
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclotribenzylene alkynylgold(I) phosphine complexes: synthesis, chirality, and exchange of phosphine†
Jing
Zhang
a,
Nathalie
Zorn
 b,
Emmanuelle
Leize-Wagner
*b,
Marion
Jean
c,
Nicolas
Vanthuyne
*c,
Enrique
Espinosa
d,
Emmanuel
Aubert
*d,
Bruno
Vincent
a and
Jean-Claude
Chambron
*a
b,
Emmanuelle
Leize-Wagner
*b,
Marion
Jean
c,
Nicolas
Vanthuyne
*c,
Enrique
Espinosa
d,
Emmanuel
Aubert
*d,
Bruno
Vincent
a and
Jean-Claude
Chambron
*a
aInstitut de Chimie de Strasbourg, UMR 7177 CNRS, Université de Strasbourg, 4, rue Blaise Pascal, F-67070 Strasbourg, France. E-mail: jcchambron@unistra.fr
bChimie de la Matière Complexe, UMR 7140 CNRS, Université de Strasbourg, 4, rue Blaise Pascal, F-67070 Strasbourg, France
cAix-Marseille Univ., CNRS, Centrale Marseille, iSm2, Marseille, France
dUniversité de Lorraine, CNRS, CRM2, F-54000 Nancy, France
First published on 29th February 2024
Abstract
Two different alkynyl-substituted C3-symmetric cyclotribenzylenes (CTB) were synthesized in racemic and enantiomerically pure forms, and six gold(I) phosphine complexes differing by the nature of the CTB and the phosphine were prepared and characterized, in particular by NMR spectroscopy, DOSY, electronic circular dichroism (ECD), and electrospray ionization mass spectrometry (ESI-MS). Their ECD patterns depended on the substitution of the starting CTBs and were shifted bathochromically by comparison with the latter. ESI-MS in the presence of HCO2H allowed us to detect the complexes as proton adducts. The intensities of the signals were stronger when the phosphine was more electron-rich. This technique was also used to investigate the exchange of phosphine betweeen pairs of CTB complexes. The scrambling reaction was demonstrated by the higher intensity of the signals of the complexes subjected to the exchange of a single phosphine ligand by comparison with the intensity of the signals of the starting complexes.
Introduction
Self-assembly under thermodynamic control is an efficient and expeditious tool for the synthesis of highly symmetrical cage-like molecular structures from simple components.1 As a matter of fact, it has been used extensively for the construction of molecular containers and nanoreactors.2,3 This synthetic approach takes advantage of bonding interactions that are reversible in certain conditions, such as hydrogen bonding,4 coordination bonds,5 dynamic covalent bonds,6,7 and hydrophobic interactions.8 Among other reversible but less common interactions, the Au(I)⋯Au(I) aurophilic bonding interaction is worth mentioning because it has an energy similar to that of a standard hydrogen bond.9 This attractive interaction can occur, in the absence of steric hindrance, between linearly two-coordinate Au+ ions at distances between 2.50 and 3.50 Å, that is, below the sum of their van der Waals radii (>4 Å).10 The occurrence of aurophilic interactions in gold complexes stems from the low coordination geometry of Au(I) and the maximum relativistic contraction of gold orbitals. Therefore, among the identified M⋯M contacts, those involving Au(I) are the strongest. Aurophilic interactions have been identified, in particular, by single crystal X-ray crystallography analysis of several aryl acetylide gold(I) phosphine complexes, whether they were unsupported11–15 or supported by covalent bonds,16–19 and the so-called topological bonds.20We have been interested for several years in the self-assembly and study of chiral metallacages based on tritopic cyclotribenzylene (CTB) organic ligands and [ML]2+ assembling metal complex fragments (M = Pd, Pt; L = phosphine or diphosphine).21,22 The first members of this family of compounds were reported in 2001 by Shinkai and coworkers.23 Further developments in the last few decades were performed by the group of Hardie.24–28 The structure, the stereochemistry, and the properties of CTB-based metallacages make them coordination complex analogues of the cryptophanes.29–32 These latter compounds, which were designed four decades ago by Collet and coworkers, are macropolycyclic receptors. They feature a cavity lined by aromatic walls, and are able to host a great variety of substrates. The prototypical members of this family of compounds were shown to strongly complex hydro(halo)carbons, quaternary ammoniums, and the xenon atom. More recently, functionalized cryptophanes were used for the encapsulation of soft metal cations in aqueous solutions.33,34 An organometallic cryptophane, obtained by external grafting of [Cp*Ru]+ complex fragments onto the CTB subunits of an organic cryptophane was shown to host lipophilic anions such as BF4−, PF6−, and CF3SO3−.35
In a previous study, we reported on the preparation of a C3-symmetric CTB bearing three ethynyl functions.36 This compound was used as a precursor for the synthesis of three trinuclear CTB-based acetylide gold(I) phosphine complexes differing by the nature of the terminal phosphine (PPh3, PCy3, and PEt3), which showed interesting luminescence properties. In CHCl3 solution, the complexes exhibited a long-lived blue phosphorescence and a weak fluorescence in the UV. In addition, in MeOH-rich solvent mixtures, their UV emission switched to a green emission. As we observed the aggregation phenomenon concomitantly, we qualified this new emission as an aggregation induced emission (AIE). In this paper, we shall examine the consequences of a systematic variation of the nature of the phosphine and of the CTB on the solution and gas-phase structure of the complexes, their electronic and chiroptical properties, and their intermolecular interactions in the gas phase. In particular, the possibility of the alkynylgold(I) CTBs to form metallocryptophanes in solution by dimerization templated by Au⋯Au bonding will be scrutinized.
Results and discussion
Preparation and spectroscopic characterization
The chemical structures of the six alkynylgold(I) phosphine complexes C1–C6 of this study and their cyclotribenzylene (CTB) precursors are shown in Fig. 1. The complexes derive from the ethynyl γ′-substituted cyclotribenzylenes CTB(H,C2H)36 and CTB(OMe,C2H),37 both displaying the C3 symmetry, and the latter differing from the former by the methoxy substituents in γ position. Two groups of complexes were therefore synthesized and investigated, depending on the precursor CTB: C1, C2, and C3 derive from CTB(H,C2H), and C4, C5, and C6 derive from CTB(OMe,C2H). In each group, the complexes differed by the terminal phosphine ligand, with systematic variation of the balance between the steric congestion and the electrodonor character: PPh3 for C1 and C4, PPh2Me for C2 and C5, and PPhMe2 for C3 and C6. | ||
| Fig. 1 Chemical structures of the CTBs (ligand precursors and complexes). | ||
The complexes were prepared using either of the following methods of the literature: the first (method A) consists in the nucleophilic substitution reaction of the [AuCl(PPhnMe3−n)] complex by CTB(H,C2H) or CTB(OMe,C2H) in the presence of sodium methoxide and requires gentle heating,38,39 whereas the second (method B) involves the cleavage of the presynthesized organometallic coordination polymer [CTB(R,C2Au)]n (R = H or OMe) by a stoichiometric amount of the PPhnMe3−n phosphine at room temperature.40,41 The yields were moderate to good (Table S1†). Method B showed higher simplicity than method A; it operates in milder conditions, and the product can be isolated without recrystallization.
The alkynylgold(I) CTB complexes were characterized by 1H, 13C, and 31P NMR spectroscopies (Fig. S4–S33†), ESI-TOF mass spectrometry, IR spectroscopy, and elemental analysis. 1D NMR acquisitions were completed by 2D 1H/1H ROESY, 13C/1H HSQC, and 13C/1H HMBC experiments in order to assign the 1H and the 13C NMR spectra. The resulting NMR data including the complexation induced shifts (CIS) are collected in Tables S2 (1H and 31P) and S3 (13C).†
Upon metallation of CTB(H,C2H) and CTB(OMe,C2H), the singlet of the alkynyl proton, at 3.02 and 3.24 ppm respectively, disappeared, providing a convenient method for monitoring the reaction by 1H NMR spectroscopy. The typical CTB patterns were observed in the 1H NMR spectra of the ligand precursors and correspond to molecules of C3 symmetry: two pairs of doublets for the diastereotopic axial (a) and equatorial (e) protons of the methylene bridges; a doublet for each of the α and α′ protons and a doublet of doublets for the γ protons of CTB(H,C2H); a singlet for each of the α and α′ protons of CTB(OMe,C2H). The same patterns were observed in the 1H NMR spectra of the corresponding gold complexes, which, in addition, showed the signals of the phosphine ligands. Noticeably, the signals of o-H (o′-H), which are the most deshielded of the aromatic protons, shifted downfield by +0.1 ppm increments upon going from C1 to C2 and C3, as a result of the stepwise replacement of a phenyl by a methyl substituent. A similar observation could be done within the series C4, C5, C6. Characteristic features of the methyl-substituted phosphine ligands were the doublets at ca. 2.05 ppm for PPh2Me and ca. 1.75 ppm for PPhMe2 with 2JHP ranging between 9 and 11 Hz. Doublets were also observed for the carbon atoms of the same methyl substituents at ca. 14.2 ppm for PPh2Me and ca. 15.7 ppm for PPhMe2 with 1JPC of ca. 35 Hz, and for the signals of the ipso (i-C), ortho (o and o′-C), and para (p-C) carbon atoms of the phenyl substituents with heteronuclear coupling constants of 1JCP = 55.3 Hz, 2JCP = 13.8, and 3JCP = 11.3 Hz (these values precisely for C2), respectively. The chemical shift of the phosphorus atom of the phosphine terminal ligand was independent of the CTB but depended on the nature of the phosphine: 43.0 ppm for PPh3, 27.2 ppm for PPh2Me, and 14.2 ppm for PPhMe2. With regard to the 13C NMR spectra the signatures of the two series differed because of the presence of the methoxy substituent, which significantly impacted the chemical shifts of γ′-C (by ca. −11 ppm), α-C (by ca. −18 ppm), and γ-C (by ca. 28 ppm).
An interesting issue is the complexation-induced shifts (Δδ = CIS, see Tables S2 and S3†). In the case of C1, the protons α′-H were deshielded by +0.026 ppm, but α-H, γ-H, a-H and e-H were shielded by values ranging from −0.042 (γ-H) to −0.088 ppm (e-H) by comparison with CTB(H,C2H). In the series C1–C3 the CIS of α′-H and γ-H (protons ortho to the alkynyl substituents) were the most affected upon going from C1 to C2, but showed only small changes when comparing C2 and C3, indicating that the replacement of the first phenyl substituent by a methyl substituent had the strongest effect. In the case of the 13C NMR spectra, only did the signals of the carbon atoms proximal to the metal, that is γ′-C, δ′-C and ε′-C, showed changes, moving downfield by +2.3 and +21 ppm, respectively. Comparison of the CTBs of the series C4–C6 to their homologues of the series C1–C3 showed that the chemical shifts of the CTB protons in C4–C6 were less affected by metallation and by the nature of the phosphine co-ligand, α′-H excepted, the CIS of which decreased from +0.060 in C4 to +0.036 in C6. The observations pertaining to the 13C NMR spectra of complexes C1–C3 hold true in the case of C4–C6 as well.
The IR spectra of the complexes all showed the characteristic very weak (vw) absorption at ca. 2100 cm−1 corresponding to the ethynyl triple bond stretch. An absorption of medium (m) intensity at 1265 cm−1 was visible only in the spectra of the complexes C4–C6, which indicates that it corresponds to the C–O bond stretch of the arylether. Medium to strong (s) absorptions that were shared by all the spectra were found at ca. 1490 (m), 1435 (s), 1385 (weak for C1, but strong or medium for the others), 1100 (s or m), 735–745 (s or m), and 688–693 cm−1 (s or m).
Investigations of the nuclearities of the gold complexes
 | ||
| Fig. 2 Evolution of the intensities of the ESI-MS signals of the following ions: [Au(PPhnMe(3−n))2]+ (blue), [M-Au(PPhn)Me(3−n) + 2H]+ (green), [M + H]+ (red dashes) [2M + 2H]2+ (red dots). The sum of the signals of [M + H]+ and [2M + 2H]2+ is represented by the red lines. | ||
The plots in Fig. 2 also show the separate evolutions of the intensities of the signals of the doubly charged species associated with the molecular signal, [2M + 2H]2+ (see also Table S4†). The intensity of the signal of [2M + 2H]2+ is usually maximal at Ecoll = 5 eV, excepted in the case of C4, for which it is maximal at Ecoll = 10 eV. At the maximum, it increases in the order C1 < C2 < C3 < C4 < C5 ≤ C6. Noticeably, experiments at higher concentration (10−3 mol L−1) gave similar monomer to dimer ratios as those measured for C2 (Ecoll = 5 and 10 eV), C5 (Ecoll = 5, 10, and 20 eV), and C6 (Ecoll = 2 and 5 eV).
 | ||
| Fig. 3 Schematical views in simplified form of a CTB complex with (a) small and (c) large phosphine ligands and the corresponding (b and d) interpenetrated CTB dimers. The ellipsoid and cylinder shapes are marked with blue and black lines, respectively in (b) and (d). | ||
| Compound | Concentration (×10−3 mol L−1) | T/K | Imaginary or real dimer | Solvent Dt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Compound hydrodynamics | v H/vm | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensionsa | Ellipsoid vm/Å3 | Cylinder vm/Å3 |
D
tb |
r H/Åc |
v
H/Å3d |
||||||
| a/Å | b/Å | 4/3(πab2) | 2πab2 | ||||||||
| L/Å | d/Å | ||||||||||
a
a and b are respectively the radii of the polar and the equatorial axes of an oblate ellipsoid and are related to the height (L) and the diameter (d) of a cylinder by the relationship: d = 2b and L = 2a.
b Diffusion coefficient. Unit: 1010 m2 s−1.
c Hydrodynamic radius (Å), see footnote of Table S5† for the details of calculation.
d Hydrodynamic volume  .
e Ref. 36. .
e Ref. 36.
|
|||||||||||
| CTB(H,C2H)e | — | 298 | 5.31 | 6.69 | 995 | — | 30.30 | 8.64 | 6.17 | 980 | 0.98 |
| C1 | 0.9 | 298 | 5.781 | 15.088 | 5510 | — | 16.50 | 4.23 | 10.40 | 4650 | 0.84 |
| 13.910 | 30.176 | — | 9948 | 4.23 | 10.60 | 4980 | 0.50 | ||||
| 10 | 298 | 5.781 | 15.088 | 5510 | — | 16.90 | 3.89 | 11.10 | 5760 | 1.05 | |
| 13.910 | 30.176 | — | 9948 | 3.89 | 11.40 | 6180 | 0.62 | ||||
| C3 | 0.2 | 298 | 5.816 | 12.868 | 4030 | — | 23.80 | 5.01 | 9.16 | 3220 | 0.80 |
| 11.632 | 25.736 | — | 6051 | 5.01 | 9.18 | 3240 | 0.54 | ||||
| 10 | 298 | 5.816 | 12.868 | 4030 | — | 20.60 | 4.82 | 9.46 | 3550 | 0.88 | |
| 11.632 | 25.736 | — | 6051 | 4.82 | 9.48 | 3570 | 0.59 | ||||
| C4 | 0.1 | 298 | 5.882 | 14.965 | 5520 | — | 23.30 | 4.15 | 10.60 | 4930 | 0.89 |
| 13.988 | 29.930 | — | 9841 | 4.15 | 10.80 | 5240 | 0.53 | ||||
| 10 | 298 | 5.882 | 14.965 | 5520 | — | 22.00 | 4.06 | 10.80 | 5210 | 0.94 | |
| 13.988 | 29.930 | — | 9841 | 4.06 | 11.00 | 5550 | 0.56 | ||||
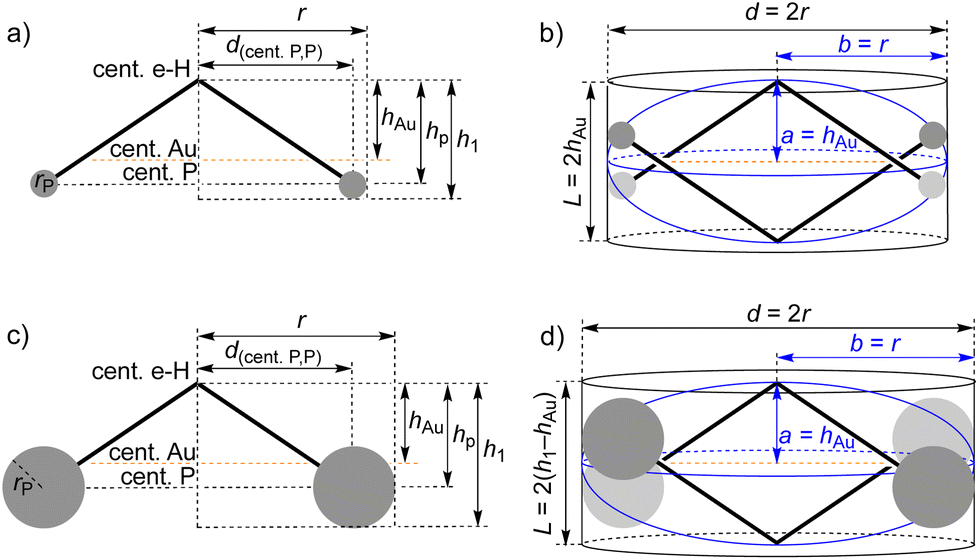
Surprisingly, the vH/vM ratio obtained for the virtual dimer of CTB(H,C2H) is very close to 1 (0.98), in spite of the fact that this dimer cannot exist! This result shows that the hydrodynamic behavior of the cone-shaped CTB is incidentally mimicked by the oblate ellipsoid, the metric parameters a and b of which correspond to a = h1 and b = r of Fig. S47.† In the case of the complexes, the ratios vH/vM calculated for the ellipsoid model varied between 0.84 and 1.05. However, given the results obtained in the case of the parent CTB, it is likely that the ellipsoid model is not suitable for the complexes as well. In the case of the cylinder model, the vH/vM ratios are all comprised between 0.50 and 0.62. They increase for C1 by 24% (for a 11 × concentration increase), for C3 by 10% (for a 50 × concentration increase), and for C4 by ca. 6% (for a 100 × concentration increase). The cylinder model seems to be better adapted to complexes with large phosphine co-ligands, which envelop the CTB cone. Given the values of the vH/vM ratios obtained for C1 and C4 in the frame of the cylinder model, these complexes are likely to be monomeric at lower concentration. The highest rate of increase of the vH/vM ratio with concentration observed for C1 (2.16) by comparison with C4 (0.06) indicates that we can consider that 10 mM solutions of the former contain a small fraction of dimer in fast exchange with the monomer. Therefore, whereas ESI-MS provided evidence for dimeric species in the gas phase for all six Cn complexes, solution DOSY experiments confirmed our earlier conclusion36 that only C1 was prone to dimerize in pure chloroform solution, albeit to a very limited extent. This was confirmed by the evolution of the 31P and 1H NMR spectra of solutions of C1 in CDCl3 in the concentration range 0.055–1.06 × 10−1 mol L−1 (Fig. S56†). As the concentration of C1 increased, the signal of the PPh3 phosphorus atom was gradually shifted to higher field and broadened, the maximum chemical shift variation in this concentration range being −0.17 ppm. The contrasted observations between NMR and ESI-MS are outward discrepancies, as the conditions of the experimental techniques (solvent, concentrations, state of the matter) are different. Moreover, the recognized risk to detect unspecific aggregates by ESI-MS must not be overlooked. This would concern, in the present case, dimers held by other interactions than those expected (here Au⋯Au bonds).
Investigations on the chirality of the complexes
The resolution of the CTB complexes by chiral HPLC was not as straightforward, because in most cases the compounds were prone to partial decomposition upon elution through the column. Separation of the enantiomers by chiral HPLC was restricted to C1, affording (+)-C1 and (−)-C1, as reported earlier;36C5, which afforded (−)-C5 then (+)-C5; and C6, which afforded (+)-C6 then (−)-C6. However, the enantiopurity of (+)-C6 was not satisfactory. The enantiomers of the remaining complexes (C2, C3, and C4) were prepared separately from the enantiopure CTB precursors using the mild (short reaction time, room temperature) reaction conditions of method B. Accordingly, (−)-C2 and (+)-C2 were obtained in 79 and 41% yields, (−)-C3 in 73% yield, and (−)-C4 and (+)-C4 in 86 and 56% yields, respectively. Additionally, (−)-C5 on the one hand, (−)-C6 and (+)-C6 on the other hand were also prepared by method B and obtained in 80, 70, and 61% yields, respectively (Table S1†).
700 mol−1 cm−1) and a sharp maximum at 257 nm (ε = 57800 mol−1 cm−1), and a weak broad band in the 265–310 nm region with maxima at 282 nm (ε = 2500 mol−1 cm−1) and 293 nm (ε = 2050 mol−1 cm−1). The two bands correspond to the B1u and B2u forbidden transitions of the benzene ring, respectively. They are shifted bathochromically by ca. 10 nm with respect to m-ethynyltoluene (Fig. S57a†). The magnitude and sign of the spectroscopic moment sm of the ethynyl substituent for the so-called 2600A electronic transition of benzene was estimated to +19 (pp. S5–S6†). The ECD spectrum of CTB(H,C2H) (Fig. 4a and S57b†) shows a bisignate Cotton effect in the B1u region, with a broad and prominent band up to 257.9 nm and a weaker band extending between 257.9 and 266.3 nm. In the B2u region, the ECD spectrum, extending between 266.3 nm to ca. 303 nm shows a singly signed band and is even weaker. Simulation of the ECD of (M)-CTB(H,C2H) by TD-DFT calculations at the CAM-B3LYP-D3 6-311+G(d,p) level of theory (Fig. S59†) indicated that this enantiomer was the second eluted, and thus corresponded to (+)-CTB(H,C2H); therefore (P)-CTB(H,C2H) is (−)-CTB(H,C2H).
 | ||
| Fig. 4 Plots of the ECD spectra of (a) CTB(H,C2H) in CH2Cl2 and (b) CTB(OMe,C2H) in CH3CN between 225 and 345 nm, which covers the regions of the B1u and B2u transitions. The ECD spectrum of CTB(OMe, C2H) in the full range is shown in Fig. S64b.† The spectra of the first eluted enantiomers are in green, those of the second eluted enantiomers are in red. In (c) and (e) are represented the directions of polarization (angle θ1) of the B1u transitions of (M)-CTB(H,C2H) and (M)-CTB(OMe,C2H). Two are possible in the case of the latter compounds. They are discussed in the text. In (e) is also represented the direction of polarization (angle θ2) of the B2u transition of (M)-CTB(OMe,C2H). It is lacking for the B2u transition of (M)-CTB(H,C2H), because this transition does not show an exciton couplet. (d) The orientations of the dipole moments of the transitions B1u and B2u of benzene are shown in blue and pink, respectively. The expected shapes of the exciton couplets of the B1u and B2u transitions as a function of the location of the direction of polarization of the transition dipole in quadrants I (0° < θ < 45°), II (45° < θ < 90°), III (90° < θ < 135°), and IV (135° < θ < 180°) are also shown. See ref. 43 and 46 for details. | ||
The electronic absorption spectrum of CTB(OMe,C2H) in acetonitrile (Fig. S63a†) shows three bands, a high energy band with a maximum at 216 nm (ε = 68700 mol−1 cm−1), an intermediate energy band with a maximum at 247 nm (ε = 48500 mol−1 cm−1) and a shoulder at 253 nm (ε = 46300 mol−1 cm−1), and a low energy band with a shoulder at 303 nm (ε = 15300 mol−1 cm−1), a maximum at 310 nm (ε = 17100 mol−1 cm−1), and a tail extending from ca. 320 nm down to ca. 400 nm. The high energy band corresponds to the allowed E1u transition of the benzene rings, and the intermediate energy bands are located in the B1u region, whereas the low energy band is located in the B2u region. By contrast with CTB(H,C2H), the ECD spectrum of CTB(OMe,C2H) in acetonitrile (Fig. 4b and S64b†) shows three bisignate exciton patterns. The highest energy one is centered on the isotropic absorption of the E2u transition, and the lowest energy ones are centered on the isotropic absorptions of the B1u and B2u transitions. In the B1u region, a bisignate Cotton effect between 229.5 and 276 nm with sign inversion at 258.4 nm, is followed, in the B2u region, by a bisignate Cotton effect showing the opposite sequence of signs from 276 to ca. 350 nm with sign inversion at 302.2 nm. Simulation of the ECD of (P)-CTB(OMe,C2H) by TD-DFT calculations at the CAM-B3LYP-D3 6-311+G(d,p) level of theory (Fig. S65†) indicated that this enantiomer was the second and thus corresponded to (+)-CTB(OMe,C2H); therefore the absolute configuration of (+)-CTB(OMe,C2H) is (P)-CTB(OMe,C2H), and the one of (−)-CTB(OMe,C2H) is (M)-CTB(OMe,C2H).
The simulation of the ECD spectra of CTBs has been, in the past, undertaken by Collet, Gottarelli and coworkers43–45 in the frame of the Kuhn-Kirkwood coupled-oscillator theory, in which the exciton model for molecules incorporating chromophores interacting through their electric transition dipoles generated in a radiation field was developed (see Fig. 4d). The CTBs contain three benzene chromophores that are tilted around a C3 symmetry axis. Each benzene ring bears four substituents, invariably two methylene bridges in β and β′ positions, and two variable substituents R and R′ in γ and γ′ positions. The lowest energy transitions in the UV are labelled B1u (230–250 nm) and B2u (255–310 nm); if R = R′ their dipole moments are polarized along the long and short axes of the benzene ring, respectively. If R ≠ R′, the transition dipole moments are tilted by an angle of θ1 (for B1u) or θ2 (for B2u), as shown in Fig. 4c and e. The sign and magnitude of θ2 depends on the values of the spectroscopic moments sm of R and R′. θ1 often equals θ2 + 90° but this turned out not to be always the case.46,47 Three coupling modes for each of the B2u and B1u transitions operate in C3-symmetric CTBs, the symmetrical A-coupling and the two degenerate non-symmetrical E-couplings. In A the electric (μ) and magnetic (m) moments are aligned along the C3 axis whereas in E they are polarized along the direction perpendicular to the C3 axis. Each of the A and E couplings generate one of the two oppositely signed Cotton bands of the exciton CD, the sign of which directly depends on the sign of the rotational strength μ × m, and the energy of which depends on the nature of the interactions between the dipoles (higher if repulsive, lower if attractive).
The ECD spectra of CTB(H,C2H) and CTB(OMe,C2H) were examined in the light of the exciton coupling theory. The former showed a bisignate pattern only in the B1u region with a positive-negative sequence from high to low energy (negative Cotton effect) for the M enantiomer. It corresponds therefore to the E–A sequence of couplings of quadrant III, which is in agreement with our finding that sm(C2H) = 19 > sm(H) = 0 M−1/2 cm−1/2. CTB(OMe,C2H) showed bisignate patterns for the B1u (positive Cotton effect) and the B2u transitions (negative Cotton effect) for the M enantiomer. The pattern of the B2u transition corresponds to the A–E sequence of couplings of quadrant I, which is in agreement with the fact that sm(C2H) = 19 < sm(OMe) ≈ 31 M−1/2 cm−1/2.48 By contrast, the B1u transition should show a negative Cotton effect (quadrant III) if the relationship: θ1 = θ2 + 90° applied. As a positive Cotton effect is observed, the B1u and B2u transitions are not polarized at 90°. Therefore, the direction of polarization of the B1u transition is either located in quadrant I (B1u(II) of Fig. 4e), or in quadrant IV (B1u(IV) of Fig. 4e) as they both account for the observed positive Cotton effect. This confirms the observation by Collet and coworkers that only the B2u transition can be used safely in the case of CTB(OMe,C2H) for the absolute configuration assignment.46
The electronic absorption and chiroptical properties ([α]25λ, electronic circular dichroism – ECD) of the enantiopure complexes, i.e., (−)-C2 and (+)-C2 (Fig. S62†), (−)-C3 and (+)-C3 (Fig. S63†), (−)-C4 and (+)-C4 (Fig. S66†), (−)-C5 and (+)-C5 (Fig. S68†), and (−)-C6 and (+)-C6 (Fig. S69†) were recorded using solutions in CH2Cl2, and the data are collected in Tables S6–S8† together with those of (−)-C1 and (+)-C1 (Fig. S60†), which were available from a previous study.36 The electronic absorption spectra of C1–C3 in CH2Cl2 all show the same pattern, a strong absorption band at high energy, which is characterized by a shoulder at ca. 236 nm (ε = 112600 M−1 cm−1), and a broad structured band extending between 255 and 315 nm, which, in the case of C1, shows a shoulder at 263 nm (ε = 56500 M−1 cm−1), maxima at 275 (82000), 285 (97500), and 297 nm (ε = 83900 M−1 cm−1), and a shoulder at 306 nm (ε = 39200 M−1 cm−1). There is a weak residual absorption extending down to ca. 350 nm. The spectra undergo a hypochromic (C1 > C2 ∼ C3) and a slight hypsochromic (ca. 1 nm) shift upon going from C1 to C3, the differences being the highest in the high energy region.
The ECD spectra of C1–C3 (Fig. 5a) show similar patterns: from high to low energy, in the B1u region, a bisignate non-symmetrical band, the highest energy and absorbance component extending down to ca. 264 nm, followed by an intermediate energy and weak absorbance component across 10 nm; in the B2u region, a singly signate band between 274 and 315 nm tailing down to 350 nm. The broadest bands are structured, which is particularly clear in the case of C3. Upon going from C1 to C3, these maxima (in absolute value) undergo hypsochromic (ca. 1–3 nm) and hypochromic (C1 > C2 ∼ C3) shifts. The ECD spectrum of (M)-C1 was simulated by TD-DFT calculations (Fig. S61†). Comparison of the simulation shows that it reproduces the sequence of signs and number of bands of the experimental spectrum of the second eluted enantiomer, (+)-C1. Therefore, (P)-C1 is (−)-C1.
 | ||
| Fig. 5 Plots of the ECD spectra of (a) complexes C1–C3 and (b) complexes C4–C6 in CH2Cl2. | ||
The electronic absorption spectra of C4–C6 show three bands as for the parent CTB(OMe,C2H). For example, in the case of C4, from the highest to the lowest energy: the foot of an intense band, then an intermediate energy structured band with three local maxima of similar absorbance (263, 268, and 275 nm), and peaking at 284 nm (ε = 59100 M−1 cm−1), followed by a low energy structured band with a shoulder at 315 nm (ε = 48600 M−1 cm−1) and a maximum at 324 nm (ε = 58800 M−1 cm−1), with a tail extending between 340 and 400 nm. As observed in the case of the series C1–C3, upon going from C4 to C6, the maxima slightly shift to lower energy, and the absorbance of C4 is much higher than those of C5 and C6.
The ECD spectra of the complexes C4–C6 (Fig. 5b) show the same sequence of bands as for C1–C3: in the case of C4, from high to low energy, a broad structured band between 229.8 and 294.1 nm, then a band of opposite sign with a single maximum at 304 nm extending down to ca. 312 nm, and in the low energy region, after sign inversion, a band peaking at 327 nm, showing the highest differential absorbance. The local maxima show a slight hypsochromic shift within the sequence C4–C6. The hypochromic shifts are less marked than in the C1–C3 series. The ECD spectrum of (P)-C4 was simulated by TD-DFT calculations (Fig. S67†). Comparison of the theoretical and experimental data shows that the calculated spectrum of (P)-C4 coincides with the experimental spectrum of (+)-C4. Therefore (P)-C4 is (+)-C4 and (M)-C4 is (−)-C4. Given the invariance of the Cotton effects within the homogeneous series C1–C3 and C4–C6 each, (M)-Cn ⇔ (+)-Cn and (P)-Cn ⇔ (−)-Cn for n = 1–3, and (M)-Cn ⇔ (−)-Cn and (P)-Cn ⇔ (+)-Cn for n = 4–6. It is worth noticing that the same relationships hold true for CTB(H,C2H) and CTB(OMe,C2H).
General interpretation of the electronic absorption and ECD spectra of C1–C6
The high energy absorptions (230–265 nm for C1–C3, 230–320 nm for C4–C6) arise from intraligand transitions of the coordinated alkynyl CTB and phosphine ligands. In particular, the decrease of the absorption between 230 and 260 nm upon going along both series is due to the decrease of the number of phenyl substituents in the series PPh3 > PPh2Me > PPhMe2. The lower energy absorption bands (265–350 nm for C1–C3, 300–400 nm for C4–C6) were attributed in previous studies to gold(I)-perturbed intraligand π → π*(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CAr) transitions of the alkynyl units49,50 and σ(Au–P) → π*(CC) transitions.51 Vibrational progressional spacings between 1276 and 1659 cm−1 (aromatic C
CAr) transitions of the alkynyl units49,50 and σ(Au–P) → π*(CC) transitions.51 Vibrational progressional spacings between 1276 and 1659 cm−1 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches)52 were calculated in the 260–300 nm regions of C1 and C4, whereas a vibrational progressional spacing of ca. 2100 cm−1 (CC stretch) was calculated in the same region for C4.53 The three chromophores in C1 are independent, as the molar extinction coefficient of this complex at 285 nm (94100 M−1 cm−1) is three times the one measured for the mononuclear complex Ph3PAuCCPh.50 The slight blue shift of the absorption maxima upon moving along the series C1–C3 on the one hand, C4–C6 on the other hand, confirms that these absorption features may involve the σ(Au–P) orbital, as its stabilization by the gradual replacement of a phenyl by a methyl substituent makes transitions originating therefrom highest in energy.
C stretches)52 were calculated in the 260–300 nm regions of C1 and C4, whereas a vibrational progressional spacing of ca. 2100 cm−1 (CC stretch) was calculated in the same region for C4.53 The three chromophores in C1 are independent, as the molar extinction coefficient of this complex at 285 nm (94100 M−1 cm−1) is three times the one measured for the mononuclear complex Ph3PAuCCPh.50 The slight blue shift of the absorption maxima upon moving along the series C1–C3 on the one hand, C4–C6 on the other hand, confirms that these absorption features may involve the σ(Au–P) orbital, as its stabilization by the gradual replacement of a phenyl by a methyl substituent makes transitions originating therefrom highest in energy.
The ECD pattern of the spectra of C1–C3 is similar to, but red shifted by ca. 5–10 nm with reference to the one of CTB(H,C2H), with a bisignate Cotton effect in the B1u region between 225 and 275 nm, and a considerably enhanced singly signed Cotton effect in the B2u region. The sequence of signs in the B1u region is the same along the series CTB(H,C2H), C1–C3, which implies that the metallation of the alkynyl substituent does not change its sm (90 < θ1 < 135). The ECD pattern of the spectra of C4–C6 differs, at first glance, from the one of CTB(OMe,C2H), unless we consider that the low energy Cotton band of the B1u couplet has merged with the high energy band of the B2u couplet, due to a stronger red shift of the former with respect to the latter. If this hypothesis is correct, the low energy Cotton band of the B2u couplet is considerably enhanced, as in the case of the former series, and its sequence of signs is preserved, which allows us to draw the same conclusions about the sm of the metallated alkynyl substituents.
Study of the phosphine exchange in the gas phase in C1–C6
Exchange of phosphine ligands of mononuclear phosphinegold ethynyl complexes has been investigated in solution by 31P NMR spectroscopy and shown to be fast.38 Inspired by reports on the use of ESI-MS to study the exchange of components in metallo-supramolecular macrocycles54 and cages,55 we examined how this technique can bring some information on the phosphine exchange reactions within the series of alkynylgold(I) phosphine complexes C1–C6. As each species is characterized by a single signal, the spectra of the mixtures are quite simple, making quantifications straightforward. The equilibrium pertaining to the phosphine exchange reaction between two complexes Cp and Cq is the following (eqn (1)):| Cp + Cq ⇄ Cp′ + Cq′ | (1) |
 | (2) |
The nine following pairs {Cp, Cq} of CTBs were examined: {C1, C2}, {C1, C3}, {C1, C4}, {C2, C3}, {C2, C5}, {C3, C6}, {C4, C5}, {C4, C6}, and {C5, C6}. Except for the pairs {C1, C4}, {C2, C5}, and {C3, C6}, in which the Cp and Cq complexes contain the same phosphine ligand, that is, PPh3 for {C1, C4}, PPh2Me for {C2, C5}, and PPhMe2 for {C3, C6}, and give rise to degenerate exchange reactions, these experiments allowed us to observe new signals that could not be detected when a single complex was examined, that is, signals resulting from the exchange of a phosphine, e.g. [Cp′ + H]+ = [Cp-PPhmMe(3−m) + PPhnMe(3−n) + H]+, and signals arising from the diprotonated heterodimer, e.g. [Cp + Cq + 2H]2+. Therefore, for a given {Cp, Cq} pair, we monitored the signal intensities of the following species as a function of Ecoll: the singly charged species [Cp + H]+, [Cq + H]+, [Cp′ + H]+, [Cq′ + H]+, and the doubly charged species [2Cp + 2H]2+, [2Cq + 2H]2+, [Cp + Cq + 2H]2+, [2Cp′ + 2H]2+, [2Cq′ + 2H]2+, [Cp + Cp′ + 2H]2+ and [Cq + Cq′ + 2H]2+. The spectra and the corresponding plots of the relative proportion of each species as a function of Ecoll are collected in Fig. S70–S87,† the spectra of {C4, C5} being also reproduced in Fig. 6.
 | ||
| Fig. 6 Evolution of the ESI-MS spectra of a 1:1 mixture of 10−4 M solutions of complexes C4 and C5 in CHCl3/i-PrOH/HCO2H 50:49.5:0.5. | ||
It must be emphasized that the relative proportions of the different species are those of the protonated CTBs, which are detected in this form. Therefore they a priori depend on the relative basicities (electron richness) of the complexes. We expect that it is stronger in the case of the methoxy-substituted series (C4–C6) and, within each series, it increases with the degree of methyl-substitution of the phosphine, which was roughly observed in the case of the experiments run on each complex alone (see Fig. 2). Moreover, the basicity of Cp and Cp′ (respectively Cq and Cq′) should differ in the following way: if p < q, the basicity of Cp′ should be higher than the basicity of Cq′, and the basicity of Cq′ should be lower than the basicity of Cq, and vice versa if p > q.
Examination of the nine plots shows that the populations of the doubly charged, dimer species decrease as Ecoll increases, while the populations of the singly charged, monomer species, that is, [Cp′ + H]+, [Cq′ + H]+, [Cp + H]+, and [Cq + H]+, increase concomitantly. The Ecoll at which the former approach their minimum roughly coincides with the Ecoll at which the latter approach their maximum, between 10 and 15 eV. Another general trend is the highest proportion of species containing two different phosphines, e.g. [Cp′ + H]+vs. [Cp + H]+ and [2Cp′ + 2H]2+vs. [2Cp + 2H]2+. These differences result from the decrease of the concentrations of Cp and Cq, as Cp′ and Cq′ are generated, as explicited in the equilibrium (1).
The differences between the signals of Cp and Cq on the one hand, and of Cp′ and Cq′ on the other hand, are usually minimal at low Ecoll (5 eV), then they increase with Ecoll. This is particularly the case of the pairs {C1, C3}, {C4, C5}, {C4, C6}, and {C5, C6}. The other non-degenerate pairs {C1, C2} and {C2, C5} illustrate two opposite behaviors, as for the former these differences are low, whatever Ecoll, and for the latter they are high, whatever Ecoll.
We consider first in detail the case of the pairs of complexes {C1, C4}, {C2, C5}, and {C3, C6}, giving degenerate exchange reactions (Fig. S74, S75 and S78–S81†). The relative proportions within each pair follow the order [C1 + H]+ > [C4 + H]+, [C2 + H]+ ≈ [C5 + H]+, and [C3 + H]+ ≫ [C6 + H]+, which are opposite the expected trends, at least for the pairs {C1, C4} and {C3, C6}. Meanwhile, in the case of the pairs {C1, C4} and {C2, C5}, at Ecoll < 10 eV, the signals of highest intensity originate from the dimer species [C1 + C4 + 2H]2+ > [2C4 + 2H]2+ ≫ [2C1 + 2H]2+ for the former pair, and [C2 + C5 + 2H]2+ > [2C5 + 2H]2+ ≫ [2C2 + 2H]2+ for the latter pair, the intensities of the signals of [2C1 + 2H]2+ and [2C2 + 2H]2+ being the weakest of all the species detected. Therefore, in the case of the homodimers, the order of the signal intensities follows the expected CTB basicities. The plots of Fig. S75 and S79† nicely illustrate the fact that the dimers [2C4 + 2H]2+ and [2C5 + 2H]2+ play the role of reservoir of C4 and C5, respectively, and that [2C4 + 2H]2+ ≫ [2C1 + 2H]2+ compensates for [C1 + H]+ > [C4 + H]+, while [2C5 + 2H]2+ ≫ [2C2 + 2H]2+ compensates for [C2 + H]+ ≈ [C5 + H]+ at least at Ecoll ≤ 15 eV. Taking into account the dimers restores the order expected on basicity grounds. In the case of the {C3, C6} pair, the observed [C3 + H]+ ≫ [C6 + H]+ order is consistent with the ranking deduced from subjecting separately C3 and C6 to the ESI-MS experiments, as shown in Fig. 2, which could suggest that other factor(s) than basicity could contribute to the singular behavior of C6.
We now turn to the six pairs giving non-degenerate exchange reactions. In the case of {C1, C2} (Fig. S70 and S71†) the differences between the apparent proportions of [C1 + H]+ and [C2 + H]+ (and of [C1′ + H]+ and [C2′ + H]+) are small and in keeping with the observations done on the individual complexes (Fig. 2). As a consequence, the ratios [Cp′ + H]+/[Cp + H]+ are similar, and at 20 eV, where only these four species remain, we can calculate the equilibrium constant as shown in eqn (3):
 | (3) |
Switching to the homologous system in the other series, that is, {C4, C5} (Fig. S82 and S83†) shows substantial differences between the responses of C4 and C5. However, they are now in agreement with the expected basicity order, the signals arising from C5 being more intense than those arising from C4. However, as the ratios [Cp′ + H]+/[Cp + H]+ differ by 30% at 20 eV, it is not possible to determine an equilibrium constant for this system. Back to the unsubstituted series, we examine the two systems {C1, C3} (Fig. S72 and S73†) and {C2, C3} (Fig. S76 and S77†). The responses of the CTBs are fully consistent with the basicity order C3 > C2 > C1, but the differences in the ratios [Cp′ + H]+/[Cp + H]+ for each pair again preclude their use for the calculation of equilibrium constants. Finally, we examined the cases of the systems homologous of {C1, C3} and {C2, C3} in the methoxy-substituted series, that is, {C4, C6} (Fig. S84 and S85†) and {C5, C6} (Fig. S86 and S87†). C4′ and C6′ are not distinguished from each other whatever the Ecoll value, but the response of C4 is stronger than the response of C6 at Ecoll > 2 eV, that is opposite the one expected from their basicity order. Nevertheless, C4 and C6 give signals of relatively close intensities at 20 eV, which allowed us to estimate the equilibrium constant as shown in eqn (4):
 | (4) |
The differences between the responses of C5 and C6 on the one hand, and of C5′ and C6′ on the other hand, are also opposite the expected basicity order C6 > C5, but they are too high to allow us to calculate an equilibrium constant. The fact remains that the observed inversions affect all three CTB pairs involving C6, that is {C3, C6}, {C4, C6}, and {C5, C6}, which also confirms the singularity of C6.
Conclusions
Organometallic cyclotribenzylenes, in particular those containing gold, are novel chiral compounds, the properties of which deserve being investigated further. In this work, we have investigated methods and selected the best for their preparation in enantiomeric pure form. Their electronic absorption and ECD bands, shifted to the red by gold complexation, can be tuned by the nature of the substituents of the CTB platform. The complexes are mainly monomeric in solution, according to DOSY experiments. We have shown, by ESI-MS, that the ability of the CTB complexes to form proton adducts was dependent upon the nature of the phosphine and CTB substitution, the more basic the complex, the stronger the signal of the protonated species. ESI-MS was also used to probe phosphine exchange reactions. Future work will explore the emission properties of these alkynylgold complexes. In addition, as they encompass a cavity lined by phosphine termini of different sizes, we plan to screen potential guests and study the kinetics and thermodynamics of their interaction with the guests discovered.Experimental
Materials, methods and instrumentation
The following abbreviations were used: THF (tetrahydrofuran) and DMF (N,N-dimethylformamide). Unless otherwise stated, all reactions were performed under argon using the following solvents and liquid reagents, which were dried and distilled under argon: DMF (anhydrous magnesium sulfate), THF (Na/benzophenone), dichloromethane (P2O5), methanol (Mg), triethylamine (KOH), and acetone (K2CO3). Other commercially available solvents and reagents were used as received. Separations by flash column chromatography used silicagel (Geduran Si 60, 40–63 μm from Merck). CTB(H,C2H),36C1, (+)-C1 and (−)-C1,36 CTB(OMe,C2H),37 and [Au(PPhnMe3−n)Cl], n = 1, 2, and 3,56 were obtained according to the literature. [CTB(H,C2Au)]n and [CTB(OMe,C2Au)]n were prepared in racemic and enantiomeric pure forms by using reported procedures.41 NMR spectra were recorded on Bruker Avance III 400 MHz and Avance II 500 MHz spectrometers. Chemical shifts were reported using the residual solvent 1H signal (7.26 and 77.16 ppm for CHCl3) as internal reference for 1H NMR and 13C NMR, respectively, and the phosphorus signal (set at 0 ppm) of 85% H3PO4 placed inside an insert for 31P NMR. IR spectra were recorded using a Bruker Alpha II FT-IR spectrometer. CAUTION: Some gold acetylide have been reported to be shock sensitive. These compounds should be handled in small quantities using protective equipment.Analytical and preparative chiral HPLC
Chiral HPLC analyses were performed using the Agilent 1260 Infinity unit (pump G1311B, autosampler G1329B, DAD G1315D) with Igloo-Cil ovens, monitored by SRA Instruments Seleccol software. The chiroptical detection was performed with Jasco OR-1590 and CD-2095 polarimetric and circular dichroism detectors, respectively. The signs given by the on-line circular dichroïsm at 254 nm and the polarimeter are the signs of the compound in the solvent used for the chromatographic separation. HPLC grade heptane and isopropanol were degassed and filtered on a 0.45 μm millipore membrane before use. Retention times Rt (min), retention factors ki = (Rti − Rt0)/Rt0, enantioselectivity factors α = k2/k1, and resolution Rs = 1.18(Rt2 − Rt1)/(w1 + w2) were determined (Rt0 was measured by injection of 1,3,5-tri(tert-butyl) benzene and wi is the peak width at half-height). Preparative chiral HPLC separation were performed using an Agilent 1260 Infinity unit (pump G1311C, autosampler G1329B, DAD G1365D and fraction collector G1364C), monitoring with an Agilent OpenLAB CDS Chemstation LC.Mass spectrometry
Mass data were obtained using a MicrOTOF-Q II (Bruker Daltonics, Bremen, Germany) mass spectrometer fitted with Z-spray electrospray ion source and with a mass range of 2–4000 Thomson. The mass spectrometer was operated in positive ion mode, with a potential of 3500 V applied to the electrospray probe body and the source temperature set to 180 °C. MS data were acquired from 300 to 3000 m/z. The ISCID energy was kept at 0 eV, while energy of the collision cell (Ecoll) was varied from 2 to 20 eV in order to dissociate potential dimers. Mass calibration was performed using a mix of Tune Low (Agilent Technologies G1969-8500) prior to analysis. Data were processed with Data Analysis software (Bruker Daltonics). CTB solutions at a concentration of 10−4 mol L−1 in CHCl3 were diluted to a final concentration of 5 × 10−5 mol L−1 with an equal volume of isopropanol acidified with HCO2H 1%, so that the composition of the sample mixture was CHCl3/iPrOH/HCO2H 50:49.5:0.5 prior to injection into the mass spectrometer.
Preparation and characterization of CTB(R,C2AuPPhnMe3−n) C2–C6
:1, v/v) at 40 °C. After stirring the reaction mixture for several days, the solvents were removed by rotary evaporation. The residue was taken up in dichloromethane, and clarified by filtration, which afforded a yellow solution. The latter was concentrated to a volume of 1–2 mL and treated by dropwise addition of cyclohexane (1.5–2 mL) until the product started to precipitate. The mixture was stored in the refrigerator overnight and filtered. A yellow powder was obtained after drying in vacuum.
C2. (Method A) Prepared from CTB(H,C2H) (0.005 g; 0.0146 × 10−3 mol), [AuCl(PPh2Me)] (0.019 g; 0.0439 × 10−3 mol), and sodium methoxide (0.012 g; 0.222 × 10−3 mol) in MeOH (4 mL) and THF (4 mL), 3 days stirring. Yield: 0.0095 g (0.0063 mol, 43%). (Method B) Prepared from PPh2Me (0.016 mL, 0.0173 g; 0.0862 × 10−3 mol) and [CTB(H,C2Au)]n (0.026 g; 0.0279 × 10−3 mol), in CH2Cl2 (3 mL) and THF (1 mL), 1.5 h stirring. Yield: 0.036 g (0.0235 × 10−3 mol, 84%). Mp 195 °C; 1H NMR (500.13 MHz, CDCl3, 295 K): δ (ppm) = 7.636 (m, 12 H; o- and o′-H), 7.490 (d, 4JH,H = 1.5 Hz, 3 H; α′-H), 7.453 (m, 6 H; p-H), 7.439 (m, 12 H; m- and m′-H), 7.247 (d, 3JH,H = 8.0 Hz, 3 H; α-H), 7.186 (dd, 4JH,H = 1.5 Hz, 3JH,H = 7.5 Hz, 3 H; γ-H), 4.742 (d, 2JH,H = 13.5 Hz, 3 H; a-H), 3.624 (d, 2JH,H = 13.5 Hz, 3 H; e-H), 2.052 (d, 2JH,P = 9.0 Hz; CH3); 13C NMR (125.77 MHz, CDCl3, 295 K): δ (ppm) = 138.82 (s; β′-C), 138.15 (s; β-C), 134.14 (s; α′-C), 132.97 (d, 2JC,P = 13.8 Hz; o- and o′-C), 131.72 (d, 1JC,P = 55.3 Hz; i-C), 131.58 (s; p-C), 131.00 (s; γ-C), 130.25 (s; α-C), 129.27 (d, 3JC,P = 11.3 Hz; m- and m′-C), 123.40 (s; γ′-C), 104.71, 104.48 (2 s; δ′- and ε′-C), 37.04 (s; CH2), 14.24 (d, 1JC,P = 35.2 Hz; CH3); 31P NMR (161.98 MHz, CDCl3, 298 K): δ (ppm) = 27.19 (s); IR (KBr): ν (cm−1) = 3053 (w), 3019 (w), 2958 (sh), 2922, 2854 (m), 2352 (w), 2319 (w), 2103, 1968, 1892 (w), 1600 (m), 1545 (w), 1486 (m), 1434 (s), 1417, 1385, 1334, 1310 (m), 1294 (w), 1267, 1239 (m), 1204 (sh), 1189, 1162 (m), 1102 (s), 1075 (sh), 1030 (w), 1002 (m), 970 (sh), 957 (m), 890 (s), 834 (m), 807 (w), 737 (s), 690 (s), 635 (m), 483, 475, 427 (w), 511 (s), 478 (m), 453 (sh), 431 (sh); HR-MS (+ESI): m/z calcd for C66H55Au3P3, 1531.251 [M + H]+; found, 1531.249; elemental analysis: calcd for C66H54Au3P3·½H2O (1539.99), C 51.48, H 3.60; found, C 51.45, H 3.66.
C3. (Method B) Prepared from [CTB(H,C2Au)]n (0.073 g; 0.0785 × 10−3 mol) and PPhMe2 (0.035 mL, 0.034 g; 0.246 × 10−3 mol) in a mixture of CH2Cl2 (12 mL) and THF (3 mL), 4 h stirring. Yield: 0.081 g (0.0602 × 10−3 mol, 77%). Mp 187.5 °C; 1H NMR (500.13 MHz, CDCl3, 295 K): δ (ppm) = 7.724 (m, 4JH,H = 1.5 Hz, 3JH,H = 8.0 Hz, 6 H; o- and o′-H), 7.480 (d, 4JH,H = 1.5 Hz, 3 H; α′-H), 7.460 (br m, 9 H; p-, m-, and m′-H), 7.244 (d, 3JH,H = 7.5 Hz, 3 H; α-H), 7.177 (dd, 4JH,H = 1.5 Hz, 3JH,H = 7.5 Hz, 3 H; γ-H), 4.743 (d, 2JH,H = 13.5 Hz, 3 H; a-H), 3.624 (d, 2JH,H = 13.5 Hz, 3 H; e-H), 1.747 (d, 2JP,H = 9.5 Hz; CH3); 13C NMR (125.76 MHz, CDCl3, 295 K): δ (ppm) = 138.83 (s; β′-C), 138.13 (s; β-C), 134.12 (s; α′-C), 132.56 (d, 1JC,P = 56.9 Hz; i-C), 132.16 (d, 2JC,P = 13.8 Hz; o- and o′-C), 131.75 (s; p-C), 130.98 (s; γ-C), 130.22 (s; α-C), 129.29 (d, 3JC,P = 11.6 Hz; m- and m′-C), 123.47 (s; γ′-C), 104.87, 104.84 (2 s; δ′- and ε′-C), 37.03 (s; CH2), 15.66 (d, 1JC,P = 35.2 Hz; CH3); 31P NMR (161.98 MHz, CDCl3, 298 K): δ (ppm) = 14.24 (s); IR (KBr): ν (cm−1) = 3054, 2959 (w), 2920 (s), 2852, 2353, 2331, 2321, 2102, 1750, 1733, 1716, 1696, 1682, 1645 (w), 1600 (m), 1579 (w), 1560 (m), 1543, 1523, 1509 (w), 1487 (s), 1473 (sh), 1457 (w), 1434 (s), 1418 (m), 1396 (w), 1384 (s), 1363, 1340, 1313, 1302, 1108, 1274, 1265, 1237, 1199, 1187, 1161, 1136 (w), 1107 (m), 1095 (sh), 1071 (sh), 1058 (sh), 1047 (sh), 1033 (w), 1022 (w), 1006 (w), 986 (w), 952 (s), 915 (s), 906 (sh), 893 (sh), 840, 805 (m), 739 (s), 718 (m), 688 (s), 633 (m), 612, 590, 572, 561, 548, 536, 521, 513, (w), 483 (s), 461 (w), 446 (sh), 435 (m), 426, 413 (sh); HR-MS (+ESI): m/z calcd for C51H49Au3P3 [M + H]+, 1344.197; found, 1345.202; elemental analysis: calcd for C51H48Au3P3·½H2O (1353.77), C 45.25, H 3.65; found, C 45.23, H 3.74.
C4. (Method A) Prepared from CTB(OMe,C2H) (0.008 g; 0.0185 × 10−3 mol), [AuCl(PPh3)] (0.030 g; 0.061 × 10−3 mol), and sodium methoxide (0.025 g; 0.463 × 10−3 mol) in MeOH (5 mL) and THF (5 mL), 4 h stirring. Yield: 0.025 g (0.0138 × 10−3 mol, 75%). (Method B) Prepared from [CTB(OMe,C2Au)]n (0.070 g; 0.0686 × 10−3 mol) and PPh3 (0.058 g; 0.2212 × 10−3 mol) in CH2Cl2 (12 mL) and THF (4 mL), 5 h stirring. Yield: 0.082 g (0.0454 × 10−3 mol, 66%). Mp 181 °C; 1H NMR (500.13 MHz, CDCl3, 295 K): δ (ppm) = 7.553 (dd, 3JH,H = 7.8 Hz, 4JH,H = 1.5 Hz, 9 H; o-H); 7.528 (dd, 3JH,H = 7.8 Hz, 4JH,H = 1.5 Hz, 9 H; o′-H); 7.485 (s, 3 H; α′-H), 7.474 (dd, 3JH,H = 7.8 Hz, 4JH,H = 1.5 Hz, 9 H; p-H), 7.429 (dd, 3JH,H = 7.8 Hz, 9 H; m-H), 7.425 (dd, 3JH,H = 7.8 Hz, 9 H; m′-H), 6.789 (s, 3 H; α-H), 4.679 (d, 2JH,H = 13.5 Hz, 3 H; a-H), 3.898 (s, 9 H; OCH3), 3.550 (s, 2JH,H = 13.5 Hz, 3 H; e-H); 13C NMR (125.76 MHz, CDCl3, 295 K): δ (ppm) = 159.43 (s; γ-C), 139.92 (s, β-C), 135.83 (s; α′-C), 134.49 (d, 2JC,P = 13.8 Hz; o- and o′-C), 131.56 (d, 4JC,P = 2.5 Hz; p-C), 130.61 (s; β′-C), 130.08 (d, 1JC,P = 55.3 Hz; i-C), 129.18 (d, 3JC,P = 11.3 Hz; m- and m′-C), 112.67 (s; γ′-C), 111.91 (s; α-C), 99.93, 99.72 (2 s; δ′- and ε′-C), 56.30 (s; OCH3), 29.83 (s; CH2); 31P NMR (161.98 MHz, CDCl3, 298 K): δ (ppm) = 42.89; IR (KBr): ν (cm−1) = 3448 (br, m), 3049 (w), 2947 (w), 2843 (w), 2099 (w), 1967 (w), 1892 (w), 1812 (w), 1600 (m), 1493 (s), 1481 (s), 1435 (vs), 1385 (s), 1308 (m), 1265 (s), 1217 (m), 1186 (m), 1142 (m), 1099 (s), 1080 (s), 997 (m), 897 (m), 841 (m), 744 (vs), 711 (m), 692 (vs), 626 (w), 536 (vs), 509 (s); HR-MS (+ESI): m/z calcd for C84H67Au3O3P3, 1807.33; found, 1807.33 [M + H]+; elemental analysis: calcd for C84H66Au3O3P3·3/2CH2Cl2 (1934.66), C 53.08, H 3.60; found, C 52.96, H 3.74.
C5. (Method A) Prepared from CTB(OMe,C2H) (0.0129 g; 0.0298 × 10−3 mol), [AuCl(PPh2Me)] (0.040 g; 0.0925 × 10−3 mol), and sodium methoxide (0.032 g; 0.592 × 10−3 mol), overnight stirring. Yield: 0.040 g (0.0247 × 10−3 mol, 83%). (Method B) Prepared from [CTB(OMe,C2Au)]n (0.055 g; 0.0539 × 10−3 mol) and PPh2Me (0.032 mL, 0.0346 × 10−3 g; 0.173 × 10−4 mol) in CH2Cl2 (6 mL) and THF (1 mL), 4 h stirring. Yield: 0.074 g (0.0456 × 10−3 mol, 85%). Mp 177.2 °C; 1H NMR (300.13 MHz, CDCl3, 298 K): δ (ppm) = 7.642 (m, 4JH,H = 1.8 Hz, 12 H; o- and o′-H), 7.468 (s, 3 H; α′-H), 7.451 (d, 4JH,H = 1.8 Hz, 12 H; m- and m′-H), 7.430 (t, 4JH,H = 1.8 Hz, 6 H; p-H), 6.683 (s, 3 H; α-H), 4.679 (d, 2JH,H = 13.5 Hz, 3 H; a-H), 3.888 (s, 9 H; OCH3), 3.591 (d, 2JH,H = 13.5 Hz, 3 H; e-H), 2.032 (d, 3JH,P = 9.0 Hz, 9 H; CH3); 13C NMR (125.77 MHz, CDCl3, 295 K): δ (ppm) = 159.44 (s; γ-C), 139.89 (s; β-C), 135.75 (s; α′-C), 132.99 (d, 2JC,P = 13.8 Hz; o- and o′-C), 131.85 (d, 1JC,P = 55.3 Hz; i-C), 131.52 (s; p-C), 130.60 (s; β′-C), 129.21 (d, 3JC,P = 11.3 Hz; m- and m′-C), 112.72 (s; γ′-C), 111.92 (s; α-C), 100.50 (2 s; δ′- and ε′-C), 55.28 (s; OCH3), 37.08 (s; CH2); 14.25 (d, 1JC,P = 34.0 Hz); 31P NMR (161.98 MHz, CDCl3, 298 K): δ (ppm) = 27.15; IR (KBr): ν (cm−1) = 3444 (br, vs), 3074 (sh), 3053 (w), 3022, 3004, 2988, 2959 (sh), 2918, 2849 (m), 2826 (sh), 2353, 2321, 2222, 2104 (w), 1603 (m), 1577, 1561 (w), 1545 (sh), 1494 (m), 1475 (sh), 1458 (d; m), 1435 (s), 1417 (m), 1385 (s), 1333 (w), 1309 (m), 1298 (sh), 1265, 1217, 1182 (m), 1163 (w), 1140, 1103, 1078 (m), 1049, 1029, 1013 (sh), 999 (m), 978, 965 (sh), 948 (w), 893 (s), 857, 841 (m), 813 (sh), 735 (s), 690 (s), 647, 627 (w), 611, 587, 571 (sh), 558 (w), 543 (m), 511 (s), 484, 472, 455, 441, 427, 413 (m); HR-MS (+ESI): m/z calcd for C69H61Au3O3P3 [M + H]+, 1621.283; found, 1621.283; elemental analysis: calcd for C69H60Au3O3P3·2CH2Cl2 (1790.91), C 47.62, H 3.60; found, C 47.66, H 3.62.
C6. (Method A) Prepared from CTB(OMe,C2H) (0.010 g; 0.0231 × 10−3 mol), AuCl(PPhMe2) (0.0944 × 10−3 mol), and NaOMe (0.555 × 10−3 mol) in MeOH (7 mL) and THF (5 mL), 24 h stirring. Yield: 0.018 g (0.0125 × 10−3 mol, 54%). (Method B) Prepared from [CTB(OMe,C2Au)]n (0.055 g; 0.054 × 10−3 mol) and PPhMe2 (0.023 g; 0.166 × 10−3 mol) in a mixture of CH2Cl2 (12 mL) and THF (3 mL), 5 h stirring. Yield: 0.048 g (0.0335 × 10−3 mol; 62%). Mp 182.5 °C; 1H NMR (400.13 MHz, CDCl3, 298 K): δ (ppm) = 7.738 (m, 4JH,H = 2.0 Hz, 6 H; o- and o′-H), 7.477 (d, 4JH,H = 2.0 Hz, 9 H; m-, m′-, and p-H), 7.461 (s, 3 H; α′-H), 6.785 (s, 3 H; α-H), 4.681 (d, 2JH,H = 13.6 Hz, 3 H; a-H), 3.889 (s, 9 H; OCH3), 3.548 (s, 2JH,H = 13.6 Hz, 3 H; e-H), 1.737 (d, 3JH,P = 9.6 Hz, 18 H; CH3); 13C NMR (125.77 MHz, CDCl3, 295 K): δ (ppm) = 159.47 (s; γ-C), 139.87 (s; β-C), 135.72 (s; α′-C), 132.69 (d, 1JC,P = 54.1 Hz; i-C), 132.19 (d, 2JC,P = 12.6 Hz; o- and o′-C), 131.70 (s; p-C), 130.61 (s, β′-C), 129.25 (d, 3JC,P = 11.3 Hz; m- and m′-C), 112.79 (s; γ′-C), 111.95 (s; α-C), 100.23 (2 s; δ′- and ε′-C), 56.28 (s; OCH3), 37.07 (s; CH2), 15.70 (d; 1JC,P = 35.2 Hz; CH3); 31P NMR (121.49 MHz, CDCl3, 299 K): δ (ppm) = 14.16; IR (KBr): ν (cm−1) = 3445 (v br, s), 3057, 2961 (w), 2919 (m), 2850 (m), 2360, 2341 (w), 2221 (w), 2100 (w), 1600 (m), 1558 (w), 1491 (s), 1462 (w), 1435 (s), 1422 (w), 1385 (s), 1308 (m), 1263 (s), 1217, 1190, 1147, 1107, 1080 (m), 1042 (w), 1017, 1000, 952 (m), 909 (s), 862 (w), 840 (m), 823, 802, 782 (sh), 742 (s), 722 (m), 691 (s), 645, 623, 608, 583, 561 (w), 542, 515, 488, 447, 430, 408 (m); HR-MS (+ESI): m/z calcd for C54H55Au3O3P3 [M + H]+, 1435.236; found, 1435.236; elemental analysis: calcd for C54H54Au3O3P3·½C6H12 (1476.92), C 46.35, H 4.09; found, C 46.19, H 4.04.
(−)- C2. (Method B) Prepared from [(−)-CTB(H,C2Au)]n (0.010 g; 0.01074 × 10−3 mol) and PPh2Me (0.0088 mL, 0.0095 g; 0.0474 × 10−4 mol) in a mixture of CH2Cl2 (3 mL) and THF (1 mL), 20 min stirring. Yield: 0.013 g (0.0849 × 10−4 mol; 79%).
(+)- C2. (Method B) Prepared from [(+)-CTB(H,C2Au)]n (0.030 g; 0.0322 × 10−3 mol) and PPh2Me (0.024 mL, 0.0259 g; 1.292 × 10−4 mol) in CH2Cl2 (5 mL). Yield: 0.020 g (0.0131 × 10−3 mol; 41%).
(−)- C3. (Method B) Prepared from [(−)-CTB(H,C2Au)]n (0.016 g; 0.0172 × 10−3 mol) and PPhMe2 (0.010 mL, 0.0097 g; 0.0702 × 10−3 mol), in CH2Cl2 (2.5 mL). Yield: 0.017 g (0.0126 × 10−3 mol; 73%).
(+)- C3. (Method B) Prepared from [(+)-CTB(H,C2Au)]n (0.040 g; 0.43 × 10−4 mol) and PPhMe2 (25 × 10−3 mL, 0.0243 × 10−3 g; 1.757 × 10−4 mol) in CH2Cl2 (6 mL).
(−)- C4. (Method B) Prepared from [(−)-CTB(OMe,C2Au)]n (0.015 g; 0.0147 × 10−3 mol) and PPh3 (0.0116 g; 0.0442 × 10−3 mol) in CH2Cl2 (3 mL) and THF (1 mL). Yield: 0.023 g (0.0127 × 10−3 mol; 86%).
(+)- C4. (Method B) Prepared from [(+)-CTB(H,C2Au)]n (0.016 g; 0.0157 × 10−3 mol) and PPh3 (0.014 g; 0.0534 × 10−3 mol) in CH2Cl2 (4 mL) and THF (2 mL) for 5 h. Yield: 0.016 g (0.00885 × 10−3 mol; 56%).
(−)-
C5
and (+)-
C5. (Method B) (−)-C5 was prepared from [(−)-CTB(OMe,C2Au)]n (0.0125 g; 0.0123 × 10−3 mol) and PPh2Me (0.007 mL, 0.0076 g; 0.038 × 10−3 mol) in CH2Cl2 (2.5 mL) and THF (1 mL), overnight stirring. Yield: 0.016 g (0.010 × 10−3 mol; 80%). (Chromatographic separation) Resolution of the C5 racemate (0.004 g dissolved in 1.2 mL of a mixture of CH2Cl2/EtOH/hexane, 50:25:25) was performed by chiral HPLC using Chiralpak IB column (250 × 4.6 mm), eluting with hexane/EtOH/CH2Cl2 40:40:20 at a flow rate of 1 mL min−1 with UV detection at 254 nm. 24 injections of 50 μL each every 10 min were necessary. The first eluted pure enantiomer was (−)-C5 (1.55 mg) and the second eluted pure enantiomer was (+)-C5 (1.4 mg).
(−)-
C6. (Method B) Prepared from [(−)-CTB(OMe,C2Au)]n (0.0135 g; 0.0132 × 10−3 mol) and PPhMe2 (0.007 mL, 0.0068 g; 0.0492 × 10−3 mol) in CH2Cl2 (5 mL) and THF (1 mL). Yield: 0.0133 g (0.00927 × 10−3 mol, 70%); or: resolution of a sample of the racemate prepared according to method A (0.006 g dissolved in 3 mL of a mixture of CH2Cl2/hexane, 67:33) was performed by chiral HPLC using Chiral Art Cellulose-SJ column (250 × 4.6 mm), eluting with hexane/EtOH/CH2Cl2 40:20:40 at a flow rate of 1 mL min−1 and UV detection at 230 nm. 40 injections of 50 μL each every 6.4 min were necessary. The first eluted enantiomer was (+)-C6 (1 mg) but its optical purity was not satisfactory, and the second eluted enantiomer was pure (−)-C6 (1.4 mg).
(+)- C6. (Method B) Prepared from [(+)-CTB(OMe,C2PPhMe2)]n (0.014 g; 0.0137 × 10−3 mol) and PPhMe2 (0.0071 mL, 0.0069 g; 0.0499 × 10−3 mol) in THF (1 mL) and CH2Cl2 (6 mL). Yield: 0.012 g (0.00836 × 10−3 mol; 61%).
Calculation of the structures and of the ECD spectra
Molecular structures of CTB(H,C2H), CTB(OMe,C2H), C1, C3 and C4 were optimized at the DFT level of theory using the Gaussian09 software.57 The B3LYP functional was used and completed with D3 empirical dispersion correction, together with Def2-TZVPP basis set for light atoms and Def2-TZVPD basis set for Au, including the associated pseudo-potential taken from the Basis Set Exchange library.58 Frequency calculations were performed in order to check that true energy minima were obtained.The molecular structures of CTB(H,C2H), CTB(OMe,C2H), C1 and C4 were also optimized at the DFT level of theory (Gaussian09 software57) using the CAM-B3LYP functional together with the 6-311+G(d,p) basis set for light atoms and Def2-TZVPD basis set for Au, including the associated pseudo-potential taken from the Basis Set Exchange library.58 The solvent effects (CH2Cl2) were taken into account through a PCM model. Frequency calculations were performed in order to check that true energy minima were obtained. ECD spectra were calculated by TD-DFT using the same functional and basis sets. The half-widths at half height were adjusted for the comparison of the simulated ECD spectra with the experimental ones.
Author contributions
J. Z. was in charge of the syntheses of the CTBs and their complexes; N. Z. and E. L.-W. carried out the ESI-MS experiments; M. J. and N. V. performed the chiral separations, and recorded the electronic and ECD spectra; E. E. and E. A. were in charge of the DFT calculations. J.-C. C. was responsible for the conceptualization and the methodology of the work, the supervision of J. Z., and the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the French National Research Agency (ANR) through the “Programme d'Investissement d'Avenir” under contract 17-EURE-0016 (PhD fellowship to J. Z.), the CNRS, and the University of Strasbourg. Dr Martine Heinrich and Noémie Schneider are thanked for the elemental analyses. E. A. and E. E. are grateful to the EXPLOR mesocenter for providing access to its computing facility (project 2021CPMXX2483).References
- Y. Domoto and M. Fujita, Coord. Chem. Rev., 2022, 466, 214605 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed.
- T. Murase and M. Fujita, Chem. Rec., 2010, 10, 342 CrossRef CAS PubMed.
- K. Kobayashi and M. Yamanaka, Chem. Soc. Rev., 2015, 44, 449 RSC.
- E. G. Percásregui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480 CrossRef PubMed.
- M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042 CrossRef CAS PubMed.
- G. Montà-González, F. Sancenón, R. Martínez-Máñez and V. Martí-Centelles, Chem. Rev., 2022, 122, 13636 CrossRef PubMed.
- M. Yoshizawa and L. Catti, Acc. Chem. Res., 2019, 52, 2392 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS.
- Z. Chen, L. Yang, Y. Hu, D. Wu, J. Yin, G.-A. Yu and S. H. Liu, RSC Adv., 2015, 5, 93757 RSC.
- V. Cámara, N. Barquero, D. Bautista, J. Gil-Rubio and J. Vicente, Chem. – Eur. J., 2015, 21, 1992 CrossRef PubMed.
- J. R. Shakirova, M. Shimada, D. A. Olisov, G. L. Starova, H. Nishihara and S. P. Tunik, Z. Anorg. Allg. Chem., 2018, 644, 308 CrossRef CAS.
- A. Belyaev, I. Kolesnikov, A. S. Melnikov, V. V. Gurzhiy, S. P. Tunik and I. O. Koshevoy, New J. Chem., 2019, 43, 13741 RSC.
- M. Głodek, S. Pawlędzio, A. Makal and D. Plażuk, Chem. – Eur. J., 2019, 25, 13131 CrossRef PubMed.
- W. J. Hunks, M.-A. MacDonald, M. C. Jennings and R. J. Puddephatt, Organometallics, 2000, 19, 6063 CrossRef.
- V. W.-W. Yam, K.-L. Cheung, S.-K. Yip and K.-K. Cheung, J. Organomet. Chem., 2003, 681, 196 CrossRef CAS.
- X. He, E. C.-C. Chen, N. Zhu and V. W.-W. Yam, Chem. Commun., 2009, 4016 RSC.
- L. Rodríguez, M. Ferrer, R. Crehuet, J. Anglada and J. C. Lima, Inorg. Chem., 2012, 51, 7636 CrossRef PubMed.
- C. P. McArdle, S. Van, M. C. Jennings and R. J. Puddephatt, J. Am. Chem. Soc., 2002, 124, 3959 CrossRef CAS PubMed.
- A. Schaly, Y. Rousselin, J.-C. Chambron, E. Aubert and E. Espinosa, Eur. J. Inorg. Chem., 2016, 832 CrossRef CAS.
- A. Schaly, M. Meyer, J.-C. Chambron, M. Jean, N. Vanthuyne, E. Aubert, E. Espinosa, N. Zorn and E. Leize-Wagner, Eur. J. Inorg. Chem., 2019, 2691 CrossRef CAS.
- Z. Zhong, A. Ikeda, S. Shinkai, S. Sakamoto and K. Yamaguchi, Org. Lett., 2001, 3, 1085 CrossRef CAS PubMed.
- J. J. Henkelis, C. J. Carruthers, S. E. Chambers, R. Clowes, A. I. Cooper, J. Fisher and M. J. Hardie, J. Am. Chem. Soc., 2014, 136, 14393 CrossRef CAS PubMed.
- V. E. Pritchard, D. Rota Martir, S. Oldknow, S. Kai, S. Hiraoka, N. J. Cookson, E. Zysman-Colman and M. J. Hardie, Chem. – Eur. J., 2017, 23, 6290 CrossRef CAS PubMed.
- T. Kojima, F. L. Thorp-Greenwood, M. J. Hardie and S. Hiraoka, Chem. Sci., 2018, 9, 4104 RSC.
- S. Oldknow, D. Rota Martir, V. E. Pritchard, M. A. Blitz, C. W. G. Fishwick, E. Zysman-Colman and M. J. Hardie, Chem. Sci., 2018, 9, 8150 RSC.
- E. Britton, R. J. Ansell, M. J. Howard and M. J. Hardie, Inorg. Chem., 2021, 60, 12912 CrossRef CAS PubMed.
- A. Collet, Tetrahedron, 1987, 43, 5725 CrossRef CAS.
- A. Collet, J.-P. Dutasta, B. Lozach and J. Canceill, Top. Curr. Chem., 1993, 165, 103 CrossRef CAS.
- T. Brotin and J.-P. Dutasta, Chem. Rev., 2009, 109, 88 CrossRef CAS PubMed.
- G. El-Ayle and K. T. Holman, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, 2017, p. 199 Search PubMed.
- T. Brotin, R. Montserret, A. Bouchet, D. Cavagnat, M. Linares and T. Buffeteau, J. Org. Chem., 2012, 77, 1198 CrossRef CAS PubMed.
- T. Brotin, P. Berthault, D. Pitrat and J.-C. Mulatier, J. Org. Chem., 2020, 85, 9622 CrossRef CAS PubMed.
- R. M. Fairchild and K. T. Holman, J. Am. Chem. Soc., 2005, 127, 16364 CrossRef CAS PubMed.
- J. Zhang, A. Schaly, J.-C. Chambron, B. Vincent, N. Zorn, E. Leize-Wagner, M. Jean and N. Vanthuyne, Chem. – Eur. J., 2022, 28, e202103759 CrossRef CAS PubMed.
- B. M. Schulze, D. L. Watkins, J. Zhang, I. Ghiviriga and R. K. Castellano, Org. Biomol. Chem., 2014, 12, 7932 RSC.
- L. Peyrard, M.-L. Dumartin, S. Chierici, S. Pinet, G. Jonusauskas, P. Meyrand and I. Gosse, J. Org. Chem., 2012, 77, 7023 CrossRef CAS PubMed.
- R. J. Cross and M. F. Davidson, J. Chem. Soc., Dalton Trans., 1996, 411 Search PubMed.
- W. Wu, N. Zhu and C.-M. Che, J. Organomet. Chem., 2003, 670, 11 CrossRef.
- G. E. Coates and C. J. Parkin, J. Chem. Soc., 1962, 3220 RSC.
- G. Jia, R. J. Puddephatt, J. D. Scott and J. J. Vittal, Organometallics, 1993, 12, 3565 CrossRef CAS.
- A. Collet and G. Gottarelli, J. Am. Chem. Soc., 1981, 103, 204 CrossRef CAS.
- J. Canceill, A. Collet, J. Gabard, G. Gottarelli and G. P. Spada, J. Am. Chem. Soc., 1985, 107, 1299 CrossRef CAS.
- A. Collet and G. Gottarelli, Croat. Chem. Acta, 1989, 62, 279 CAS.
- J. Canceill and A. Collet, New J. Chem., 1986, 10, 17 CAS.
- C. Garcia and A. Collet, Bull. Soc. Chim. Fr., 1995, 132, 52 CAS.
- J. R. Platt, J. Chem. Phys., 1951, 19, 263 CrossRef CAS.
- D. Li, X. Hong, C.-M. Che, W.-C. Lo and S.-M. Peng, J. Chem. Soc., Dalton Trans., 1993, 2929 RSC.
- F. K.-W. Hau, K.-L. Cheung, N. Zhu and V. W.-W. Yam, Org. Chem. Front., 2019, 6, 1205 RSC.
- H.-S. Lo, N. Zhu, V. K.-M. Au and V. W.-W. Yam, Polyhedron, 2014, 83, 178 CrossRef CAS.
- T. E. Müller, S. W.-K. Choi, D. M. P. Mingos, D. Murphy, D. J. Williams and V. W.-W. Yam, J. Organomet. Chem., 1994, 484, 209 CrossRef.
- Y.-P. Zhou, E.-B. Liu, J. Wang and H.-Y. Chao, Inorg. Chem., 2013, 52, 8629 CrossRef CAS PubMed.
- Y.-R. Zhenga and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 3487 CrossRef PubMed.
- Z. Qi, T. Heinrich, S. Moorthy and C. Schalley, Chem. Soc. Rev., 2015, 44, 515 RSC.
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR, ESI-MS, electronic absorption, and ECD (experimental and calculated) spectra; views of the calculated structures, ESI-MS spectra of the CTB mixtures, and the corresponding graphs. Tables of all spectroscopic data. See DOI: https://doi.org/10.1039/d3dt04279k |
| This journal is © The Royal Society of Chemistry 2024 |