
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-defined Cu(I) complexes based on [N,P]-pyrrole ligands catalyzed a highly endoselective 1,3-dipolar cycloaddition†
Miguel A.
Alvarado-Castillo
ab,
Salvador
Cortés-Mendoza
a,
José E.
Barquera-Lozada
 a,
Francisco
Delgado
*b,
Ruben A.
Toscano
a,
M. Carmen
Ortega-Alfaro
c and
José G.
López-Cortés
*a
a,
Francisco
Delgado
*b,
Ruben A.
Toscano
a,
M. Carmen
Ortega-Alfaro
c and
José G.
López-Cortés
*a
aInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Coyoacán, C.P. 04360 CdMx, Mexico. E-mail: jglcvdw@unam.mx
bDepartamento de Química Organica, Escuela Nacional de Ciencias Biológicas Instituto Politécnico Nacional Prol. Carpio y Plan de Ayala, S/N, CdMx 11340, Mexico
cInstituto de Ciencias Nucleares, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Coyoacán, C.P. 04510 CdMx, Mexico
First published on 22nd December 2023
Abstract
We herein report the synthesis and catalytic application of a new family of dinuclear Cu(I) complexes based on [N,P]-pyrrole ligands. The Cu(I) complexes (4a–d) were obtained in good yields and their catalytic properties were evaluated in the1,3-dipolar cycloaddition of azomethine ylides and electron-deficient alkenes. The air-stable complexes 4a–d exhibited high endo-diasteroselectivity to obtain substituted pyrrolidines, and the catalytic system showed excellent reactivity and wide substitution tolerance.
Introduction
Nowadays, copper(I) complexes have acquired great importance in the design of new materials for organic light-emitting devices (OLEDs)1 and photosensitizers.2 Likewise, the catalytic properties of these complexes have been largely explored. In the literature, we can find some recent examples of using Cu(I) in combination with [N,P] ligands in important catalytic transformations. These included conjugated additions of boron to α,β-unsaturated compounds,3 asymmetric alkylation of indoles,4 direct asymmetric alkynylation of α-ketoesters,5 and asymmetric A3 couplings,6 among others. Particularly, the 1,3-dipolar cycloaddition (1,3-DC) of azomethine ylides to electron-deficient alkenes constitutes one of the most powerful and versatile methods to obtain highly substituted 5-membered heterocycles.7,8 This reaction can be accomplished using several metal complexes such as zinc(II),9 nickel(II),10 silver(I),11 copper(I),12 and copper(II).13 Particularly, in the case of copper(I) catalyzed reactions, the use of hemilabile ferrocenyl [N,P]-ligands appears as an interesting approach that allows the synthesis of exo-aducts12b with good to excellent diastereo- and enantio-selective results. On the other hand, the synthesis of endo-adducts is favored when [N,N]-, [S,S]- and [P,S]-ligands are used in combination with different sources of copper(I) precursors.12j–m Similar results were obtained with the catalytic systems of Zn(II)/N,O-ligands9a and Ag(I)/chiral BINOL-derived phosphoric acids.11dPreviously, we reported an efficient synthesis of pyrrole-based [N,P] ligands with a dimethylamine group and a phosphine moiety as hard and soft donors, respectively.14 Moreover, we have demonstrated that their palladium complexes catalyzed some C–C coupling reactions,15 their gold(I)-complexes catalyzed the cycloisomerization of enynes,16 and their ruthenium counterparts promoted the transfer hydrogenation of ketones (Fig. 1).17 As part of our continuous interest in developing catalytic applications of [N,P]-pyrrole phosphines, in this work we address the synthesis of new copper(I)-[N,P] dinuclear complexes, which were tested in 1,3-dipolar cycloaddition, obtaining pyrrolidines with high endo-selectivity.
 | ||
| Fig. 1 Catalytic applications of [N,P] ligands based on pyrrole. | ||
Results and discussion
Initially, we synthesized the ligands 2a–d in good yields using the methodology previously described by our group.14,15 In order to know about the σ-donor behavior of these phosphines, we reacted 2a–d with elemental selenium overnight in an NMR tube at 0 °C using CDCl3 as the solvent (Scheme 1). | ||
| Scheme 1 Preparative route to [N,P] ligands based on pyrrole 2a–d. | ||
Thereafter, we obtained 31P {1H} NMR spectra. The values obtained for the coupling constant JP–Se are shown in Table 1. In general, all phosphine selenides with an alkyl group (Table 1, entries 2–4) exhibit lower JP–Se values compared to PPh3Se (Table 1, entry 5) indicating a stronger σ-donor behavior but less than the corresponding trialkylphosphines (Table 1, entries 6–8). In contrast, 3a has the greatest JP–Se value due to this best π-acceptor character.19
| Entry | Phosphine | R | δ 31Pligandb | δ 31PP–Seb | 1JP–Sec |
|---|---|---|---|---|---|
| a 31P{1H} NMR: 121.65 MHz, CDCl3, 298 K. b ppm. c Hz. d 31P{1H} NMR: 121.65 MHz, CDCl3, 323 K. | |||||
| 1 | 2a 15 | Ph | −29.2 | 16.2 | 736 |
| 2 | 2b 14 | t-Bu | 1.5 | 72.4d | 707 |
| 3 | 2c 15 | i-Pr | −17.6 | 56.4 | 704 |
| 4 | 2d 16 | cPent | −25.6 | 50.2 | 705 |
| 5 | PPh318 | — | −4.7 | 35.2 | 730 |
| 6 | t-Bu3P18 | — | 61.1 | 92.9 | 687 |
| 7 | i-Pr3P18 | — | 19 | 70.5 | 686 |
| 8 | cPent3P18 | — | 4.7 | 63.4 | 685 |
We prepared the air-stable complexes 4a–d by reacting 2a–d with CuBr in CH2Cl2. In all cases, a pale-yellow powder was isolated after recrystallization from diethyl ether (Scheme 2). As expected, in 1H NMR, the proton signals were shifted at higher frequencies relative to the values assigned for free ligands. The same behavior occurs in 31P NMR where the coordinated phosphine signals resonate at higher frequencies, due to the electron-withdrawing effect of the Cu atom. These spectroscopic data lead us to suggest that these ligands behave as bidentate donors (Table 2).
 | ||
| Scheme 2 Synthesis of dinuclear copper complexes 4a–d. | ||
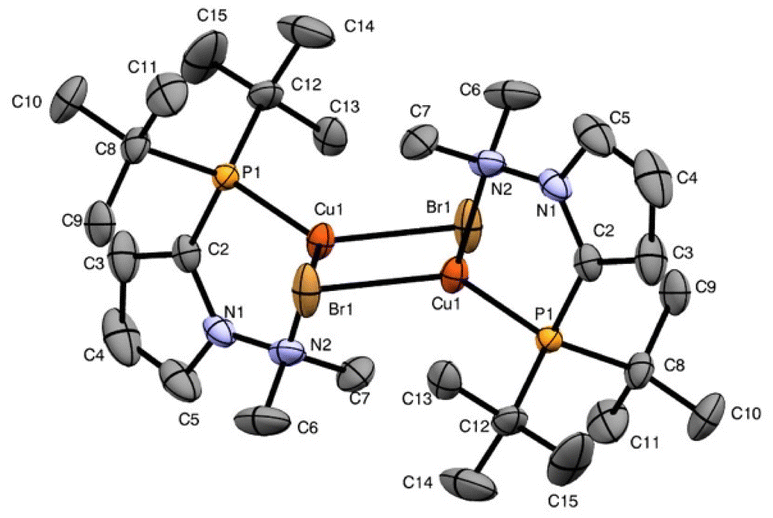
Suitable crystals for the X-ray analysis of these complexes were obtained from a solution of Et2O. The solid-state molecular geometries of complexes 4a–d were unequivocally confirmed by X-ray diffraction analysis (Fig. 2, 3 and S3, S4†). In a simple view, these complexes exhibit a tetrahedral geometry around the copper atom, with the [N,P] pyrrole-based ligands bonded in a κ2-NP coordination mode. The complexes 4a–d are dimers containing bromine atoms as a μ-bridge and have a five-membered metallacycle, in which the ligand has bite angles of P(1)–Cu(1)–N(2), 80.63°(6) for the complex 4a, P(1)–Cu(1)–N(2), 84.97° (12) for 4b, P(5)–Cu(5)–N(82), 81.81° (17) for 4c and P(1)–Cu(1)–N(2), 84.56° (3) for 4d.
 | ||
| Fig. 2 ORTEP representation of ligand 4a. Ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and bond angles (°): Br(1)–Cu(1), 2.4374(9); Br(1)–Cu(1), 2.4437(8); Cu(1)–P(1), 2.1910(9); Cu(1)–N(2), 2.559(2); P(1)–C(2), 1.808(3); P(1)–C(8), 1.822(2); P(1)–C(14), 1.836(2); N(1)–C(5), 1.371(3); N(1)–C(2), 1.373(3); N(1)–N(2), 1.422(3); N(2)–C(7), 1.480(4); N(2)–C(6), 1.481(4); Cu(1)–Br(1)–Cu(1), 71.28(2); P(1)–Cu(1)–Br(1), 126.82(3); P(1)–Cu(1)–Br(1), 122.39(3); Br(1)–Cu(1)–Br(1), 108.72(2); P(1)–Cu(1)–N(2), 80.63(6); Br(1)–Cu(1)–N(2), 100.31(6); Br(1)–Cu(1)–N(2), 105.38(6); 112.42(6); C(2)–P(1)–C(8), 105.70(11); C(2)–P(1)–C(14), 100.56(11); C(8)–P(1)–C(14), 103.72(11); C(2)–P(1)–Cu(1), 104.20(8); C(8)–P(1)–Cu(1), 116.57(8); C(14)–P(1)–Cu(1), 123.67(8); C(5)–N(1)–C(2), 110.3(2); C(5)–N(1)–N(2), 127.4(2); C(2)–N(1)–N(2), 122.3(2); N(1)–N(2)–C(7), 110.3(2); N(1)–N(2)–Cu(1), 105.55(14); N(1)–C(2)–P(1), 121.09(18). | ||
 | ||
| Fig. 3 ORTEP representation of ligand 4b. Ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and bond angles (°): Br(1)–Cu(1), 2.4460(8); Br(1)–Cu(1), 2.4591(8); Cu(1)–P(1), 2.1888(13); Cu(1)–N(2), 2.368(4); P(1)–C(2), 1.809(5); P(1)–C(8), 1.880(5); P(1)–C(12), 1.886(6); N(1)–C(5), 1.350(7); N(1)–C(2), 1.388(7); N(1)–N(2), 1.412(6); N(2)–C(7), 1.462(7); N(2)–C(6), 1.475(8). Cu(1)–Br(1)–Cu(1), 77.35(3); P(1)–Cu(1)–N(2), 84.97(12); P(1)–Cu(1)–Br(1), 124.61(4); N(2)–Cu(1)–Br(1), 106.04(11); P(1)–Cu(1)–Br(1), 127.33(5); N(2)–Cu(1)–Br(1), 104.59(12); Br(1)–Cu(1)–Br(1), 102.65(3); C(2)–P(1)–C(8), 104.6(2); C(2)–P(1)–C(12), 103.9(3); C(8)–P(1)–C(12), 114.5(3); C(2)–P(1)–Cu(1), 103.22(19); C(8)–P(1)–Cu(1), 114.74(19); C(12)–P(1)–Cu(1), 114.0(2); C(5)–N(1)–C(2), 110.6(6). | ||
All complexes have the expected tetra-coordinated environment around the copper metal center. To determine how closely these complexes could adopt a perfectly tetrahedral geometry Td (τ4 = 1.0) or a perfectly trigonal pyramidal geometry C3v (τ4 = 0.85),20 we calculated the angular structural parameter (τ4) for 4a–d, obtaining the following values: 0.77, 0.76, 0.73 and 0.81, respectively. From these data, we concluded that all complexes show highly distorted trigonal pyramidal geometries. Furthermore, the phosphine ligands and both bromides occupy the equatorial positions, whereas the dimethylamino group occupies the apical position in all complexes. The bond distances N(2)–Cu(1) 2.559(2) Å for complex 4a, N(82)–Cu(5) 2.523(8) Å for complex 4c and N(2)–Cu(1) 2.415(15) Å for 4d, are slightly longer in comparison to that observed in complex 4b, N(2)–Cu(1) 2.368(4) Å. This behavior suggests that for complexes 4a, 4c and 4d, the phosphine group behaves as a strong σ-donor with a slight π-acceptor character, while in complex 4b, the phosphine group only behaves as a strong σ-donor. The angles between Br(1)–Cu(1)–Br(1) 102.6° (3) for 4b and Br(5)–Cu(5)–Br(6) 98.6° (4) for 4c are narrower than those observed in complexes 4a [Br(1)–Cu(1)–Br(1) 108.7° (2)] and 4d [Br(2)–Cu(2)–Br(1) 109.8° (10)]. This fact prevents the Cu–Cu interaction observed in 4a [Cu(1)–Cu(2) 2.844(10) Å] and 4d [Cu(1)–Cu(2) 2.805(3)].
The UV/Vis absorption spectra of ligands 2a–d and complexes 4a–d acquired in CH2Cl2 are shown in Fig. 4. Both the complexes and the ligands exhibit similar intense and broad absorption bands (ε = 104 M−1 cm−1) in the wavelength range from 225 to 285 nm, assigned to the allowed ligand-centered transitions (π–π* and n–π*) of 2a–d. Complexes 4a–d exhibit an intense absorption band in the 230–260 nm region, which is assigned to π–π* ligand-centered (LC) transitions of the N,P-ligand.21 In the case of 4a, we observed an additional absorption between ≈275 and ≈350 nm, which is attributed to metal-to-ligand charge-transfer (MLCT) transitions involving Cu(I) atoms and the ligand, while the complexes 4b–d exhibit these transitions in the 265–290 nm region but with less intensity. We calculated the molar extinction coefficients (ε) for 4a at 228 nm (ε = 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 090 M−1 cm−1) and 265 nm (ε = 22775 M−1 cm−1), while the complexes 4b–d showed a maximum absorption band located at 233 nm (ε = 32434 M−1 cm−1), (ε = 31018 M−1 cm−1), and (ε = 30539 M−1 cm−1), respectively.
090 M−1 cm−1) and 265 nm (ε = 22775 M−1 cm−1), while the complexes 4b–d showed a maximum absorption band located at 233 nm (ε = 32434 M−1 cm−1), (ε = 31018 M−1 cm−1), and (ε = 30539 M−1 cm−1), respectively.
 | ||
| Fig. 4 UV–vis absorption spectra of 2a–d, and 4a–d in CH2Cl2 solution at room temperature. | ||
In order to evaluate the catalytic performance of these copper complexes, we explored a 1,3-dipolar cycloaddition using the iminoester 5a and N-phenyl maleimide 6 with 5% molar CuBr in the presence of Et3N but only 19% yield of 7a was obtained (Table 3, entry 1). To evaluate the effect of the ligand in the reaction, we used CuBr (5% mol) and the ligand 2a (5.5% mol), achieving only 17% yield, suggesting that pre-formation of the catalytic complex must be essential to carry out the transformation (Table 3, entry 2). Thus, when the copper(I) salt was placed in the presence of ligand 2a with a previous preformation of 30 min, the yield increased by 61%. This indicates that the use of the ligand is essential for stabilizing the intermediate copper species that are formed during the reaction (Table 3, entry 3). When the reaction was carried out with the copper catalytic precursor 4a, we got a better result (75% yield), further confirming our assumption (Table 3, entry 4). Then, we tried to increase the yield by changing the copper precursor (Table 3, entries 5–8). The best result was obtained using Cu(OTf)2 (Table 3, entry 7) in combination with ligand 2a, reaching a 65% yield; however, the use of complex 4a gave a slightly better yield (Table 3, entry 4). Regarding the organic base effect, we tested Et3N and DIPEA; we determined that the nature of these two bases does not generate an evident change (Table 3, entries 4 and 11), but we need a stoichiometric amount of it, to have a higher yield (Table 3, entries 9–13).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Copper precursor [M] | Ligand | Base (eq.) | Solvent | 7a Yieldb [%] | endo/exo |
| a All reactions were performed with 1.0 mmol phenyl maleimide, 1.5 mmol imino ester 5a and 2 mL of solvent at room temperature. b Isolated yield after SiO2 column chromatography. c Without previous activation [Cu] and ligand. d Previous activation [Cu] and ligand 30 min. e HFP is 1,1,1,3,3,3-hexafluoro-2-propanol. | ||||||
| 1 | CuBr | — | Et3N (1) | THF | 19 | — |
| 2c | CuBr | 2a | Et3N (1) | THF | 17 | — |
| 3d | CuBr | 2a | Et3N (1) | THF | 61 | — |
| 4 | 4a | — | Et3N (1) | THF | 75 | — |
| 5d | Cu(OTf) | 2a | Et3N (1) | THF | 54 | — |
| 6d | Cu(OAc) | 2a | Et3N (1) | THF | 53 | — |
| 7d | Cu(OTf)2 | 2a | Et3N (1) | THF | 65 | — |
| 8d | Cu(OAc)2 | 2a | Et3N (1) | THF | 33 | — |
| 9 | 4a | — | Et3N (0.5) | THF | 65 | — |
| 10 | 4a | — | Et3N (0.25) | THF | 55 | — |
| 11 | 4a | — | DIPEA (1) | THF | 73 | — |
| 12 | 4a | — | DIPEA (0.5) | THF | 60 | — |
| 13 | 4a | — | DIPEA (0.25) | THF | 54 | — |
| 14 | 4a | — | Et3N (1) | AcOEt | 83 | — |
| 15 | 4a | — | Et 3 N (1) | EtOH | 95 | Only endo |
| 16 | 4a | — | Et3N (1) | 1,4-Dioxane | 64 | — |
| 17 | 4a | — | Et3N (1) | CH3CN | 56 | — |
| 18 | 4a | — | Et3N (1) | Toluene | 78 | — |
| 19 | 4a | — | Et3N (1) | HFPe | 18 | — |
| 20 | 4b | — | Et3N (1) | EtOH | 75 | Only endo |
| 21 | 4c | — | Et3N (1) | EtOH | 80 | Only endo |
| 22 | 4d | — | Et3N (1) | EtOH | 85 | Only endo |

Next, several experiments were carried out with different solvents, and the best result corresponds to EtOH reaching 95% yield (Table 3, entry 15). In general, using polar solvents such as AcOEt and acetonitrile, we obtained good yields (Table 3, entries 14 and 17). This behavior suggests that the dimeric complex 4a is in equilibrium with its monomeric form in solution, forming 4a′ (vide infra). Finally, we studied the Sigma donor effect on the phosphine group, and for this, we tested 4a–d (Table 3, entries 15 and 20–22) in this cycloaddition reaction. All complexes catalyzed the reaction in good to excellent yields; the best result was obtained with 4a (Table 3, entry 15). The endo/exo ratio diastereoselectivity in product 7a was determined by 1H NMR, and in all cases, only the endo adduct was observed (Table 3, entries 15 and 20–22).
The previous results allowed us to expand the scope of the reaction, obtaining pyrrolidines with substituents in position 2. This was done using imines derived from essential amino acids such as methionine and serine, in addition to those already reported derivatives of alanine and phenylglycine (see the ESI†). Once the imines 5a–i were successfully obtained,22 they were exposed to the best reaction conditions, furnishing in all cases the endo-adducts 7, in good to excellent yields (Table 4, entries 1–5). This confirms that our methodology is effective for obtaining new pyrrolidines with different substituents at position 2 in a diastereoselective manner.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Product | Yieldb [%] |
| a All reactions were performed with 1.0 mmol phenyl maleimide 6, 1.5 mmol imino ester 5, 1.0 mmol Et3N, 2 mL of EtOH and 5%mol [Cu] 4a at room temperature. b Isolated yield after SiO2 column chromatography. | |||||
| 1 | Me | Et | p-Cl-C6H4 | 7a | 95 |
| 2 | Ph | Me | p-Cl-C6H4 | 7b | 75 |
| 3 | CH2CH2SCH3 | Et | p-Cl-C6H4 | 7c | 85 |
| 4 | CH2OH | Me | p-Cl-C6H4 | 7d | 80 |
| 5 | Me | Et | p-F-C6H4 | 7e | 74 |
| 6 | Me | Et | Ferrocenyl | 7f | 56 |
| 7 | Me | Et | 2-Thienyl | 7g | 89 |
| 8 | Me | Et | p-MeO-C6H4 | 7h | 79 |
| 9 | Me | Et | p-CF3-C6H4 | 7i | 85 |
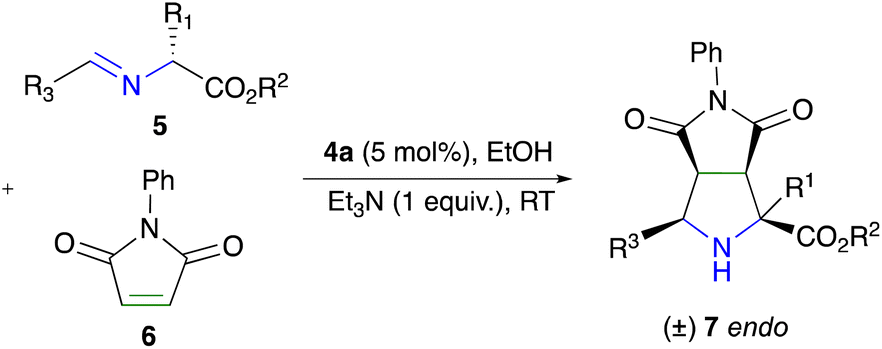
At this point, our interest focused on observing the behavior of the dinuclear complex 4a in obtaining endo-selectivity when the imine had a different electronic contribution at the R3 position. The results obtained had a pleasant trend in all cases, with yields ranging from moderate to good and only the endo adduct was observed (Table 4, entries 6–9).
The high endo-diastereoselectivity could be explained in two possible ways; a π interaction between the benzene ring in maleimide and the copper atom, or an N–Cu interaction between maleimide and the complex within the endo transition state. Subsequently, in order to gain insights into the origin of endo diastereoselectivity, we used N-methyl maleimide 8 as a dipolarophile, avoiding the possible Cu–π interaction. The reaction proceeded successfully in excellent yield (90%), furnishing only the endo diastereomer 9 (Scheme 3).
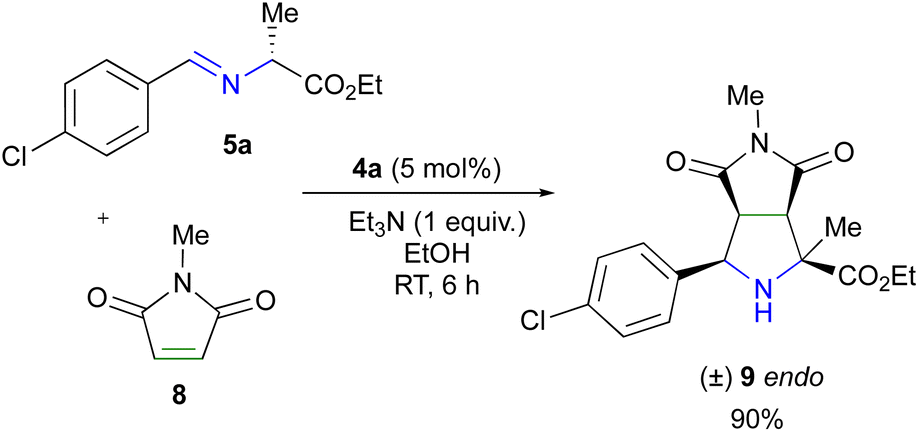 | ||
| Scheme 3 Study of the origin of endo diastereoselectivity. | ||
This finding suggests that the N–Cu interaction is responsible for the endo diastereoselectivity by anchoring the endo approximation between the azomethine ylide and the dipolarophile in the transition state.
Based on these results, we could propose the following reaction pathway to explain the endo product (Scheme 4). Initially, the metal complex 4a undergoes a cleavage, induced by the solvent, forming the complex 4a′. To probe this, we conducted a follow-up of the cleavage of 4a in the presence of EtOH by 31P-NMR (Fig. S152†). A similar behavior was observed in other copper(I) complexes.23 We found that the addition of one drop of EtOH to a solution of 4a in CD2Cl2 provokes a slight color change (colorless to pale-yellow) and the apparition of a new signal at −34.45 ppm. Likewise, we screened the UV-visible behavior of the solution of 4a by the successive addition of EtOH, detecting a slight bathochromic shift (∼10 nm) of the main absorption band placed at 238 nm (Fig. S153†). These results suggest the plausible cleavage of dimer 4a in the presence of ethanol. Then, the imine is complexed in two stages (A and B); the base takes a proton, forming the catalytically active species C. In this step, the presence of a π-acceptor phosphine as 4a (Table 1) in the catalytic precursor becomes critical to stabilize the formation of C. The follow-up of the reaction by 31P NMR reveals a new signal at −32.77 ppm. The Cu(I)-coordinated iminoester initiates the catalytic cycle, interacting with the dipolarophile forming the transition state D, which is responsible for the formation of the endo-7 product. Finally, we recorded a spectrum at the end of the reaction, detecting the presence of free ligand 2a and its corresponding oxide (18.57 ppm).
 | ||
| Scheme 4 Proposed pathway for the 1,3-cycloaddition catalyzed by complexes 4a′. | ||
DFT calculations were performed to find out if the proposed species D can explain the observed diasteroselectivity. The converged geometry of D also has a distorted trigonal pyramidal structure as in complex 4 but τ4 is significantly lower (0.59), due to the bite angle of the imine group. According to this calculation, the negative charge is delocalized between the imino group and the oxygen bonded to the copper, which could enhance its reactivity towards the maleimide group. Before the 3 + 2 addition, this charge distribution and the pyramidal structure of species C allow a strong intermolecular interaction (18.1 kcal mol−1) in an endo fashion between the π system of the imide and the base of the pyramid. The pyramid includes the imine, the metal center, and the substituents of phosphine.
The reduced density gradient (RDG) intermolecular isosurface that has been used to study non-covalent interactions,24–26 shows that there are three main interactions between the complex C and the maleimide group (blue regions in Fig. 5a). The most prominent is the dipolar-π staking interaction between the reactive carbons. The second most important interaction occurs between the imide moiety and the Cu atom. The last relevant interaction is between the phenyl group of the maleimide and the groups of phosphine. Non-interactions between the carbonyl groups of maleimide and copper were observed as suggested by Hu et al.,12j and were responsible for the endo-selectivity.
 | ||
| Fig. 5 (a) Intermolecular RDG surface of the interaction between species C and maleimide. The virial field [V(r)] is plotted over the isosurface. V(r) color code: ≤−5.0 × 10−3 a.u. (dark blue), −2.5 × 10−3 a.u. (green) and 0.0 a.u. (red). Endo (solid line) and exo (dashed line) reaction paths of the [3 + 2] dipolar cycloaddition for (b) phenyl and (c) methyl maleimide. Relative energies in kcal mol−1. | ||
This result was also confirmed from the bond paths that were obtained using the quantum theory of atoms in molecules (QTAIM); see the ESI.†
We searched for an energy minimum of the metal complex-maleimide dimer with an exo arrangement, but all the attempts ended in the endo geometry, which indicates that the imide preferably approaches species C with an endo orientation. After this approach, it is still possible to form an exo transition state (TS), but according to the calculation, this TS is significantly more energetic than the endo TS (8.1 kcal mol−1, Fig. 5b). Moreover, the endo product is also less energetic than the exo product. Changing the phenyl group of the maleimide for a methyl group, as was performed experimentally does not change the energetic order of the endo and exo TS, which coincides with the experimental observations. However, the energy difference between these two TSs is reduced (4.4 kcal mol−1, Fig. 5c), which confirms that the intermolecular interactions of the phenyl group are important but are not the only relevant ones. The calculations show that the pyramidal structure of the Cu complex allows the formation of several intermolecular interactions that are responsible for the endo-diasteroselectivity.
With these results in our hands, we decided to conduct a reaction using the nitrogen-free substrate 10 under the same reaction conditions standardized for this work (Scheme 5). The reaction was completed after one hour in an 88% yield, and the endo/exo ratio diastereoselectivity was determined by 1H NMR, observing a 50:50 ratio of endo–exo adducts. These results agree with the mechanistic proposal since they put into evidence the role of the nitrogen atom included in the dipolarophile.
 | ||
| Scheme 5 Synthesis of pyrrolidines 11. | ||
Comparing the catalytic performance of 4a in this reaction with the results obtained for other catalytic systems (Table 5), we observed that our catalytic system exhibits a similar behavior to that of other catalytic systems involving [P,S]- or [N,N]-ligands. This catalytic system is robust and air-moisture stable.
| Entry | Ligand | Conditions | endo-Adduct | Time | Examples | Ref. |
|---|---|---|---|---|---|---|
| a Pre-formation of the copper complex in situ, 1 h. b Copper complex 4a synthesized and isolated. | ||||||
| 1 |
|
Cu(MeCN)4ClO4 (3 mol%) |

|
15–60 min | 6 | 12m |
| (R)-L1 (3 mol%) | ||||||
| Et3N (18%), CH2Cl2 | ||||||
| 4 Å MS, −10 °C | ||||||
| 2 |

|
Cu(MeCN)4ClO4 (5 mol%) |

|
24 h | 10 | 12l |
| L2 (5.5 mol%)a | ||||||
| iPrNEt (10 mol), CH2Cl2 | ||||||
| −40 °C | ||||||
| 3 |

|
CuBF4 (3 mol%) |

|
10 min | 13 | 12k |
| L3 (3 mol%)a | ||||||
| Et3N (15%), CH2Cl2 | ||||||
| 0 °C | ||||||
| 4 |

|
4a (5 mol%)b |

|
0.5–1 h | 10 | This work |
| Et3N (1 eq.), EtOH, RT | ||||||
Experimental
General considerations
All operations were carried out under an inert atmosphere of argon gas using standard Schlenk techniques. Column chromatography was performed using 70–230 mesh silica gel. All reagents and solvents were obtained from commercial suppliers and used without further purification. NMR spectra were obtained with a Bruker Avance III at 300 MHz for 1H NMR, 75 MHz for 13C{1H} NMR, and 121 MHz for 31P NMR using CDCl3 as the solvent. Chemical shifts are expressed in ppm (δ), relative to TMS. All compounds were characterized by IR spectroscopy recorded on a PerkinElmer Spectrum 100 FT-IR equipped with an ATR accessory, and all data are expressed in wavenumbers (cm−1). Melting points were obtained on a Melt-Temp II apparatus and were left uncorrected. The MS-FAB spectra were obtained on a JMS-SX102A using nitrobenzyl alcohol and polyethylene glycol matrices. MS-DART spectra were obtained on an AccuTOF JMS-T100LC; the values of the signals are expressed in mass/charge units (m/z).UV-Vis absorption spectra were recorded using a Thermo Scientific Evolution 60S UV-visible spectrophotometer, using spectrophotometric grade CH2Cl2 solvent purchased from Sigma-Aldrich Co. and a 1 cm quartz cell.
Structure determination by X-ray crystallography
Suitable X-ray-quality crystals of 4a–d were grown by slow evaporation of a mixture of diethyl ether/hexane at −5 °C, respectively. The crystals of each compound were mounted on a glass fiber at room temperature. The crystals of 4a–d were then placed on a Bruker D8 Venture (4a–b) or APEX-II CCD (4c–d) with Mo-Kα radiation; the decay was negligible in all cases. Details of the crystallographic data collected for compounds 4a–d are provided in Table 6. Systematic absences and intensity statistics were used in space group determinations. The structure was solved using direct methods.27 Anisotropic structure refinements were achieved using the full matrix least-squares technique on all non-hydrogen atoms. All hydrogen atoms were placed in idealized positions based on hybridization, with isotropic thermal parameters fixed at 1.2 times the value of the attached atom. Structural solutions and refinements were performed using SHELXTL V6.10.28 Crystallographic data for 4c and 4d are available in the CIF format in the ESI.† The X-ray crystallographic structures of compounds 4a–d are available in the CCDC.| Compound | 4a | 4b | 4c | 4d |
|---|---|---|---|---|
| Empirical formula | C36 H38 Br2 Cu2 N4 P2 | C28H54Br2Cu2N4P2 | C24 H46 Br2 Cu2 N4 P2 | C32 H54 Br2 Cu2 N4 P2 |
| Formula weight (g mol−1) | 875.54 | 795.59 | 739.49 | 843.63 |
| Crystal size (nm) | 0.414 × 0.382 × 0.222 | 0.377 × 0.354 × 0.184 | 0.408 × 0.378 x0.336 | 0.405 × 0.362 × 0.238 |
| Color | Colorless | Colorless | Colorless | Colorless |
| Crystal system | Triclinic | Monoclinic | orthorhombic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | Pc | P21/c |
| a (Å) | 8.817(3) | 14.1029(8) | 48.617(2) | 8.7269(3) |
| b (Å) | 9.727(3) | 9.0827(4) | 12.8598(5) | 16.8301(7) |
| c (Å) | 12.564(4) | 15.0051(7) | 15.4227(6) | 24.7526(10) |
| α (°) | 94.751(12) | 90 | 90 | 90 |
| β (°) | 105.422(11) | 108.427(2) | 90 | 93.2040(10) |
| γ (°) | 99.772(12) | 90 | 90 | 90 |
| V (Å3) | 1014.3(6) | 1823.49(16) | 9642.3(7) | 3629.8(2) |
| Z | 1 | 2 | 12 | 4 |
| Temperature (K) | 298 | 298 | 100 | 100 |
| D calc (g cm−3) | 1.433 | 1.449 | 1.528 | 1.544 |
| Number of collected reflections | 25914 |
20959 |
116095 |
48837 |
| Number of independent reflections (Rint) | 4839, Rint = 0.0655 | 5114, Rint = 0.0623 | 30479, Rint = 0.0637 |
12070, Rint = 0.036 |
| Maximum and minimum transmission | 0.4091 and 0.7460 | 0.5405 and 0.746 | 0.572 and 0.7462 | 0.6068 and 0.7464 |
| Data/parameters | 4839/210 | 20959/180 |
30479/1023 |
12070/383 |
| Final R indices | R = 0.0368 | R = 0.0632 | R = 0.0617 | R = 0.0289 |
| [I > 2σ(I)] | wR2 = 0.0864 | wR2 = 0.1499 | wR2 = 0.1357 | wR2 = 0.0668 |
| R indices (all data) | R = 0.0679, wR2 = 0.076 | R = 0.1278, wR2 = 0.1277 | R = 0.0744, wR2 = 0.1312 | R = 0.0403, wR2 = 0.0636 |
| GoF(F2) | 1.029 | 1.022 | 1.149 | 1.022 |
| Absorption correction method | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| Clave | 2295705 | 2295706 | 2295707 | 2295708 |
Theoretical methods
All geometry optimizations were performed in Gaussian 1629 with the M06/def2-TZVP level of theory. A frequency calculation was performed for all the structures to confirm that the TS and the energy minimum have 1 or 0 imaginary frequencies, respectively. The obtained wave functions from these calculations were used to calculate all the density properties with the AIMALL software.30 The RDG isosurface of the non-covalent interaction between species D and the maleimide moiety was plotted with the ParaView program. Over this surface, besides the V(r) discussed in the main text, was also plotted the density multiplied by the sign of the second eigenvalue of its hessian (sign(λ2)ρ), which is the property that is usually used in the NCI method.31 However, this plot does not show which are the most important interactions (see the ESI†). Moreover, recently it has been proved that sign (λ2)ρ is not appropriate for studying the strength of π–π interactions,32 because of that we used V(r) instead of essential experimental procedures/data.Synthesis of ligands 2a–d
The synthesis of ligands 2a–d was conducted following the procedures reported in the literature.14,15Synthesis of complex 4a–d
In a 50 mL Schlenk flask was placed the corresponding ligand 2 (0.5 mmol, 1 equiv.), followed by the addition of CuBr (0.5 mmol, 1 equiv.). The flask was then purged with N2, and anhydrous CH2Cl2 (5 mL) was added. The reaction mixture was stirred for approximately 1 hour at room temperature. Upon completion of the reaction, the solvent was removed under vacuum, leaving behind a white residue, which was then washed with hexane to obtain the pure complex 4.General procedure for the 1,3 dipolar cycloaddition reaction
To a solution of the appropriate imino ester (1.2 mmol) in 3.0 mL of EtOH were added 0.17 mL of Et3N (1.2 mmol) and N-phenyl maleimide (174 mg, 1 mmol). Then, 0.03 mmol (27 mg) of 4a were added under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 3 hours. Then, the reaction mixture was evaporated under vacuum and the crude was redissolved with 5 mL of CH2Cl2 and washed with H2O (3 × 15 mL). The organic phase was dried with anhydrous sodium sulfate and then filtered through a pad of Celite and alumina, using 5.0 mL of CH2Cl2. Subsequently, the solvent was removed under reduced pressure. The crude was further purified through silica gel chromatography (eluent: 7:3 hexanes/EtOAc) to obtain the corresponding adducts.
Conclusions
In summary, we have developed a new family of Cu(I) dinuclear complexes based on [N,P]-pyrrole ligands. These complexes are air-stable dimers that contain bromine atoms as μ-bridges and exhibit a five-membered metallacycle with a highly distorted trigonal pyramidal geometry in all cases.The developed catalytic system allows us to obtain substituted pyrrolidines with high endo-diastereoselectivity under mild conditions, with yields ranging from good to excellent. Likewise, theoretical experiments showed that the diastereoselectivity arises from a strong N–Cu interaction between the maleimide substrate and the copper catalytic species in the cycloaddition transition state, resulting in the formation of the endo adduct in all cases. The development of the chiral version of these ligands is in progress in our laboratories. In this way, the catalytic system can be used in the synthesis of natural and pharmaceutical products that require diastereoselective conditions with a high degree of reliability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the technical assistance provided by M. Carmen García González, M. Paz Orta, Isabel Chávez, and César I. Sandoval Chávez and Diego Martínez Otero. We also thank the projects PAPIIT IN216323, and DGTIC (project LANCAD-UNAM-DGTIC-304) for the computer time. Miguel A. Alvarado-Castillo also thanks CONACYT for the Ph.D. grant.References
- For selected reviews on OLED uses see: (a) C. E. Hosecroft and E. C. Constable, J. Mater. Chem. C, 2022, 10, 4456–4482 RSC; (b) V. K.-M. Au, Energy Fuels, 2021, 35, 18982–18999 CrossRef CAS; (c) H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508–3535 CrossRef CAS PubMed.
- For selected references see: (a) F. Doettinger, Y. Yang, M. Karnahl and S. Tschierli, Inorg. Chem., 2023, 62, 8166–8178 CrossRef CAS PubMed; (b) F. Doettinger, M. Obermeir, V. Caliskanyürek, L. E. Burmeister, C. Kleeberg, M. Karnahl, M. Schwalbe and S. Tschierli, ChemCatChem, 2023, 15, e202300452 CrossRef CAS; (c) F. Sueyoshi, X. Zhang, K. Yamauchi and K. Sakai, Angew. Chem., Int. Ed., 2023, 62, e202217807 CrossRef CAS PubMed; (d) B. Ma, Q. Xia, D. Wang, J.-K. Jin, Z. Li, Q.-J. Liang, M.-Y. Sun, D. Liu, L.-J. Liu, H.-X. Shu, J. Yang, D. Li and J. He, Angew. Chem., Int. Ed., 2023, 62, e202300233 CrossRef CAS PubMed; (e) C. Dragonetti, M. Magni, A. Colombo, F. Fagnani, D. Roberto, F. Melchiorre, P. Biagni and S. Fantacci, Dalton Trans., 2019, 48, 9703–9711 RSC.
- J.-B. Xie, S. Lin, S. Qiao and G. Li, Org. Lett., 2016, 18, 3926–3929 CrossRef CAS PubMed.
- A. Buchcic, A. Zawisza, S. Lésniak and M. Rachwalski, Catalysts, 2020, 10, 971 CrossRef CAS.
- M. C. Schwarzer, A. Fujioka, T. Ishii, H. Ohmiya, S. Mori and M. Sawamura, Chem. Sci., 2018, 9, 3484–3493 RSC.
- B. V. Rokade and P. J. Guiry, J. Org. Chem., 2019, 84, 5763–5772 CrossRef CAS PubMed.
- For selected reviews on 1,3-dipolar cycloadditions, see: (a) S. Dubey, A. Pal, S. Roy, S. Sasmal, A. Tamrakar, R. Jana and T. Das, New J. Chem., 2023, 47, 8997–9034 RSC; (b) S. V. Kumar and P. J. Guiry, Chem. – Eur. J., 2023, 29, e202300296 CrossRef CAS PubMed; (c) J. Adrio and J. C. Carretero, Chem. Commun., 2019, 55, 11979–11991 RSC; (d) M. Ríos-Gutiérrez and L. R. Domingo, Eur. J. Org. Chem., 2019, 267–282 CrossRef; (e) J. Adrio and J. C. Carretero, Chem. Commun., 2011, 47, 6784–6794 RSC; (f) A. Cózar and F. P. Cossío, Phys. Chem. Chem. Phys., 2011, 13, 10858–10868 RSC.
- (a) Z.-H. Wang, J.-H. Liu, Y.-P. Zhang, J.-Q. Zhao, Y. You, M.-Q. Zhou, W.-Y. Han and W.-C. Yuan, Org. Lett., 2022, 24, 4052–4057 CrossRef CAS PubMed; (b) S. Furuya, K. Kanemoto and S.-I. Fukuzawa, Chem. – Asian J., 2022, 17, e202200239 CrossRef CAS PubMed; (c) A. G. Taha, E. E. Elboray, Y. Kobayashi, T. Furuta, H. H. Abas-Temirek and M. F. Aly, J. Org. Chem., 2021, 86, 547–558 CrossRef CAS PubMed; (d) W.-R. Zhu, Q. Su, N. Lin, Q. Chen, Z.-W. Zhang, J. Weng and G. Lu, Org. Chem. Front., 2020, 7, 3452–3458 RSC; (e) J. Otero-Fraga, S. Suárez-Pantiga, M. Montesinos-Magraner, D. Rhein and A. Mendoza, Angew. Chem., Int. Ed., 2017, 56, 12962–12966 CrossRef CAS PubMed.
- (a) S. V. Kumar and P. J. Guiry, Angew. Chem., Int. Ed., 2022, 61, e202205516 CrossRef CAS PubMed; (b) Y. Yi, Y.-Z. Hua, H.-J. Lu, L. T. Liu and M.-C. Wang, Org. Lett., 2020, 22, 2527–2531 CrossRef CAS PubMed; (c) I. N. Chaithanya Kiran, K. Fujita, S. Tanaka and M. Kitamura, ChemCatChem, 2020, 12, 5613–5617 CrossRef CAS.
- Z. T. Gugkaeva, M. V. Panova, A. F. Smol'yakov, M. G. Medvedev, A. T. Tsaloev, I. A. Godovikov, V. I. Maleev and V. A. Larionov, Adv. Synth. Catal., 2022, 364, 2395–2402 CrossRef CAS.
- (a) Q. Hou, Y. You, X. Song, Y. Wang, K. Chen and H. Wang, Catalysts, 2020, 10, 28 CrossRef; (b) Y.-P. Zhang, Y. You, J.-Q. Zhao, X.-J. Zhou, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Org. Chem. Front., 2019, 6, 1879–1884 RSC; (c) M. Zhi, Z. Gan, R. Ma, H. Cui, E.-Q. Li, Z. Duan and F. Mathey, Org. Lett., 2019, 21, 3210–3213 CrossRef CAS PubMed; (d) A. Cayuelas, O. Larranaga, V. Selva, C. Nájera, T. Akiyama, J. M. Sansano, A. Cozar, J. I. Miranda and F. P. Cossio, Chem. – Eur. J., 2018, 24, 8092–8097 CrossRef CAS PubMed; (e) X. Zheng, Q. Deng, Q. Hou, K. Zhang, P. Wen, S. Hu and H. Wang, Synthesis, 2018, 50, 2347–2358 CrossRef CAS.
- (a) B.-R. Wang, Y.-B. Li, Q. Zhang, D. Gao, P. Tian, Q. Li and L. Yin, Nat. Chem., 2023, 14, 4688 CAS; (b) X. Chang, X.-T. Liu, F. Li, Y. Yang, L. W. Chung and C.-J. Wang, Chem. Sci., 2023, 14, 5460–5469 RSC; (c) X. Xu, L. Bao, L. Ran, Z. Yang, D. Yan, C.-J. Wang and H. Teng, Chem. Sci., 2022, 13, 1398–1407 RSC; (d) H. Cui, K. Li, Y. Wang, M. Song, C. Wang, D. Wei, E.-Q. Li, Z. Duan and F. Mathey, Org. Biomol. Chem., 2022, 18, 3740–3746 RSC; (e) H. Deng, T. T. Liu, Z. D. Ding, W. L. Yang, Y. Luo and W. P. Deng, Org. Chem. Front., 2020, 7, 3247–3252 RSC; (f) S. N. Greszler, G. Zhao, M. Buchman, X. B. Searle, B. Liu and E. A. Voight, J. Org. Chem., 2020, 85(11), 7620–7632 CrossRef CAS PubMed; (g) G. S. Caleffi, O. Larrañaga, M. Ferrándiz-Sapewras, P. R. R. Costa, C. Nájera, A. Cozar, F. P. Cossío and J. M. Sansano, J. Org. Chem., 2019, 84, 10593–10605 CrossRef CAS PubMed; (h) H. Deng, R. Jia, W. L. Yang, Z. Yu and W. P. Deng, Chem. Commun., 2019, 55, 7346–7349 RSC; (i) F. Tian, F.-S. He, H. Deng, W.-L. Yang and W.-P. Deng, Org. Lett., 2018, 20, 3838–3842 CrossRef CAS PubMed; (j) F.-Z. Han, S.-B. Yu, C. Zhang and X.-P. Hu, Tetrahedron, 2016, 72, 2616–2622 CrossRef CAS; (k) C.-J. Wang, G. Liang, Z.-Y. Xue and F. Gao, J. Am. Chem. Soc., 2008, 130, 17250–17251 CrossRef CAS PubMed; (l) M. Shi and J. W. Shi, Tetrahedron: Asymmetry, 2007, 18, 645–650 CrossRef CAS; (m) S. Cabrera, R. Gómez Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2005, 127, 16394–16395 CrossRef CAS PubMed.
- (a) F. Cheng, S. J. Kalita, Z.-N. Zhao, X. Yang, Y. Zhao, U. Schneider, N. Shibat and Y.-Y. Huang, Angew. Chem., Int. Ed., 2019, 58, 16637–16643 CrossRef CAS PubMed; (b) I. Rivilla, A. Cózar, T. Schäfer, F. J. Hernandez, A. M. Bittner, A. Eleta-Lopez, A. Aboudzadeh, J. I. Dsantos, J. I. Miranda and F. Cossío, Chem. Sci., 2017, 8, 7038–7043 RSC; (c) F.-F. Tang, W.-L. Yang, X. Yu and W.-P. Deng, Catal. Sci. Technol., 2015, 5, 3568–3575 RSC; (d) L. M. Castelló, C. Nájera, J. M. Sansano, O. Larrañaga, A. Cózar and F. P. Cossío, Synthesis, 2015, 47, 934–943 CrossRef.
- J. V. Súarez-Meneses, E. Bonilla-Reyes, E. A. Blé-González, M. C. Ortega-Alfaro, R. A. Toscano, A. Cordero-Vargas and J. G. López-Cortés, Tetrahedron, 2014, 70, 1422–1430 CrossRef.
- (a) J. V. Suarez-Meneses, A. Oukhrib, M. Gouygou, M. Urrutigoïty, J.-C. Daran, A. Cordero-Vargas, M. C. Ortega-Alfaro and J. G. López-Cortés, Dalton Trans., 2016, 23, 9621–9630 RSC; (b) S. Cortés-Mendoza, D. Adamczyk, J. Badillo-Gómez, M. Urrutigoity, M. C. Ortega-Alfaro and J.-G. López-Cortés, Adv. Synth. Catal., 2022, 364, 2837–2845 CrossRef.
- E. P. Sánchez-Rodríguez, S. Cortés-Mendoza, J.-C. Daran, M. C. Ortega-Alfaro, J. G. López-Cortés and M. Gouygou, Appl. Organomet. Chem., 2020, 34, e5709 CrossRef.
- E. P. Sánchez-Rodríguez, A. J. Fragoso-Medina, E. Ramírez-Meneses, M. Gouygou, M. C. Ortega-Alfaro and J. G. López-Cortés, Catal. Commun., 2018, 115, 49–54 CrossRef.
- Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610–617 CrossRef CAS PubMed.
- W. D. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51–54 RSC.
- (a) A. W. Addison and T. Nageswara Rao, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC; (b) S. R. Harutyunyan, F. López, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A. Meetsma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 9103–9118 CrossRef CAS PubMed.
- (a) R. Czerwieniec, K. Kowalski and H. Yersin, Dalton Trans., 2013, 42, 9826–9830 RSC; (b) M. Knorn, T. Rawner, R. Czerwieniec and O. Reiser, ACS Catal., 2015, 5, 5186–5193 CrossRef CAS.
- S. Xu, Z.-M. Zhang, B. Xu, B. Liu, Y. Liu and J. Zhang, J. Am. Chem. Soc., 2018, 140, 2272–2283 CrossRef CAS PubMed.
- W. Liang, K. Nakajima, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2019, 58, 1168–1173 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- G. Saleh, C. Gatti and L. Lo Presti, Comput. Theor. Chem., 2015, 1053, 53–59 CrossRef CAS.
- Y. Cornaton and J.-P. Djukic, Acc. Chem. Res., 2021, 54, 3828–3840 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Canalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- T. A. Keith, AIMAll 2019, TK Gristmill Software, Overland Park KS, USA, https://aim.tkgristmill.com Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- D. Morales-Pumarino and J. E. Barquera-Lozada, Int. J. Quantum Chem., 2023, 123, e27051 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of all compounds. CCDC 2295705–2295708 (4a–b). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03692h |
| This journal is © The Royal Society of Chemistry 2024 |