
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Half-sandwich Ni(II) complexes bearing enantiopure bidentate NHC-carboxylate ligands: efficient catalysts for the hydrosilylative reduction of acetophenones†
Jorge
Sanz-Garrido
 ,
Avelino
Martin
,
Camino
González-Arellano
* and
Juan C.
Flores
*
,
Avelino
Martin
,
Camino
González-Arellano
* and
Juan C.
Flores
*
Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Río”, Universidad de Alcalá, Campus Universitario, 28871 Alcalá de Henares, Madrid, Spain. E-mail: camino.gonzalez@uah.es; juanc.flores@uah.es
First published on 15th December 2023
Abstract
Chiral nickel complexes containing NHC-carboxylate chelate ligands derived from the (S)-isomeric form of amino acids have been synthesised from the corresponding imidazolium salt and nickelocene. The presence of the carboxylate on the N-side arm of the heterocycle results in the competing formation of mixtures of mono- and bis-NHC complexes (i.e., [Ni(η5-Cp)(κ2-C,O-NHC)] and [Ni(κ2-C,O-NHC)2]), both of which retain the (S)-configuration of the stereogenic center and which can be separated by chromatography. Both the 18e− and 16e− complexes are found to be very stable and cannot be interconverted. The composition of the resulting mixtures depends mainly on the entity of the amino acid residue and, of more practical interest, on the reaction conditions. Thus, microwave heating and MeCN as a solvent favor the formation of the half-sandwich nickel complexes, rather than the bis-NHC compounds. Some of the [Ni(η5-Cp)(κ2-C,O-NHC)] complexes turn out to be among the best nickel catalysts for the hydrosilylative reduction of p-acetophenones described to date, although without chiral induction, in the absence of activating additives and under mild catalytic conditions.
Introduction
The superb expansion of N-heterocyclic carbene (NHC) chemistry that followed the seminal isolation of a stable crystalline carbene by Arduengo three decades ago,1–3 continues to provide new breakthroughs at a remarkable rate, with great recent advances of interest for a broad research domain.4 Indeed, their impact in organometallic chemistry means that this type of ligand can be considered to form part of the select and small group of versatile and broadly catalytically useful ligands that also includes cyclopentadienyls or phosphines.Amongst other major fields in which the properties of NHCs have found great practical importance,2–4 catalysis stands out as perhaps the most well-known.4,5 The widespread use of second- and third-row transition metals in many catalytic processes, ranging from C–H activation and cross-coupling reactions to olefin metathesis, has concentrated the development of NHC chemistry on platinum group and heavier coinage metals, thus resulting in new generations of improved catalysts for many of these reactions.3,6 More recently, and coinciding with the increasing awareness regarding the preferred use of 3d earth-abundant metals instead of less sustainable precious metals, much attention has been focused on the study of NHC complexes of metals such as Fe, Co, Ni or Cu.7 Given that they are more readily available and less toxic than their heavier counterparts, the specific features of first-row metals (stabilization of lower oxidation states and coordination numbers, frequent higher reactivity and tendency to form radicals, etc.) open up novel mechanistic possibilities that allow new synthetic transformations. In particular, nickel has offered interesting perspectives in recent years, for instance by exhibiting an extraordinary ability to bind and activate unsaturated molecules or to promote challenging cross-coupling reactions.8
Within the realm of NHC-nickel compounds,7b,9 NHC/cyclopentadienyl hybrid complexes of formula [Ni(Cp)(NHC)X] comprise a predominant class. The first examples thereof were reported by Cowley and Jones,10 followed by others from the groups of Shen and Nolan, who started to explore their use as catalysts.11 Pietrzykowsk, Royo, and Buchowicz, and coworkers,12 have also published widely in this field, with the research team of Ritleng and Chetcuti being perhaps the most prolific.13 This topic was reviewed by Buchowicz in 2019.14
Despite the considerable number of [Ni(Cp)(NHC)X] complexes reported to date, to the best of our knowledge only a few have been obtained as either racemic mixtures of chiral compounds containing Cp-NHC chelate ligands,12c,15 or enantiopure compounds tethering stereogenic centers on the N-side arm (Chart 1).16 We have recently detected the formation of a complex of this type, bearing a chiral κ2-C,O bidentate NHC-carboxylate ligand with an arm derived from (S)-valine, as a byproduct (Chart 1).17
 | ||
| Chart 1 | ||
After deciding to investigate complexes of a similar nature, herein we disclose the synthesis and characterization of a family of nickel complexes containing NHC-carboxylate chelate ligands, namely [Ni(η5-Cp)(κ2-C,O-NHC)], which have been obtained enantiomerically pure by using NHCs based on the (S)-isomeric form of amino acids, and the catalytic behavior thereof in the hydrosilylation of phenones.
Results and discussion
NHC-ligand precursors 1a–h were obtained as zwitterionic forms by adapting the multicomponent and straightforward synthesis reported by Baslé and Mauduit for 1b (Scheme 1). The one-pot procedure is described to be efficient for a non-symmetrical N,N-substitution pattern of NHCs tethering a carboxylic function, which are otherwise inaccessible, in which the use of cheap and available enantiopure amino acids gives access to chiral imidazolium derivatives, as demonstrated with the isolation of hexafluorophosphate salts of protonated 1a–c, the corresponding for R = tBu, and dipolar ion 1b.18 Gratifyingly, we found this strategy to be successful beyond the involvement of alkyl α-functionalized amino acids (R = Me (a), iPr (b), iBu (c), sBu (d)), and widened the scope to aromatic (R = Ph (e), Bn(f)) and sulfide (R = CH2SMe (g), (CH2)2SMe (h)) functional groups. The selectivity for the desired hetero-disubstituted salts 1a–h, as opposed to formation of their two possible homo-disubstituted counterparts, is above the statistically expected value (63–79%; established by 1H NMR inspection of the crude reaction mixture) and in the range of those reported by Baslé and Mauduit, with some general improvement in yield (up to 70%) after workup to isolate the zwitterions. | ||
| Scheme 1 Synthesis of chiral zwitterionic imidazolium salts 1a–h, and their corresponding selectivity (isolated yield) %. | ||
The procedure first described by Cowley10b was tested for the synthesis of the nickel complexes (method A, scheme in Table 1). Thus, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acid-base reaction between zwitterions 1 and nickelocene in warm acetonitrile led to monocarbene complexes 2 when starting from alanine (1a), phenylglycine (1e), methylcysteine (1g) and methionine (1h) derivatives (entries 1, 11, 17 and 19, respectively; Table 1). In contrast, imidazolium salts containing valine (1b), leucine (1c) and isoleucine (1d) residues gave the excepted compounds 2b–d together with different amounts (10–32%, according to 1H-NMR spectra of the crude reaction mixtures) of the corresponding biscarbenes 3b–d (entries 3, 6 and 9), whilst no reaction was observed with the phenylalanine compound 1f after several days (entry 12). The synthesis was also tested with 1b, 1c and 1f in THF as solvent. In this case, the selectivity changed to give 3b as a single product (entry 4) and a greater proportion of 3c (entry 7 vs. 6), whereas 1f remained unreactive in this solvent (entry 13).
:1 acid-base reaction between zwitterions 1 and nickelocene in warm acetonitrile led to monocarbene complexes 2 when starting from alanine (1a), phenylglycine (1e), methylcysteine (1g) and methionine (1h) derivatives (entries 1, 11, 17 and 19, respectively; Table 1). In contrast, imidazolium salts containing valine (1b), leucine (1c) and isoleucine (1d) residues gave the excepted compounds 2b–d together with different amounts (10–32%, according to 1H-NMR spectra of the crude reaction mixtures) of the corresponding biscarbenes 3b–d (entries 3, 6 and 9), whilst no reaction was observed with the phenylalanine compound 1f after several days (entry 12). The synthesis was also tested with 1b, 1c and 1f in THF as solvent. In this case, the selectivity changed to give 3b as a single product (entry 4) and a greater proportion of 3c (entry 7 vs. 6), whereas 1f remained unreactive in this solvent (entry 13).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | R | Solvent | Method | Selectivity 2:3b |
Yield (%) 2/3c |
|
a Reactions 1:1 with 1 mmol of 1 and NiCp2 in 40 mL of solvent for Method A, or 0.8 mmol of 1 and NiCp2 in 4 mL for Method B.
b Determined by 1H NMR spectroscopy of the crude reaction mixture and based on 1.
c Purified/separated by column chromatography using AcOEt as eluent and based on 1.
d Selectivity 2:3 = 39:91 at r.t.
e Selectivity 2b:3b = 75:25 in 100 instead of 40 mL of solvent.
f Selectivity 2b:3b = 85:15 adding 1b in three portions.
g Reaction time 1 h.
h Purified by washing with cold Et2O.
|
||||||
| 1 | 1a | Me | MeCN | A | 100:0 |
43/— |
| 2 | 1a | Me | MeCN | B | 100:0 |
68/— |
| 3d,e,f | 1b | iPr | MeCN | A | 68:32 |
37/25 |
| 4 | 1b | iPr | THF | A | 0:100 |
—/44 |
| 5 | 1b | iPr | MeCN | B | 85:15 |
40/19 |
| 6 | 1c | iBu | MeCN | A | 72:28 |
56/0 |
| 7 | 1c | iBu | THF | A | 67:33 |
29/20 |
| 8 | 1c | iBu | MeCN | B | 100:0 |
86/— |
| 9 | 1d | sBu | MeCN | A | 90:10 |
41/5 |
| 10 | 1d | sBu | MeCN | B | 81:19 |
54/15 |
| 11 | 1e | Ph | MeCN | A | 100:0 |
30/— |
| 12 | 1f | CH2Ph | MeCN | A | —:— |
—/— |
| 13 | 1f | CH2Ph | THF | A | —:— |
—/— |
| 14 | 1f | CH2Ph | MeCN | B | 63:37 |
nd |
| 15g | 1f | CH2Ph | MeCN | B | 87:13 |
46/12 |
| 16 | 1f | CH2Ph | THF | B | 25:75 |
25/30 |
| 17 | 1g | CH2SMe | MeCN | A | 100:0 |
68/—h |
| 18 | 1g | CH2SMe | MeCN | B | 100:0 |
72/—h |
| 19 | 1h | (CH2)2SMe | MeCN | A | 100:0 |
69/—h |
| 20 | 1h | (CH2)2SMe | MeCN | B | 100:0 |
51/—h |

The group of Nolan has considered that the strong Ni–Cl bond formed in the reaction of NiCp2 with imidazolium chlorides for the synthesis of [Ni(Cp)(NHC)Cl] complexes, must act as a significant driving force leading to product.11b For complexes 2 and 3 the thermodynamics must comprise the formation of strong Ni–O linkages instead, and the additional stabilization provided by the formation of six-membered metallocycles after the chelating coordination of the bidentate NHC ligands.
At this point, it should be noted that an excess of 1 or NiCp2, or the addition of NaX (X = Cl, Br; 1–5 equiv.) as a source of anionic halide ligand in an attempt to form Na[Ni(Cp)(NHC−)Cl] (or Na2[Ni(NHC−)2Cl2]),19 barely affect the 2:3 ratio of the product mixture. Moreover, and unexpectedly, no intermolecular ligand exchange leading to 3b and NiCp2 is observed upon heating isolated 2b in MeCN, and no reaction between 2b (or 2c) and 1b (or 1c) to form the biscarbene takes place in MeCN or THF at 65 °C after several days.
The formation of biscarbene complexes by abstraction of the acidic proton of two imidazolium species by both cyclopentadienyl ligands in NiCp2 has been documented to occur only in the presence of a second equivalent or excess of carbene precursor.20 The tendency to form biscarbene complexes 3 must be attributed to the presence of the carboxylate group in 1 and its coordinating and chelating capabilities. The set of outcomes observed here suggests the participation of transient species such as I and II depicted in Scheme 2. Thus, deprotonation of 1 by a η5-Cp ligand and bonding of the carbene carbon to the metal center results in 18-electron species with a coordination site occupied by solvent S or η2-cyclopentadiene (I), or by a second ion 1 coordinated as an anionic ligand via the carboxylate moiety (II). Chelation by intramolecular substitution of S in I, or double chelation accompanied by deprotonation of the κ-O-imidazolium ligand and elimination of the second C5H6 ring in II, must irreversibly lead to complexes 2 or 3, respectively. More-branched R groups in 1b, 1c and 1d hinder chelation, thereby favoring the shift to species II, whereas the better coordinating properties and higher polarity of acetonitrile compared to THF are likely to stabilize species I and hinder that shift. The scarce effect of an excess of 1 or NiCp2 in the products ratio 2:3 is explained by the fact that none of the reactants are fully soluble under the reaction conditions and by the irreversible formation of the complexes. Moreover, the reaction of entry 3 carried out in solution by using 100 mL of MeCN rises the selectivity 2b:3b from 68:32 (entry 3) to 75:25 (entry 3e), and reaches the maximun towards the formation of 2b (85%, entry 3f) by adding 1b in portions every 24 h to the nickelocene suspension.
 | ||
| Scheme 2 Transient species proposed for the formation of 2 and 3. | ||
We tentatively ascribe the lack of reactivity of 1f to conformational issues, with the α-benzyl group deterring deprotonation at the 2-position of the heterocycle by the η5-Cp ring. Indeed, clear correlations between that C–H group and the benzyl protons are observed in the NOE spectrum of 1f (see ESI†).
In an attempt to improve these syntheses, we applied the protocol reported by Navarro replacing conventional heating with microwave heating at 110 °C and using shorter reaction times (method B, scheme in Table 1).21 Most of the reactions went to completion in just 30 min, resulting in the formation of monocarbenes 2 and providing cleaner reaction mixtures from which higher yields were generally attained (entries 2, 5, 8, 10, 14–16, 18 and 20). To our delight, even the elusive imidazolium 1f reacts under these conditions, although the conversion (50% for entry 14) required a longer time to complete (60 min, entry 15), and biscarbene 3f is again favored in THF (entry 16).
Salts 1 are soluble in water, dmso, CH2Cl2, MeOH, to some extent in MeCN, and insoluble in Et2O, toluene or alkanes. In addition, 1g,h are slightly soluble in THF. The metal complexes 2 and 3 are all soluble in polar solvents, in Et2O, THF, and toluene, insoluble in alkanes, and only 2g,h are soluble in water (and sparingly soluble in THF). Zwitterionic compounds 1 are stable when exposed to air in solution and in the solid state, although they must be stored under an inert atmosphere given their hygroscopic nature. The 18- (2) and 16-electron (3) nickel complexes are stable as solids under air for months, but noticeable decomposition is observed in solutions exposed to air after a month.
Experiments with DCl have been carried out to test the robustness of the monocarbene complexes. The instantaneous and quantitative acidolysis and substitution of the Cp ligand by acetonitrile molecules has been reported for related half-sandwich κ2-C,C-alkyl-NHC nickel complexes.22 In contrast, the same test, comprising the addition of one equivalent of DCl to a solution of 2a and KPF6 in CD3CN, hardly affects the mixture, and only traces of free monodeuterated cyclopentadiene are detected by 1H NMR spectroscopy after 1 h, irrespective of whether the addition is performed at low (−78 °C) or at room temperature (Scheme 3). Moreover, with two equivalents of DCl, only a 12% yield of Cp-D is observed after 1 h, thus confirming the stability of the complex.
 | ||
| Scheme 3 Resistance of 2a to acidolysis by DCl. | ||
Compounds 1d–h exhibit the NMR spectroscopic features observed for [(1a–c)H]+ BF4− or 1b,18 with different resonances due to the different R substituents in the acetate side arm. Thus, the 1H/13C signals for the CH at the 2-,4-, and 5-Imz positions are observed at around 9.4/137, 7.9/123, and 7.8/122 ppm, respectively, and in the range 4.3–4.9/64–71 ppm for the corresponding nuclei at the stereogenic center, although that proton is more deshielded (δ = 5.9) for 1e with R = Ph. The NMR spectra show the peak corresponding to the carbene carbon at around 156 ppm for complexes 2, except for 2f (δ = 137), which is shielded by the benzyl group, and at 160 ppm for bis-NHC compounds 3, whereas for [Ni(Cp)(NHC)Cl] complexes this signal appears above 166 ppm.10b,11b The resonances for most of the characteristic nuclei also shift upon complex formation and coordination of the carbene ligands to nickel (e.g., δ for 1H/13C for 4- and 5-Imz at around 7.8/124 and 7.3/123 ppm for 2, and δ 7.5/122 and 7.2/123 for 3, respectively, but at 7.1/130 and 7.0/129 for 2f, thus pointing to the proximity of the Bn and the Imz rings). The proton and carbon at the chiral center are observed in the range (δ 4.3–4.7/60–71 for 2 and 3.9–4.6/62–70 for 3), with the proton more deshielded for 2e (δ 5.9) with R = Ph. The cyclopentadienyl ring signals appear at 4.7 (1H) and 91 (13C) ppm for complexes 2, and the carboxylic carbon shifts to 171 ppm for 2 and 3. It should also be noted that the methylthio group in 2g,h resonates at 2.2 (1H) and 15 (13C) ppm, almost the same values found for precursors 1g,h (δ 2.1 and 14 respectively), thus denoting the absence of an interaction between this donor group and the metal center in solution.
The HRMS (ESI/TOF+) spectra of the new compounds 1 show the ion [M + H]+ as the most intense peak in all cases, and this peak is often accompanied by cation [M + Na]+ always matching the calculated isotopic patterns. Most of the monocarbene complexes 2 afford very high specific rotation values in ethanol (Table 2).
| x= | a | b | c | d | e | f | g | h |
|---|---|---|---|---|---|---|---|---|
| a [α]20D determined in EtOH; C ≃ 0.1 g/100 mL. | ||||||||
| 1x | +35 | −16 | +43 | +61 | +1 | −60 | −10 | +20 |
| 2x | −169 | −553 | −450 | −598 | −7 | −574 | −8 | −423 |
| 3x | −220 | −106 | −284 | −278 | ||||
Further structural insights have been gained by X-ray analysis of single crystals for complexes 2a–h and biscarbenes 3b–d (Table 3). The NHC ligands coordinate to the nickel center in a κ2-C,O-chelating mode, retaining the (S)-configuration of the starting amino acids. In other words, no epimerization takes place during the formation of heterocycles 1 or during metalation to form enantiopure complexes 2 and 3. The absolute configuration for 2g is unique because, given this retention, the positions at the stereogenic center (H atom and R group) are exchanged relative to the other structures, as corresponds to the (S)-enantiomer and the CIP descriptors with R = CH2SMe. The coordination geometry of all monocarbenes 2 is trigonal planar considering the Cp ring centroid, the carbene and the coordinated oxygen atom (sum of bond angles 360°), with a narrow bond angle for the chelating donor atoms (ca. 92°) in the range (90–96°) found in the crystal structure of neutral and cationic [Ni(Cp)(NHC)] moieties bearing a κ2-E,C-carbene six-membered chelate ligand (E = S,23 C,24 or N25). The Ni–carbene bond lengths are in the lower limit of the range found for [Ni(Cp)(NHC)Cl] and related complexes, whereas the C–Ni–Cp(c) bond angles are, in general, wider.10b,11b The structures of 3b–d resemble that found for the palladium equivalent of 3c, namely square-planar complexes with a trans arrangement of the carbene ligands.17 The trans influence of the carbenes can be seen from the longer Ni–C bond lengths than for monocarbenes 2. The NHC moiety is tilted to the coordination plane of the metal in all cases (27–33° for 2a–h, 35–9° for 3b–d), and is almost coplanar in the biscarbene complexes (deviations of 5–15°).
|
|
||||||
|---|---|---|---|---|---|---|
| Ni–C | Ni–O | Ni–Cp(c) | C–Ni–O | C–Ni–Cp(c) | O–Ni–Cp(c) | |
| a Only the hydrogen atom at the stereogenic centers is included; the remainder are omitted for clarity. b Averaged values. | ||||||
| 2a | 1.851(3) | 1.887(2) | 1.753 | 92.6(1) | 137.0 | 130.4 |
| 2b | 1.860(3) | 1.882(2) | 1.767 | 92.8(1) | 137.0 | 130.2 |
| 2c | 1.858(4) | 1.882(3) | 1.758 | 92.1(1) | 138.4 | 129.6 |
| 2d | 1.860(4) | 1.876(3) | 1.767 | 91.6(2) | 137.9 | 130.3 |
| 2e | 1.858(5) | 1.873(4) | 1.766 | 92.1(2) | 139.5 | 128.4 |
| 2f | 1.860(3) | 1.888(2) | 1.758 | 92.6(1) | 136.4 | 130.9 |
| 2g | 1.851(3) | 1.889(2) | 1.757 | 91.9(1) | 134.6 | 133.5 |
| 2h | 1.857(3) | 1.889(2) | 1.757 | 92.6(1) | 137.9 | 129.5 |
| Ni–C | Ni–O | C–Ni–O | C–Ni–C′ | O–Ni–O′ | ||
| 3b | 1.90(1)b | 1.87(1)b | 90(2)b | 177.3(1) | 178.8(1) | |
| 3c | 1.91(1)b | 1.855(3)b | 90(1)b | 176.9(1) | 177.3(1) | |
| 3d | 1.90(2)b | 1.869(4)b | 90(2)b | 178.8(2) | 178.5(2) | |

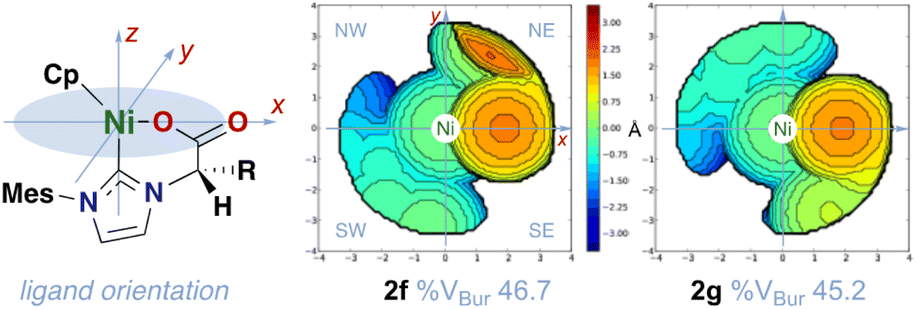
Topographical analysis of the steric maps of the ligands, obtained by uploading the crystal structures data to Cavallo's SambVCa application,26 reveals a higher steric pressure of the NHC in 2a–h (%VBur = 44–47, Table 4) than for the IMes ligand in the reference compound [Ni(Cp)(IMes)Cl] (36%, calculated using the same set of structural parameters).27 The average %VBur for NHCs in biscarbenes 3b–d lies below the lower limit of that range (43 ± 2%), in agreement with the slightly longer Ni–C(NHC) bond distances. The asymmetry of the carbene ligands results in high disparities in quadrant occupancies, although they are relatively constant for all structures. According to the defined coordinates for complexes 2 (Table 4), the difference in %VBur between the least- (NW = 25 ± 3%) and the most-encumbered (NE = 69 ± 4%) quadrants contrasts with the uniform steric map for IMes in the reference compound (36 ± 3% in all quadrants).27 Similar asymmetric pockets can be seen for the ligands in complexes 3, and the least- and most-hindered quadrants for 2g turn to be SW (23%) and SE (67%), respectively, due to the absolute configuration thereof (compare the figures for 2f and 2g in Table 4).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| %VBur | 2a | 2b | 2c | 2d | 2e | 2f | 2g | 2h |
| a %VBur values were calculated with the metal at the center of the sphere with a radius of 3.5 Å. Bondi radii scaled by 1.17, mesh spacing of 0.1. H atoms were excluded.26 | ||||||||
| Total | 44.1 | 45.0 | 45.2 | 45.2 | 46.8 | 46.7 | 45.2 | 46.2 |
| SW | 39.2 | 41.4 | 41.9 | 40.3 | 40.5 | 40.5 | 23.4 | 40.5 |
| NW | 22.4 | 24.8 | 23.8 | 23.0 | 28.5 | 24.1 | 39.1 | 23.3 |
| NE | 65.2 | 65.8 | 66.2 | 66.8 | 67.4 | 73.0 | 50.9 | 71.6 |
| SE | 49.7 | 48.0 | 48.9 | 50.8 | 50.8 | 49.2 | 67.2 | 49.4 |
Having successfully synthesized the nickel compounds, we envisioned their use in catalytic reduction reactions of ketones, namely acetophenone derivatives, by hydrosilylation followed by hydrolysis. This procedure complements the traditional reduction of carbonyl functionalities and avoids the hazards and often poor selectivity of classic reducing reagents (e.g., alkali metals and metal hydrides),28 the use of catalysts based on expensive precious metals,29 and the need for high-pressure hydrogenation reactors. Indeed, efficient first-row transition metal catalysts for hydrosilylative reductions have emerged recently,30 although, for nickel, only a limited number of catalysts for this reaction are known,31 most of which are either of the pincer32 or the [Ni(Cp)(NHC)X] type.15b,16b,23,25b,33,34
Good performance in the hydrosilylative reduction of aldehydes has been reported for all these catalysts, although most of them showed low (or no) catalytic activity with ketones. Inspired by the results reported by Royo,15b Chetcuti and Ritleng16b,23,25b,33 and Albretch34 in the hydrosilylation of carbonyl compounds with [Ni(Cp)(NHC)X] complexes, we decided to test ketones using complexes 2 and 3 as catalysts. After some preliminary testing to optimize the reaction with acetophenone as a model substrate (see ESI†), the conditions selected were those summarized in Table 5. Conversions to the secondary alcohol (i.e., 1-phenylethanol) were high in 0.5–1.5 h (Table 5, entries 1, 4, 7, 10, 13 and 16), except for complexes 2g,h, which contain a methylthio group (entries 19 and 22), and the biscarbene 3b (entry 25), for which significant conversions required much longer reaction times (>20 h).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | R | R′ | t (h) | Conv. (%)b |
| a All reactions were carried out with 1.5 mmol of acetophenone derivative, 0.15 mmol of 2 (10 mol%) and 1.8 mmol of PhSiH3 in dry THF (7.5 mL, [Ni] = 0.02 m) at 50 °C. The catalytic reactions were performed at least in duplicate and the reproducibility of the conversions measured is estimated at ±2%. b Conversions determined by GC-MS after methanolysis. | |||||
| 1 | 2a | Me | H | 0.5 | 97 |
| 2 | Cl | 1.5 | 97 | ||
| 3 | MeO | 1.5/24 | 76/97 | ||
| 4 | 2b | iPr | H | 1.5 | 94 |
| 5 | Cl | 1.5 | 92 | ||
| 6 | MeO | 1/22.5 | 58/90 | ||
| 7 | 2c | iBu | H | 0.5 | 99 |
| 8 | Cl | 0.5 | 96 | ||
| 9 | MeO | 1 | 94 | ||
| 10 | 2d | sBu | H | 1.5 | 96 |
| 11 | Cl | 2.5 | 96 | ||
| 12 | MeO | 1/20.5 | 24/95 | ||
| 13 | 2e | Ph | H | 1 | 98 |
| 14 | Cl | 2.5 | 94 | ||
| 15 | MeO | 1/21.5 | 55/98 | ||
| 16 | 2f | CH2Ph | H | 0.5 | 99 |
| 17 | Cl | 1.0 | 96 | ||
| 18 | MeO | 1.5 | 99 | ||
| 19 | 2g | CH2SMe | H | 1.5/21 | 2/38 |
| 20 | Cl | 1.5/21 | 3/62 | ||
| 21 | MeO | 1.5/21 | 0/18 | ||
| 22 | 2h | (CH2)2SMe | H | 1.5/24 | 23/93 |
| 23 | Cl | 1.5/23 | 26/98 | ||
| 24 | MeO | 1.5/24 | 3/31 | ||
| 25 | 3b | iPr | H | 22 | 87 |

The activities found for 2a–f, with an average TOF (TOFav) of up to 20 h−1 (entries 7 and 16), are better than those reported for the reference catalysts in the same transformation: [Ni(Cp*-NHCMe)OtBu] (85% conversion, 2 mol%, 24 h, 100 °C in toluene, TOFav = 2 h−1),15b [NiCp(IMes)Cl] (97%, 5 mol%, NaHBEt3 5 mol%, 17 h, 25 °C in THF, TOFav = 1 h−1),33 [NiCp(NHCpy)]Br (99%, 2 mol%, KOtBu 2 mol%, 24 h, 100 °C in toluene, TOFav = 2 h−1);25b or for the most active pincer nickel complexes reported to date [Ni(L)H] (L = bis(phosphino)boryl PBP-ligand) (99%, 5 mol%, 6 h, 70 °C in C6D6, TOFav = 3 h−1),32d [Ni{NHC-(CH2PPh2)2}(SPh)2] (98%, 2.5 mol%, 8 h, 70 °C in toluene, TOFav = 5 h−1).32f It should be noted that the significant conversions obtained in shorter reaction times with complexes 2a–f are obtained at a relatively low temperature and without the need for any additive/activator. For instance, the reaction catalyzed by 2c (2 mol%) in toluene at 100 °C gives 99% conversion in 1 h, with an increased TOFav of 48 h−1 (see ESI†).
The reduction of para-chloro and -methoxy acetophenone was also tested. Complexes 2a,d,e,f are somewhat less active in the presence of the electron-withdrawing group, requiring longer reaction times for similar conversions (entries 2, 11, 14 and 17 vs. 1, 10, 13 and 16), whereas the other catalysts (2b,c) show similar activities as with acetophenone (entries 5 and 8 vs. 4 and 7). Lower activities are observed with p-methoxyacetophenone in all cases, with significant conversions only after reaction for 21–24 h (entries 3, 6, 12 and 15 vs. 1, 4, 10 and 13) except for complexes 2c,f (entries 9 and 18 vs. 7 and 16), which complete the reaction in only 1.5 h.
Given the solubility of complexes containing the thioether functionality in water, we checked the hydrosilylative reduction of p-chloroacetophenone with 2h in water (rest of conditions as in Table 5, entry 23) and compared the outcome with the performance of 2c in aqueous solution (other conditions as in entry 8). Despite the very low water-solubility of the latter, the conversion is, once again, higher (93%) than for 2h (72%) after reaction for 7 h. As such, the behavior summarized in Table 5 for complexes 2g,h cannot be attributed to their poor solubility in THF. The presence of hemilabile groups based on N- or S-coordinated donor atoms in related NiCp(NHC) complexes has been associated with high TOF values in the hydrosilylation of aldehydes as a result of a protecting effect on the catalytic species in the resting state.23,34 In this case, the presence of the additional pendant donor group in complexes 2g,h appears to hamper the catalysis.
Chiral-HPLC determinations indicate a racemic composition for the resulting secondary alcohols. We also observed the darkening of all catalytic solutions with time. Ritleng and Chetcuti have explained the lack of chiral induction in this reaction using a D-menthyl-functionalized [Ni(Cp)(NHC)Cl] complex, suggesting a distant location away from the metal center of the chiral group.16b The same authors have found evidence for the formation of nickel nanoparticles (NiNPs) under the harsh reaction conditions necessary for a [Ni(Cp-NHCpy)]Br complex to be active against acetophenone.25b We have ruled out the formation of NiNPs during the catalysis under our milder conditions because none were observed in any field inspected by TEM in samples from solutions of the reactions performed with 2c. Moreover, the catalytic reaction with 2f was found to be unchanged by the addition of a few drops of mercury at t = 40 min (conv. = 60% and 97% at t = 45 and 60 min, respectively) compared to the experiment without Hg (conv. = 62% and 96% at t = 45 and 60 min, respectively). Therefore, the total absence of enantioselectivity with complexes 2 is likely to be due to decoordination of the carboxylate from the 18 e− metal center under the catalytic conditions, rather than to the participation of NiNPs with non-selective surfaces, thus resulting in a dangling NHC side-arm that is free to rotate. As such, in line with a recent proposal for a pincer nickel dithiolate complex,32f one possible reaction pathway involves silylation of the κ-O-carboxylate before it enters the catalytic cycle. Nevertheless, no detectable changes are observed in the 1H NMR spectra of complex 2a in the presence of p-chloroacetophenone or PhSiH3 (1 equiv.) after 1 h at 50 °C in THF-d8.
Since the catalysts do not degrade to nanoparticles, apparently operating under homogeneous conditions with discrete species, we tested the durability of 2c by reloading with fresh p-chloroacetophenone and PhSiH3 at the end of the reaction (Table 5, entry 8). Some catalyst depletion was observed, with the initial conversion (97% in 1.5 h, TOFav = 6.5 h−1) decreasing in the first (92% in 2.5 h, TOFav = 3.5 h−1) and second (85% in 4.5 h, TOFav = 2 h−1) reruns of the reaction.
Conclusions
In addition to expanding the procedure reported by Baslé and Mauduit for the preparation of chiral imidazolium derivatives by using amino acids functionalized with aromatic or thiolate groups, we have managed to synthesize, to the best of our knowledge, the first family of nickel complexes containing NHC-carboxylate chelate ligands. Moreover, since no epimerization takes place during either formation of the starting imidazolium salts 1 or during their nickelation, the complexes have been obtained as enantiomerically pure compounds. The reactions between 1 and nickelocene tend to form mixtures of mono- and bis-NHC complexes (i.e., [Ni(η5-Cp)(κ2-C,O-NHC)] and [Ni(κ2-C,O-NHC)2]) due to the competing coordination capability of the carboxylate group, in ratios that depend on the reaction conditions. Thus, microwave heating accelerates the reactions and favors formation of the half-sandwich compounds in a polar and good coordinating solvent such as acetonitrile. All complexes were found to be very stable to air and moisture. In fact, mono-NHC complexes catalyze the hydrosilylation of chloroacetophenone in water. Some of the [Ni(η5-Cp)(κ2-C,O-NHC)] complexes turn out to be among the best nickel catalysts for the hydrosilylative reduction of p-acetophenones described to date. Moreover, no activating additives are required, and catalysis takes place under mild operational conditions. The lack of chiral induction during formation of the secondary alcohol points to decoordination of the κ-O-carboxylate before it enters the catalytic cycle.The features of the new complexes demonstrate the potential of suitable ligands to impart improved catalytic performance to earth-abundant metal compounds.
Author contributions
J. S.-G.: investigation, conceptualization, validation, data curation. A. M.: resources, formal analysis, data curation. C. G.-A.: methodology, conceptualization, supervision, project administration, funding acquisition. J. C. F.: conceptualization, supervision, project administration, funding acquisition, writing – original draft. All authors contributed to the writing – review and editing process of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministerio de Ciencia e Innovación (PID2020-114637GB-I00), and the Comunidad de Madrid (EPU-INV/2020/013) are gratefully acknowledged.References
- A. J. Arduengo III, R. L. Harlow and M. J. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- (a) H. Amouri, Chem. Rev., 2023, 123, 230–270 CrossRef CAS PubMed; (b) W. K. Liu and R. Gust, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS; (c) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- See for instance: (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) M. C. Jahnke and F. E. Hahn, in Transition Metal Complexes of Neutral η1-Carbon Ligands, ed. R. Chauvin and Y. Canac, Topics in Organometallic Chemistry 30, Springer, Berlin, 2010, pp. 95–129 Search PubMed; (c) L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- (a) V. A. Voloshkin, N. V. Tzouras and S. P. Nolan, Dalton Trans., 2021, 50, 12058–12068 RSC; (b) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS PubMed; (c) S. Bai and Y.-F. Han, Acc. Chem. Res., 2023, 56, 1213–1227 CrossRef CAS PubMed.
- (a) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (b) E. Peris, Chem. Rev., 2018, 118(19), 9988–10031 CrossRef CAS PubMed; (c) N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer-Verlag, Heidelberg, 2011 Search PubMed; (d) N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, RSC Catalysis Series No. 6, The Royal Society of Chemistry, 2011 Search PubMed; (e) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (f) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, WILEY-VCH, Weinheim, 2006 Search PubMed.
- (a) S. P. Nolan, Acc. Chem. Res., 2011, 44, 91–100 CrossRef CAS PubMed; (b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed.
- (a) J. Cheng, L. J. Wang, P. Wang and L. Deng, Chem. Rev., 2018, 118, 9930–9987 CrossRef CAS PubMed; (b) A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed.
- (a) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- (a) V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef CAS; (b) V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2015, 5, 1283–1302 CrossRef; (c) B. C. Lee, C.-F. Liu, L. Q. H. Lin, K. Z. Yap, N. Song, C. H. M. Ko, P. H. Chan and M. J. Koh, Chem. Soc. Rev., 2023, 52, 2946–2991 RSC.
- (a) C. D. Abernethy, J. A. C. Clyburne, A. H. Cowley and R. A. Jones, J. Am. Chem. Soc., 1999, 121, 2329–2330 CrossRef CAS; (b) C. D. Abernethy, A. H. Cowley and R. A. Jones, J. Organomet. Chem., 2000, 596, 3–5 CrossRef CAS.
- (a) H. M. Sun, Q. Shao, D. M. Hu, W. F. Li, Q. Shen and Y. Zhang, Organometallics, 2005, 24, 331–334 CrossRef CAS; (b) R. A. Kelly III, N. M. Scott, S. Díez-González, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 3442–3447 CrossRef.
- For leading references, see: (a) A. Włodarska, A. Kozioł, M. Dranka, A. Gryff-Keller, P. Szczeciński, J. Jurkowski and A. Pietrzykowski, Organometallics, 2015, 34, 577–581 CrossRef; (b) R. Lopes, M. M. Pereira and B. Royo, ChemCatChem, 2017, 9, 3073–3077 CrossRef CAS; (c) W. Buchowicz, Ł. Banach, R. Kamiński and P. Buchalski, Dalton Trans., 2017, 46, 3805–3808 RSC.
- For leading references, see: (a) F. Ulm, Y. Cornaton, J.-P. Djukic, M. J. Chetcuti and V. Ritleng, Chem. – Eur. J., 2020, 26, 8916–8925 CrossRef CAS PubMed; (b) S. Shahane, B. de P. Cardoso, M. J. Chetcuti and V. Ritleng, Catalysts, 2019, 9, 76 CrossRef; (c) B. de P. Cardoso, J.-M. Bernard-Schaaf, S. Shahan, L. F. Veiros, M. J. Chetcuti and V. Ritleng, Dalton Trans., 2018, 47, 1535–1547 RSC.
- Ł. Banach, P. A. Guńka, J. Zachara and W. Buchowicz, Coord. Chem. Rev., 2019, 389, 19–58 CrossRef.
- (a) H.-M. Sun, D.-M. Hu, Y.-S. Wang, Q. Shen and Y. Zhang, J. Organomet. Chem., 2007, 692, 903–907 CrossRef CAS; (b) L. Postigo and B. Royo, Adv. Synth. Catal., 2012, 354, 2613–2618 CrossRef CAS; (c) L. Postigo, R. Lopes and B. Royo, Dalton Trans., 2014, 43, 853–858 RSC.
- (a) J. S. E. Ahlin, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 13229–13233 CrossRef CAS PubMed; (b) M. Rocquin, V. Ritleng, S. Barroso, A. M. Martins and M. J. Chetcuti, J. Organomet. Chem., 2016, 808, 57–62 CrossRef CAS; (c) J. Diesel, A. M. Finogenova and N. Cramer, J. Am. Chem. Soc., 2018, 140, 4489–4493 CrossRef CAS PubMed.
- A. Sánchez, J. Sanz-Garrido, C. J. Carrasco, F. Montilla, E. Álvarez, C. Gonzalez-Arellano, J. C. Flores and A. Galindo, Inorg. Chim. Acta, 2022, 537, 120946 CrossRef.
- (a) C. Jahier-Diallo, M. S. T. Morin, P. Queval, M. Rouen, I. Artur, P. Querard, L. Toupet, C. Crévisy, O. Baslé and M. Mauduit, Chem. – Eur. J., 2015, 21, 993–997 CrossRef CAS PubMed; (b) J. Thongpaen, T. E. Schmid, L. Toupet, V. Dorcet, M. Mauduit and O. Baslé, Chem. Commun., 2018, 54, 8202–8205 RSC.
- E. A. Baquero, G. F. Silbestri, P. Gómez-Sal, J. C. Flores and E. de Jesús, Organometallics, 2013, 32, 2814–2826 CrossRef CAS.
- (a) Z.-H. Liu, Y.-C. Xu, L.-Z. Xie, H.-M. Sun, Q. Shen and Y. Zhang, Dalton Trans., 2011, 40, 4697–4706 RSC; (b) F. P. Malan, E. Singleton, P. H. van Rooyen, J. Conradie and M. Landman, J. Mol. Struct., 2017, 1147, 235–243 CrossRef CAS.
- B. Landers and O. Navarro, Inorg. Chim. Acta, 2012, 380, 350–353 CrossRef CAS.
- M. Henrion, A. M. Oertel, V. Ritleng and M. J. Chetcuti, Chem. Commun., 2013, 49, 6424–6426 RSC.
- F. Ulm, A. I. Poblador-Bahamonde, S. Choppin, S. Bellemin-Laponnaz, M. J. Chetcuti, T. Achard and V. Ritleng, Dalton Trans., 2018, 47, 17134–17145 RSC.
- A. M. Oertel, J. Freudenreich, J. Gein, V. Ritleng, L. F. Veiros and M. J. Chetcuti, Organometallics, 2011, 30, 3400–3411 CrossRef CAS.
- (a) L. B. Junquera, F. E. Fernández, M. C. Puerta and P. Valerga, Eur. J. Inorg. Chem., 2017, 2547–2556 CrossRef CAS; (b) F. Ulm, S. Shahane, L. Truong-Phuoc, T. Romero, V. Papaefthimiou, M. Chessé, M. J. Chetcuti, C. Pham-Huu, C. Michon and V. Ritleng, Eur. J. Inorg. Chem., 2021, 3074–3082 CrossRef CAS.
- (a) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed, (https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html). (b) A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679–2681 CrossRef CAS.
- %VBur for IMes in [NiCp(IMes)Cl] determined using the data from: (a) A. R. Martin, Y. Makida, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 6265–6270 CrossRef CAS; (b) O. R. Luca, B. A. Thompson, M. K. Takase and R. H. Crabtree, J. Organomet. Chem., 2013, 730, 79–83 CrossRef CAS.
- (a) F. A. Carey and R. J. Sundberg, Reduction of Carbonyl and Other Functional Groups, in Advanced Organic Chemistry. Springer, Boston, 1977, pp. 129–161 Search PubMed; (b) G. W. Gribble, Chem. Soc. Rev., 1998, 27, 395–404 RSC.
- (a) A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef; (b) I. Ojima, Z. Li and J. Zhu, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, 1998, vol. 2, pp. 1687–1792 Search PubMed; (c) I. Ojima, in The Chemistry of Organic Silicon Compounds, ed. S. Patei and Z. Rappoport, John Wiley & Sons, 1989, vol. 1, pp. 1479–1526 Search PubMed; (d) B. Marciniec, Comprehensive Handbook on Hydrosilylation, Pergamon, Oxford, 1992 Search PubMed.
- (a) M. Bhunia, P. Sreejyothi and S. K. Mandal, Coord. Chem. Rev., 2020, 405, 213110 CrossRef CAS; (b) B. Royo, Adv. Organomet. Chem., 2019, 72, 59–102 CrossRef CAS; (c) S. Chakraborty and H. R. Guan, Dalton Trans., 2010, 39, 7427–7436 RSC.
- (a) F.-G. Fontaine, R.-V. Nguyen and D. Zargarian, Can. J. Chem., 2003, 81, 1299–1306 CrossRef CAS; (b) B. L. Tran, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 2234–2243 CrossRef CAS; (c) S. N. MacMillan, W. H. Harman and J. C. Peters, Chem. Sci., 2014, 5, 590–597 RSC; (d) C. L. Rock, T. L. Groy and R. J. Trovitch, Dalton Trans., 2018, 47, 8807–8816 RSC; (e) S. Bertini and M. Albrecht, Chimia, 2020, 74, 483–488 CrossRef CAS PubMed.
- (a) S. Chakraborty, J. A. Krause and H. Guan, Organometallics, 2009, 28, 582–586 CrossRef CAS; (b) S. Kundu, W. W. Brennessel and W. D. Jones, Inorg. Chem., 2011, 50, 9443–9453 CrossRef CAS PubMed; (c) K. A. Gudun, M. Segizbayev, A. Adamov, P. N. Plessow, K. A. Lyssenko, M. P. Balanay and A. Y. Khalimon, Dalton Trans., 2019, 48, 1732–1746 RSC; (d) J. A. Fernández, J. M. García, P. Ríos and A. Rodríguez, Eur. J. Inorg. Chem., 2021, 2993–2998 CrossRef; (e) K. Kobayashi and H. Nakazawa, Inorg. Chim. Acta, 2021, 523, 120403 CrossRef CAS; (f) A. Kumar, R. Gupta and G. Mani, Organometallics, 2023, 42, 732–744 CrossRef CAS.
- L. P. Bheeter, M. Henrion, L. Brelot, C. Darcel, M. J. Chetcuti, J.-B. Sortais and V. Ritleng, Adv. Synth. Catal., 2012, 354, 2619–2624 CrossRef CAS.
- Y. Wei, S.-X. Liu, H. Mueller-Bunz and M. Albrecht, ACS Catal., 2016, 6, 8192–8200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and catalysis details, characterization data, and NMR spectra. CCDC 2302830–2302840. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03739h |
| This journal is © The Royal Society of Chemistry 2024 |