
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar CO2 reduction using a molecular Re(I) catalyst grafted on SiO2via amide and alkyl amine linkages†
Thomas
Fenton
,
Esraa
Ahmad
and
Gonghu
Li
 *
*
Department of Chemistry, University of New Hampshire, Durham, New Hampshire 03824, USA. E-mail: gonghu.li@unh.edu
First published on 4th January 2024
Abstract
Heterogenized molecular catalysts have shown interesting activities in different chemical transformations. In our previous studies, a molecular catalyst, Re(bpy)(CO)3Cl where bpy is 2,2′-bipyridine, was covalently attached to silica surfaces via an amide linkage for use in photocatalytic CO2 reduction. Derivatizing the bpy ligand with electron-withdrawing amide groups led to detrimental effects on the catalytic activity of Re(bpy)(CO)3Cl. In this study, an alkyl amine linkage is utilized to attach Re(bpy)(CO)3Cl onto SiO2 in order to eliminate the detrimental effects of the amide linkage by breaking the conjugation between the bpy ligand and the amide group. However, the heterogenized Re(I) catalyst containing the alkyl amine linkage demonstrates even lower activity than the one containing the amide linkage in photocatalytic CO2 reduction. Infrared studies suggest that the presence of the basic amine group led to the formation of a photocatalytically inactive Re(I)-OH species on SiO2. Furthermore, the amine group likely contributes to the stabilization of a surface Re(I)-carboxylato species formed upon light irradiation, resulting in the low activity of the heterogenized Re(I) catalyst containing the alkyl amine linkage.
Introduction
Photocatalytic CO2 reduction has been extensively investigated in recent years as a sustainable way to utilize CO2 as a renewable C1 feedstock.1–3 Different systems, including homogeneous and heterogeneous photocatalysts, have been developed to convert the thermodynamically stable CO2 molecule to less stable molecules such as CO and HCOOH.4–8 In homogeneous systems, some molecular catalysts can function as photocatalysts by themselves since they can harvest light and activate CO2, while others require the use of additional photosensitizers for light harvesting.9–12 Diimine-tricarbonyl Re(I) complexes, in particular Re(bpy)(CO)3Cl where bpy is 2,2′-bipyridine, have demonstrated excellent reactivity and selectivity towards CO formation in both photocatalytic and electrocatalytic CO2 reduction.13,14While molecular catalysts are often highly efficient in mediating CO2 reduction, many of them suffer from poor stability under photochemical conditions. Different strategies have been designed to enhance the robustness and recyclability of molecular catalysts. For instance, heterogenized molecular catalysts can be obtained by attaching molecular catalysts onto solid-state surfaces.15–19 Such photocatalysts generally show enhanced activity and improved photostability. Many studies utilized Re(bpy)(CO)3Cl as a model complex to obtain heterogenized molecular catalysts on photoactive surfaces20–23 for enhanced CO2 reduction under solar irradiation.
We have employed SiO2 as a model support for Re(bpy)(CO)3Cl to carry out spectroscopic investigations since SiO2 is almost “transparent” in studies using many spectroscopic techniques,24 including diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). In our previous studies, covalent attachment of Re(bpy)(CO)3Cl on SiO2 involved derivatization of the bpy ligand with amide groups.25–27 However, such ligand derivatization resulted in significant changes in the photophysical, electrochemical, and photocatalytic properties of Re(bpy)(CO)3Cl.26,27 For instance, the metal-to-ligand charge transfer (MLCT) transition of Re(bpy)(CO)3Cl was shifted from ∼370 nm to ∼400 nm upon covalent attachment on SiO2via the amide linkage. Derivatizing the bpy ligand with the electron-withdrawing amide groups also shifted the redox potentials of Re(bpy)(CO)3Cl to more positive values. Consequently, activity of the amide-derivatized Re(I) complex was found to be much lower than Re(bpy)(CO)3Cl in photocatalytic CO2 reduction.26 Derivatizing Re(bpy)(CO)3Cl with amide groups also showed detrimental effects in electrocatalytic CO2 reduction. In particular, covalent linkages containing the amide groups were shown to be unstable under highly reducing conditions, while alkyl linkages were more stable.28
In addition to ligand derivatization, some surface features could also affect the structure, complexation, and behavior of molecular catalysts grafted on surfaces. For example, it was shown that the binding structure of surface attached Re(bpy)(CO)3Cl can be significantly influenced not only by the nature and length of the covalent linkages, but also by the crystallographic facet of the surface.29 Our studies using DRIFTS spectroscopy showed that the presence of surface water had profound effects on the complexation of Re(bpy)(CO)3Cl attached onto SiO2 surfaces. In particular, three infrared absorptions associated with the formation of a photochemically inactive Re(I)-OH species were observed in the DRIFTS spectrum of a heterogenized Re(bpy)(CO)3Cl in the presence of triethanolamine (TEOA) and water.30 This Re(I)-OH species was formed upon replacing the Cl− ligand of Re(bpy)(CO)3Cl with –OH, which could be produced in the reaction between the basic TEOA and water on the surface.
In this current study, an alkyl amine linkage is utilized to covalently attach Re(bpy)(CO)3Cl onto SiO2 to break the conjugation between the bpy ligand and the amide group. Surprisingly, the resulting heterogenized Re(I) catalyst demonstrates lower activity than the one containing the amide linkage in photocatalytic CO2 reduction. We utilize DRIFTS spectroscopy to explore possible reasons for this observation, including the formation of the Re(I)-OH species and the stabilization of a surface Re(I)-carboxylato species generated upon light irradiation.
Experimental
Materials
Aerosil 200 silica, hydrochloric acid, triethylamine (TEA), triethanolamine (TEOA), toluene, diethyl ether, dichloromethane (DCM), dimethylformamide (DMF), thionyl chloride, 2,2′-bipyridine-4,4′-dicarboxylic acid, pentacarbonyl chlororhenium(I) (98%), N-bromosuccinimide, azobisisobutyronitrile, and tris(2,2′-bipyridine) dichlororuthenium(II) hexahydrate (99%) were obtained from Sigma-Aldrich. Two silane coupling agents, p-aminophenyl trimethoxysilane and m-aminophenyl trimethoxysilane, were obtained from Gelest. All chemicals were used as received without any further purification.Synthesis of aminophenyl functionalized SiO2
100 mg of Aerosil 200 silica (SiO2) was dried at 100 °C for 2 hours before being dispersed in 50 mL dry toluene under an inert atmosphere, to which 0.189 g of p-aminophenyl trimethoxysilane was added. The resulting mixture was refluxed for 24 hours under a nitrogen atmosphere. The product was recovered via centrifugation and washed three times with toluene, diethyl ether, and DCM, respectively. The resulting brown powder was then dried under vacuum.Covalent attachment of Re(bpy)(CO)3Cl on SiO2via an amide linkage
The molecular Re(I) complex was attached to SiO2 surface via an amide linkage following our previous studies (Scheme 1).26,27 In a typical synthesis, 100 mg of 4,4′-dicarboxylic-2,2′-bipyridine was dispersed in 15 mL of thionyl chloride. This solution was heated to 85 °C under an inert atmosphere and refluxed overnight. Rotary evaporation was used to isolate the acylated yellow powder. The freshly synthesized 4,4′-bis(chlorocarbonyl)-2,2′-bipyridine was then dispersed in 15 mL DCM and added dropwise to 100 mg of p-aminophenyl functionalized SiO2 in 40 mL DCM under N2 flow. The solution was then heated to 70 °C and stirred overnight. The product was retrieved via centrifugation and washed three times with DCM, diethyl ether, and DCM again, respectively. A light yellow powder was isolated via drying under vacuum. Next, 150 mg of the resulting material was dispersed in 50 mL dry toluene, to which 50 mg of pentacarbonyl chlororhenium(I) was added under stirring. The mixture was then heated to 95 °C under an inert atmosphere and was stirred overnight while covered with foil to reduce light exposure. The product was then retrieved via centrifugation and washed three times with toluene, diethyl ether, and then repeatedly with DCM, respectively. A yellow powder was isolated and dried under vacuum. The final product bearing an amide linkage is denoted “Re-1-SiO2” (Scheme 1). | ||
| Scheme 1 Synthesis of Re-1-SiO2 featuring an amide linkage. | ||
Synthesis of 4-(bromomethyl)-4′-methyl-2,2′-bipyridine
In our study, 4-(bromomethyl)-4′-methyl-2,2′-bipyridine was synthesized following a method reported by Joseph and co-workers.31 In this synthesis, 1 g of 4,4′-dimethyl-2,2′-bipyridine and 1 g of N-bromosuccinimide were dissolved in 50 mL of carbon tetrachloride at which point a catalytic amount of azobisisobutyronitrile (5 mg) was added. The reaction mixture was refluxed at 77 °C for 16 hours under nitrogen. The hot reaction mixture was then filtered and the filtrate was concentrated using rotary evaporation. The resulting residue was purified using column chromatography (∼30% yield).Covalent attachment of Re(bpy)(CO)3Cl on SiO2via an alkyl amine linkage
Following a method similar to Scheme 1, a surface Re(I) catalyst containing an alkyl amine linkage, denoted “Re-2-SiO2”, was synthesized using 4-(bromomethyl)-4′-methyl-2,2′-bipyridine instead of 4,4′-bis(chlorocarbonyl)-2,2′-bipyridine (Scheme 2). | ||
| Scheme 2 Synthesis of Re-2-SiO2 featuring an alkyl amine linkage. | ||
Following the approaches shown in Schemes 1 and 2, two isomeric Re(I) catalysts, Re-3-SiO2 and Re-4-SiO2, were also synthesized starting with SiO2 functionalized with m-aminophenyl trimethoxysilane (see Fig. S1† for their structures).
Characterization
Elemental analysis of the metal content in the synthesized materials was performed using a Varian Vista AX inductively coupled plasma atomic emission spectrometer. The loadings of Re were determined to be 1.8 and 3.4 μmol per 10 mg of Re-1-SiO2 and Re-2-SiO2, respectively. Optical spectra of the samples were obtained using a Cary 50 Bio spectrophotometer outfitted with a Barrelino diffuse reflectance probe. DRIFTS spectra were collected on a Nicolet 6700 FTIR spectrometer equipped with a Harrick Praying Mantis diffuse reflectance accessory.Photocatalytic testing
In a typical photocatalytic test, 10 mg of a heterogenized catalyst sample was dispersed in 4 mL of a DMF : TEOA (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution in a quartz test tube. A molecular photosensitizer, tris(2,2′-bipyridine) dichlororuthenium(II) hexahydrate, was added to the reaction solution (3 mg for each test). The test tube was sealed with a septum and parafilm, then bubbled with CO2 (99.999% Airgas) for 30 minutes in the dark before being exposed to light irradiation provided by a Fiber-Light series 180 lamp fitted with a 420 nm long-pass optical filter (see Fig. S2† for the output spectrum). The light intensity was fixed at 100 mW cm−2. While the reaction solution was stirred under light, the head space was sampled using a gas tight syringe for analysis using an Agilent 7820 GC equipped with a thermal conductivity detector and a 60/80 Carboxen 1000 column. Possible products in the solution phase were monitored using nuclear magnetic resonance spectroscopy.
:1 v/v) solution in a quartz test tube. A molecular photosensitizer, tris(2,2′-bipyridine) dichlororuthenium(II) hexahydrate, was added to the reaction solution (3 mg for each test). The test tube was sealed with a septum and parafilm, then bubbled with CO2 (99.999% Airgas) for 30 minutes in the dark before being exposed to light irradiation provided by a Fiber-Light series 180 lamp fitted with a 420 nm long-pass optical filter (see Fig. S2† for the output spectrum). The light intensity was fixed at 100 mW cm−2. While the reaction solution was stirred under light, the head space was sampled using a gas tight syringe for analysis using an Agilent 7820 GC equipped with a thermal conductivity detector and a 60/80 Carboxen 1000 column. Possible products in the solution phase were monitored using nuclear magnetic resonance spectroscopy.
Results and discussion
The heterogenized Re(I) catalysts synthesized following Schemes 1 and 2 display slightly different colors. Their optical spectra were collected using a UV-visible spectrophotometer fitted with a diffuse reflectance accessory. Fig. 1 shows the spectra of Re-1-SiO2 and Re-2-SiO2, which feature MLCT bands around 400 nm and 370 nm, respectively. In our previous study,26 the MLCT band of the homogeneous Re(bpy)(CO)3Cl complex was recorded to be at 369 nm, and was shifted to 396 nm upon derivatizing the bpy ligand with amide groups. Therefore, the 30 nm shift in the MLCT band of Re-1-SiO2 compared to Re-2-SiO2 originates from the amide derivatization on the bpy ligand. More specifically, this shift most likely occurs due to stabilization of the bpy π* based lowest unoccupied molecular orbital of the complex upon derivatization with the electron withdrawing amide group. The MLCT transition of Re-2-SiO2 is similar to that of the homogeneous Re(bpy)(CO)3Cl complex, indicating that ligand derivatization with the alkyl amine group has minimal effect on the MLCT band. | ||
| Fig. 1 Diffuse reflectance UV-vis spectra of (a) Re-1-SiO2 and (b) Re-2-SiO2. Barium sulfate was used as the background. | ||
In our study, two isomeric heterogenized Re(I) catalysts, Re-3-SiO2 and Re-4-SiO2, were synthesized starting with SiO2 functionalized with m-aminophenyl trimethoxysilane (Fig. S1†). The same difference was observed in the MLCT bands of Re-3-SiO2 and Re-4-SiO2, which contain amide and alkyl amine linkages, respectively (Fig. S3†).
The carbonyl groups of the heterogenized Re(I) catalysts can be used as a sensitive molecular probe to investigate the complexation and structures of the Re(I) centers. The infrared spectrum of Re(bpy)(CO)3Cl in the molecular state contains three carbonyl stretches in the 1900–2100 cm−1 range, corresponding to one high-energy, fully symmetric mode and two nearly degenerate lower-energy modes.20,32 In the DRIFTS spectra of Re-1-SiO2 and Re-2-SiO2, the high-energy stretch is seen at 2025 cm−1 while the two lower-energy modes coalesce to one broad feature at 1905 cm−1 (Fig. 2), similar to our previous observations of heterogenized Re(I) catalysts.25,26,30 This coalescence is attributed to peak broadening due to surface heterogeneity rather than any change in symmetry at the Re center. Additional features in the DRIFTS spectrum of Re-1-SiO2 include absorptions associated with amide groups at 1664, 1524, and 1400 cm−1 (Fig. 2a), similar to those observed in our prior studies.25 Furthermore, an absorption at 1726 cm−1 is present in the spectrum of Re-1-SiO2 shown in Fig. 2a, corresponding to the –COOH group26 on the bpy ligand (see the structure of Re-1-SiO2 in Scheme 1). Both spectra shown in Fig. 2 also contain an absorption around 1600 cm−1, which is attributed to the phenyl group in both Re-1-SiO2 and Re-2-SiO2.
 | ||
| Fig. 2 DRIFTS spectra of (a) Re-1-SiO2 and (b) Re-2-SiO2. Potassium bromide was used as the background. | ||
The two lower-energy modes of the Re(I)-carbonyl stretches can be resolved by exposing the surface Re(I) complexes to TEOA and CO2.24,26,33 In this study, the heterogenized Re(I) catalysts in the powder form were diluted with KBr and then mixed with a few drops of TEOA. The resulting mixture was then placed in the DRIFTS cell. Prior to spectrum collection, the mixture was purged with CO2 in the dark. Fig. 3 shows the DRIFTS spectra of Re-1-SiO2 and Re-2-SiO2 collected under CO2. In the spectrum of Re-1-SiO2 (Fig. 3a), three peaks at 2021, 1917, and 1895 cm−1 are present, indicating the formation of a CO2-bound Re(I) adduct, Re(I)–OOC–OCH2CH2NR2 where R is CH2CH2OH, in the presence of TEOA and CO2.34 The Re(I)–OOC–OCH2CH2NR2 species was suggested to be the real catalyst in photocatalytic CO2 reduction using diimine-tricarbonyl Re(I) catalysts and TEOA.34
 | ||
| Fig. 3 DRIFTS spectra of (a) Re-1-SiO2 and (b) Re-2-SiO2 in the presence of TEOA and CO2. | ||
Under the same experimental conditions, three additional Re(I)-carbonyl stretches at 1995, 1875, and 1850 cm−1 are seen in the DRIFTS spectrum of Re-2-SiO2 (Fig. 3b). These three peaks are associated with a photochemically inactive Re(I)-OH species according to our previous study30 and studies by Gibson and co-workers.35 Since these three peaks are not present in the spectrum of Re-1-SiO2, it can be deduced that the formation of the Re(I)-OH species in Re-2-SiO2 is due to the presence of the basic amine group, which likely produced –OH to replace the Cl− ligand on Re-2-SiO2. The same difference was observed in the DRIFTS spectra of Re-3-SiO2 and Re-4-SiO2 (Fig. S4 and S5†), further confirming the observed effect of the basic amine group.
The synthesized samples demonstrated poor activity in photocatalytic CO2 reduction under simulated solar irradiation (Fig. S6†). In this study, the heterogenized Re(I) catalysts were coupled with a molecular Ru(II) photosensitizer in visible-light CO2 reduction in the presence of TEOA as a sacrificial electron donor. Diimine-tricarbonyl Re(I) complexes are known for their activity in selective reduction of CO2 to CO, though formation of CH4 was recently reported in photocatalytic CO2 reduction using a molecular Re(I) complex.36 Under the experimental conditions employed in this study, CO and a small amount of H2 were detected as the only products; no appreciable production of HCOOH, HCHO, CH3OH, or CH4 was detected. Turnover numbers (TONs) for CO production of 13.6 and 4.5 were obtained using Re-1-SiO2 and Re-2-SiO2, respectively, after photocatalysis for 120 minutes (Fig. 4). Similar comparison was observed in photocatalysis using Re-3-SiO2 and Re-4-SiO2 (Fig. S7†).
 | ||
| Fig. 4 CO production in photocatalytic CO2 reduction using (a) Re-1-SiO2 and (b) Re-2-SiO2 under visible light using a molecular Ru(II) photosensitizer. Light intensity was 100 mW cm−2. | ||
The photocatalysis results shown in Fig. 4 are surprising since the alkyl amine linkage in Re-2-SiO2 was expected to alleviate the detrimental effect of the electron-withdrawing amide group in Re-1-SiO2. One of the reasons for the relatively lower activity of Re-2-SiO2 is the formation of photocatalytically inactive Re(I)-OH species in the presence of TEOA, as indicated by its DRIFTS spectrum shown in Fig. 3b. As discussed earlier, the formation of Re(I)-OH species in Re-2-SiO2 is due to the presence of the basic amine group.
Additional effects of the amine group were discovered using in situ DRIFTS experiments. In such experiments, the heterogenized Re(I) catalysts were mixed with a few drops of TEOA and purged with CO2. For each sample, DRIFTS spectra were collected prior to and after exposing the samples to light irradiation. Difference spectra shown in Fig. 5 were obtained by subtracting the spectra after light irradiation for 2, 10, 30 and 60 min from the spectrum collected prior to irradiation. In the difference spectra, positive peaks are associated with the formation of new species, while negative peaks are associated with the consumption of species formed prior to light irradiation. In the difference DRIFTS spectra of both Re-1-SiO2 and Re-2-SiO2 (Fig. 5), negative peaks at 2025 and 1925 cm−1 are present and the peaks became more intense over time, indicating the consumption of the CO2-bound Re(I) adduct, Re(I)–OOC–OCH2CH2NR2, upon light irradiation. Positive peaks around 2003 and 1873 cm−1 are seen in the spectra shown in Fig. 5. These positive peaks are associated with the formation of a reduced species similar to [Re(bpy)(CO)3Cl]˙−.27 Such negative and positive peaks, representing changes in the spectra upon light irradiation, are also seen in the difference DRIFTS spectra of Re-3-SiO2 and Re-4-SiO2 (Fig. S8†).
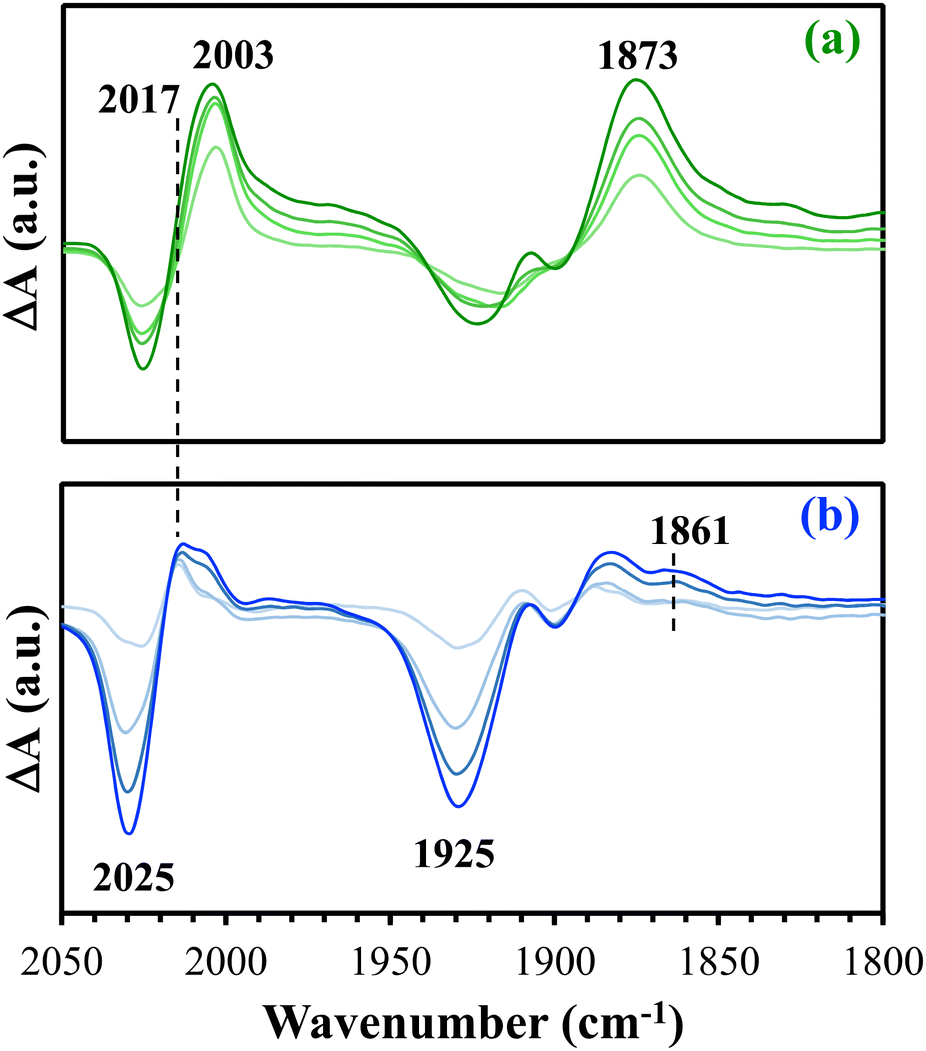 | ||
| Fig. 5 Difference DRIFTS spectra of (a) Re-1-SiO2 and (b) Re-2-SiO2 upon light irradiation in the presence of TEOA and CO2. The spectra were obtained by subtracting spectra collected before light irradiation (t = 0) from corresponding spectra collected after light irradiation for different times (2, 10, 30, and 60 min). | ||
A unique feature in the difference spectra of Re-2-SiO2 is the presence of a peak around 2017 cm−1, which is absent from the spectra of Re-1-SiO2 (Fig. 5). This peak, together with a less obvious absorption around 1861 cm−1, suggests the formation of a Re(I)-carboxylato complex on Re-2-SiO2 upon light irradiation.37 These two peaks are also seen in the difference DRIFTS spectra of Re-4-SiO2 but not in the difference spectra of Re-3-SiO2 (Fig. S8†). A viable intermediate in CO2 reduction using diimine-tricarbonyl Re(I) catalysts, the Re(I)-carboxylato species requires additional protons to be converted into CO.35,38 The comparison shown in Fig. 5 and S7† implies that the photochemically produced Re(I)-carboxylato species is stabilized in Re-2-SiO2 and Re-4-SiO2 that contain the alkyl amine linkage. In the presence of the alkyl amine group, electrostatic interactions may be hindering the Re(I)-carboxylato intermediate from participating in C–OH bond cleavage and releasing CO as the product.
Discussions on the spectra shown in Fig. 3 and 5 suggest that the alkyl amine linkage contributed to (i) formation of the Re(I)-OH species and (ii) stabilization of the Re(I)-carboxylato species generated upon light irradiation. Our previous studies30 demonstrated that the Re(I)-OH species is photochemically inactive. Stabilization of the Re(I)-carboxylato species prevented its further conversion to CO, the final product of CO2 reduction. These two consequences of using the alkyl amine linkage in catalyst heterogenization likely contributed to the observed relatively low activity of Re-2-SiO2 (Fig. 4) and Re-4-SiO2 (Fig. S7†).
Due to the presence of the phenyl group, the alkyl amine in Re-2-SiO2 and Re-4-SiO2 should be less basic than TEOA, although it is more basic than the amide group in Re-1-SiO2 and Re-3-SiO2. Since the spectra shown in Fig. 3 and 5 are collected in the presence of TEOA, our results suggest that local basicity of the alkyl amine group could have some impact on the fate of the heterogenized Re(I) catalysts. In Re-2-SiO2 and Re-4-SiO2, the basic amine group is in close proximity with the Re(I) center, and led to the formation of Re(I)-OH and stabilization of Re(I)-carboxylato. In our study, these Re(I) species were not detected by DRIFTS in Re-1-SiO2 or Re-3-SiO2, even in the presence of the more basic TEOA. There are other possible reasons that could account for the observed difference in photocatalytic activities. For instance, the aniline unit in Re-2-SiO2 and Re-4-SiO2 could potentially alter the photophysical properties of the reaction system under the experimental conditions employed in this study. Further studies using time-resolved spectroscopic techniques are needed to reveal possible differences in photophysical properties.
Conclusions
We have utilized an amide linkage and an alkyl amine linkage to obtain heterogenized molecular catalysts by covalently attaching Re(bpy)(CO)3Cl onto silica surface. Since derivatizing the bpy ligand with the amide group was previously shown to have detrimental effects on the photocatalytic activity of the Re(I) catalyst, using the alkyl amine linkage was expected to achieve improved photocatalysis. However, the opposite trend was observed in photocatalytic CO2 reduction using these heterogenized Re(I) catalysts. Infrared studies suggest that the basic amine group led to the formation of a photocatalytically inactive Re(I)-OH species and contributed to the stabilization of a surface Re(I)-carboxylato species formed upon light irradiation. These spectroscopic observations were used to explain the low activity of the heterogenized Re(I) catalyst containing the alkyl amine linkage relative to the one containing the amide linkage. Our studies provide useful insights on the design of covalent linkages for the heterogenization of molecular catalysts for photocatalytic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. National Science Foundation through grants 1352437 (materials synthesis, characterization, and photocatalysis) and 2247575 (analysis of infrared spectra).References
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, The Teraton Challenge. A Review of Fixation and Transformation of Carbon Dioxide, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Recent advances in hybrid photocatalysts for solar fuel production, Energy Environ. Sci., 2012, 5, 5902–5918 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Photochemical and photoelectrochemical reduction of CO2, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Light-Driven Heterogeneous Reduction of Carbon Dioxide: Photocatalysts and Photoelectrodes, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- Y. Tamaki and O. Ishitani, Supramolecular Photocatalysts for the Reduction of CO2, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- Y. Wang, D. He, H. Chen and D. Wang, Catalysts in electro-, photo- and photoelectrocatalytic CO2 reduction reactions, J. Photochem. Photobiol., C, 2019, 40, 117–149 CrossRef.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- E. Fujita, Photochemical carbon dioxide reduction with metal complexes, Coord. Chem. Rev., 1999, 185–186, 373–384 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- C. D. Windle and R. N. Perutz, Advances in molecular photocatalytic and electrocatalytic CO2 reduction, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Molecular catalysis of CO2 reduction: recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes, Chem. Soc. Rev., 2020, 49, 5772–5809 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Efficient Photochemical Reduction of CO2 to CO by Visible Light Irradiation of Systems Containing Re(bipy)(CO)3X or Ru(bipy)32+-Co2+ Combinations as Homogeneous Catalysts, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- G. Sahara and O. Ishitani, Efficient Photocatalysts for CO2 Reduction, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- C. D. Windle and E. Reisner, Heterogenised Molecular Catalysts for the Reduction of CO2 to Fuels, Chimia, 2015, 69, 435–441 CrossRef CAS PubMed.
- M. E. Louis, T. G. Fenton, J. Rondeau, T. Jin and G. Li, Solar CO2 Reduction Using Surface-Immobilized Molecular Catalysts, Comments Inorg. Chem., 2016, 36, 38–60 CrossRef CAS.
- K. Maeda, Metal-Complex/Semiconductor Hybrid Photocatalysts and Photoelectrodes for CO2 Reduction Driven by Visible Light, Adv. Mater., 2019, 31, 1808205 CrossRef PubMed.
- A. Perazio, G. Lowe, R. Gobetto, J. Bonin and M. Robert, ight-driven catalytic conversion of CO2 with heterogenized molecular catalysts based on fourth period transition metals, Coord. Chem. Rev., 2021, 443, 214018 CrossRef CAS.
- P. Huang, E. Shaaban, E. Ahmad, A. St. John, T. Jin and G. Li, Well-defined surface catalytic sites for solar CO2 reduction: heterogenized molecular catalysts and single atom catalysts, Chem. Commun., 2023, 59, 9301–9319 RSC.
- N. M. Orchanian, L. E. Hong, J. A. Skrainka, J. A. Esterhuizen, D. A. Popov and S. C. Marinescu, Surface-Immobilized Conjugated Polymers Incorporating Rhenium Bipyridine Motifs for Electrocatalytic and Photocatalytic CO2 Reduction, ACS Appl. Energy Mater., 2019, 2, 110–123 CrossRef CAS.
- M. Abdellah, A. M. El-Zohry, L. J. Antila, C. D. Windle, E. Reisner and L. Hammarström, Time-Resolved IR Spectroscopy Reveals a Mechanism with TiO2 as a Reversible Electron Acceptor in a TiO2–Re Catalyst System for CO2 Photoreduction, J. Am. Chem. Soc., 2017, 139, 1226–1232 CrossRef CAS PubMed.
- C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Improving the Photocatalytic Reduction of CO2 to CO through Immobilization of a Molecular Re Catalyst on TiO2, Chem. – Eur. J., 2015, 21, 3746–3754 CrossRef CAS PubMed.
- C. L. Anfuso, R. C. Snoeberger III, A. M. Ricks, W. Liu, D. Xiao, V. S. Batista and T. Lian, Covalent Attachment of a Rhenium Bipyridyl CO2 Reduction Catalyst to Rutile TiO2, J. Am. Chem. Soc., 2011, 133, 6922–6925 CrossRef CAS PubMed.
- K. D. Dubois, A. Petushkov, E. G. Cardona, S. C. Larsen and G. Li, Adsorption and Photochemical Properties of a Molecular CO2 Reduction Catalyst in Hierarchical Mesoporous ZSM-5: An In Situ FTIR Study, J. Phys. Chem. Lett., 2012, 3, 486–492 CrossRef CAS PubMed.
- K. D. Dubois, H. He, C. Liu, A. S. Vorushilov and G. Li, Covalent attachment of a molecular CO2-reduction photocatalyst to mesoporous silica, J. Mol. Catal. A: Chem., 2012, 363–364, 208–213 CrossRef CAS.
- C. Liu, K. D. Dubois, M. E. Louis, A. S. Vorushilov and G. Li, Photocatalytic CO2 Reduction and Surface Immobilization of a Tricarbonyl Re(I) Compound Modified with Amide Groups, ACS Catal., 2013, 3, 655–662 CrossRef CAS.
- T. G. Fenton, M. E. Louis and G. Li, Effect of ligand derivatization at different positions on photochemical properties of hybrid Re(I) photocatalysts, J. Mol. Catal. A: Chem., 2016, 411, 272–278 CrossRef CAS.
- X. Jia, H. S. Nedzbala, S. R. Bottum, J. F. Cahoon, J. J. Concepcion, C. L. Donley, A. Gang, Q. Han, N. Hazari, M. C. Kessinger, M. R. Lockett, J. M. Mayer, B. Q. Mercado, G. J. Meyer, A. J. Pearce, C. L. Rooney, R. N. Sampaio, B. Shang and H. Wang, Synthesis and Surface Attachment of Molecular Re(I) Complexes Supported by Functionalized Bipyridyl Ligands, Inorg. Chem., 2023, 62, 2359–2375 CrossRef CAS PubMed.
- A. Ge, B. Rudshteyn, P. E. Videla, C. J. Miller, C. P. Kubiak, V. S. Batista and T. Lian, Heterogenized Molecular Catalysts: Vibrational Sum-Frequency Spectroscopic, Electrochemical, and Theoretical Investigations, Acc. Chem. Res., 2019, 52, 1289–1300 CrossRef CAS PubMed.
- H. He, C. Liu, M. E. Louis and G. Li, Infrared studies of a hybrid CO2-reduction photocatalyst consisting of a molecular Re(I) complex grafted on Kaolin, J. Mol. Catal. A: Chem., 2014, 395, 145–150 CrossRef CAS.
- K. L. V. Joseph, J. Lim, A. Anthonysamy, H.-i. Kim, W. Choi and J. K. Kim, Squaraine-sensitized composite of a reduced graphene oxide/TiO2 photocatalyst: π–π stacking as a new method of dye anchoring, J. Mater. Chem. A, 2015, 3, 232–239 RSC.
- J. M. Smieja and C. P. Kubiak, Re(bipy-tBu)(CO)3Cl-improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- M. E. Louis and G. Li, Infrared studies of surface carbonate binding to diimine-tricarbonyl Re(I) and Mn(I) complexes in mesoporous silica, J. Coord. Chem., 2019, 72, 1336–1345 CrossRef CAS.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, CO2 Capture by a Rhenium(I) Complex with the Aid of Triethanolamine, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- D. H. Gibson, X. Yin, H. He and M. S. Mashuta, Synthesis and reactions of fac-[Re(dmbpy)(CO)3X] (dmbpy = 4,4′-dimethyl-2,2′-bipyridine; X = COOH, CHO) and their derivatives, Organometallics, 2003, 22, 337–346 CrossRef CAS.
- J. K. Nganga, L. M. Wolf, K. Mullick, E. Reinheimer, C. Saucedo, M. E. Wilson, K. A. Grice, M. Z. Ertem and A. M. Angeles-Boza, Methane Generation from CO2 with a Molecular Rhenium Catalyst, Inorg. Chem., 2021, 60, 3572–3584 CrossRef CAS PubMed.
- D. H. Gibson and H. He, Synthesis and properties of fac-Re(dmbpy)(CO)3CHO (dmbpy = 4,4′-dimethyl-2,2′-bipyridine), a possible intermediate in reductions of CO2 catalyzed by fac-Re(dmbpy)(CO)3Cl, Chem. Commun., 2001, 2082–2083 RSC.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, Mechanistic Contrasts between Manganese and Rhenium Bipyridine Electrocatalysts for the Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structures, UV-vis spectra, DRIFTS spectra and photocatalytic activities of two isomeric Re(I) catalysts. See DOI: https://doi.org/10.1039/d3dt03623e |
| This journal is © The Royal Society of Chemistry 2024 |