
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen reduction reaction (ORR) in alkaline solution catalysed by an atomically precise catalyst based on a Pd(II) complex supported on multi-walled carbon nanotubes (MWCNTs). Electrochemical and structural considerations†
Valeria
Monini
a,
Marco
Bonechi
 *a,
Carla
Bazzicalupi
*a,
Antonio
Bianchi
ab,
Pietro
Gentilesca
a,
Walter
Giurlani
a,
Massimo
Innocenti
ab,
Arianna
Meoli
a,
Giammarco Maria
Romano
a and
Matteo
Savastano
*bc
*a,
Carla
Bazzicalupi
*a,
Antonio
Bianchi
ab,
Pietro
Gentilesca
a,
Walter
Giurlani
a,
Massimo
Innocenti
ab,
Arianna
Meoli
a,
Giammarco Maria
Romano
a and
Matteo
Savastano
*bc
aDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3-13, 50019 Sesto Fiorentino, Italy. E-mail: marco.bonechi@unifi.itCarla; carla.bazzicalupi@unifi.it
bNational Interuniversity Consortium of Materials Science and Technology (INSTM), Research Unit of Florence, Via G. Giusti 9, 50121 Florence, Italy. E-mail: matteo.savastano@uniroma5.it
cDepartment of Human Sciences for the Promotion of Quality of Life, University San Raffaele Roma, Via di Val Cannuta 247, 00166 Rome, Italy
First published on 2nd January 2024
Abstract
A new atomically precise, single-ion catalyst (MWCNT-LPd) for ORR (oxygen reduction reaction), consisting of a Pd(II) complex of a tetraazacycloalkane anchored on multiwalled carbon nanotubes, has been prepared through a supramolecular approach ensuring a uniform distribution of catalytic centres on the support surface. A tetraazacycloalkane was chosen to saturate the four coordination sites of the typical square planar coordination geometry of Pd(II) with the aim of ascertaining whether the metal ion must have free coordination sites to function effectively in the ORR or whether, as predicted by quantum mechanical calculations, the catalytic effect can be originated from an interaction of O2 in the fifth coordinative position. The results clearly demonstrated that tetracoordination of Pd(II) does not influence its catalytic capacity in the ORR. Electrodes based on this catalyst show ORR performance very close to that of commercial Pt electrodes, despite the low Pd(II) content (1.72% by weight) in the catalyst. The onset potential (Eon) value and the half-wave potential (E1/2) of the catalyst are, respectively, only 53 mV and 24 mV less positive than those observed for the Pt electrode and direct conversion of O2 to H2O reaches 85.0%, compared to 89% of the Pt electrode. Furthermore, a preliminary galvanostatic test (simulating a working fuel cell at a fixed potential) showed that the catalyst maintains its efficiency continuing to produce water throughout the process (the average number of electrons exchanged over time per O2 molecule remains close to 4).
Introduction
The oxygen reduction reaction (ORR) is one of the most important reactions both in life processes (e.g. aerobic respiration) and in artificial energy conversion systems such as fuel cells.1,2 The ORR that occurs at the fuel cell cathode is a sluggish process, so it needs to be catalysed for profitable use and Pt-based electrocatalysts are the primary choice for this purpose due to their high catalytic performance.3–6Unfortunately, the scarcity and the high price of this precious metal make Pt-based electrocatalysts very expensive, too expensive to find a daily application in support of human activities that require an energy supply. Nonetheless, fuel cells are an important clean alternative to internal combustion engines that burn fossil fuels and generate greenhouse gases, in addition to a multitude of pollutants,7 and accordingly the search for alternative catalysts is garnering strong consideration. To replace platinum and other platinum group metals of similar cost, several strategies are being studied that make use of electrocatalysts based on less expensive non-noble transition metals8–12 or employ non-metallic12–16 or atomically precise17–20 electrodes. Atomically precise electrocatalysts are of special interest due to their high atomic utilization efficiency, special electronic structure and homogeneous distribution of active centres.17–20
Two main pathways are possible for ORR in aqueous solutions: (i) a 4-electron (4e) reduction of O2 to H2O (or OH−) in acidic (or alkaline) solutions and (ii) a 2-electron (2e) reduction of O2 to H2O2, (or HO2−) in acidic (or alkaline) solutions. Although the electrocatalytic production of hydrogen peroxide (H2O2) is gaining increasing interest, as H2O2 is considered a green oxidant,21–23 the formation of H2O2 in fuel cells is deleterious as this undesired side reaction leads to loss of energy and can cause serious damage to cell catalysts and membranes.24,25 Nevertheless, H2O2 can be converted into water via another 2e reaction.
We recently reported that two atomically precise, single-ion Pd(II) catalysts constructed via supramolecular (non-covalent) decoration of multiwalled carbon nanotubes (MWCNTs) with Pd(II) tetraaza-macrocyclic complexes have demonstrated great potential to accelerate the ORR.26 These catalysts, having a very low Pd load (5%), exhibited excellent half-cell PGM mass activity, exceeding state-of-the-art reference goals of the time,27 onset potential (Eon) values comparable to or better than that of a commercial Pt electrode and a nearly unique (90% or above) 4e process, leading to direct production of H2O. The macrocyclic ligands employed in this study26 and in successive studies28,29 to append Pd(II) ions to MWCNTs were selected with the dual goal of ensuring robust metal ion coordination, to prevent loss of catalytic centres during electrode operation, and involving a metal ion in the coordination of up to three nitrogen atoms of the ligand, thus avoiding saturation of the square planar coordination sphere typical of Pd(II) with macrocycle's donor atoms, in the belief that the presence of at least one labile donor is beneficial for the proper functioning of the catalysts.30–32 Accordingly, our Pd(II) complexes contained one labile Cl− ligand coordinated to the metal ion. While this fast exchangeable ancillary ligand (Cl−) was fundamental for Sonogashira cross-coupling reactions,33,34 which requires the simultaneous coordination to Pd(II) of two different reagent molecules, theoretical calculations35 showed that this is not mandatory for ORR, as O2 is able to form an apical bond with the metal centre.
To find an experimental verification of these theoretical results, which would greatly simplify the design of atomically precise catalysts based on d8 metal ions by eliminating the need to build unsaturated environments around them, we prepared a new catalyst containing Pd(II) ions blocked into the N4 square planar coordination environment offered by a cyclen-based (cyclen = 1,4,7,10-tetraaza-cyclododecane) ligand (H2L, Fig. 1). The Pd(II) complex of H2L was anchored on MWCNTs via spontaneous adsorption under environmentally friendly conditions (water, room temperature, unprotected atmosphere) and the resulting MWCNT-LPd hybrid material was tested toward the ORR in alkaline media.
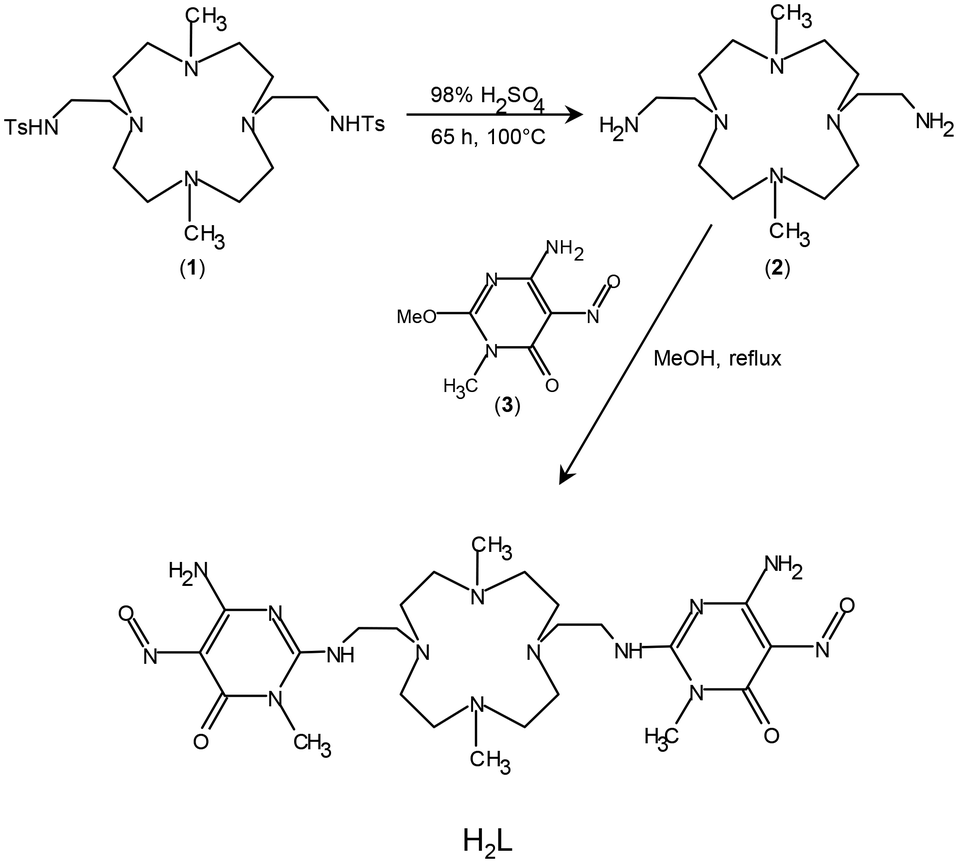 | ||
| Fig. 1 The synthetic procedure adopted to prepare the H2L ligand. | ||
Experimental
Materials
All starting materials were high purity compounds purchased from commercial sources and were used without further purification. 6-Amino-3,4-dihydro-3-methyl-2-methoxy-5-nitroso-4-oxopyrimidine ((3), Fig. 1) was prepared according to a literature procedure.36 Graphitized MWCNTs (99.9% C, outer diameter 8–15 nm, inner diameter 3–5 nm, specific surface area 117 m2 g−1) were purchased from NanoAmor (Texas, USA).0.50 g (1.75 mmol) of (2) were dissolved in 150 cm3 of methanol and 0.67 g (3.67 mmol) of (3) were added to the resulting solution in small portions, under stirring, at room temperature. The mixture was then refluxed for 3 hours to obtain a solution that was kept at room temperature overnight. The solid formed was filtered, washed with methanol and dried under reduced pressure at room temperature. Another batch of the same compound was obtained from the mother liquor. The mother liquor was treated with 10 drops of 37% NH3 aqueous solution to convert the excess of (3) into its insoluble derivative 2,4-diamino-1-methyl-5-nitroso-6-oxopyrimidine. The solid formed upon standing in a fridge overnight was removed by filtration, and the resulting solution was evaporated to dryness under reduced pressure. The remaining solid compound was dried at room temperature under reduced pressure. Overall yield 0.70 g (1.12 mmol), 64%. 1H NMR (400 MHz, CDCl3): δ 2.16 (s, 6H), 2.99 (m, 8H), 3.06 (m, 4H), 3.15 (m, 4H), 3.28 (m, 4H), 3.34 (m, 3H), 3.47 (m, 4H), 3.80 (t, 4H). Elem Anal. Calcd (%) for C24H42N14O4: C 48.80, H 7.17, N 33.20. Found: C 48.68, H 7.13, N 33.08.
Potentiometric measurements
The protonation constants of H2L were determined by means of potentiometric (pH-metric) titrations in 0.1 M NMe4Cl aqueous solution at 298.1 ± 0.1 K using an automated apparatus and a procedure previously described.38 The acquisition of the emf data was performed with the computer program PASAT.39,40 The combined electrode (Metrohm 6.0262.100) was calibrated as a hydrogen-ion concentration probe by titration of known amounts of HCl with CO2-free NaOH solutions and determining the equivalent point by Gran's method,41 which gives the standard potential, E°, and the ionic product of water (pKw = 13.83(1) in 0.1 M NMe4Cl at 298.1 K). The stability constants were calculated from the potentiometric data by means of the computer program HYPERQUAD.42 The concentration of H2L was about 1 × 10−3 M in all experiments and the studied pH range was 2.0–11.5. Three measurements were performed and used to determine the protonation constants.Preparation of the catalyst
The MWCNT-LPd catalyst was prepared in two steps according to a previously reported successful procedure:43 (i) adsorption of H2L onto the surface of MWCNTs in water via a π–π interaction of the hydrophobic pyrimidine residue of the ligand with the graphene surface of the nanotubes and (ii) decoration of the macrocyclic ligand moiety with coordinated Pd(II) ions.First, 100 mg of MWCNTs were suspended in 50 cm3 of a 1 mM solution of the ligand at pH 8.5. The suspension was sonicated for a few minutes and successively kept under stirring for 6 days at room temperature. The solid was then recovered by filtration and dried under reduced pressure, at room temperature, and in the presence of a desiccant (NaOH) until constant weight. The amount of ligand adsorbed (0.174 mmol g−1) was determined by measuring the concentration of ligand remaining in the solution by means of UV spectroscopy (Fig. S1†). The resulting solid was successively suspended in water (1 mg cm−3 of water, pH 5), and the suspension was kept under stirring at room temperature for 6 days. The solid was recovered and dried under reduced pressure, at room temperature, and in the presence of a desiccant (NaOH) until constant weight. The amount of ligand remaining on the MWCNTs (0.162 mmol g−1) was determined by measuring the concentration of the ligand desorbed into the solution by means of UV spectroscopy (Fig. S1†).
In the second step, 50 mg of the MWCNT-L material, obtained in the first step, was suspended in 50 cm3 of a solution, at pH 5, containing one equivalent of K2PdCl4 (relative to the ligand) and the resulting suspension was kept under stirring at room temperature. After 6 days, all metal ions had been adsorbed, as shown by the UV spectrum of the filtered suspension, which corresponds to the formation of a MWCNT-LPd catalyst containing 1.72% of Pd by weight. The MWCNT-LPd catalyst was recovered by filtration, washed with water and dried under reduced pressure, at room temperature, and in the presence of a desiccant (NaOH) until constant weight.
Electrochemical measurement and modified electrodes’ preparation
All the electrochemical measurements, cyclic voltammetry (CV) and linear sweep voltammetry (LSV) experiments, were performed with an AUTOLAB PGSTAT12 using NOVA software from Metrohm Autolab and a modulated speed rotator (MSR) 636A from Pine Instrument.Electrochemical measurements were performed using a three-electrode cell. The reference electrode was an Ag/AgCl/sat. KCl electrode with a potential of +197 mV with respect to the normal hydrogen electrode (NHE). In the following, all potential values will be referred to the Ag/AgCl/saturated KCl reference electrode (RE), unless otherwise specified. The counter electrode was a platinum wire, and the working electrode was a ring-disk electrode from Pine Instrument Co. consisting of a glassy carbon (GC) disk insert (∅ = 5.5 mm; A = 0.238 cm2) and a Pt ring (∅inner = 6.50 mm, ∅outer = 8.50 mm, A = 0.236 cm2) with a theoretical collection efficiency (N) of 38.3%.
Rotating ring disk electrode (RRDE) experiments were performed in a glass cell filled with 0.1 M KOH solution. Nitrogen was used to achieve an oxygen-free solution (15 minutes of bubbling) while oxygen was used to obtain an oxygen-saturated electrolyte solution (20 min).
RRDE working electrodes were mechanically polished with 0.05 μm alumina and sonicated for five minutes in milliQ water. The Pt ring electrode was electropolished by scanning the potential at 100 mV s−1 between −0.20 and +1.15 V in a 0.5 M H2SO4 solution until a reproducible curve was obtained.44 Activation of the working electrode was confirmed in a 1 mM K3Fe(CN)6 solution in 0.1 M KCl by recording CVs in the −0.3 to +0.7 V potential range at 10 mV s−1 scan rate; the typical ΔEp for an activated electrode is very close to 60 mV.45
Immobilization of the catalysts onto the GC disk working electrode was achieved by preparing an ink consisting of MWCNT-LPd dispersed in a polymeric membrane (Nafion®). Samples were prepared according to reported procedures.28,29,46 An appropriate amount of samples (2 mg) was dispersed in 52 μL of water, 32 μL of ethanol and 23 μL of a 5% Nafion® solution. The composition of wet ink was as follows: catalyst 2%, water 52%, EtOH 26% and Nafion® 20%. The resulting ink was sonicated for 30 min and drop-cast onto the glassy carbon disk electrode (10 μL). The modified GC electrode was then dried at room temperature.
Cyclic voltammograms were obtained by scanning the potential between +0.2 and −0.75 V at a potential scan rate of 5 mV s−1. Linear sweep voltammograms were obtained at a rotation rate of 1600 rpm and a scan rate of 5 mV s−1, in the potential range from +0.1 to −0.70 V for the modified GC electrode while the Pt ring electrode was held at +0.50 V. The ring current was recorded simultaneously with the disk current.
RRDE galvanostatic analysis at 1600 rpm and a selected potential value of −0.6 V (for the modified GC disk electrode) and +0.50 V for the Pt ring was performed within a 600 s framework.
RRDE calibration experiment was performed to determine the experimental collection efficiency (N = Iring/Idisk) of the ring-disk, recording LSV at 1600 rpm of a oxygen-free K3Fe(CN)6 solution (1 mM) in 0.1 M KCl at a scan rate of 5 mV s−1 in the potential range from +0.60 V to −0.60 V (for the modified GC disk) and applying a constant potential of +0.6 V at the ring.47 The electron transfer number exchanged per O2 molecule (n) occurring during the ORR process was calculated using the equation n = 4Idisk/(Idisk + Iring/N).48,49
A commercial Pine Instrument electrode consisting of a Pt disk insert (∅ = 5.5 mm; A = 0.238 cm2) and a Pt ring (∅inner = 6.50 mm, ∅outer = 8.50 mm, A = 0.236 cm2) with a theoretical collection efficiency (N) of 38.3% was employed to benchmark the performance of the materials presented in this report.
Spectrophotometric measurements
UV-vis absorption spectra were recorded at 298 K by using a Jasco V-670 spectrophotometer. The UV (vis) spectra of H2L were recorded in the pH range of 0.80–12.01 (0.78–12.15) with an H2L concentration of 1.80 × 10−5 M (1.95 × 10−3 M). The UV-vis spectra of the Pd(II) complex were recorded in the pH range of 2.32–9.12 with H2L and Pd(II) (as K2PdCl4) concentrations of 1.80 × 10−5 M. Mother solutions containing the ligand and the metal ions (concentrations given above) were prepared at pH 4, to avoid formation of metal hydroxides/oxides, and allowed to equilibrate for one day. Aliquots of these solutions were separated and their pH was adjusted to different values. Equilibration of these solutions was checked daily by means of UV-vis spectroscopy until invariance of the recorded spectra was reached (6 days). The recorded pH values of solutions were measured after equilibrium was reached.NMR spectroscopy
1H NMR spectra (400 MHz) in D2O solution were recorded at 298 K on a 400 MHz Bruker Avance III spectrometer.XPS analysis
MWCNT-LPd was fixed on a sample holder dropcasting a water dispersion onto a gold substrate and dried under a flow of N2. The samples were then introduced into the ultra-high vacuum (UHV) chamber of the instrument. X-ray photoelectron spectroscopy (XPS) spectra were recorded while using Al Kα radiation and a pass energy of 44 eV for XPS survey and 22 eV for high resolution XPS spectra, using previously reported instrumentation and methods, namely using a model TA10 X-ray source and a model HA100 hemispherical analyser (VSW Scientific Instrument Limited, Manchester, UK) with a 12 channel detector. The C 1s transition at 284.8 eV was used as a reference for obtaining the heteroatom binding energies.50,51 The collected data were analysed using the dedicated software CasaXPS (version 2.3.16Dev52). The background was subtracted using Shirley's method52 and the peaks were fitted using mixed Gaussian–Lorentzian components.SEM analysis
SEM micrographs were obtained with a Hitachi SU3800 scanning electron microscope equipped with an UltimMax Oxford instrument detector (Oxford Instruments, Wiesbaden, Germany) and AZtecLive software (Oxford Instruments NanoAnalysis, Abingdon, United Kingdom). X-ray elemental mapping was carried out at 5 kV acceleration voltage and 10 mm working distance. MWCNT samples were fixed on the surface of conductive tape for SEM analysis.Crystal structure determination of (H5L)(ClO4)3·2H2O, (H3L)(H4L)(ClO4)3·11H2O, [Cu(H2L)(H2O)](ClO4)2·10.46H2O, [Cu(H2L)Cl]Cl·3H2O and [Pd(1)]Cl2·4H2O
A yellow crystal ((H5L)(ClO4)3·2H2O), a violet crystal ((H3L)(H4L)(ClO4)3·11H2O), a blue crystal ([Cu(H2L)(H2O)](ClO4)2·10.46H2O), a dark blue crystal ([Cu(H2L)Cl]Cl·3H2O) and a yellow crystal ([Pd(1)]Cl2·4H2O) were used for X-ray diffraction analyses. A summary of the crystallographic data is reported in Table S1.† In the case of the compound [Pd(1)]Cl2·4H2O, reflections were recorded up to 2θ = 98° (1 Å), due to the significant lowering of intensity with increasing θ value. However, the reached resolution and the observed reflections/refined parameter ratio, about 8, ensure that the overall geometries of the complexes are confidently defined. The integrated intensities were corrected for Lorentz and polarization effects and an empirical absorption correction (SADABS)53 was applied. Crystal structures were resolved by direct methods (SHELXT)54 and refinements were performed by means of full-matrix least-squares using SHELXL Version 2019/3.55 Non-H atoms were anisotropically refined. H atoms were introduced as riding atoms with the thermal parameter calculated in agreement with the linked atom. Some of the water hydrogen atoms and the ionisable hydrogen atoms, such as the amidic hydrogens of [Cu(H2L)(H2O)](ClO4)2·10.46H2O, [Cu(H2L)Cl]Cl·3H2O and [Pd(1)]Cl2·4H2O or the acidic hydrogen on the nitroso group of ((H5L)(ClO4)3·2H2O), were located in the Fourier difference maps, included in the calculations and freely refined. The amidic nitrogen atoms that were not localized in the Fourier difference map were always introduced as riding atoms with the thermal parameter calculated in agreement with the linked atom, with the exception of one of the Pd(II) complexes. The not localized water H atoms were not introduced in the calculations. Molecular plots were obtained using the software CCDC Mercury.56Results and discussion
The choice of 6-amino-3,4-dihydro-3-methyl-5-nitroso-4-oxopyrimidine as the anchor group to fix the Pd(II) complex on the surface of the MWCNTs was made considering the ability of pyrimidine residues to adhere with the arene centres of graphene surfaces, through the formation of strong π–π stacking interactions, giving rise to very stable, non-covalently assembled, hybrid materials which are suitable for successive functionalization with metal ions.57 This route has already been proven to be effective for the preparation of robust single-ion Pd(II) catalysts.26,28,29,33,34Nevertheless, before proceeding to verify the efficiency of a new catalyst, it is advisable to verify its ability to resist the conditions under which it will be used. The robustness of our single-ion catalyst (MWCNT-LPd) is principally determined by its resistance toward the loss of catalytic metal centres that might occur through two main processes: demetallation of the complex unit and detachment of the metal complex linked to MWCNTs. For similar catalysts, some demetallation of the complex unit is sometimes observed during the processes for which they are intended. However, this often occurs by reduction of Pd(II) to Pd(0), with the formation of Pd nanoparticles which can have their own catalytic activity and therefore the catalyst continues to be efficient.28,33,34,43–45 Instead, detachment of the metal complex from the catalyst causes the loss of catalytic activity. The use of a macrocyclic ligand to bind Pd(II) ensures good resistance of the complex toward demetallation, both thermodynamically and kinetically. This is particularly true when the macrocycle fulfils the square-planar environment of Pd(II), as in our case, but there is a risk that the absence of readily available coordination positions on Pd(II) could be deleterious for the catalyst efficiency. The verification of this possibility is the main objective of the present study.
A screening of complex stability in solution over a wide pH range (see below) is preliminary to any other study to rule out pH-promoted demetallation effects that might occur, in acidic solutions, due to competition of ligand protonation with metal coordination and, in alkaline media, for the formation of insoluble metal hydroxides or oxides. A spectrophotometric study (UV-vis) was performed to address this aspect (see below).
Another crucial point is the effectiveness of the anchor group in ensuring a stable attachment of the complex to the surface of the carbon support (MWCNTs). The pyrimidine group used for our catalyst has proven effective for various catalytic processes, despite the non-covalent nature of the interaction with the carbon surface.28,33,34,44–47 In this respect, the presence of two such anchor groups in H2L should provide greater stability to the catalyst.29 Nevertheless, for a stable attachment on the support surface, it is important that the ligand and the complex have an appropriate conformation for a correct “landing” on the support surface during the absorption processes. This means that, in the most stable conformations, both anchor groups should remain on the same side of the macrocyclic ring to avoid energy costs for conformational rearrangements or the possibility of only one anchor group adhering to the support surface. Since the construction of the catalyst occurs in two steps, first the adsorption of the ligand and subsequently the coordination of Pd(II), it is important to address this conformational issue both for the free ligand and for the metal complex. Concerning the free (not complexed) ligand, encouraging information was obtained from crystal structures of various protonated ligand forms (see below). Instead, despite our efforts, we were not able to obtain crystals of the Pd(II) complex suitable for XRD analysis. Crystallographic information was however obtained for the Cu(II) complex of H2L and for the Pd(II) complex of the di-tosylated precursor (1) (Fig. 1) which is structurally rather similar to H2L.
As we will see later, the results demonstrate that both the ligand and its metal complex have the requisites for the formation of a robust catalyst, as indeed it has been proven to be a property which usefully complements its high catalytic efficiency in the ORR.
Ligand protonation and Pd(II) complexation in solution
H2L contains two acidic groups that can undergo deprotonation (the NH groups of the bridges connecting the pyrimidine residues to the macrocycle) and six basic, protonable groups (the 4 macrocyclic N atoms and the two pyrimidine nitroso groups). Potentiometric (pH-metric) titrations performed in aqueous solution (in 0.1 M NMe4Cl, 298.1 ± 0.1 K) accounted for only one deprotonation and four protonation equilibria involving H2L in the pH range of 2–11.5. The relevant equilibrium constants are reported in Table 1.| Equilibrium | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K K |
|---|---|
| HL− + H+ = H2L | 11.58(2) |
| H2L + H+ = H3L+ | 10.50(2) |
| H3L+ + H+ = H4L2+ | 6.40(8) |
| H4L2+ + H+ = H5L3+ | 2.31(8) |
| H5L3+ + H+ = H6L4+ | 2.14(8) |
Information on the protonation/deprotonation sites was obtained from the UV-vis spectra recorded at different pHs (Fig. 2a and b) as the pyrimidine chromophore is very sensitive to these processes. As can be seen, significant spectral variations are observed below pH 3 and above pH 11 when H5L3+ and H6L4+ (pH < 3) and HL− (pH > 11) are formed (Fig. 2c). Consequently, we can deduce that the formation of H5L3+ and H6L4+ involves, respectively, the protonation of one and two nitroso groups of the pyrimidine residues, while the formation of HL− takes place with deprotonation of one NH group of a bridge connecting a pyrimidine residue to the macrocycle. In the intermediate pH region (3–11), the spectra are poorly affected by the protonation equilibria (Fig. 2), indicating that they do not involve the pyrimidine chromophores but the macrocyclic unit. That is, in H3L+ and H4L2+, one and two N atoms of the macrocycle are respectively protonated. This protonation/deprotonation pattern is in agreement with the behaviour previously found for similar molecules,28,33,34,48,49 and it is also consistent with the crystal structures of (H5L)(ClO4)3·2H2O and (H3L)(H4L)(ClO4)3·11H2O, determined by XRD, which show that single- and double-protonation of the macrocyclic ring in H3L+, H4L2+ and H5L3+ occur at the methylated nitrogen atoms and one nitroso group is protonated in H5L3+ (see below). In summary, in the pH range of 2–11.5, only one NH group of the bridges connecting the pyrimidine residues to the macrocycle undergoes deprotonation (the second one is expected to deprotonate at higher pH), both nitroso groups protonate and the macrocycle unit is involved in only two protonation equilibria (two of the amine groups remain unprotonated).
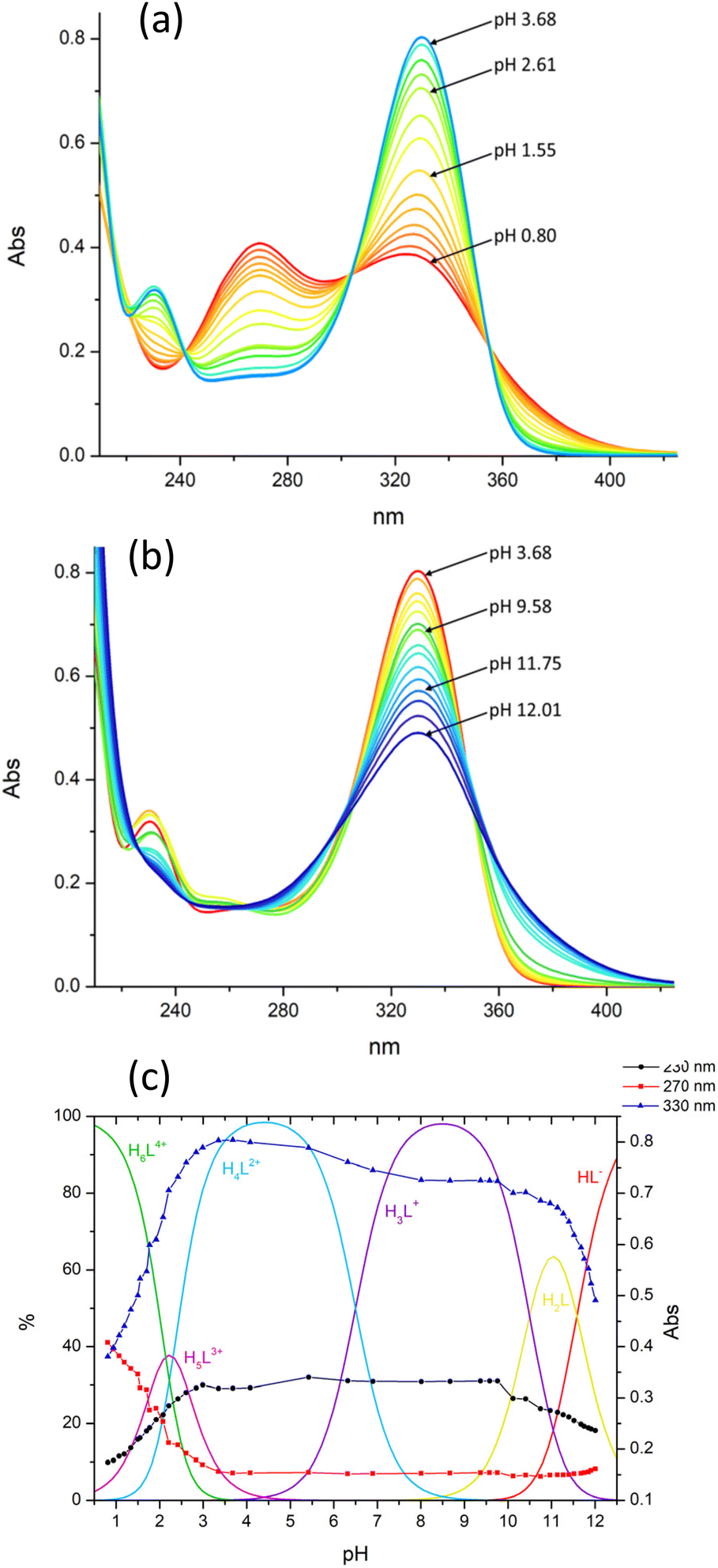 | ||
| Fig. 2 UV-Vis absorption spectra of H2L in the pH ranges 0.8–3.68 (a) and 3.68–12.01 (b). (c) Variation with pH of the 230 nm, 270 nm and 330 nm absorbances superimposed to the distribution diagram of the species formed by H2L. [H2L] = 1.8 × 10−5 M. | ||
The formation of Pd(II) complexes with H2L was followed by means of UV-vis absorption spectroscopy. The complexation reaction is slow; it takes 6 days to reach the invariance of the recorded spectra at pH 4. The UV-vis spectra at different pH values (Fig. 3a and b) were recorded using a solution of the complex formed at pH 4 after changing its pH and monitoring the achievement of equilibrium by UV-vis spectroscopy. The pH attributed to each spectrum was measured once equilibration was achieved. A comparison of these spectra with the spectra of the metal-free ligand (Fig. 3c) shows that the Pd(II) complex is already formed in very acidic solutions (pH 2) and is preserved in alkaline media. The spectra of the complex remained unchanged for several weeks providing evidence of the long-lasting robustness of the complex under different pH conditions.
 | ||
| Fig. 3 UV-Vis absorption spectra of the H2L/Pd(II) system in the pH ranges 1.7–6.2 (a) and 6.2–9.1 (b). (c) Variation with pH of the 230 nm and 330 nm absorbances. [H2L] = [Pd(II)] = 1.8 × 10−5 M. | ||
Crystallographic study
Four crystal structures for the ligand, H2L ((H5L)(ClO4)3·2H2O, (H3L)(H4L)(ClO4)3·11H2O, [Cu(H2L)(H2O)](ClO4)2·10.46H2O and [Cu(H2L)Cl]Cl·3H2O), and one crystal structure for the di-tosylated molecule (1) ([Pd(1)]Cl2·4H2O) were obtained and analysed.The ORTEP-like drawings for the two protonated ligand forms and for the two Cu(II) complexes are shown in Fig. 4. The small macrocyclic moiety is quite rigid and in all structures assumes the same [3333] conformation,58 which features all nitrogen atoms in endo conformation, with convergent lone pairs, and gauche dihedrals for the C–C ethylenic bonds. As a consequence, the acidic hydrogen atoms of the two metal-free structures (Fig. 4a and b), which were localized on the methylated nitrogen atoms using the Fourier difference maps, point towards a water molecule, with which they form H-bonds, located approximately above the centre of the macrocyclic ring. Further H-bonds are formed by this water molecule with the arms of symmetry related ligand molecules. Notably, macrocyclic rigidity prevents conformational changes when one H+ ion is added to H3L+ to give H4L2+, as observed in (H3L)(H4L)(ClO4)3·11H2O. Overall, the two cations (H3L+ and H4L2+) assume the same conformation in the crystal, thus resulting in symmetry equivalence and making it impossible to assign a different degree of protonation. On the other hand, the [3333] macrocyclic conformation preorganizes the donor atoms in a square planar geometry suitable for metal ion coordination and in the two Cu(II) complexes, the cation has an almost perfect square pyramidal coordination environment with the apical position occupied by a water oxygen atom ([Cu(H2L)(H2O)](ClO4)2·10.46H2O) or a chloride anion ([Cu(H2L)Cl]Cl·3H2O) (Table S2†).
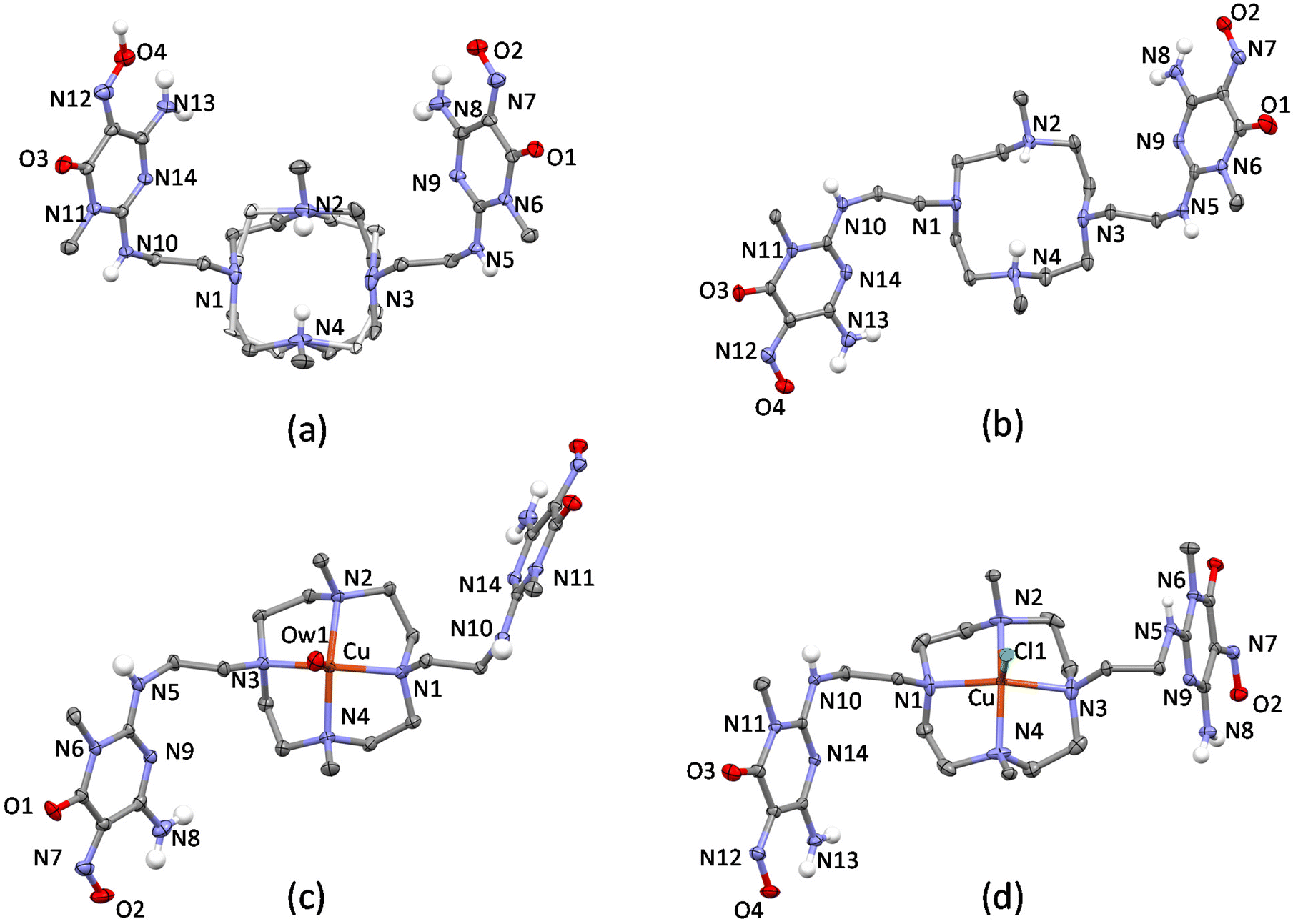 | ||
| Fig. 4 H2L ligand or Cu(H2L)2+ complex in (a) (H5L)(ClO4)3·2H2O (both disordered conformers are shown), (b) (H3L)(H4L)(ClO4)3·11H2O, (c) [Cu(H2L)(H2O)](ClO4)2·10.46H2O and (d) [Cu(H2L)Cl]Cl·3H2O crystal structures (non-polar H atoms are omitted for clarity). | ||
Notably, the [3333] conformation determines the position of the methyl carbon atoms and of the first carbon atoms in the pendant arms, which occupy almost the same position above the mean plane defined by the four nitrogen atoms in all structures. The conformational rigidity given by the small and preorganized macrocycle also partially affects the pendant arms, which have the first C–C dihedrals in the more stable trans conformation. Nevertheless, as shown in Fig. S2,† the rotatable torsions can be affected by environment and packing forces so that while in the more hydrated (H3L)(H4L)(ClO4)3·11H2O and [Cu(H2L)(H2O)](ClO4)2·10.46H2O, H2L assumes very similar conformations, in (H5L)(ClO4)3·2H2O and [Cu(H2L)Cl]Cl·3H2O, the nitroso pyrimidine groups are differently oriented.
In fact, the molecular symmetry of H2L can be analysed in terms of the N–C–C–Namide torsion angular values along the pendant arms. In (H3L)(H4L)(ClO4)3·11H2O and [Cu(H2L)(H2O)](ClO4)2·10.46H2O, the N–C–C–Namide features a ±gauche/trans/−gauche sequence, which is associated with the presence of a 2-fold rotational axis passing through the centre of the N4 plane and normal to it. In (H5L)(ClO4)3·2H2O, the molecular symmetry is better described as Cs, the mirror plane being normal to the N4 plane and containing the two methyl C–N bonds. The N–C–C–Namide dihedral sequence is ±gauche/trans/+gauche, resulting in convergent nitroso groups and in the formation of a supramolecular macrocycle held together by two strong N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–H⋯ON H-bonds. On the other hand, in [Cu(H2L)Cl]Cl·3H2O only one of the pendant arms has the C–C bond in trans conformation, while in the other, the C–C bond is of the gauche type (Fig. 5). These findings evidence that the complexed ligand can have enough mobility to rearrange itself and place the nitroso pyrimidine groups in contact with the surface of carbon nanotubes, while at the same time having the metal centre exposed to the solvent and available for binding or interaction with exogenous species. Interestingly, preliminary molecular dynamics simulations (empirical forcefield method) carried out to study the interaction of the [(H2L)Pd]2+ complex with a graphene slice, as a model for the MWCNT, gave both the nitroso pyrimidine groups stacked on the graphene and gauche/gauche/gauche sequence for the N–C–C–Namide dihedrals (Fig. S3†).
O–H⋯ON H-bonds. On the other hand, in [Cu(H2L)Cl]Cl·3H2O only one of the pendant arms has the C–C bond in trans conformation, while in the other, the C–C bond is of the gauche type (Fig. 5). These findings evidence that the complexed ligand can have enough mobility to rearrange itself and place the nitroso pyrimidine groups in contact with the surface of carbon nanotubes, while at the same time having the metal centre exposed to the solvent and available for binding or interaction with exogenous species. Interestingly, preliminary molecular dynamics simulations (empirical forcefield method) carried out to study the interaction of the [(H2L)Pd]2+ complex with a graphene slice, as a model for the MWCNT, gave both the nitroso pyrimidine groups stacked on the graphene and gauche/gauche/gauche sequence for the N–C–C–Namide dihedrals (Fig. S3†).
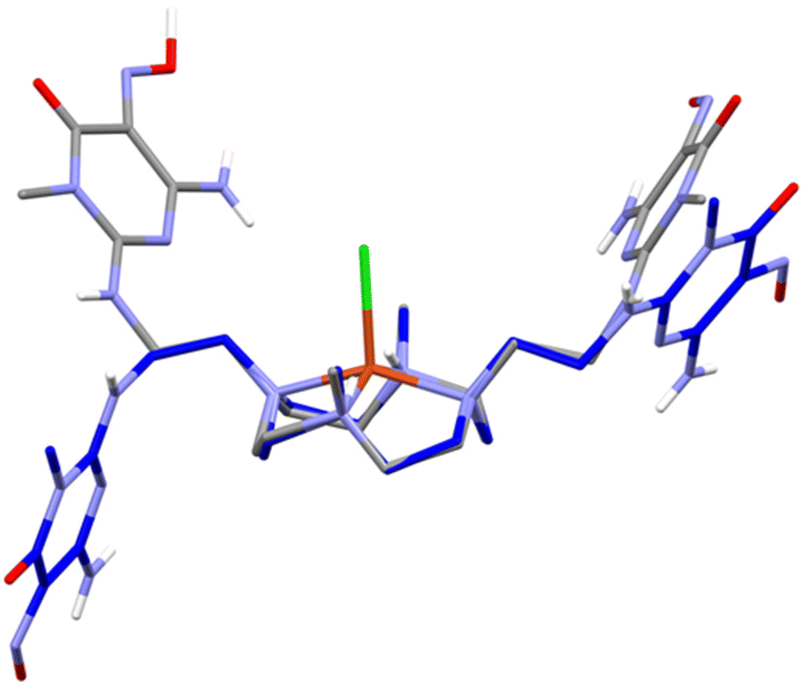 | ||
| Fig. 5 Superposition of the H2L ligands in the crystal structure of (H5L)(ClO4)3·2H2O (gray, only the prevalent conformer is shown) and [Cu(H2L)Cl]Cl·3H2O (blue). | ||
Concerning the crystal structure of [Pd(1)]Cl2·4H2O, the asymmetric unit contains two not symmetry equivalent [Pd(1)]2+ complexes, which differ from each other only for the relative arrangement of the sulfonamide moieties of the pendant arms (Fig. S4†). The ORTEP-like drawing of the two Pd(II) complexes (Fig. 6) clearly shows that the cyclen unit assumes the same [3333] conformation found in the crystals of protonated and Cu(II)-complexed H2L and that Pd(II) and Cu(II) ions are coordinated by the tetraazamacrocycle in an almost equivalent manner. The deviation from planarity of the four nitrogen atoms is at most 0.025 Å in the four metal complexes (two Cu(II) complexes and two not symmetry equivalent Pd(II) complexes). The Cu(II) ions are shifted from the basal plane toward the apical position by 0.4882(6) Å in [Cu(H2L)(H2O)](ClO4)2·10.46H2O and by 0.5417(4) Å in [Cu(H2L)Cl]Cl·3H2O, while in the case of the Pd(II) complexes, the deviations from the mean planes are 0.3835(9) Å (Pd1) and 0.378(1) Å (Pd2). It is worth noting that in both Pd(II) complexes, a pendant arm places its nitrogen atom at about 3 Å from the metal ion (Table S3†), suggesting long-distance occupation of the fifth position of a strongly distorted square pyramid. This hypothesis is confirmed by the position of the amidic hydrogen atom found in the Fourier difference map for one of the two complexes and by the corresponding C–N–S bond angle (116.2(8)°, Fig. 6b), which are in agreement with the presence of a sp3 nitrogen atom with the lone pair available for coordination. This finding is in line with the theoretical calculations35 already mentioned showing that O2 is able to form an apical bond with Pd(II) centres. Indeed, despite the known propensity of Pd(II) to square planar coordination, it is possible to recover almost 150 structures from the Cambridge Structural Database showing a possibly five-coordinated Pd(II) ion, almost half of them showing the metal in a distorted square pyramidal environment. For those having a nitrogen atom in the apical position, the Pd–N bond distances range from 2.2 to 2.9 Å.59
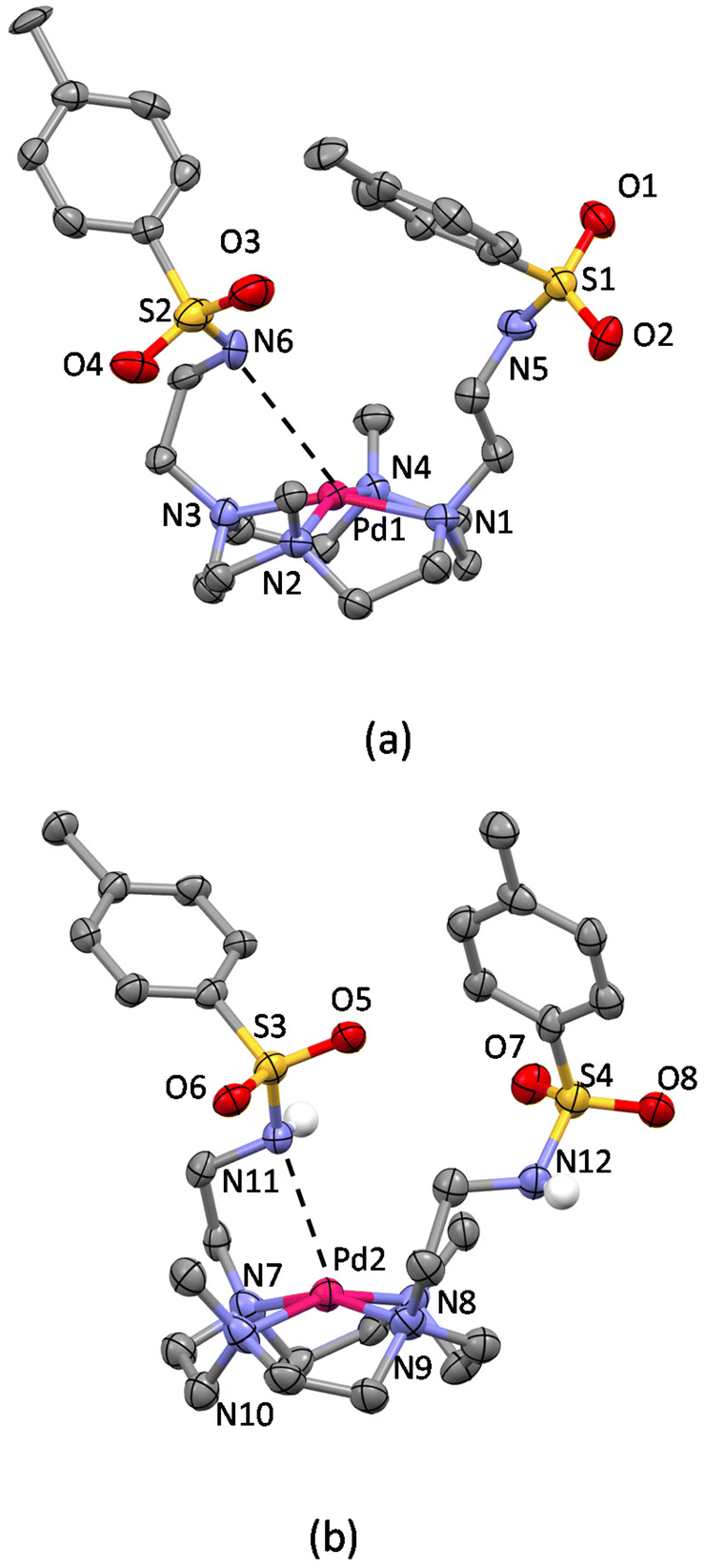 | ||
| Fig. 6 Not symmetry equivalent [Pd(1)]2+ complexes in the asymmetric unit of the [Pd(1)]Cl2·4H2O crystal structure. (a) Mol A and (b) mol B (non-polar hydrogen atoms are omitted for clarity). | ||
SEM and XPS characterization of the catalyst
The SEM images of MWCNT, MWCNT-L and MWCNT-LPd (Fig. S5†) show homogeneous networks of carbon nanotubes with no marked differences in the morphology of the three samples. In the relevant EDS (SEM-energy-dispersive spectroscopy) spectra (Fig. S6†), pristine MWCNTs show only the C signal and a minimal presence of oxygen, in accordance with the specifications provided by the supplier, whereas in the case of MWCNT-L, there is also the N signal, due to the presence of the ligand, and the MWCNT-LPd catalyst shows a Pd loading of about 1.5% by weight in good agreement with the catalyst preparation data (1.71%) reported above. The EDS map of MWCNT-LPd (Fig. 7) shows that Pd is uniformly distributed over the sample, evidencing that the supramolecular approach adopted for the preparation of the catalyst successfully leads to a homogeneous distribution of the catalytic centres. Chloride, which is expected to be present as the counter-ion of coordinated Pd(II) ions, is also detected in the EDS analysis (Fig. 7d) as well as in the XPS spectra (Fig. S7†). | ||
| Fig. 7 EDS mapping of MWCNT-LPd: (a) EDS layered image, (b) C signals, (c) Pd signals, and (d) Cl signals. | ||
XPS analysis of MWCNT-LPd, focused on the Pd region and Pd oxidation state (3d transition of palladium, binding energy from 360 eV to 325 eV), shows that the metal is mainly present as Pd(II). Peak fitting reveals that the sample contains a small amount (ca. 12%) of PdO/C. In Fig. S7b,† the Pd1 component corresponds to Pd(II)/H2L (binding energy 3d5/2 = 338.8 eV, 3d3/2 = 344.1 eV)60 while the Pd2 component corresponds to PdO/C (binding energy 3d5/2 = 336.3 eV, 3d3/2 = 341.5 eV).61,62 No component of the reduced metal Pd(0) was found. Similar behaviour was shown by analogous systems,28 and it is perhaps connected to the preparation method and/or reducing character of the leftover C impurities or Pd interactions with surface defects.34
Electrochemical evaluation of the catalytic performance in the ORR
The evaluation of the catalytic activity of electrocatalysts in the ORR was carried out with reference to the three parameters: the onset potential (Eon), the half-wave potential (E1/2) and the number of exchanged electrons (n).63 The onset potential represents the limit value at which the ORR starts to evolve in significant current density values. In this work, the onset potential is defined as the potential at which the current density reaches a value of −0.1 mA cm−2 (black dotted line, Fig. 8). Note that a more positive potential corresponds to a lower barrier for the ORR.64 The half-wave potential (E1/2) is the potential to reach half the limiting current density obtained from the LSV curve, and it is a widely accepted indicator for evaluating the ORR performance.65,66 Indeed, the E1/2 in the mixed controlled region of LSV curves reflects the coupling behaviour of surface reaction and mass diffusion.63 The third important parameter, the number of exchanged electrons (n), allows the assessment of the principal reaction pathway in the ORR. A catalyst should preferably promote a four-electron exchange leading to the formation of H2O and prevent a two-electron exchange producing H2O2 to avoid the negative influence of the latter on the yields of the ORR process and the corrosive effects it has on the membrane used in fuel cells.67,68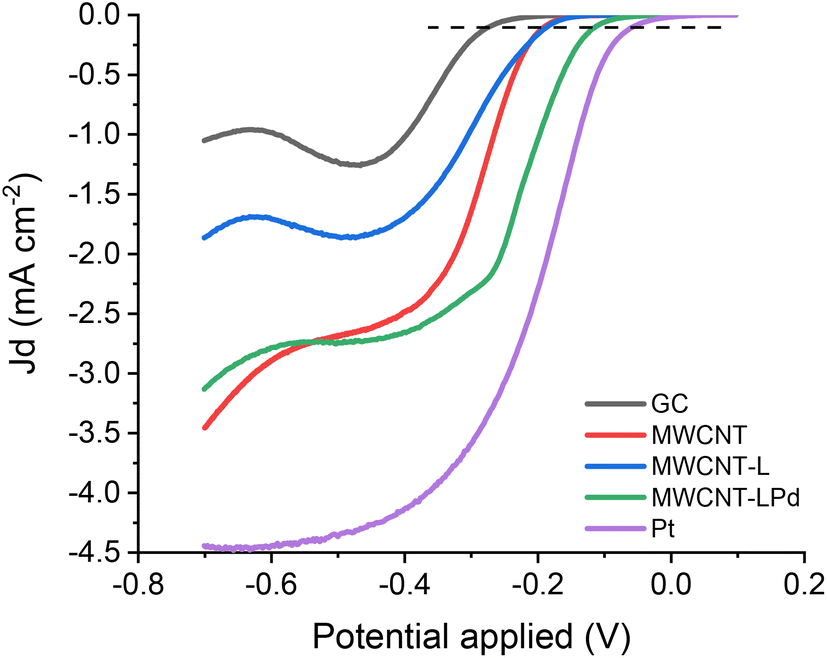 | ||
| Fig. 8 ORR electrocatalytic performance, LSV 1600 rpm rotation rate, and 5 mV s−1 scan rate in KOH 0.1 M O2 saturated solution. RRDE disk current densities of the modified GC electrode and the benchmark electrode. The horizontal dotted line at a current density of −0.1 mA cm−2 shows the onset potential of samples. | ||
We studied the electrochemical behaviour of MWCNT-LPd by cyclic voltammetry in 0.1 M KOH purged with either N2 or O2. Cyclic voltammograms (CVs) of the modified electrode are shown in Fig. S8.† In all cases, a sharp and irreversible peak of oxygen reduction is observed in the presence of O2, which is not present when the environment is saturated with N2. The ORR peak current signal shows a higher density peak current with Pd-containing catalysts, confirming the catalytic activity of this complexed metal ion. CVs of MWCNT-L and MWCNT-LPd also show a redox couple at the potential of −0.3 V and −0.4 V, respectively, which is present both in O2 and N2 purged solutions, while only capacitive currents are observed in the same potential range for MWCNTs and the benchmark GC electrode (compare Fig. S8a–d†). The redox couple of the H2L ligand may play an important role in the ORR electron transfer phenomena,35 and the influence of the redox couple on the electron transfer involved in the ORR process is still being studied.
Fig. 8 shows the ORR polarization curves recorded at 1600 rpm for each sample at a potential scan rate of 5 mV s−1 in a KOH 0.1 M oxygen saturated solution. The polarization curves for the bare GC electrode, Pt electrode, as well as the GC electrode functionalized with pristine MWCNTs and MWCNT-L (same functionalization method) are also reported. The horizontal dotted line at the current density of −0.1 mA cm−2 shows the onset potential of the samples.
Table 2 provides the electrochemical results for all systems.
| Electrode |
E
ona (V) |
E
1/2b (V) |
n | % H2O2d |
|---|---|---|---|---|
| a Onset potential vs. Ag/AgCl KCl sat. b Half-wave potential calculated at the half of the limiting current density obtained from the LSV curve. c Number of exchanged electrons per O2 molecule, the value at −0.35 V calculated with an experimentally determined collection efficiency number (reported in Fig. S10†). d Percentage of hydrogen peroxide calculated from the value of n. | ||||
| GC | −0.276 | −0.354 | 2.16 | 91.7 |
| MWCNT | −0.191 | −0.284 | 2.43 | 78.31 |
| MWCNT-H2L | −0.187 | −0.298 | 2.07 | 96.6 |
| MWCNT-H2L-Pd(II) | −0.112 | −0.222 | 3.70 | 15.0 |
| Pt | −0.059 | −0.198 | 3.79 | 10.7 |
As displayed in Fig. 8, the catalytic activities, in terms of both Eon and current values, increase depending on the catalyst nature in the order: bare GC < pristine MWCNTs ≈ MWCNT-L < MWCNT-LPd < bare Pt electrode. This evidence rules out significant catalytic properties of the MWCNT-supported substrate and ligands, while highlighting the prominent role of Pd(II) in enhancing the catalytic activity. The Eon values observed for MWCNT-LPd are close to the benchmark values obtained under the same conditions with a commercial Pt working electrode: the Eon value for MWCNT-LPd is only 53 mV less positive than that observed for the Pt electrode (Table 2). The same trend is followed by the half-wave potentials (E1/2, Table 2).
In order to provide the number of exchanged electrons and investigate the reaction pathway of the ORR during the RRDE LSV measurements, the Pt ring electrode was held at a potential of +0.50 V to ensure the complete oxidation of H2O2 eventually produced at the modified GC disk electrode. The ring disk current densities in N2 and O2 saturated KOH solutions are shown in Fig. S9.† Background currents (under N2 saturated conditions) have been used to account for capacitive current contribution.
RRDE calibration with K3Fe(CN)6 was used to determine the experimental collection efficiency (N) for each modified GC electrode, so that accurate values for the number of exchanged electrons could be derived.69 Fig. S10† shows the LSV ring-disk current; the collection efficiency values are very close to the theoretical value (38.3%). The number of exchanged electrons per O2 molecule (n) as a function of the potential applied at the RRDE working electrode is presented in Fig. 9. As can be seen in this figure, this number (n) is very close to 4 for the MWCNT-LPd catalyst, especially in the onset potential region, signifying an almost exclusive conversion of O2 into H2O.
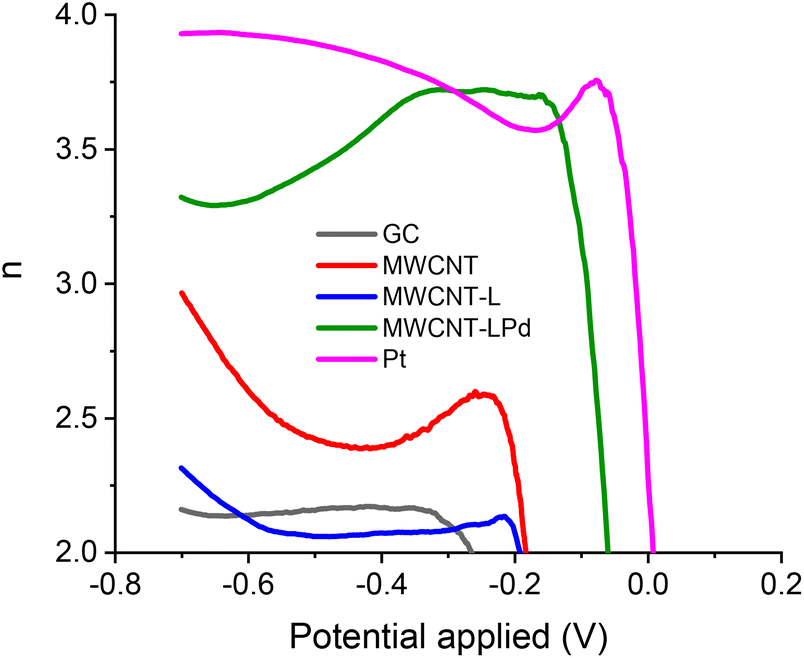 | ||
| Fig. 9 Dependency of the RRDE number of exchanged electrons per O2 molecule vs. the LSV scanning potential values. | ||
The measured ring and disk currents for MWCNT-LPd at the suggested potential of −0.35 V demonstrate that, under the said conditions, the direct conversion to H2O reaches 85.0%, compared to 89% of the Pt electrode (Table 2).
Quite different results are instead obtained in the case of the GC electrode used as a benchmark, MWCNT and MWCNT-L, the latter giving an exchanged electron number close to 2, thus a predominant formation of hydrogen peroxide.
Further characterization of the half-cell with MWCNT-LPd was performed by using the RRDE in galvanostatic mode to simulate a working fuel cell at a fixed potential. The modified GC disk electrode potential was held at −0.60 V and the ring electrode was held at +0.50 V, respectively, for 600 s at 1600 rpm. The galvanostatic results shown in Fig. S11† also provide a short-time stability test. The ring-disk current density values remain constant during the analysis, indicating the robustness of the catalyst. The time-averaged number of exchanged electrons per O2 molecule shows that direct conversion to water is the main process during the evolution of time. Nevertheless, further investigation is needed to evaluate the long-term stability of the catalyst for fuel-cell applications.
Conclusions
The main objective of this work was to ascertain whether the ability of Pd(II) to function as a catalyst in the oxygen reduction reaction (ORR) in alkaline aqueous solutions is strictly connected to the presence of readily available coordination sites in the square planar coordination geometry typical of this metal ion. This objective is intended as an experimental verification of previous theoretical results showing that Pd(II) can still function as an efficient catalyst for ORR through the axial interaction of O2 with metal centres.35 Beyond its conceptual value, this objective has great relevance for the design and synthesis of ligands used to anchor Pd(II) in heterogeneous catalysts. The possibility of using macrocyclic ligands that saturate the square planar coordination environment typical of this metal ion, without the need to preserve coordination sites occupied by labile donors, greatly facilitates the preparation of these ligands and allows for stronger coordination of the metal ion with consequent enhancement of the catalyst robustness.The results obtained in the present work using a Pd(II) complex anchored on MWCNTs in which the square planar coordinative geometry of the metal ion is saturated by the four nitrogen atoms of a tetraazacycloalkane leave no room for doubt about the ability of Pd(II) to function very well as a catalyst in the ORR even under similar conditions. Indeed, the new MWCNT-LPd catalyst has an onset potential (Eon) which is only 53 mV less positive than that of a commercial Pt electrode, and the same trend is followed by the half-wave potentials (E1/2 is only 24 mV less positive than that observed for the Pt electrode). The number of exchanged electrons per O2 molecule is very close to 4, especially in the onset potential region, denoting an almost exclusive conversion of O2 into H2O. Ring and disk currents measured at the potential of −0.35 V demonstrate that, under the said conditions, the direct conversion to H2O reaches 85.0%, compared to 89% of the Pt electrode. Furthermore, a short-term test (600 s) conducted on the half-cell with MWCNT-LPd in galvanostatic mode to simulate a fuel cell operating at a fixed potential demonstrated that the catalyst is very robust in this time interval: the current density remained constant during the analysis and the time-averaged number of electrons exchanged per O2 molecule showed that the direct conversion to water was maintained throughout. Remarkably, these results were obtained with a heterogeneous catalyst containing only 1.72% by weight of the precious metal (Pd) homogeneously distributed over the surface of the solid support (MWCNTs).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Project 2022NW4P2T CUP B53D23013890006 “From metal nanoparticles to molecular complexes in electrocatalysis for green hydrogen evolution and simultaneous fine chemicals production” PRIN2022 Progetti di Ricerca di Rilevante Interesse Nazionale funded by the Italian Ministry of University and Research and NextGenerationEU is acknowledged.References
- Fuel Cell Technology Handbook, ed. G. Hoogers and F. Kreith, CRC Press, Boca Raton, 2002 Search PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- X. He, S. Minelli, A. Vertova and A. Minguzzi, Curr. Opin. Electrochem., 2022, 36, 101166 CrossRef CAS.
- J. Hou, M. Yang, C. Ke, G. Wei, C. Priest, Z. Qiao, G. Wu and J. Zhang, EnergyChem, 2020, 2, 100023 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- F. Nosheen, T. Anwar, A. Siddique and N. Hussain, Front. Chem., 2019, 7, 456 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- M. Kiani, X. Q. Tian and W. Zhang, Coord. Chem. Rev., 2021, 441, 213954 CrossRef CAS.
- X. Huang, Y. Wang, W. Li and Y. Hou, Sci. China: Chem., 2017, 60, 1494–1507 CrossRef CAS.
- H. A. Miller, M. Bevilacqua, J. Filippi, A. Lavacchi, A. Marchionni, M. Marelli, S. Moneti, W. Oberhauser, E. Vesselli, M. Innocenti and F. Vizza, J. Mater. Chem. A, 2013, 13337–13347 RSC.
- P. Jiao, D. Ye, C. Zhu, S. Wu, C. Qin, C. An, N. Hu and Q. Deng, Nanoscale, 2022, 14, 14322–14340 RSC.
- M. D. Bhatt and J. Y. Lee, Energy Fuels, 2020, 34, 6634–6695 CrossRef CAS.
- X. Li, G. Liu, H. Zheng, K. Sun, L. Wan, J. Cao, S. Asif, Y. Cao, W. Si, F. Wang and A. Bokhari, Energies, 2023, 16, 128 CrossRef CAS.
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, Chem, 2018, 4, 106–123 CAS.
- G. Tuci, C. Zafferoni, A. Rossin, A. Milella, L. Luconi, M. Innocenti, L. Truong Phuoc, C. Duong-Viet, C. Pham-Huu and G. Giambastiani, Chem. Mater., 2014, 26, 3460–3470 CrossRef CAS.
- Z. Yan, L. Gao, C. Dai, M. Zhang, X. Lv and P. K. Shen, Int. J. Hydrogen Energy, 2018, 43, 3705–3715 CrossRef CAS.
- (a) L. Yan, P. Li, Q. Zhu, A. Kumar, K. Sun, S. Tian and X. Sun, Chem, 2023, 9, 280–342 CrossRef CAS; (b) A. Kumar, S. Ibraheem, T. A. Nguyen, R. K. Gupta, T. Maiyalagan and G. Yasin, Coord. Chem. Rev., 2021, 446, 214122 CrossRef CAS; (c) A. Kumar, Y. Zhang, W. Liu and X. Sun, Coord. Chem. Rev., 2020, 402, 213047 CrossRef CAS; (d) A. Kumar, V. K. Vashistha and D. K. Das, Coord. Chem. Rev., 2021, 431, 213678 CrossRef CAS.
- M. Wang, H. Zhang, Y. Liu and Y. Pan, J. Energy Chem., 2022, 72, 56–72 CrossRef CAS.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- B. Singh, V. Sharma, R. P. Gaikwad, M. B. Gawande, P. Fornasiero, R. Zboril and R. Zboril, Small, 2021, 17, e2006473 CrossRef PubMed.
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 CrossRef.
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Catal. Today, 2015, 248, 3–9 CrossRef CAS.
- A. Goti and F. Cardona, Hydrogen peroxide in green oxidation reactions: Recent catalytic processes, in Green Chemical Reactions, ed. P. Tundo and V. Esposito, Springer, Berlin/Heidelberg, 2008, pp. 191–212 Search PubMed.
- A. Gómez-Marín, J. Feliu and T. Edson, ACS Catal., 2018, 8, 7931–7943 CrossRef.
- L. Gubler, S. M. Dockheer and W. H. Koppenol, J. Electrochem. Soc., 2011, 158, B755 CrossRef CAS.
- M. Passaponti, M. Savastano, M. P. Clares, M. Inclán, A. Lavacchi, A. Bianchi, E. García-España and M. Innocenti, Inorg. Chem., 2018, 57, 14484–14488 CrossRef CAS PubMed.
- US Department of Energy: Office of Energy Efficiency and Renewable Energy. https://www.energy.gov/eere/fuelcells/doe-technical-targets-polymer-electrolyte-membrane-fuel-cell-components.
- M. Savastano, M. Passaponti, W. Giurlani, L. Lari, A. Bianchi and M. Innocenti, Energies, 2020, 13, 5539 CrossRef CAS.
- (a) M. Savastano, M. Passaponti, W. Giurlani, L. Lari, N. Calisi, E. Delgado-Pinar, E. S. Serrano, E. Garcia-España, M. Innocenti, V. K. Lazarov and A. Bianchi, Inorg. Chim. Acta, 2021, 518, 120250 CrossRef CAS; (b) M. Savastano, C. Zoppi, A. Bianchi and C. Bazzicalupi, Inorg. Chim. Acta, 2020, 511, 119793 CrossRef CAS.
- Q. An, S. Bo, J. Jiang, C. Gong, H. Su, W. Cheng, Q. Liu and W. Cheng, Adv. Sci., 2023, 10, 2205031 CrossRef CAS PubMed.
- R. Majee, S. Parvin, Q. A. Islam, A. Kumar, B. Debnath, S. Mondal, S. Bhattacharjee, S. Das, A. Kumar and S. Bhattacharyya, Chem. Rec., 2022, 22, 202200070 CrossRef PubMed.
- W. Xu, Y. Sun, J. Zhou, M. Cao, J. Luo, H. Mao, P. Hu, H. Gu, H. Zhai, H. Shang and Z. Cai, Nano Res., 2023, 16, 2294–2301 CrossRef CAS.
- M. Savastano, P. Arranz-Mascarós, C. Bazzicalupi, M. P. Clares, M. L. Godino-Salido, M. D. Gutiérrez-Valero, M. Inclán, A. Bianchi, E. García-España and R. López-Garzón, J. Catal., 2017, 353, 239–249 CrossRef CAS.
- (a) A. M. Valbuena-Rus, M. Savastano, P. Arranz-Mascarós, C. Bazzicalupi, M. P. Clares, M. L. Godino-Salido, M. D. Gutiérrez-Valero, M. Inclán, A. Bianchi, E. García-España and R. López-Garzón, Inorg. Chem., 2022, 61, 12610–12624 CrossRef CAS PubMed; (b) M. Savastano, P. Arranz-Mascarós, M. P. Clares, R. Cuesta, M. L. Godino-Salido, L. Guijarro, M. D. Gutiérrez-Valero, M. Inclán, A. Bianchi, E. García-España and R. López-Garzón, Molecules, 2019, 24, 2714 CrossRef CAS PubMed.
- M. Bonechi, W. Giurlani, M. Vizza, M. Savastano, A. Stefani, A. Bianchi, C. Fontanesi and M. Innocenti, Catalysts, 2021, 11, 764 CrossRef CAS.
- N. J. Low, M. D. López, P. Arranz, J. Cobo, M. L. Godino, R. López, M. D. Gutiérrez, M. Melguizo, G. Ferguson and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 882–892 CrossRef PubMed.
- M. Savastano, V. Monini, C. Bazzicalupi and A. Bianchi, Inorganics, 2022, 10, 12 CrossRef CAS.
- C. Bazzicalupi, A. Bianchi, T. Biver, C. Giorgi, S. Santarelli and M. Savastano, Inorg. Chem., 2014, 53, 12215–12224 CrossRef CAS PubMed.
- M. Fontanelli and M. Micheloni, Proceedings of the Ist Spanish-Italian Congress on Thermodynamics of Metal Complexes. Diputación de Castellón, Castellón, Spain, 1990, pp. 41–43.
- M. Savastano, M. Fiaschi, G. Ferraro, P. Gratteri, P. Mariani, A. Bianchi and C. Bazzicalupi, Molecules, 2020, 25, 1355 CrossRef CAS PubMed.
- G. Gran, Analyst, 1952, 77, 661–671 RSC.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- J. García-Martín, R. López-Garzón, M. L. Godino-Salido, M. D. Gutiérrez-Valero, P. Arranz-Mascarós, R. Cuesta and F. Carrasco-Marín, Langmuir, 2005, 21, 6908–6914 CrossRef.
- Experiments in Analytical Electrochemistry The Care and Feeding of Electrodes. https://www.semanticscholar.org/paper/Experiments-in-Analytical-Electrochemistry-The-Care/1d6b28acd37f31f270fdf6da081a369b7bb9c176.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2020 Search PubMed.
- M. Passaponti, L. Rosi, M. Savastano, W. Giurlani, H. A. Miller, A. Lavacchi, J. Filippi, G. Zangari, F. Vizza and M. Innocenti, J. Power Sources, 2019, 427, 85–90 CrossRef CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, ed. X. Wei, G. Yin and J. Zhang, Elsevier, 2014 Search PubMed.
- C. Song and J. Zhang, Electrocatalytic Oxygen Reduction Reaction, in PEM Fuel Cell Electrocatalysts and Catalyst Layers, ed. J. Zhang, Springer, London, 2008 Search PubMed.
- G. Maccaferri, C. Zanardi, Z. Y. Xia, A. Kovtun, A. Liscio, F. Terzi, V. Palermo and R. Seeber, Carbon, 2017, 120, 165–175 CrossRef CAS.
- C. Borri, N. Calisi, E. Galvanetto, N. Falsini, F. Biccari, A. Vinattieri, G. Cucinotta and S. Caporali, Nanomaterials, 2020, 10, 60 CrossRef CAS PubMed.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- SADABS, Bruker AXS Inc., Madison (WI), USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- R. López-Garzón, M. L. Godino-Salido, M. D. Gutiérrez-Valero, P. Arranz-Mascarós, M. Melguizo, C. García, M. Domingo-García and F. J. López-Garzón, Inorg. Chim. Acta, 2014, 417, 208–221 CrossRef.
- J. Dale, Acta Chem. Scand., 1973, 27, 1115–1129 CrossRef CAS.
- (a) T. J. Hubin, N. W. Alcock and D. H. Busch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1404–1406 CrossRef; (b) M. Rimoldi, F. Ragaini, E. Gallo, F. Ferretti, P. Macchi and N. Casati, Dalton Trans., 2012, 41, 3648–3658 RSC; (c) M. Kujime, S. Hikichi and M. Akita, Chem. Lett., 2003, 32, 486–487 CrossRef CAS; (d) J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Angew. Chem., Int. Ed., 2011, 50, 5532–5536 CrossRef CAS PubMed; (e) K. Wieghardt, E. Schoffmann, B. Nuber and J. Weiss, Inorg. Chem., 1986, 25, 4877–4883 CrossRef CAS; (f) T. Sakai, Z. Taira, S. Yamazaki and T. Ama, Polyhedron, 1989, 8, 1989–1993 CrossRef CAS; (g) E. C. Constable, S. M. Elder, J. Healy, M. D. Ward and D. A. Tocher, J. Am. Chem. Soc., 1990, 112, 4590–4592 CrossRef CAS; (h) J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Inorg. Chem., 2014, 53, 13112–13129 CrossRef PubMed; (i) M. Kujime, S. Hikichi and M. Akita, Organometallics, 2001, 20, 4049–4060 CrossRef CAS; (j) K. Yamamoto, J. Li, J. A. O. Garber, J. D. Rolfes, G. B. Boursalian, J. C. Borghs, C. Genicot, J. Jacq, M. van Gastel, F. Neese and T. Ritter, Nature, 2018, 554, 511–514 CrossRef CAS PubMed; (k) J. Luo, N. P. Rath and L. M. Mirica, Organometallics, 2013, 32, 3343–3353 CrossRef CAS; (l) M. Broring and C. D. Brandt, Chem. Commun., 2003, 2156–2157 RSC.
- NIST https://srdata.nist.gov/xps/XPSDetailPage.aspx?AllDataNo=28665#Citation.htm.
- NIST https://srdata.nist.gov/xps/XPSDetailPage.aspx?AllDataNo=29100.
- NIST https://srdata.nist.gov/xps/XPSDetailPage.aspx?AllDataNo=66751.
- J. Wang, C.-X. Zhao, J.-N. Liu, D. Ren, B.-Q. Li, J.-Q. Huang and Q. Zhang, Nano Mater. Sci., 2021, 3, 313–318 CrossRef CAS.
- Y.-F. Xia, P. Guo, J.-Z. Li, L. Zhao, X.-L. Sui, Y. Wang and Z.-B. Wang, iScience, 2021, 24, 103024 CrossRef CAS PubMed.
- A. Brouzgou, S. Song, Z.-X. Liang and P. Tsiakaras, Catalysts, 2016, 6, 159 CrossRef.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2021, 143, 9423–9428 CrossRef CAS PubMed.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 17832–17852 CrossRef CAS PubMed.
- M. Passaponti, L. Lari, M. Bonechi, F. Bruni, W. Giurlani, G. Sciortino, L. Rosi, L. Fabbri, M. Vizza, V. K. Lazarov, C. Fontanesi and M. Innocenti, Energies, 2020, 13, 5646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data, refinement parameters, and selected bond distances and angles; superimposition of ligands and complexes in crystal structures; MD simulation of the graphene–[Pd(H2L)]2+ interaction; SEM, SEM-EDS and XPS data; CV and LSV diagrams; galvanostatic RRDE data. CCDC 2309459–2309463. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03947a |
| This journal is © The Royal Society of Chemistry 2024 |