
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ordering by cation replacement in the system Na2−xLixGa7†
Chia-Chi
Yu
a,
Yurii
Prots
a,
Alim
Ormeci
a,
Mitja
Krnel
 a,
Marcus
Schmidt
a,
Lev
Akselrud
ab,
Frank R.
Wagner
a,
Yuri
Grin
a and
Michael
Baitinger
*a
a,
Marcus
Schmidt
a,
Lev
Akselrud
ab,
Frank R.
Wagner
a,
Yuri
Grin
a and
Michael
Baitinger
*a
aMax-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Str. 40, 01187 Dresden, Germany. E-mail: Michael.Baitinger@cpfs.mpg.de
bIvan Franko Lviv National University, Kyryla i Mefodia St. 6, 29005 Lviv, Ukraine
First published on 15th December 2023
Abstract
Samples of the pseudo-binary system Na2−xLixGa7 (x ≤ 1) were synthesized from the elements at 300 °C in sealed Ta ampoules or by the reaction of Na2Ga7 with LiCl. The peritectic formation temperature decreases with increasing Li content from 501(2) °C (x = 0) to 489(2) °C (x = 1). The boundary compositions Na2Ga7 and Na1Li1Ga7 crystallize with different structure types related by a group–subgroup relation. While the Na-rich compositions (x ≤ 0.5) represent a substitutional solid solution (space group Pnma), the Li-rich compositions feature an unconventional replacement mechanism in which Li atoms occupying interstitial positions induce vancancies at the Na positions (space group Cmce). The crystal structure of Na1Li1Ga7 (a = 8.562(1) Å, b = 14.822(2) Å, c = 11.454(2) Å; Z = 8) was determined from X-ray single-crystal diffraction data, and reveals an anionic framework comprising 12-bonded Ga12 icosahedra and 4-bonded Ga atoms, with alkali–metal atoms occupying channels and cavities. The arrangement of cations makes NaLiGa7 a new structure type within the MgB12Si2 structure family. Band structure calculations for the composition NaLiGa7 predict semiconducting behavior consistent with the balance [Na+]2[Li+]2[(Ga12)2−][Ga–]2, considering closo Wade clusters [(12b)Ga12]2− and Zintl anions [(4b)Ga]−. Susceptibility measurements indicate temperature-independent diamagnetic behavior.
Introduction
Inorganic framework compounds composed of p-elements and encapsulated metal cations are frequently studied.1,2 Their crystal structures show the characteristic bonding features of the framework atoms involved. While clathrate-type structures are commonly observed among frameworks based on 4-bonded group 14 elements,1 frameworks of group 13 elements lack valence electrons to solely form two-center, two-electron bonds between all framework atoms.2 They typically adopt polyhedral Wade clusters, such as closo [B6]2− in CaB6,3 or form multiatomic bonds, like in BaAl4,4 which may be considered as a framework of 2D edge-condensed nido clusters. An example of the first type is the recently discovered Na2Ga7,5 which exhibits a framework of 12-bonded [Ga12]2−closo clusters and 4-bonded [Ga]– Zintl anions. The electronic balance is achieved by the Na cations according to [Na+]4[(Ga12)2−][Ga–]2. The structure of this family can also be formed with heteroatomic frameworks and varying cation contents, as evidenced by the boride silicides MgB12Si26 and Li2B12Si2,7 which are composed of [B12]2−closo clusters and 4-bonded Si atoms. In contrast to the boride silicides, the higher number of cations in Na2Ga7 (i.e. Na4Ga12Ga2) leads to their disorder in the Ga channels. In this study, we investigate the pseudo-binary system Na2Ga7/NaLiGa7 to determine the response of the Ga framework on the cation substitution and the possibility to obtain new members of the MgB12Si2 family.Experimental details
Preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and LiCl (Chempur, 99.9%) were mixed with molar ratios of 1:1 or 1:1.5. These mixtures were pressed into pellets (d = 10 mm, h = 2 mm), sealed in a Ta crucible under an Ar atmosphere, and placed in an evacuated glass vessel. The vessel was annealed at 300 °C in a tube furnace for one week and then cooled down to room temperature by switching off the furnace.
5 and LiCl (Chempur, 99.9%) were mixed with molar ratios of 1:1 or 1:1.5. These mixtures were pressed into pellets (d = 10 mm, h = 2 mm), sealed in a Ta crucible under an Ar atmosphere, and placed in an evacuated glass vessel. The vessel was annealed at 300 °C in a tube furnace for one week and then cooled down to room temperature by switching off the furnace.
Characterization
Results and discussion
Preparation of Na2−xLixGa7 (x ≤ 1)
When a stoichiometric mixture of the elements at composition NaLiGa7 was melted and cooled down to room temperature within a few minutes, the resulting product predominantly consisted of NaLiGa7, along with smaller amounts of Li3Ga1412 and an unidentified phase, as determined by PXRD analysis. This behaviour notably differs from the preparation of Na2Ga7, which does not form directly from the melt.5 However, the presence of foreign phases indicates that NaLiGa7 forms incongruently as well. Subsequent annealing at 300 °C for one week led to the formation of single-phase products for all compositions Na2−xLixGa7, with x = 0.2, 0.5, 0.8 and 1 as confirmed by PXRD analysis (Fig. 1). For the sample with x = 1.5, the majority phase was Na1Li1Ga7, accompanied by foreign phases (Table 1). During annealing, the sample melted partially, indicating that the identified phases, Na1Li1Ga7, Li3Ga14, LiGa2 and LiGa6,13 were not in equilibrium upon cooling. Therefore, we conclude that the upper limit for substitution of Na atoms is reached at x = 1. Furthermore, a homogeneity range Na1Li1+δGa7 with a higher Li content was not detected by refinement of the lattice parameters. The product with nominal composition Na1Li1.2Ga7 consisted of Na1Li1Ga7 and exhibited weak reflections of an unknown phase.
 | ||
| Fig. 1 PXRD patterns of Na2−xLixGa7 (x = 0, 0.5, 1) after annealing at 300 °C (Cu Kα1). The calculated patterns of Na2Ga7 (space group Pnma, red lines) and NaLiGa7 (space group Cmce, blue lines) are based on single-crystal diffraction data. For Na1.5Li0.5Ga7 and Na2Ga7, some characteristic reflections breaking the C-centered lattice are indexed. In NaLiGa7, symmetry inequivalent reflections overlap due to the hexagonal axis ratio b/a (Table 1). | ||
| Nominal composition | x | Lattice parameters | Space group | b/a | |||
|---|---|---|---|---|---|---|---|
| a/Å | b/Å | c/Å | V/Å3 | ||||
| Na2Ga7 | 0 | 8.6766(6) | 14.8580(6) | 11.6105(5) | 1497.1(1) | Pmnb | 1.7124(1) |
| Na1.8Li0.2Ga7 | 0.2 | 8.646(2) | 14.847(3) | 11.558(3) | 1483.7(6) | Pmnb | 1.7172(5) |
| Na1.5Li0.5Ga7 | 0.5 | 8.5956(9) | 14.834(1) | 11.488(2) | 1464.8(3) | Pmnb | 1.7257(2) |
| Na1.2Li0.8Ga7 | 0.8 | 8.5692(9) | 14.836(2) | 11.466(2) | 1457.7(4) | Cmce | 1.7313(3) |
| Na1.0Li1.0Ga7 | 1 | 8.562(1) | 14.822(2) | 11.454(2) | 1453.6(4) | Cmce | 1.7311(3) |
| Na0.5Li1.5Ga7 | 1.5 | 8.584(7) | 14.80(2) | 11.444(6) | 1453(4) | Cmce | 1.724(3) |
| Na1.0Li1.2Ga7 | — | 8.558(1) | 14.820(2) | 11.453(2) | 1452.6(3) | Cmce | 1.7317(3) |
Other than from direct synthesis, Na1Li1Ga7 can be formed through a metathesis reaction of Na2Ga7 and LiCl (eqn (1)). The reaction is supported by the higher stability of NaCl compared to LiCl increasing with temperature.14 When Na2Ga7 and LiCl were reacted in a 1:1 molar ratio at 300 °C, the product consisted of Na1Li1Ga7 and NaCl as confirmed by PXRD analysis (Table 2).
| Method | Lattice parameters | Space group | |||
|---|---|---|---|---|---|
| a/Å | b/Å | c/Å | V/Å3 | ||
| Cast product | 8.554(2) | 14.826(3) | 11.457(2) | 1453.0(5) | Cmce |
| Annealing at 300 °C | 8.562(1) | 14.822(2) | 11.454(2) | 1453.6(4) | Cmce |
| 1 Na2Ga7 + 1 LiCl | 8.5622(9) | 14.828(2) | 11.458(2) | 1454.7(4) | Cmce |
No residual LiCl was detected, and the refined lattice parameter of NaCl (a = 5.6400(7) Å) matched the reported value.15 This indicates that the cation substitution was quantitative.
| 1 Na2Ga7 + 1 LiCl → 1 Na1Li1Ga7 + 1 NaCl | (1) |
When Na2Ga7 was reacted with 1.5 equivalents of LiCl at 300 °C, a mixture of Na2−xLixGa7, Li3Ga14 and NaCl was observed in PXRD. The overlapping reflections lowered the precision of the lattice parameter refinement, but the obtained values are consistent with the composition NaLiGa7. This finding confirms that Li can substitute up to one equivalent of Na in Na2Ga7. With an excess amount of LiCl, Na1Li1Ga7 was not formed. Reacting an equimolar mixture of NaLiGa7 and LiCl resulted in the formation of binary gallides Li3Ga14, LiGa2 and NaGa4.16
Thermal stability of the composition range Na2−xLixGa7
The binary phase Na2Ga7 forms peritectically at 501(2) °C from Na7Ga1317 and the melt.5 Regarding the ternary system Na2−xLixGa7, the thermal stability decreases with increasing Li content. A single-phase sample of Na1.8Li0.2Ga7 revealed a slightly lower decomposition temperature of T = 498(2) °C. At the maximum Li-content Na1Li1Ga7, the formation temperature is further reduced to T = 489(2) °C. At all compositions of Na2−xLixGa7, the presence of two signals in the DTA experiments indicates that the samples undergo incongruent melting (Table S6†).
The thermal decomposition of Na1Li1Ga7 was investigated on bulk material to examine the possibility of the formation of a new ternary phase through the evaporation of Na. However, no such phase was observed. Under static vacuum conditions at 300 °C, no reaction was observed, and the initial material remained unchanged for a period of 1 week. At 400 °C, after 1 day, sodium droplets were observed in the cooler region of the glass reactor. PXRD patterns of the solid residue consisted of NaLiGa7, minor amounts of Li3Ga14 and amorphous Ga, as indicated by its characteristic background. When the reaction time was prolonged to 3 days, only Li3Ga14 was detected along with contributions of amorphous Ga (eqn (2)).
| 3 Na1Li1Ga7 → 1 Li3Ga14 + 7 Ga + 3 Na↑ | (2) |
Crystal structure of Na2−xLixGa7
5 and refined with isotropic ADPs. Subsequently, the positions of cations were determined. The partial occupancy of Na and Li positions could only be approximated.
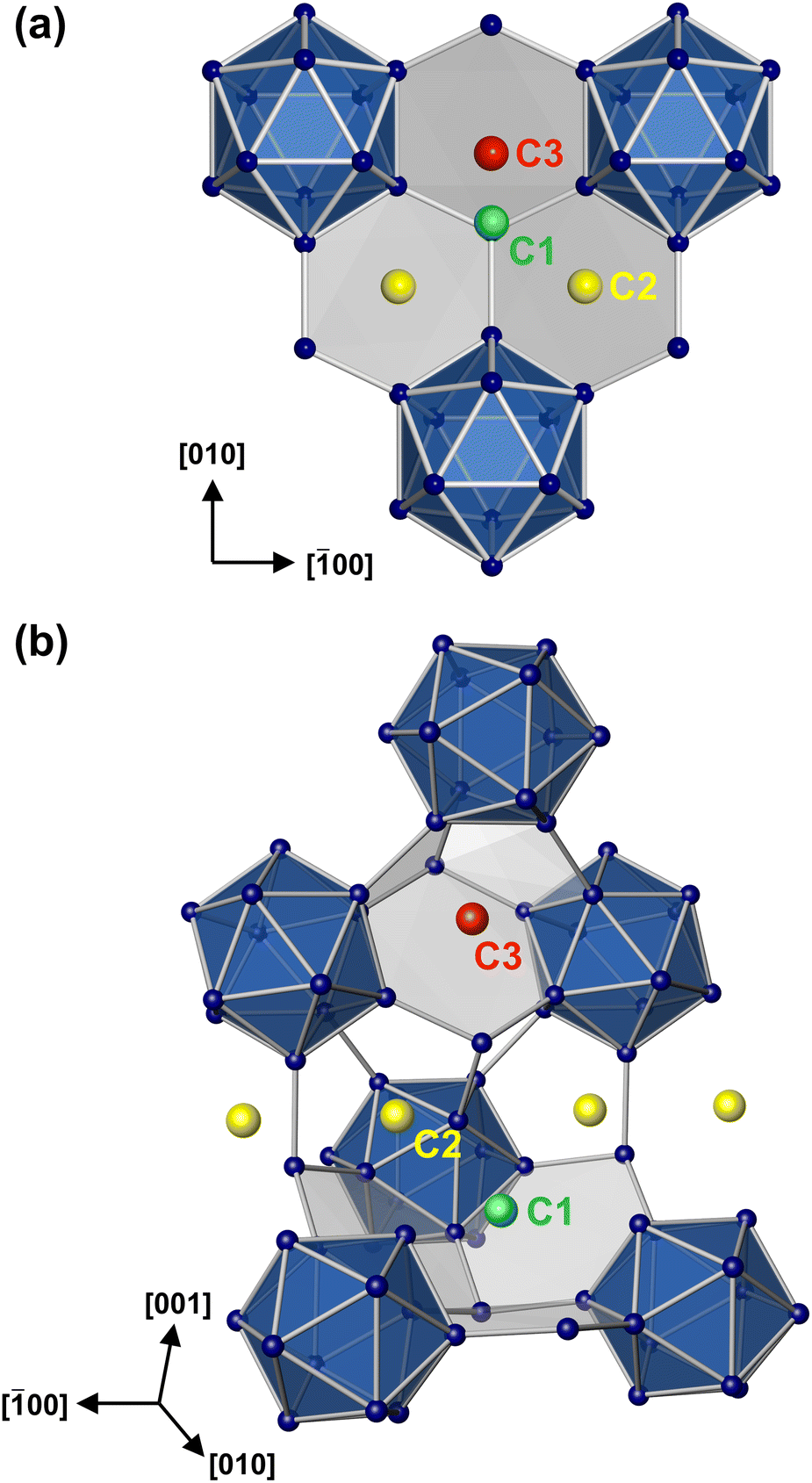 | ||
| Fig. 2 (a) Local environment of the cation positions C1, C2 and C3 in NaLiGa7. (b) Positions C1 and C3 are alternatively occupied. | ||
 | ||
| Fig. 3 Cation positions for the representatives of the MgB12Si2 family: (a) Li2B12Si2, (b) MgB12Si2, (c) Na2Ga7 and (d) NaLiGa7. | ||
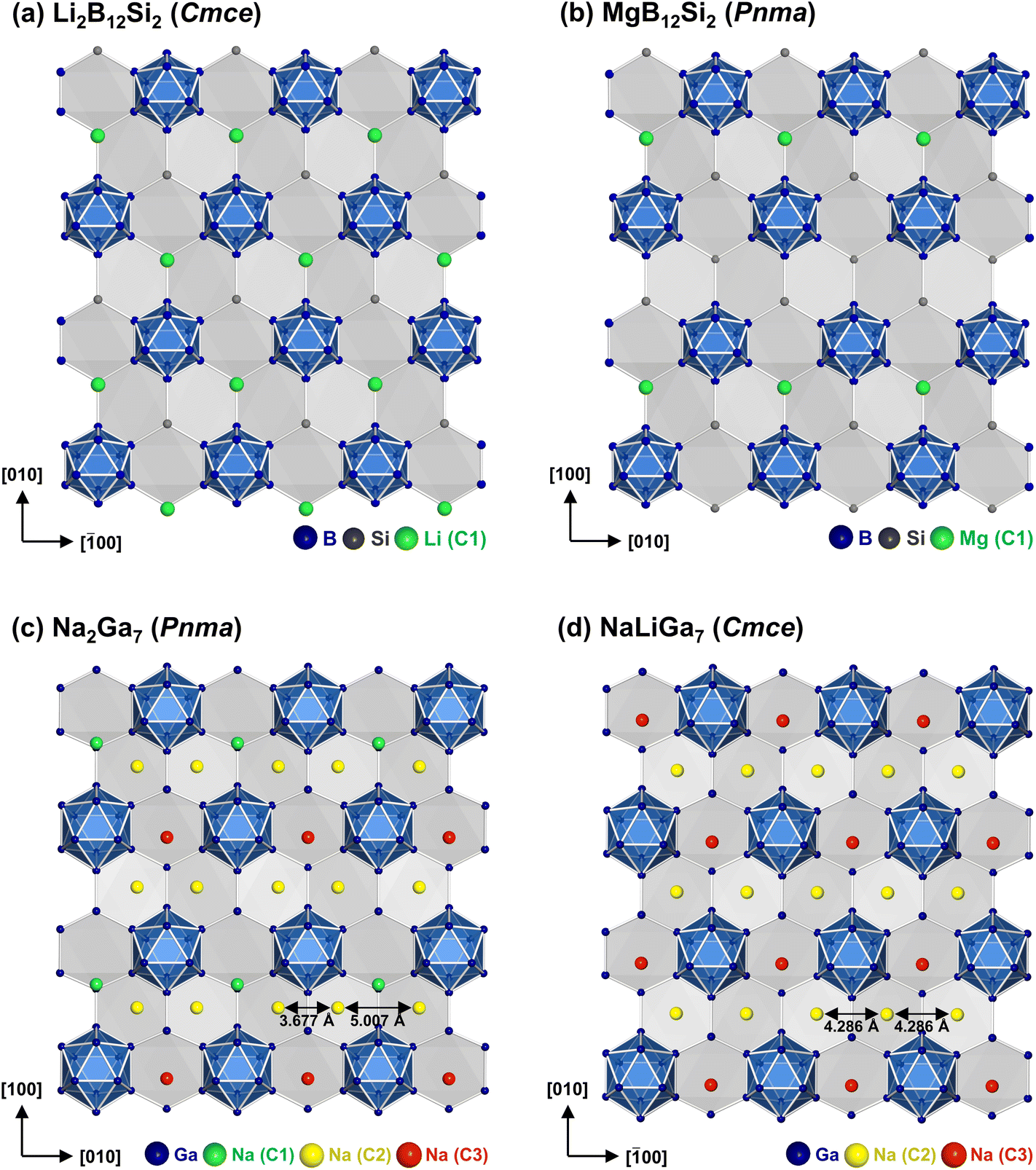 | ||
| Fig. 4 Puckered hexagon layers of (a) Li2B12Si2, (b) MgB12Si2, (c) Na2Ga7 and (d) NaLiGa7. For NaLiGa7, the mean position of the split sites Na1a/Na1b (C2 position) is shown. | ||
| Compound | Occupancy (in %) | M/F | Space group | ||
|---|---|---|---|---|---|
| C1 | C2 | C3 | |||
| MgB12Si2 | 50 | 0 | 0 | 1:14 |
Pnma |
| Li2B12Si2 | 100 | 0 | 0 | 2:14 |
Cmce |
| Na2Ga7 | 50 | 100 | 50 | 4:14 |
Pnma |
| NaLiGa7 | 0 | 100 | 100 | 4:14 |
Cmce |
In the boride silicide Li2B12Si2, all C1 positions are filled by Li atoms (Fig. 3a, 4a), while in MgB12Si2 only half of them are occupied by Mg atoms (Fig. 3b, 4b). The partial occupancy manifests in such a way that in each channel half positions are removed. In this way, the partial occupancy leads to a reduction in symmetry from Cmce in Li2B12Si2 to Pnma in MgB12Si2. The crystal structure of Na2Ga7 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Na4Ga12Ga2) contains twice as many cations as Li2B12Si2, so that the occupation of all positions C1 is not sufficient to accommodate all Na atoms. Consequently, two additional cation positions C2 and C3 are found, which have not been observed in the boride silicides (Fig. 3c, 4c). Atoms at position C2 are also located in the channels (Fig. 2b), which creates a conflict with the spatial requirements of the atoms at C1. In Na2Ga7, full occupancy of C1 and C2 in the channels by Na atoms is impossible for steric reasons. The maximum filling of the channels is achieved, when 50% of the C1 positions and 100% of C2 are occupied. The replaced C1 atoms occupy C3 positions instead, located in small cavities between three Ga12 icosahedra (Fig. 2b). However, only half of the available C3 positions are occupied by Na atoms. Always two C3 positions are arranged in close proximity (Fig. 5), resulting in a distance d(C3–C3) = 2.78 Å being too short if both neighboring positions were filled by Na. The half occupancy of C1 and C3 in Na2Ga7 along with the ordered arrangement of Na results in a symmetry reduction from Cmce to Pnma.
Na4Ga12Ga2) contains twice as many cations as Li2B12Si2, so that the occupation of all positions C1 is not sufficient to accommodate all Na atoms. Consequently, two additional cation positions C2 and C3 are found, which have not been observed in the boride silicides (Fig. 3c, 4c). Atoms at position C2 are also located in the channels (Fig. 2b), which creates a conflict with the spatial requirements of the atoms at C1. In Na2Ga7, full occupancy of C1 and C2 in the channels by Na atoms is impossible for steric reasons. The maximum filling of the channels is achieved, when 50% of the C1 positions and 100% of C2 are occupied. The replaced C1 atoms occupy C3 positions instead, located in small cavities between three Ga12 icosahedra (Fig. 2b). However, only half of the available C3 positions are occupied by Na atoms. Always two C3 positions are arranged in close proximity (Fig. 5), resulting in a distance d(C3–C3) = 2.78 Å being too short if both neighboring positions were filled by Na. The half occupancy of C1 and C3 in Na2Ga7 along with the ordered arrangement of Na results in a symmetry reduction from Cmce to Pnma.
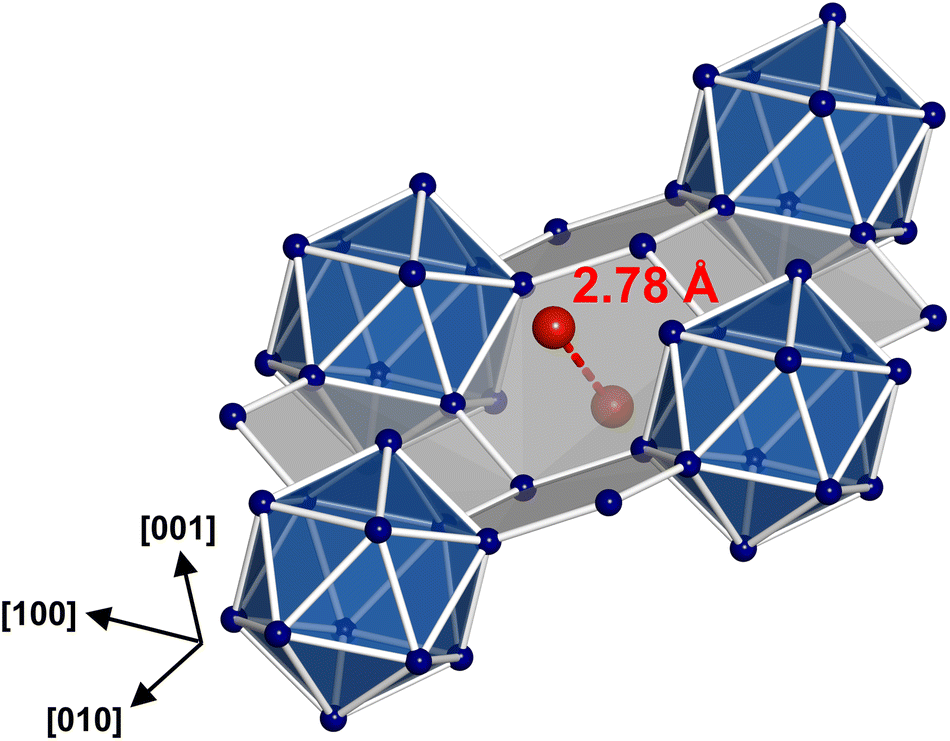 | ||
| Fig. 5 Pair of Li atoms situated in adjacent C3 cavities having a distance of 2.78 Å. | ||
In the crystal structure of NaLiGa7, the smaller Li atoms fill up all C3 positions. Simultaneous occupation of the adjacent positions C1 and C3 is excluded for their short distance of ≈1.60 Å requiring the C1 positions to remain empty. The C2 positions in the channels of NaLiGa7 are fully occupied by Na atoms (Fig. 3d, 4d), as in Na2Ga7. With full occupancy of C3, there is no symmetry reduction observed in NaLiGa7 and the space group remains Cmce (Table 3). The arrangement of cations in NaLiGa7 introduces a new structure type within the MgB12Si2 structure family.
The topology of the Ga framework in NaLiGa7 is very similar to the one in Na2Ga7. The Ga12 icosahedra are distorted with endohedral bond distances ranging from 2.601(1) Å to 2.817(1) Å. The distances between atoms in trans-position vary from d(Ga3–Ga3) = 4.902 Å to d(Ga2–Ga2) = 5.338 Å (Fig. S4 and S5†). As expected, the exohedral two-center, two-electron bonds (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) exo = 2.54 Å) are shorter than the endohedral multicenter bonds (endo = 2.67 Å). The longest Ga–Ga bond d(Ga4–Ga4) = 2.817 Å is found at the cavity position C3, which shows that the occupation of this position creates distortion in the framework. For the boride silicides, where C3 is not occupied, this distortion of the icosahedra does not occur. The 4-bonded atoms Ga5 deviate from ideal tetrahedral coordination with bond angles ranging from 97.59(1) to 123.83(4) degrees and bond distances ranging from 2.517 Å to 2.591 Å. Comparing the bond spectra of Na1Li1Ga7 and Na2Ga7, the framework in Na1Li1Ga7 shows a smaller dispersion of the Ga–Ga bond lengths (Fig. 6). The dispersion of bond lengths in Na2Ga7 is mainly caused by the accommodation of the larger Na atoms in 50% of the small C3 cavities. In agreement with the reduced cell volume, the mean Ga–Ga bond distance in Na1Li1Ga7 of 2.62 Å is shorter than 2.66 Å in Na2Ga7.
exo = 2.54 Å) are shorter than the endohedral multicenter bonds (endo = 2.67 Å). The longest Ga–Ga bond d(Ga4–Ga4) = 2.817 Å is found at the cavity position C3, which shows that the occupation of this position creates distortion in the framework. For the boride silicides, where C3 is not occupied, this distortion of the icosahedra does not occur. The 4-bonded atoms Ga5 deviate from ideal tetrahedral coordination with bond angles ranging from 97.59(1) to 123.83(4) degrees and bond distances ranging from 2.517 Å to 2.591 Å. Comparing the bond spectra of Na1Li1Ga7 and Na2Ga7, the framework in Na1Li1Ga7 shows a smaller dispersion of the Ga–Ga bond lengths (Fig. 6). The dispersion of bond lengths in Na2Ga7 is mainly caused by the accommodation of the larger Na atoms in 50% of the small C3 cavities. In agreement with the reduced cell volume, the mean Ga–Ga bond distance in Na1Li1Ga7 of 2.62 Å is shorter than 2.66 Å in Na2Ga7.
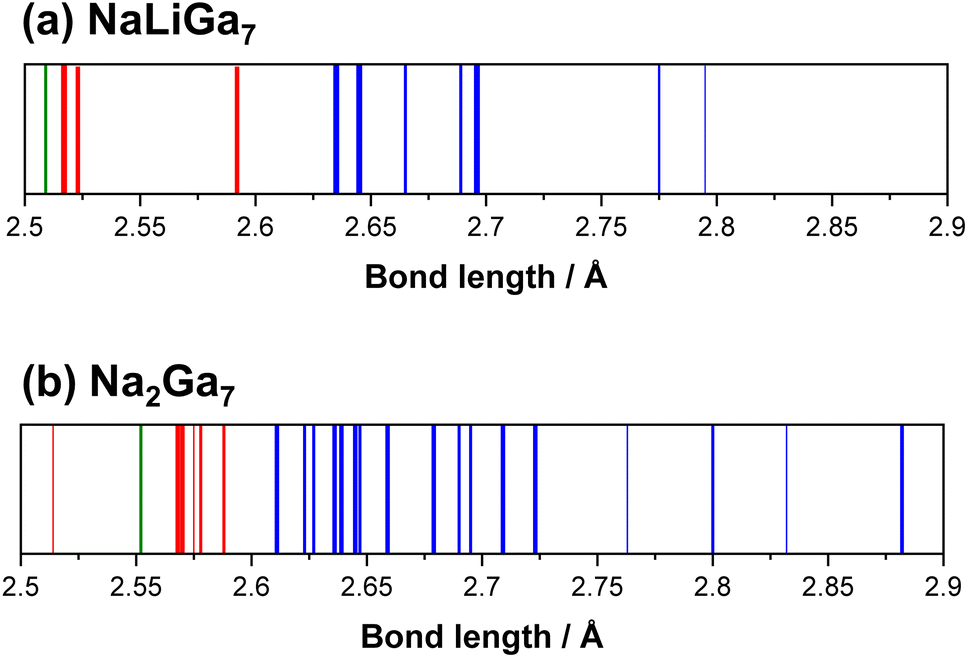 | ||
| Fig. 6 Distance spectra in the Ga framework of (a) NaLiGa7 and (b) Na2Ga7. Blue: endohedral bonds of the Ga12 icosahedra; green: exohedral bonds between icosahedra; red: exohedral bonds between the icosahedra and 4-bonded Ga atoms. | ||
• In Na2Ga7, the positions C1 and C3 are half-occupied by Na.
• In NaLiGa7, C1 is empty, while C3 is fully occupied by Li.
• In both phases, position C2 is entirely filled with Na atoms
 | ||
| Fig. 7 (a) Cell volume of Na2−xLixGa7 (x = 0, 0.2, 0.5, 0.8, 1) against Li content. (b) Substitution model (lines) and refined occupancies (red triangles: Na at C1; blue circles: Na at C3; black circles: Li at C3). (c) b/a ratio of the hexagon planes. (d) Na–Na distances at position C2. (e) Depths of the hollow at C1. Shaded areas show the assumed homogeneity ranges. Error bars for all figures are smaller than the symbols used. | ||
To determine the cation site occupancies for the compositions between 0 < x < 1, only PXRD data were available (Table S11†). The accuracy of the derived structure models was further constrained by the low scattering power of Li located in proximity to the heavier Ga atoms. Despite of these experimental limitations, the substitution mechanism can be inferred: For 0 ≤ x ≤ 0.5, the Na occupancy at C1 remains constant, while the Na occupancy at C3 clearly decreases (Fig. 7b).
Consequently, starting from Na2Ga7, the Li atoms first replace Na atoms in the small cavities at C3. This behavior corresponds to a conventional substitution alloy (Fig. 8a). Finally, at x = 0.5, all Na atoms at C3 are substituted, so that C3 is half occupied by Li. Now, for 0.5 < x ≤ 1, the Li atoms fill up the remaining vacant C3 positions, while Na atoms at C1 evanish (Fig. 7b). This exchange mechanism is unconventional as it represents an intermediate between an interstitial and a substitution alloy. The Li atoms occupy interstitial positions but simultaneously replace Na atoms located at other positions (Fig. 8b). At x = 1, C3 is completely filled by Li and C1 is completely emptied. Deviations of the experimental results from the substitution model only occur for low Na occupancies and Li positions, as expected. Seemingly, the occupancy of the narrow C3 cavities has a stronger impact on the cell volume than the occupancy of C1, sharing the large channels with C2. This is also reflected in the distortion of the six-membered ring planes (Fig. 4). While the lattice parameters a and b in NaLiGa7 nearly exhibit the ideal hexagonal ratio (Table 1), the value deviates significantly in Na2Ga7. When the Na atoms in the small C3 cavities are replaced by Li in the range of 0 ≤ x ≤ 0.5, the ratio approaches the hexagonal value (Fig. 7c). Conversely, the occupation of the C1 position by Na in the range of 0.8 ≤ x ≤ 1 has no influence on the axis ratio.
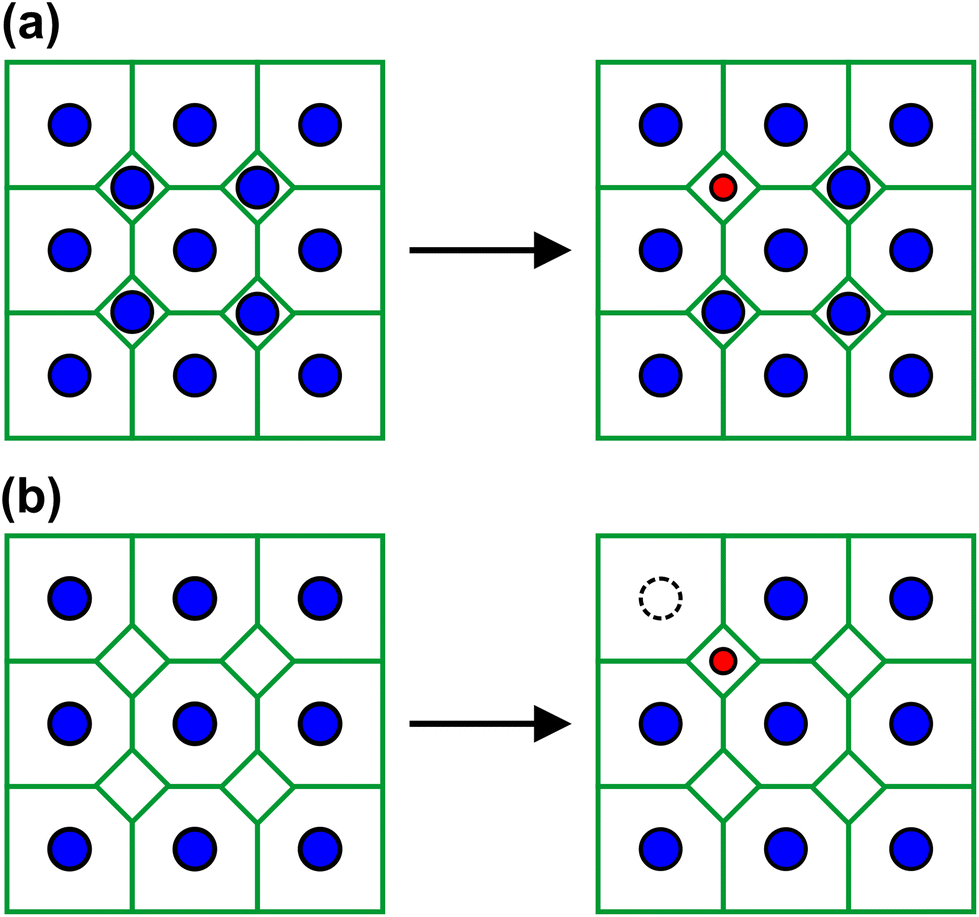 | ||
| Fig. 8 (a) Scheme of a substitution alloy. (b) The unconventional intermediate of an interstitial and a substitution alloy. Green lines symbolize the Ga framework, blue spheres Na and red spheres Li. | ||
The proposed substitution mechanism finds further support in the interatomic distances of the Na atoms at C2, located within the channels of the Ga framework. In NaLiGa7, the Na atoms at C2 show uniform distances from each other (Fig. 4d). Conversely, in Na2Ga7, the channels are shared by Na atoms at C2 and C1, resulting in mutual repulsion that leads the Na atoms at C2 to form dumbbells (Fig. 4c). Therefore, the refined distance between C2 atoms is a signature of the Na occupancy at position C1. Starting from Na2Ga7, the experimentally determined Na–Na distances at C2 remain constant for 0 ≤ x ≤ 0.5, indicating the occupation of position C1 (Fig. 7d). For x > 0.5 the distances converge to a common value, signifying vacancies at C1. Moreover, the occupancy of C1 by Na affects the adjacent Ga atoms. The Na atoms at C1 are situated in a hollow formed by three Ga six-membered rings (Fig. 2b). As C1 becomes vacant, the depth of the hollows decreases (Fig. 7e and Fig. S6†). The decrease clearly starts for x > 0.5, in agreement with the proposed substitution model.
The X-ray results in this work suggest that the two boundary phases, Na2Ga7 and Na1Li1Ga7, each have a homogeneity range. However, the two-phase region between both solid solutions was not detected in this study. In general, the ternary phase diagram Na–Li–Ga is unknown so far. A key challenge in determining the phase equilibria in future work will be the metallographic treatment of the air and moisture sensitive samples.
Electronic density of states
The computed electronic density of states (DOS) for NaLiGa7 reveals a band gap of 0.47 eV (Fig. 9). This result is in line with the expectation of an electronically balanced composition for [(12b)Ga12]2− icosahedral closo clusters and [(4b)Ga]− atoms. The Ga contributions are classified into two groups: (i) sum of the partial DOS of the Ga atoms forming the icosahedra Ga12; (ii) DOS contributions of the 4-bonded Ga. These two chemically different Ga species are expected to be distinguishable in their local DOS projections.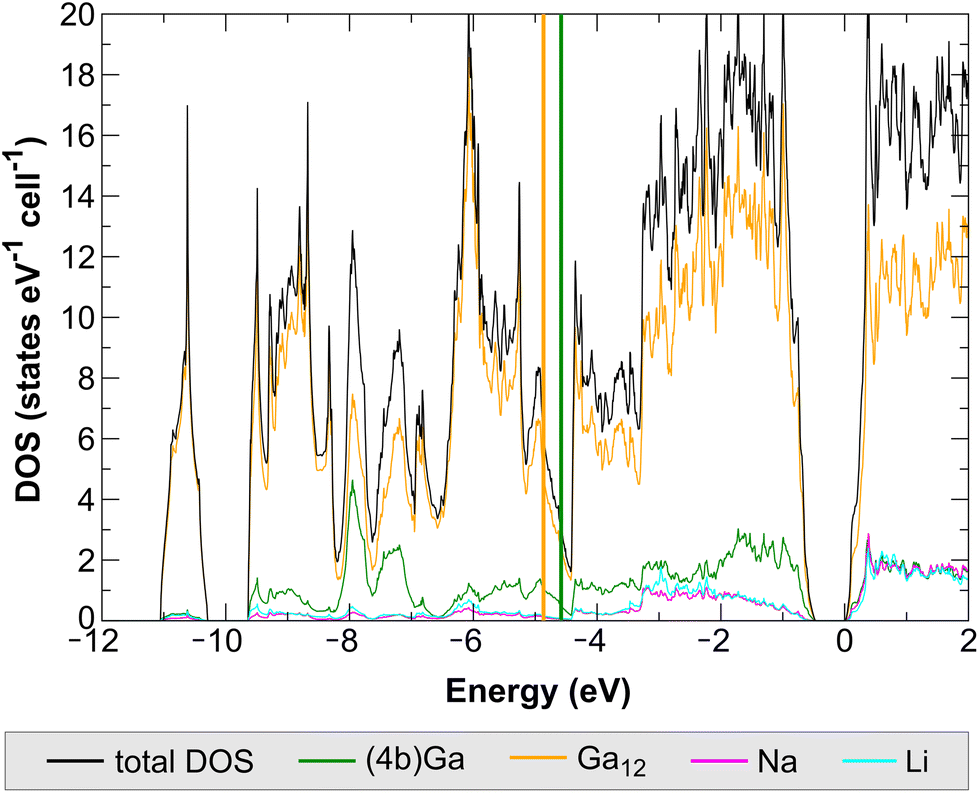 | ||
| Fig. 9 Computed electronic density of states (total DOS) for NaLiGa7 and local DOS projections of the [(4b)Ga]− species, the summed [(12b)Ga12]2− cluster species, and the cationic species. Thick vertical lines indicate energetical center of gravities of the [(4b)Ga]− local DOS (green) and the corresponding sum for [(12b)Ga12]2− species (orange). | ||
The [(4b)Ga]− species do not significantly participate in the lowest-lying cluster orbital below −10 eV, containing 2 e− per Ga12 cluster, and the computed energetical center of gravity of the (5 + 1)-fold coordinated cluster Ga species lies energetically lower than the one for the 4-fold coordinated one (Fig. 9). All these results are in good agreement with the results previously found for Na2Ga7 (calculated band gap = 0.29 eV).5 As the DOS reveals (Fig. 9), there is no fundamental difference in the contribution of Na and Li atoms to the electronic structure of NaLiGa7. We attribute the differences in the band gap between both compounds mainly to the larger distortions of the Ga icosahedra in Na2Ga7 (Fig. 6). The significant variation in the nearest neighbor distances is expected to result in a smaller intra-icosahedral band gap, which is of the bonding-antibonding type.26,27
Physical properties
The magnetization M(T) of the sample of NaLiGa7 was measured in the temperature range from T = 1.8 K to 400 K in magnetic fields μ0H = 0.002 T, 0.1 T, 3.5 T and 7 T. The magnetic moment of the sample holder was subtracted from the measured data and the magnetic susceptibility χ = M/H was calculated. The magnetic susceptibility χ(T) is temperature-independent and diamagnetic (Fig. S7†) with value χ = –1.70 × 10−4 emu mol−1 per formula unit, agreeing within 1% with the calculated value (−1.59 × 10−4 emu mol−1) from diamagnetic increments for Na+, Li+ and elemental Ga.28 The superconductivity observed below T ≈ 8 K can be attributed to the presence of elemental Ga impurities since the Meissner fraction of the signal is low (≈ 0.09%). From the reported data, the allotropes β-, γ- and δ-Ga show superconducting transitions at 5.9 K–6.2 K, 6.9 K–7.6 K and 7.85 K, respectively.29 The resistivity measurements on Na2−xLixGa7 were challenging because of the tendency to form elemental Ga segregation on the surface of the sample during the measurements caused by oxidation.Conclusions
The pseudo-binary system Na2−xLixGa7 is terminated by the compositions Na2Ga7 and Na1Li1Ga7. Both phases show a homogeneity range with different substitution mechanisms. The crystal structure of Na1Li1Ga7 belongs to the MgB12Si2 structure family, characterized by a framework of closo Ga12 icosahedra and 4-bonded Ga atoms. Within the Ga framework, the alkali metal atoms occupy distinct channels and cavities. For Na1Li1Ga7, the arrangement of cations is fully ordered, representing a new variant within the MgB12Si2 structure family. Na1Li1Ga7 was prepared by either annealing the elements or by the quantitative metathesis reaction of Na2Ga7 with LiCl. Considering the electron balance [Na+]8[Li+]8[(12b)Ga122−]4[(4b)Ga−]8, the compound can be classified as a Zintl–Wade phase. The balance is consistent with the diamagnetic susceptibility and an electronic band gap, calculated to be 0.47 eV.Author contributions
Chia-Chi Yu: investigation, formal analysis, conceptualization, methodology, writing – original draft; Yurii Prots: investigation, conceptualization, writing – review & editing; Alim Ormeci: investigation, writing – review & editing; Mitja Krnel: investigation, writing – review & editing; Marcus Schmidt: investigation; Lev Akselrud: formal analysis; Frank R. Wagner: formal analysis, writing – review & editing; Yuri Grin: formal analysis, conceptualization, methodology, writing – review & editing; Michael Baitinger: formal analysis, conceptualization, methodology, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank H. Borrmann and S. Hückmann for providing PXRD data, and S. Scharsach for DTA measurements. Open Access funding provided by the Max Planck Society. C.-C. Y. acknowledges financial support by the International Max Planck Research School for Chemistry and Physics of Quantum Materials (IMPRS-CPQM).References
- The Physics and Chemistry of Inorganic Clathrates, ed. G. S. Nolas, Springer Series in Materials Science, 2014 Search PubMed.
- B. Albert and H. Hillebrecht, Angew. Chem., Int. Ed., 2009, 48, 8640 CrossRef CAS PubMed.
- L. Pauling and S. Weinbaum, Z. Kristallogr., 1934, 87, 181 CAS.
- C. Zheng and R. Hoffmann, Z. Naturforsch., B: J. Chem. Sci., 1986, 41, 292 CrossRef.
- C.-C. Yu, A. Ormeci, I. Veremchuk, X.-J. Feng, Yu. Prots, M. Krnel, P. Koželj, M. Schmidt, U. Burkhardt, B. Böhme, L. Akselrud, M. Baitinger and Yu. Grin, Inorg. Chem., 2023, 62, 9054 CrossRef CAS PubMed.
- T. Ludwig and H. Hillebrecht, J. Solid State Chem., 2006, 79, 1623 CrossRef.
- N. Vojteer, M. Schroeder, C. Röhr and H. Hillebrecht, Chem. – Eur. J., 2008, 14, 7331 CrossRef CAS PubMed.
- L. Akselrud and Yu. Grin, J. Appl. Crystallogr., 2014, 47, 803 CrossRef CAS.
- Diamond V4.6.8, Crystal Impact, 2022 Search PubMed.
- K. Koepernik and H. Eschrig, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1743 CrossRef CAS.
- J. P. Perdew and J. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1922, 45, 13244 CrossRef PubMed.
- C. Belin and R.-G. Ling, J. Solid State Chem., 1982, 45, 290 CrossRef CAS.
- J. Sappl and C. Hoch, Inorg. Chem., 2020, 59, 6566 CrossRef CAS PubMed.
- P. W. Atkins, T. L. Overton, J. P. Rourke, M. T. Weller and F. A. Armstrong, Shriver and Atkins’ Inorganic Chemistry, W. H. Freeman and Company, New York, 5th edn, 2010 Search PubMed.
- W. T. Barrett and W. E. Wallace, J. Am. Chem. Soc., 1954, 76, 366 CrossRef CAS.
- G. Bruzzone, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1206 CrossRef CAS.
- U. Frank-Cordier, G. Cordier and H. Schäfer, Z. Naturforsch., B: J. Chem. Sci., 1982, 37, 119 CrossRef.
- K. Ghosh, A. Ovchinnikov, M. Baitinger, M. Krnel, U. Burkhardt, Yu. Grin and S. Bobev, Dalton Trans., 2023, 52, 10310 RSC.
- K. Ghosh and S. Bobev, Chem. – Eur. J., 2023, 29, e20230238 Search PubMed.
- M. Kotsch, Yu. Prots, A. Ormeci, M. Bobnar, F. R. Wagner, A. Senyshyn and Yu. Grin, Eur. J. Inorg. Chem., 2020, 29, 2842 CrossRef.
- K. Wade, J. Chem. Soc. D, 1971, 15, 792 RSC.
- K. Wade, Adv. Inorg. Chem. Radiochem., 1976, 18, 1 CrossRef CAS.
- H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1981, 20, 33 CrossRef.
- Yu. Grin, U. Schwarz and W. Steurer, Crystal Structure and Chemical Bonding. In Alloy Physics: A Comprehensive Reference, ed. W. Pfeiler, Wiley-VCH, Weinheim, 2007; pp. 19 Search PubMed.
- T. F. Fässler, Zintl Phases: Principles and Recent Developments, Springer, Berlin-Heidelberg, 2011 Search PubMed.
- H. C. Longuet-Higgins and M. De V. Roberts, Proc. R. Soc. London, Ser. A, 1955, 230, 110 CAS.
- K. Wade, Electron Deficient Compounds, Springer, New York, 1971 Search PubMed.
- Diamagnetic Susceptibility and Anisotropy of Inorganic and Organometallic Compounds, ed. R. R. Gupta, Springer, Berlin-Heidelberg, 2007 Search PubMed.
- Y. Quan, P. J. Hirschfeld and R. G. Hennig, Phys. Rev. B, 2021, 104, 075117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2304096 (NaLiGa7). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03628f |
| This journal is © The Royal Society of Chemistry 2024 |