bologna) is acknowledged for the availability of high-performance computer resources (class C project INLIGHT). We thank the referees for very useful suggestions in revising the manuscript.References
- S. Sculfort and P. Braunstein, Intramolecular d10-d10 interactions in heterometallic clusters of the transition metals, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- H. Schmidbaur and A. Schier, Aurophilic interactions as a subject of current research: an up-date, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- H. Schmidbaur, The aurophilicity phenomenon: a decade of experimental findings, theoretical concepts and emerging applications, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- H. Schmidbaur and A. Schier, A briefing on aurophilicity, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- J. Muñiz, C. Wang and P. Pyykkö, Aurophilicity: the effect of the neutral ligand L on [{ClAuL}2] systems, Chem. – Eur. J., 2011, 17, 368–377 CrossRef PubMed.
- P. Pyykkö, Strong closed-shell interactions in inorganic chemistry, Chem. Rev., 1997, 97, 597–636 CrossRef PubMed.
- Q. Zheng, S. Borsley, G. S. Nichol, F. Duarte and S. L. Cokroft, The energetic significance of metallophilic interactions, Angew. Chem., Int. Ed., 2019, 58, 12617–12623 CrossRef CAS PubMed.
- A. Das, U. Das and A. K. Das, Relativistic effects on the chemical bonding properties of the heavier elements and their compounds, Coord. Chem. Rev., 2023, 479, 215000 CrossRef CAS.
- P. Pyykkö, Theoretical chemistry of gold. III, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- S. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Understanding gold chemistry through relativity, Chem. Phys., 2005, 311, 151–161 CrossRef.
- N. Mirzadeh, H. Privér, A. J. Blake and H. Schmidbaur, Innovative Molecular Design Strategies in Materials Science Following the Aurophilicity Concept, Chem. Rev., 2020, 120, 7551–7591 CrossRef CAS PubMed.
- J. Coetzee, W. F. Gabrielli, K. Coetzee, O. Schuster, S. D. Nogai, S. Cronje and H. G. Raubenheimer, Structural Studies of Gold(I, II, and III) Compounds with Pentafluorophenyl and Tetrahydrothiophene Ligands, Angew. Chem., Int. Ed., 2007, 46, 2497–2500 CrossRef CAS PubMed.
- M. J. Katz, K. Sakai and D. B. Leznoff, The use of aurophilic and other metal-metal interactions as crystal engineering design elements to increase structural dimensionality, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC.
- P. Croizat, S. Sculfort, R. Welter and P. Braunstein, Hexa- and octanuclear heterometallic clusters with copper-, silver-, or gold-molybdenum bonds and d10-d10 interactions, Organometallics, 2016, 35, 3949–3958 CrossRef CAS.
- M. Olaru, J. F. Kögel, R. Aoki, R. Sakamoto, H. Nishihara, E. Lork, S. Mebs, M. Vogt and J. Beckmann, Tri- and tetranuclear metal-string complexes with metallophilic d10-d10 interactions, Chem. – Eur. J., 2020, 26, 275–284 CrossRef CAS PubMed.
- C. Cesari, B. Berti, F. Calcagno, C. Femoni, M. Garavelli, M. C. Iapalucci, I. Rivalta and S. Zacchini, Polymerization isomerism in Co-M (M = Cu, Ag, Au) carbonyl clusters: synthesis, structures and computational investigation, Molecules, 2021, 26, 1529 CrossRef CAS PubMed.
- B. Berti, M. Bortoluzzi, C. Cesari, C. Femoni, M. C. Iapalucci, R. Mazzoni, F. Vacca and S. Zacchini, Polymerization Isomerism in [{MFe(CO)4}n]n– (M = Cu, Ag, Au; n = 3, 4) Molecular Clusters Supported by Metallophilic Interactions, Inorg. Chem., 2019, 58, 2911–2915 CrossRef CAS PubMed.
- B. Berti, M. Bortoluzzi, C. Cesari, C. Femoni, M. C. Iapalucci, R. Mazzoni, F. Vacca and S. Zacchini, Thermal growth of Au-Fe heterometallic carbonyl clusters containing N-heterocyclic carbine and phosphine ligands, Inorg. Chem., 2020, 59, 2228–2240 CrossRef CAS PubMed.
- B. Berti, M. Bortoluzzi, C. Cesari, C. Femoni, M. C. Iapalucci, R. Mazzoni, F. Vacca and S. Zacchini, Synthesis and characterization of heterobimetallic carbonyl clusters with direct Au-Fe and Au⋯Au interactions supported by N-heterocyclic carbine and phosphine ligands, Eur. J. Inorg. Chem., 2019, 3084–3093 CrossRef CAS.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, The role of gold in transition metal carbonyl clusters, Coord. Chem. Rev., 2018, 355, 27–38 CrossRef CAS.
- M. Bortoluzzi, I. Ciabatti, C. Cesari, C. Femoni, M. C. Iapalucci and S. Zacchini, Synthesis of the highly reduced [Fe6C(CO)15]4− carbonyl carbide cluster and its reactions with H+ and [Au(PPh3)]+, Eur. J. Inorg. Chem., 2017, 3135–3143 CrossRef CAS.
- R. D. Adams and M. Chen, Synthesis and Structures of Iridium-Gold Carbonyl Cluster Compounds Containing Methyl and σ-Aryl Ligands, Organometallics, 2012, 31, 6457–6465 CrossRef CAS.
- J. W. Lauher and K. Wald, Synthesis and Structure of [FeCo3(CO)12AuPPh3]: A Trimetallic Trigonal-Bipyramidal Cluster. Gold Derivatives as Structural Analogues of Hydrides, J. Am. Chem. Soc., 1981, 103, 7648–7650 CrossRef CAS.
- P. Braunstein, J. Rosé, Y. Dusausoy and J.-P. Mangeot, Complexes à liaisons métal-métal. XXI. Synthèse et structure du “cluster” (Ph3P)AuRuCo3(CO)12: une bipyramide trigonale à trois métaux différents, C. R. Chim., 1982, 294, 967–970 CAS.
- S. Zacchini, Using Metal Carbonyl Clusters To Develop a Molecular Approach towards Metal Nanoparticles, Eur. J. Inorg. Chem., 2011, 4125–4145 CrossRef CAS.
- X. Li, B. Kiran and L.-S. Wang, Gold as Hydrogen. An Experimental and Theoretical Study of the Structures and Bonding in Disilicon Gold Clusters Si2Aun– and Si2Aun (n = 2 and 4) and Comparisons to Si2H2 and Si2H4, J. Phys. Chem. A, 2005, 109, 4366–4374 CrossRef CAS PubMed.
- P. Braunstein and J. Rosé, Gold in Bimetallic Molecular Clusters. Their Synthesis, Bonding and Catalytic Reactivities, Gold Bull., 1985, 18, 17–30 CrossRef CAS.
- P. Braunstein, H. Lehner, D. Matt, A. Tiripicchio and M. Tiripicchio-Camellini, Synthesis of the first Pt-Au cluster by an unexpected H+-substitution at trans-PtH(Cl)L2, Angew. Chem., Int. Ed. Engl., 1984, 23, 304–304 CrossRef.
- C. Cesari, J.-H. Shon, S. Zacchini and L. A. Berben, Metal carbonyl clusters of groups 8-10: synthesis and catalysis, Chem. Soc. Rev., 2021, 50, 9503–9539 RSC.
- M. Bortoluzzi, I. Ciabatti, C. Femoni, M. Hayatifar, M. C. Iapalucci, G. Longoni and S. Zacchini, Peraurated nickel carbide carbonyl clusters: the cationic [Ni6(C)(CO)8(AuPPh3)8]2+ monocarbide and the [Ni12(CO)(C2)(CO)17(AuPPh3)3]– anion containing one carbide and one acetylide unit, Dalton Trans., 2014, 43, 13471–13475 RSC.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, A. Ienco, G. Longoni, G. Manca and S. Zacchini, Intramolecular d10-d10 Interactions in a Ni6C(CO)9(AuPPh3)4 Bimetallic Nickel-Gold carbide Carbonyl Cluster, Inorg. Chem., 2013, 52, 10559–10565 CrossRef CAS PubMed.
- I. Ciabatti, C. Femoni, M. Hayatifar, M. C. Iapalucci, A. Ienco, G. Longoni, G. Manca and S. Zacchini, Octahedral Co-Carbide Carbonyl Clusters Decorated by [AuPPh3]+ Fragments: Synthesis, Structural Isomersim, and Aurophilic Interactions of Co6C(CO)12(AuPPh3)4, Inorg. Chem., 2014, 53, 9761–9770 CrossRef CAS PubMed.
- C. Cesari, B. Berti, F. Calcagno, C. Lucarelli, M. Garavelli, R. Mazzoni, I. Rivalta and S. Zacchini, Bimetallic Co-M (M = Cu, Ag, and Au) Carbonyl Complexes Supported by N-Heterocyclic Carbene Ligands: Synthesis, Structures, Computational Investigation, and Catalysis for Ammonia Borane Dehydrogenation, Organometallics, 2021, 40, 2727–2735 CrossRef.
- N. P. Mankand, Selectivity effects in bimetallic catalysis, Chem. – Eur. J., 2016, 22, 5822–5829 CrossRef PubMed.
- D. R. Pye and N. P. Mankand, Bimetallic catalysis for C-C and C-X coupling recations, Chem. Sci., 2017, 8, 1705–1718 RSC.
- H.-C. Yu and N. P. Mankad, Catalytic reactions by heterobimetallic carbonyl complexes with polar metal-metal interactions, Synthesis, 2021, 53, 1409–1422 CrossRef CAS.
- J. Campos, Bimetallic cooperation across the periodic table, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS PubMed.
- P. Buchwalter, J. Rosé and P. Braunstein, Multimetallic catalysis based on heterometallic complexes and clusters, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- J. Evans, A. C. Street and M. Webster, Reactivity and Catalytic Activity of Heteronuclear Clusters. 1. Fluxional Decapping of the Heterometallic Moiety in H3Ru4(CO)12MPR3 (M = Au, Cu) and the Crystal Structure of H3Ru4(CO)12AuPPh3, Organometallics, 1987, 6, 794–798 CrossRef CAS.
- M. J. Freeman, A. G. Orpen and I. D. Salter, The heteronucler cluster chemistry of Group 1B metals. Part 2. Synthesis, X-ray crystal structures, and dynamic behaviour of the bimetallic hexanuclear Group 1B metal cluster compounds [M2Ru4H2(CO)12(PPh3)2] (M = Cu, Ag, or Au), J. Chem. Soc., Dalton Trans., 1987, 379–390 RSC.
- M. I. Bruce and B. K. Nicholson, XXIII*. Mono-, Di- and Tri-Auration of H4Ru4(CO)12 with [{Au(PPh3)}3O][BF4]: X-ray crystal structure of HRu4Au3(CO)12(PPh3)3, J. Organomet. Chem., 1983, 252, 243–255 CrossRef CAS.
- J. A. K. Howard, I. D. Slater and F. G. A. Stone, Synthesis and molecular structure of [Au3Ru4(μ3-H)(CO)12(PPh3)3], Polyhedron, 1984, 3, 567–573 CrossRef CAS.
- Y. Li, W.-X. Pan and W.-T. Wong, The X-ray Structure, Electrochemistry and Catalytic Reactivity of Os4Au(μ-H)3(CO)12(PPh3) Towards the Oxidative Carbonylation of Aniline, J. Cluster Sci., 2002, 13, 223–233 CrossRef.
- B. F. G. Johnson, D. A. Kaner, P. R. Raithby and M. J. Taylor, Synthesis of some mixed Au-Os and Pt-Os Clusters and the X-ray Structure of [Os4H2(CO)12(AuPPh3)2], Polyhedron, 1982, 1, 105–107 CrossRef CAS.
- Y. Li and W.-T. Wong, Syntheses, Reactivity Studies and the Catalytic Properties of a Series of Tetraosmium-Gold Mixed-Metal Clusters, Eur. J. Inorg. Chem., 2003, 2651–2662 CrossRef CAS.
- M. Bortoluzzi, I. Ciabatti, C. Femoni, M. Hayatifar, M. C. Iapalucci, G. Longoni and S. Zacchini, Hydride Migration from a Triangular Face to a Tetrahedral Cavity in Tetranuclear Iron Carbonyl Clusters upon Coordination of [AuPPh3]+ Fragments, Angew. Chem., Int. Ed., 2014, 53, 7233–7237 CrossRef CAS PubMed.
- C. Cesari, M. Bortoluzzi, C. Femoni, M. C. Iapalucci and S. Zacchini, Synthesis, molecular structure and fluxional behavior of the elusive [HRu4(CO)12]3− carbonyl anion, Dalton Trans., 2022, 51, 2250–2261 RSC.
- R. D. Wilson, S. M. Wu, R. A. Love and R. Bau, Molecular Structures of H4Ru4(CO)12 and H4Ru4(CO)10(PPh3)2, Inorg. Chem., 1978, 17, 1271–1280 CrossRef CAS.
- C. Cesari, M. Bortoluzzi, C. Femoni, M. C. Iapalucci and S. Zacchini, One-pot atmospheric pressure synthesis of [H3Ru4(CO)12]–, Dalton Trans., 2021, 20, 9610–9622 RSC.
- M. McPartlin and W. J. H. Nelson, X-Ray structure analysis of the salts [N(PPh3)2][M4H3(CO)12] (M = Ru or Os); confirmation of the existence of discrete structural isomers for a hydridocarbonyl cluster in the solid state, J. Chem. Soc., Dalton Trans., 1986, 1557–1563 RSC.
- C. E. Ellul, J. P. Lowe, M. F. Mahon, P. R. Raithby and M. K. Whittlesey, [Ru3(6-NHC)(CO)10]: synthesis, characterisation and reactivity of rare 46-electron tri-ruthenium clusters, Dalton Trans., 2018, 47, 4518–4523 RSC.
- C. Lepetit, P. Fau, K. Fajerwerg, M. L. Kahn and B. Silvi, Topological analysis of the metal-metal bond: A tutorial review, Coord. Chem. Rev., 2017, 345, 150–181 CrossRef CAS.
- P. Braunstein, J. Rosé, A. Dedieu, Y. Dusausoy, J.-P. Mangeot, A. Tiripicchio and M. Tiripicchio-Camellini, Synthesis, structures, and bonding of heteropentametallic clusters [MCo3(CO)12{μ3-M′(EPh3)}] (M = Fe or Ru; M′ = Cu or Au; E = P or As): X-ray crystal structures of [RuCo3(CO)12{μ3-M′(PPh3)}] (M′ = Cu or Au), J. Chem. Soc., Dalton Trans., 1986, 225–234 RSC.
- C. Cesari, M. Bortoluzzi, F. Forti, L. Gubbels, C. Femoni, M. C. Iapalucci and S. Zacchini, 2-D Molecular Alloy Ru-M (M = Cu, Ag, and Au) Carbonyl Clusters: Synthesis, Molecular Structure, Catalysis, and Computational Studies, Inorg. Chem., 2022, 61, 14726–14741 CrossRef CAS PubMed.
- C. Kowala and J. M. Swan, Coordination compounds of Group IB metals. II. Some tertiary phosphine and phosphite complexes of gold(I), Aust. J. Chem., 1966, 19, 547–554 CrossRef CAS.
- E. Keller, SCHAKAL99, University of Freiburg, Freiburg, Germany, 1999 Search PubMed.
- G. M. Sheldrick, SADABS-2008/1-Bruker AXS Area Detector Scaling and Absorption Correction, Bruker AXS, Madison, WI, 2008 Search PubMed.
- G. M. Scheldrik, Crystal refinement with SHELXL, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, r2SCAN-3c: A “Swiss army knife” composite electronic-structure method, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed.
- H. Kruse and S. A. Grimme, Geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, Extension of the D3 dispersion coefficient model, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. A. Grimme, Generally applicable atomic-charge dependent London dispersion correction, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Software update: The ORCA program system-Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyser, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary experimental and computational figures and tables. Crystal data and collection details (PDF). DFT-optimized coordinates in the XYZ format (.xyz). CCDC 2305339 ([NEt4][3]·2CH2Cl2), 2305340 (4-b·2CH2Cl2), 2305341 (4-a), 2305342 (5·0.5CH2Cl2·solv), and 2305343 (5·solv). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04282k |
| This journal is © The Royal Society of Chemistry 2024 |