
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeyond the coupled distortion model: structural analysis of the single domain cupredoxin AcoP, a green mononuclear copper centre with original features†
Magali
Roger
a,
Philippe
Leone
b,
Ninian J.
Blackburn
c,
Sam
Horrell
de,
Tadeo Moreno
Chicano
d,
Frédéric
Biaso
a,
Marie-Thérèse
Giudici-Orticoni
a,
Luciano A.
Abriata
 f,
Greg L.
Hura
g,
Michael A.
Hough
de,
Giuliano
Sciara
*ah and
Marianne
Ilbert
*a
f,
Greg L.
Hura
g,
Michael A.
Hough
de,
Giuliano
Sciara
*ah and
Marianne
Ilbert
*a
aCNRS, Aix-Marseille University, Bioenergetic and Protein Engineering Laboratory, BIP UMR 7281, Mediterranean Institute of Microbiology, 13009 Marseille, France. E-mail: giuliano.sciara@inrae.fr; milbert@imm.cnrs.fr
bCNRS, Aix-Marseille University, Laboratoire d'Ingénierie des Systèmes Macromoléculaires, LISM UMR7255, 13009 Marseille, France
cDepartment of Chemical Physiology and Biochemistry, School of Medicine, Oregon Health and Science University, Portland, Oregon 97239, USA
dSchool of Life Sciences, University of Essex, Wivenhoe Park, Colchester, Essex CO4 3SQ, UK
eDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
fLaboratory for Biomolecular Modeling and Protein Purification and Structure Core Facility, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
gMolecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Lab, Berkeley, CA, USA
hAix Marseille Univ, INRAE, BBF UMR1163, Biodiversité et Biotechnologie Fongiques, 13288 Marseille, France
First published on 3rd January 2024
Abstract
Cupredoxins are widely occurring copper-binding proteins with a typical Greek-key beta barrel fold. They are generally described as electron carriers that rely on a T1 copper centre coordinated by four ligands provided by the folded polypeptide. The discovery of novel cupredoxins demonstrates the high diversity of this family, with variations in terms of copper-binding ligands, copper centre geometry, redox potential, as well as biological function. AcoP is a periplasmic cupredoxin belonging to the iron respiratory chain of the acidophilic bacterium Acidithiobacillus ferrooxidans. AcoP presents original features, including high resistance to acidic pH and a constrained green-type copper centre of high redox potential. To understand the unique properties of AcoP, we undertook structural and biophysical characterization of wild-type AcoP and of two Cu-ligand mutants (H166A and M171A). The crystallographic structures, including native reduced AcoP at 1.65 Å resolution, unveil a typical cupredoxin fold. The presence of extended loops, never observed in previously characterized cupredoxins, might account for the interaction of AcoP with physiological partners. The Cu-ligand distances, determined by both X-ray diffraction and EXAFS, show that the AcoP metal centre seems to present both T1 and T1.5 features, in turn suggesting that AcoP might not fit well to the coupled distortion model. The crystal structures of two AcoP mutants confirm that the active centre of AcoP is highly constrained. Comparative analysis with other cupredoxins of known structures, suggests that in AcoP the second coordination sphere might be an important determinant of active centre rigidity due to the presence of an extensive hydrogen bond network. Finally, we show that other cupredoxins do not perfectly follow the coupled distortion model as well, raising the suspicion that further alternative models to describe copper centre geometries need to be developed, while the importance of rack-induced contributions should not be underestimated.
Introduction
Cupredoxins belong to a widely occurring family of copper-binding proteins, ubiquitous in all kingdoms of life and involved in key biological processes, such as respiration, photosynthesis, the nitrogen cycle and copper homeostasis.1,2 They share a typical fold with a beta-sandwich involving 7–8 strands arranged into a Greek-key beta barrel. Depending on a slightly different arrangement of the N-terminal beta strands, two distinct sub-folds have been identified: the plastocyanin (Pc) and the rusticyanin-stellacyanin ones.3 The overall cupredoxin fold has been conserved throughout evolution3 and can be found in all domains of life. Well known proteins containing this fold are single domain proteins, such as Type 1 (T1) copper proteins (Azurin, plastocyanin, rusticyanin, …) and cupredoxin-like proteins (CupA, CopC or PmoD, …). It can also be found in multi-domain enzymes such as multicopper oxidases (MCOs including laccases, bilirubin oxidases, …), cytochrome c oxidase and copper-containing nitrite reductases (NiRs). Despite sharing a common fold, cupredoxins can accommodate various types of copper centres with different properties related to specific functions such as electron transfer, copper sequestration, or catalysis.1,4–6 For this reason, cupredoxins represent an ideal model system for understanding the structure–function relationship of copper proteins. Among the cupredoxins, T1-copper proteins provide an attractive model system and therefore have been the subject of intensive studies for decades (see reviews7–9). Indeed, T1-copper proteins are single domain cupredoxins that bind one copper atom, typically coordinated by three strong equatorial ligands which are highly conserved including one cysteine and two histidines as well as a weak axial ligand, most commonly a methionine residue. Despite their apparent simplicity, one fascinating feature of T1-copper proteins is their colour heterogeneity, from light blue to red, which results from the various geometries adopted by the copper atom and its ligands in cupredoxin copper centres (Fig. S1†) (for a comprehensive overview on Cu centre complexity, see reviews10–12). This gives rise to unique electronic and spectroscopic features in their oxidized form. Based on these features, T1 proteins have been divided into three sub-families: blue or “classical T1” (tetrahedral), green or “T1.5” (distorted tetragonal), and red or “T2-like” (tetragonal) (Fig. S1†).From detailed spectroscopic analysis, combined with structural data and theoretical calculations performed on natural or “engineered” T1-copper proteins, a model which rationalizes the variation of spectroscopic properties in different sub-families has been proposed.10 According to the coupled distortion model (CDM), blue copper centres (T1) have a tetrahedral geometry due to a short Cu2+–S(Cys) bond length (∼2.1 Å), while the Cu2+–S(Met) bond length is around 3 Å. This leads to an intense S(Cys) π → Cu ligand-to-metal charge transfer (LMCT) transition2,11 which gives rise to a strong absorption band at 600 nm. By contrast, green copper centres (T1.5) exhibit a distorted tetragonal geometry with a longer Cu2+–S(Cys) bond length (∼2.2 Å) and a shorter Cu–S2+(Met) bond length (∼2.5 Å). These differences result, in T1.5 proteins, into a weaker absorption band at 600 nm, as well as into an additional band near 450 nm attributed to S(Cys) σ → Cu LMCT, and in some cases to a S(Met) → Cu LMCT transition. This is concomitant with a rotation of the (Cys)S–Cu–S(Met) plane with respect to the (His)N–Cu–N(His) plane.11 Another sub-class of cupredoxins, called “blue-perturbed” (such as rusticyanin, stellacyanin and Cucumber Basic Protein), has intermediate features between T1 (blue) and T1.5 (green) copper proteins, resulting from a tetrahedral distorted geometry, with 2.2 and 2.8 Å (ref. 13) for the Cu2+–S(Cys) and Cu2+–S (Met) bond lengths, respectively. As of today, only one example of T2-like (or red) cupredoxin protein, called nitrosocyanin, has been found in nature.14 Its structure has been solved, the copper is coordinated by a histidine as the fourth ligand and a glutamate replaces one of the equatorial histidines. This rearrangement of residues results in a tetragonal geometry and the binding of a water molecule in the equatorial plane (Fig. S1†).
Strikingly, the redox potential (Em) of T1-copper proteins and cupredoxin domains greatly vary: from +90 mV to +680 mV vs. SHE, in auracyanin D15 and rusticyanin,16 respectively. Several studies based on protein engineering/site-directed mutagenesis attempted to rationalize the electronic and redox features of T1-copper proteins.17–20
From these studies, key factors such as the nature of the copper ligands, as well as the role of the second coordination sphere in regulating the electronic and redox properties of T1-copper centres were highlighted.17–20 Despite this, novel insight remains to be gained to intimately understand the rational link between protein structures, active centre geometries, redox potentials and spectral properties in cupredoxins. Further challenging our understanding of this relationship, novel cupredoxins with intriguing features are constantly being discovered,21–25 also illustrating broad versatility among this class of proteins, and requiring further analysis to explain their properties.
We recently discovered and characterized an original cupredoxin, the Acidithiobacillus ferrooxidans cytochrome c oxidase partner (AcoP), the first natural, single domain green T1 protein to be isolated26 and characterized in detail.27–30 AcoP is a membrane-associated cupredoxin consisting of a soluble, copper binding periplasmic domain that is anchored to the cell membrane by its N-terminal transmembrane segment. AcoP interacts with the terminal enzyme of the A. ferrooxidans respiratory chain, the cytochrome c oxidase (CcO),27,31 and seemed to play an important role in maintaining CcO activity under extreme acidic conditions in the periplasm (estimated to be around pH 3).27 AcoP might also play a physiological role in electron transfer as a periplasmic component of the iron oxidizing A. ferrooxidans respiratory chain, and we recently demonstrated intermolecular electron transfer between the high potential haem of a di-haem cytochrome c (Cyt c) and AcoP,29 suggesting the existence of an alternative electron transfer pathway from the Cyt c to CcO through AcoP.31 The spectroscopic properties of AcoP are similar to that of a T1.5 copper centre. Based on mutagenesis studies, we demonstrated that AcoP binds one copper atom coordinated by the classic cupredoxin set of copper ligands: one Cys, one Met and two His.28,30 Strikingly, AcoP exhibits an unexpectedly high redox potential (+566 mV at pH 5 vs. SHE)28 for a green copper protein. Indeed, examples of T1.5 copper proteins found in nature are rare. They include Nitrite reductases (CuNiRs) and auracyanin D, and are usually associated with “low” redox potentials (+250 mV and +90 mV vs. SHE, respectively).15,32 Based on several mutagenesis studies on NiRs and rusticyanin, it was proposed that shortening the Cu2+–S(Met) bond could result in the stabilization of the Cu2+ state, thus lowering the redox potential.32–34 Other studies, made on high redox potential, perturbed blue cupredoxins such as rusticyanin, have highlighted the importance of the second coordination sphere in determining the redox potential of the protein.35
In this study, we report the first structural characterization of a natural, single domain green cupredoxin. Structural data were obtained for wild type AcoP and two mutants of the first coordination sphere, using X-ray crystallography, SAXS and EXAFS. Our work unveils very unusual features of the AcoP copper centre geometry, reminiscent of both blue and green copper centres as defined by the CDM. The reasons for such features and the role of the first and second coordination spheres were investigated and the implications for electronic and redox properties are discussed. Finally, our data suggest that, for some cupredoxins, alternative models than the CDM need to be developed.
Experimental
Protein mutagenesis, protein purification and spectroscopic measurements
Expression and purification for wild-type and mutant AcoP was performed as described previously.28,30 UV-Vis ab-sorption spectra of purified proteins were recorded using a Cary 50 Bio (Varian) spectrophotometer. Protein purity and concentration were determined with a theoretical molar extinction coefficient (ε280) of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 440 M−1 cm−1 derived from the known AcoP amino acid sequence using Protparam.36
440 M−1 cm−1 derived from the known AcoP amino acid sequence using Protparam.36
Crystallization of AcoP wild-type and mutants
Crystals of wild-type (WT) AcoP, as well as of the M171A and H166A mutants, were grown using the sitting-drop vapour diffusion technique in MRC 96-well crystallization plates (Swissci). 100 nL of protein and precipitating solution were dispensed using a Mosquito robot (TTP Labtech) and equilibrated against 50 μL of precipitating solution. Early crystal hits were obtained starting from commercial crystallization solution kits (Molecular Dimensions Limited). In final crystallization experiments, the protein concentration was set to 8–10 mg mL−1. In the case of AcoP WT and M171A, the crystallization solution contained 100 mM potassium acetate, 10 mM potassium chloride, 50 mM buffer (MES, Hepes, or TRIS, pH 6.0 to 8.0) and either PEG 3000 (34% to 44% w/v) or PEG 3350 (29% to 39% w/v) as a precipitating agent. In the case of AcoP H166A, crystals were obtained in 0.1 M Bis-Tris pH 5.5 and 2.0 M ammonium sulphate. Plates were incubated at 20 °C in a Rock Imager station (Formulatrix), which also allowed monitoring of crystal growth during time. Well-defined rod-shaped crystals grew for AcoP WT and M171A, with dimensions of up to 50 μm thickness and 200 μm length. These crystals were stable and well-diffracting even several weeks after they appeared. Noteworthy, crystals of AcoP WT and M171A were still transparent and green, respectively, one year after they appeared. This behaviour is in agreement with the high redox potential measured for AcoP WT (which tends to remain reduced even in aerobic conditions) and with the impossibility to reduce the M171A mutant in vitro. Small, 30 μm transparent pyramidal crystals were obtained for AcoP H166A. The crystals were cryoprotected with ∼20% v/v glycerol added to the crystallization conditions. PEG cryoprotectant solutions were prepared using 150 mM sodium acetate pH 3.6 as a buffer; the final pH was however measured to be 4.6. The structures of reduced and oxidized WT AcoP were obtained from crystals flash-cooled in liquid nitrogen, after soaking them in 2 μL of cryoprotectant containing 10 mM of either sodium ascorbate or sodium hexachloroiridate(IV), respectively for 75 and 120 minutes.Diffraction data collection and processing
X-ray diffraction data were collected at synchrotrons ESRF (Grenoble, FR; beamlines ID29 and ID30B) and Soleil (Gif-sur-Yvette, FR; beamlines Proxima1 and 2). Cryo-cooled crystals were mounted under a stream of gaseous nitrogen at 100 K. All diffraction data were indexed and integrated using XDS.37 Subsequent data processing and interpretation relied on programs available in the CCP4 Suite (v 6.4.0).38 Space group determination and scaling was achieved using Pointless (v 1.8.12) and Scala (v 3.3.21).39,40 Crystals of all AcoP variants (reduced, oxidized, M171A and H166A) belonged to space group P41212. A data set collected above the Cu K-edge (1.07137 Å) was used for SAD phasing (single-wavelength anomalous dispersion). Copper site location and refinement, density modification, and preliminary model building was performed using the CRANK suite (v 1.5.46).41 Several secondary structure elements were unambiguously identified, pruned after visual inspection, and used as a poly-alanine model for phasing a native dataset at higher resolution by molecular replacement in PHASER (v 2.5.5).42 Starting from this solution, 95% of the model was assigned by automated model building in ARP/wARP (v 7.4).43 The structures of other AcoP variants (oxidized, M171A and H166A) were solved by molecular replacement with PHASER using the reduced wild type as a starting model. The same model was the reference for the set of diffraction reflections used to estimate Rfree in all models. Model refinement was carried out in Refmac (v 5.8.0267),44 using a B factor refinement protocol: isotropic B factors were used for all atoms except Cu(I) and Cu(II) ions, for which anisotropic B factors and no restraints were used in order to yield the most precise and unbiased atomic positions. TLS refinement was carried out with one group defined per chain. Data collection and refinement statistics are reported in Table 1. Model geometry was validated using MolProbity.45 Single crystal UV-Vis spectra were collected using the CryoBench microspectrophotometer mounted online at the ESRF beamline BM30.46 Errors on distances derived from three-dimensional structures were calculated based on atomic coordinate precision, estimated by the diffraction precision index (DPI).47| All data were in space group P41212.a Values in parentheses are for the highest-resolution shell. | ||||
|---|---|---|---|---|
| AcoP form | Reduced | Oxidized | H166A | M171A |
| SYNCHROTRON – beamline | SOLEIL – Proxima 1 | ESRF – ID30B | ESRF – ID29 | SOLEIL – Proxima 2 |
| Unit cell a, b, c (Å) | 72.95, 72.95, 112.82 | 73.48, 73.48, 112.90 | 73.91, 73.91, 113.87 | 73.75, 73.75, 113.08 |
| Unit cell α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolutiona (Å) | 50–1.65 (1.74–1.65) | 40.0–1.7 (1.79–1.7) | 40.0–2.1 (2.21–2.1) | 50–1.82 (1.92–1.82) |
| Unique reflectionsa | 37412 (5387) |
34624 (4976) |
18289 (2652) |
28048 (4087) |
| Redundancya | 6.9 (7.1) | 5.6 (5.5) | 7.3 (7.4) | 7.4 (7.5) |
| Completenessa (%) | 99.9 (99.9) | 99.7 (99.8) | 96.7 (98.1) | 98.3 (99.6) |
| I/σa | 15.4 (1.9) | 6.1 (1.4) | 10.4 (2.0) | 14.6 (1.8) |
|
R
measa (%) |
5.6 (111.1) | 16.9 (116.6) | 15.3 (124.6) | 8.5 (107.2) |
| CC1/2 | 0.999 (0.638) | 0.987 (0.485) | 0.996 (0.477) | 0.998 (0.616) |
| Mosaicity | 0.09 | 0.13 | 0.06 | 0.07 |
| Refinement and model quality | ||||
| Resolution (Å) | 44.6–1.65 | 38.3–1.7 | 35.8–2.1 | 47.4–1.82 |
| Reflections | 35493 |
32890 |
17368 |
26633 |
| Rfac/Rfree (%) | 16.0/19.0 | 23.6/26.0 | 19.1/24.2 | 16.0/19.8 |
| Number of atoms | ||||
| Protein/ion/ligand/water | 2242/10/18/173 | 2096/22/12/123 | 2101/2/12/109 | 2124/18/30/205 |
| B-Factors (Å2) | ||||
| Protein/ion/ligand/water | 21.6/36.1/44.3/40.7 | 20.5/37.2/42.0/35.2 | 28.1/35.7/58.5/43.3 | 23.6/34.5/43.8 |
| Rmsd | ||||
| Bond (Å) | 0.015 | 0.013 | 0.013 | 0.013 |
| Angle (°) | 1.930 | 1.689 | 1.840 | 1.681 |
| Ramachandran plot (%) | ||||
| Most favoured | 95.7 | 95.3 | 94.2 | 95.0 |
| Allowed regions | 3.5 | 3.5 | 4.7 | 4.2 |
| Outliers | 0.8 | 1.2 | 1.1 | 0.8 |
| PDB accession code | 7Z3B | 7Z3F | 7Z3G | 7Z3I |
EXAFS data collection and analysis
Samples were mixed with 20% (vol/vol) ethylene glycol and measured as frozen glasses at 10 K. Cu K edge (8.9 keV) extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES) were collected at the Stanford Synchrotron Radiation Lightsource on beamline 9–3 using a Si 220 monochromator with a φ = 90° crystal set and a Rh-coated mirror located upstream of the monochromator with a 13 keV energy cut-off to reject harmonics. Kα fluorescence was collected using a 100-element Canberra Ge array detector. A Z-1 metal oxide filter and Soller slit assembly was placed in front of the detector to attenuate the elastic scatter peak. A buffer blank was subtracted from the raw data to produce a flat pre-edge and eliminate residual Ni Kβ fluorescence of the metal oxide filter. Energy calibration was achieved by placing a Cu metal foil between the second and third ionization chamber. Data averaging, background subtraction, and normalization were performed using EXAFSPAK. The experimental energy threshold (k = 0) was chosen as 8985 eV. Spectral simulation was carried out by least-squares curve fitting, using full curved wave calculations, formulated by the program EXCURVE 9.2 as previously described.48–50Small angle X-ray scattering (SAXS)
SAXS analysis was conducted at the SIBYLS beamline at Lawrence Berkeley National Lab's Advanced Light Source. Data were collected from both size-exclusion-chromatography-coupled SAXS (SEC-SAXS) and from high-throughput SAXS (HT-SAXS).51 The data from both measurements were merged to maximize the signal to noise ratio in all regions. As protein size is 16.7 kDa, the injection volume and concentration of 100 μL at 6 mg mL−1 produced noisy results in the high q (momentum transfer q = 4π (sin(θ/2))/λ, where θ is the scattering angle and λ is the X-ray wavelength of 1.23 Å). The high q region was therefore supplemented with one HT-SAXS measurement on 30 μL at 6 mg mL−1. The sample was collected in a transmission geometry with a sample thickness of 1.5 mm. Initial analysis was conducted with the ScÅtter program. The shape determined from the SAXS results are from GASBOR.52 The comparison of the measured SAXS data with the crystal structure was done using FoXS.53 To build out the structure to include missing loops the program BilboMD54 was used.Results
X-ray crystal structure and overall fold of AcoP
The X-ray crystal structure (PDB: 7Z3B) of reduced AcoP was solved at 1.65 Å resolution (Fig. 1A). The lack of colour of native crystals and solutions of purified AcoP suggests that in aerobic (atmospheric) conditions the protein is in its reduced form, as expected from its quite high reduction potential (+566 mV). The asymmetric unit contains two molecules (A and B) (Fig. S2†). The two polypeptides do not engage in extensive interactions and are unlikely to represent a biological dimer, consistent with data obtained by small angle X-ray scattering (SAXS) that point to a monomeric species in solution (Fig. 1B). Indeed, the measured radius of gyration was 16 ± 1 Å from Guinier analysis and 16.5 Å from pair distribution function analysis, both values that are in good agreement with the crystal structure. The Porod exponent was 4.0, indicating a well folded globular protein in solution. The radius of cross-section was 13 Å, close to the radius of gyration, further supporting a globular structure. The mass determined from SAXS was 13 ± 5 kDa. The maximum dimension was 53 Å. To further connect the SAXS results with the atomic resolution crystal structure, the shape was defined by the GASBOR52 program and modelling was performed using FoXS53 and BilboMD.54 A model of the full-length sequence was made based on the crystal structure. Missing loops were added and then allowed to sample conformations, until best fit was found with the SAXS data. An excellent fit with χ2 agreement better than 1 was found. Fit of the model to SAXS data and the calculated shape is shown in Fig. 1B. As expected, AcoP displays Greek-key beta-barrel topology typical of cupredoxins, with three clear differences compared to canonical fold: (i) an alpha helical loop connecting three copper ligands of an unusual length of 6 residues between the C-terminal histidine and the cysteine ligands (CX6H); (ii) on the same side, the extended β6–β7 loop (res. 90–114); and (iii) on the opposite side a small β-hairpin connecting β8 and β9 (res. 119–128), that extends the β1-strand-containing beta-sheets (Fig. 1).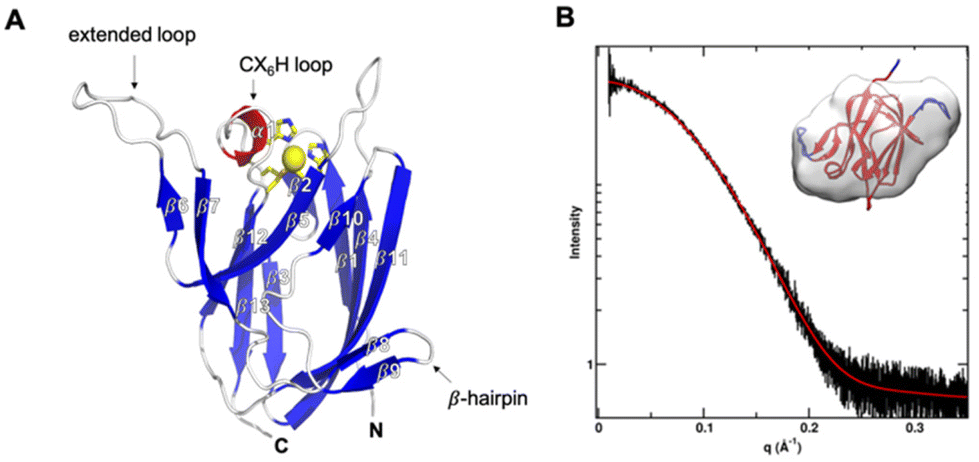 | ||
| Fig. 1 Overall structure and SAXS analysis of AcoP. (A) Chain A in the asymmetric unit of AcoP crystals. Beta strands are in blue, the alpha helical CX6H loop in red, and the copper ligand carbon as well as the copper atoms in yellow. The region from strands β6 to β9 (red dashed circles) corresponds to an amino acid sequence conserved only in AcoP and related cupredoxins from acidophiles.28 (B) SAXS data for AcoP in solution. Reciprocal space, experimental SAXS curve (black) is overlaid with the predicted scattering (red). Inset: Ab initio shape reconstruction of AcoP based on the SAXS data, overlaid with the crystal structure. | ||
The fold of AcoP is the same found in the rusticyanin/stellacyanin and not the plastocyanin subfamily.3 Structural comparison of AcoP with other well-known cupredoxins highlights the presence of a well-conserved region, corresponding to the beta sandwich that characterizes the cupredoxin fold (Fig. 2A, grey) and of a non-conserved region (residues 90–128 in AcoP) that includes the extended loop and the β-hairpins mentioned above (Fig. 2A, green). By contrast, this region is highly conserved in AcoP homologues from acidophilic bacteria.28 Hence, it can be speculated that this region might be important for specific functions or interactions with partner(s) specifically in these microorganisms. This region is a source of molecular diversity within the cupredoxin family, as shown for azurin, pseudoazurin and Cucumber Basic Protein (CBP, Fig. 2A). Strikingly, AcoP extended loop is reminiscent of protruding motifs observed in copper-loading chaperones which do not possess the cupredoxin fold, such as in the PCuAC55 and Sco56 protein families (Fig. 2B).
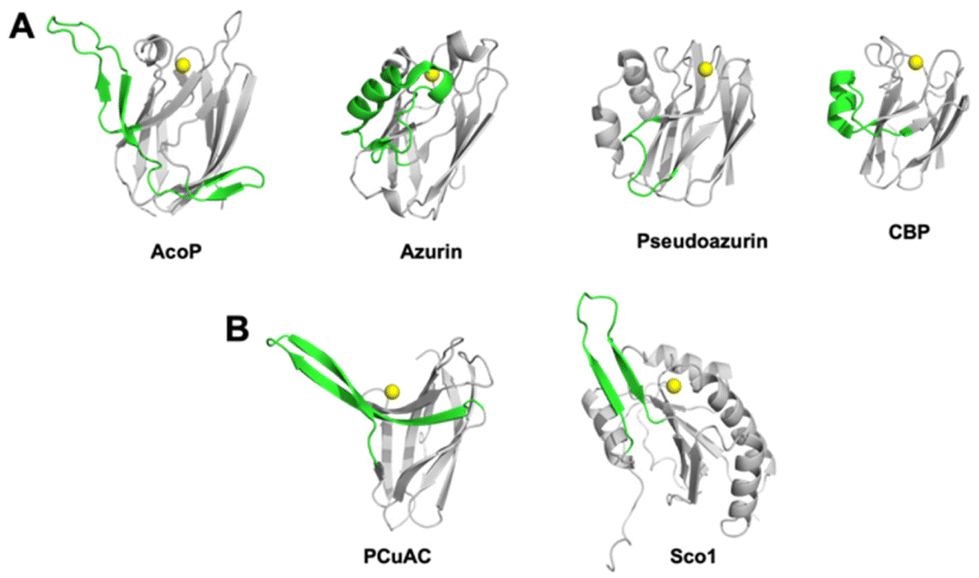 | ||
| Fig. 2 Comparison of the AcoP fold with those from other cupredoxins and copper-loading chaperones. (A) Diversity of a region coloured in green (residues 90–128 in AcoP) among the superfamily of cupredoxins: AcoP (PDB: 7Z3B), Azurin (1E5Y), Pseudoazurin (1PZA) or Cucumber Basic Protein (2CBP). (B) Comparison of the non-conserved region (coloured in green) found in AcoP, with regions from copper-bound metallochaperones such as PCuAC from Thermus thermophilus (2K70), and human Sco1 from (2GQM). | ||
Analysis of the first coordination sphere
The crystal structure of AcoP confirmed the identity of the copper ligands previously proposed based on bioinformatics analysis and mutagenesis studies.30 AcoP is, therefore, the first single domain green type cupredoxin with a set of classical ligands to be structurally characterized. The Cu–ligand distances in the reduced state AcoP structure determined by X-ray crystallography are reported in Table 2 and Fig. 3A. A more detailed description of AcoP metal centre geometries may be found in Table S1.† Bond lengths between the Cu atom and the histidine ligands are typical of T1 copper centres (expected to range from 1.9 Å to 2.2 Å). Cu–S(Cys) distance was measured to be 2.23 Å for reduced AcoP, the same value measured for AcNir (PDB 2BW4). Strikingly, the distance between the Cu atom and the Met171 axial ligand (2.92 Å) is similar to the distance observed for classic blue and perturbed blue copper sites: Amicyanin 2.91 Å (PDB 2RAC), plastocyanin 2.87 Å (5PCY), pseudoazurin 2.71 Å (8PAZ).1 This bond is much longer than what previously reported for the green copper centres in CuNiRs (2.43–2.55 Å).57–59 To measure more precisely Cu–ligand distances, EXAFS studies were performed. Spectra were recorded on purified reduced AcoP (Fig. 3B). Data were fit to a chemical model based on canonical blue copper binding motifs, namely a (His)2Cys coordination environment with potential additional coordination by a methionine thioether. Best fits were obtained using two histidine residues at Cu–N(imid) = 2.0 Å and one cysteine residue with Cu–S(Cys) = 2.20 Å. Inclusion of a Cu–S(Met) interaction at 2.73 Å improved the quality of the fit by ∼10 percent. However, given the large body of literature concerning EXAFS studies of cupredoxins in which Cu–Met interactions were not observed, coordination of a Met residue at this distance must be viewed with caution, particularly as the Debye–Waller (DW) factor for the methionine shell is large (2σ2 = 0.06 Å2). We also tried to fit the data with shorter Cu–Met distances, but the starting (shorter) Cu–S distance always refined back to 2.7 Å and to a high DW factor. Thus, we may conclude that the EXAFS data support a cupredoxin-like Cu centre with the usual caveat that the Cu–S(Met) distance is poorly defined. More importantly, they confirm the values obtained by crystallography: a long Cu–S(Cys) distance of 2.2 Å, consistent with the “green copper” classification, and a long Cu–S(Met) distance of at least 2.7 Å. This long Cu–S(Met) distance is unexpected for a green type cupredoxin, according to the CDM.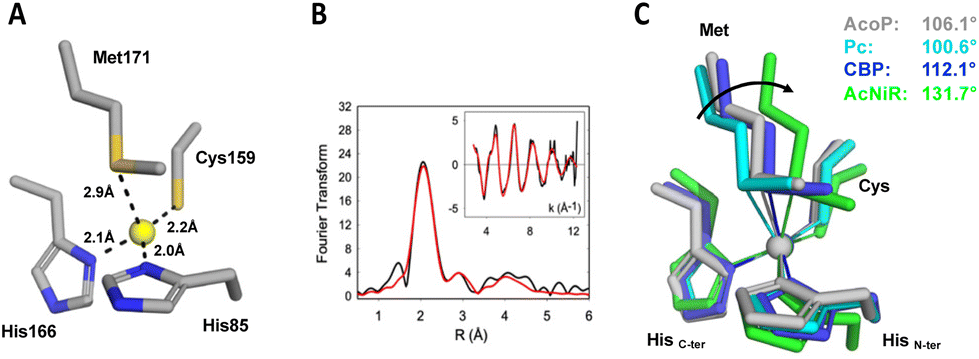 | ||
| Fig. 3 Copper centre geometry of AcoP and comparison with other copper centres. (A) Geometry and distance between Cu and the ligands in the reduced state of wildtype AcoP copper site. (B) EXAFS spectroscopy of AcoP. Fourier transform and EXAFS (inset) for AcoP as isolated protein. Black traces are experimental data, red traces are simulated ones. The best fit (shown) was used to derive metrical details, listed in Table 2. (C) Overlay of the copper sites described for plastocyanin (Pc, PDB 1PLC, light blue), Cucumber Basic Protein (CBP, PDB 2CBP, dark blue), nitrite reductase (AcNiR, PDB 1NIF, green) and AcoP (PDB 7Z3B, grey). The arrow indicates the variation of the angle (C-termHis)N–Cu–S(Met) along the T1-to-T1.5 trajectory; the angle values are indicated for each structure. | ||
| AcoP red | AcoP ox | M171A (ox) | H166A (red) | |
|---|---|---|---|---|
| 7Z3B | 7Z3F | 7Z3G | 7Z3I | |
| a Determined by X-ray crystallography (molecule A and B averaged). See Table S1† for the complete set of values. b Determined by EXAFS. Estimated values after fitting the data are listed with Debye Waller terms (2σ2) in parentheses. | ||||
| Cu–N(His85) (Å) | 2.00 ± 0.005a | 2.04 ± 0.01a | 2.03 ± 0.015a | 1.92 ± 0.03a |
| 2.00 (0.011)b | ||||
| Cu–S(Cys159) (Å) | 2.23 ± 0.005a | 2.27 ± 0.06a | 2.25 ± 0.04a | 2.15 ± 0.015a |
| 2.20 (0.009)b | ||||
| Cu–N(His166) (Å) | 2.10 ± 0.005a | 2.18 ± 0.01a | 2.05 ± 0.005a | — |
| 2.00 (0.011)b | ||||
| Cu–S(Met171) (Å) | 2.92 ± 0.06a | 2.77 ± 0.03a | — | 2.80 ± 0.015a |
| 2.73 (0.059)b | ||||
The value of the Cu–S(Cys) distance has been considered as a strong predictor of the cupredoxin sub-class (T1, T1.5 or T2) since it is dependent on the degree of covalency of the Cu–S(Cys) bond (Fig. S1†).48,60 Blue T1 copper proteins such as plastocyanin (Pc) and azurin exhibit short Cu–S(Cys) distances (2.07–2.15 Å) as the result of strong covalency associated with the dx2−y2 orbital of the copper ion to the π orbital of the sulphur atom. This bond is largely responsible for an intense S(π) to Cu(II) charge transfer band (∼600 nm) and a much weaker S(σ) to Cu(II) band (∼450 nm) in the electronic absorption spectrum. In sharp contrast, red (T2) copper proteins exemplified by nitrosocyanin61,62 and Sco60 exhibit long Cu–S(Cys) distances around 2.25 Å and dominant LMCT bands between 350 and 400 nm due to ligand-σ to Cu(II) charge transfer. Green (T1.5) copper proteins such as CuNirs have intermediate LMCT behaviour between those of T1 and T2 centres, with two bands of comparable intensity at ∼450 and 550 nm. Cu–S(Cys) distances for green cupredoxins are typically in the range 2.18 Å–2.23 Å, as described for HdNIR and AcNIR.59,63 The Cu–S(Cys) distance measured for AcoP (2.20–2.23 Å) fits with the distances expected for a green type copper centre. Indeed, several studies based on natural or engineered T1 copper centres have helped to rationalize the changes occurring in T1 copper centre from blue, green and red copper sites.33,64,65 The CDM describes the interplay between the strength of the copper–axial ligand interaction and the copper–Cys covalency, and its relationship with the spectroscopic differences observed in the T1 to T1.5 to T2 transition.10,11,58 According to this model, stretching the Cu–S(Cys) bond, such as in the T1 to T2 transitions described above, is accompanied by concomitant shortening of the Cu–axial ligand, a methionine in canonical blue copper centres. As such, the Cu–S(Met) bond distance ranges from 2.82–3.11 Å in plastocyanin – azurin (T1), to 2.71–2.61 Å in pseudoazurin – CBP (perturbed T1), to 2.53–2.48 Å in HdNIR–AcNIR (T1.5).59,63 For this reason, the Cu–S(Met) distance measured for AcoP (2.92 Å, or at least 2.73 Å as suggested by EXAFS) is unexpected for a green cupredoxin, based on the CDM. A distant methionine is expected to have little effects on system energies and spectroscopic properties. Instead, most binding-induced properties depend on the three equatorial ligands and on local electric fields.66 Confirming this, AcoP displays a green spectroscopic signature, predominantly influenced by the short Cu–S(Cys) bond. Unexpectedly, the longer Cu–S(Cys) bond of AcoP is not compensated by a shorter Cu(S) Met distance, as commonly described by the CDM. This suggests that the geometrical properties of AcoP metal centre might not be well explained by the CDM. According to the CDM, comparison of T1 and T2 sites, reveals coupling of Cu–S(Cys) and Cu–S(Met) distances together with rotation of the (Cys)S–Cu–S(Met) plane with respect to the (His)N–Cu–N(His) plane. These changes account for the transformation of tetrahedral (T1) active centre geometries into distorted tetrahedral (perturbed T1), distorted tetragonal (T1.5) and tetragonal (T2) ones. The extent of such rotation can also be quantified by measuring the (His)N–Cu–S(Met) angle involving the C-terminal copper-binding His.67 In this case, blue copper sites with tetrahedral to distorted tetrahedral geometry (such as Pc and CBP, respectively) exhibit smaller N(His)–Cu–S(Met) angles, than a green copper site with distorted tetragonal geometry, such as in CuNiRs (Fig. 3C). Strikingly, the green copper centre of AcoP exhibits a distorted tetrahedral geometry rather reminiscent of that from CBP and Pc, than that of AcNir (Fig. 3C), a result which also does not fit with the CDM.
Overall, these data demonstrate that metal centre geometries (distances and angles) in AcoP do not correlate to (green) spectroscopic features, as it should be expected by the coupled distortion theory. Notably, the Cu–S(Met) distance does not become shorter with the concomitant relaxation of the Cu–S(Cys) bond. Among characterized green cupredoxins, AcoP is the first to exhibit such a long distance between Cu and the axial ligand. Finally, independent of axial ligand effects, AcoP spectroscopic features well correlate to those of green copper sites, as expected by the CDM, based on the Cu–S(Cys) distance, but not on the Cu–S(Met) distance.
Changes in the first coordination sphere upon oxidation
Cupredoxins are well known to provide a rigidified scaffold, the polypeptide, to maintain similar geometries between reduced and oxidized copper centres, in order to facilitate electron transfer by minimizing the reorganization energy between the two oxidation states.68 This is one of the roles of the so-called entatic or rack-induced state in cupredoxins. Nevertheless, small reorganization of the metal centre occurs in order to accommodate the two different Cu(I) and Cu(II) metal oxidation states. Using crystallographic and EXAFS data, it was shown that in poplar plastocyanin (Pc), upon oxidation, the Cu ion slightly moves towards the equatorial plane defined by the (His)2Cys ligands11 (Fig. S3A†). This corresponded to decreased Cu-equatorial ligand distances (−0.06/−0.08 Å), while the angles between equatorial bonds and with the axial Met bond increased (+1/+4°) and decreased (−1/−3°) respectively. These changes are very small compared to the coordinate error and the derived values cannot be considered precise. Nonetheless they show the reorganizational trend of Pc metal centre upon oxidation, with Cu(II) closer to the equatorial plane than Cu(I). We used the same approach in this work. Obtaining the crystallographic structure of reduced AcoP (PDB 7Z3B) was very simple, because the recombinant cupredoxin is naturally in its reduced state (colourless), due to its high reduction potential.28 As such, addition of reducing agents to AcoP native crystals was an additional precaution only, because exposure to X-rays already maintained the reduced state of the cupredoxin. In fact, photoreduction of metallocentres by X-rays is a very well documented phenomenon.69 We consequently decided to use crystal microspectrophotometry to verify the validity of X-ray crystallographic data collected on oxidized AcoP. As observed in solution,28 AcoP crystals soaked into a strong oxidizing agent (Na2IrCl6) showed the expected spectroscopic features, with absorption maxima at 438 and 568 nm (Fig. S4†). These spectra confirm that crystal packing does not affect metal centre geometry to such an extent of perturbing spectroscopic features observed in solution. A typical crystallographic data collection was conducted, wholly irradiating an oxidized AcoP crystal under a large section beam, confirming full AcoP photoreduction during diffraction data collection (Fig. S4A†). We consequently decided to use, on another oxidized AcoP crystal, a helical data collection strategy with an X-ray microfocused beam to minimize photoreduction. Crystal UV/Vis spectra recorded before and after this helical data collection are directly superimposable without scaling (Fig. S4B†), showing that no photoreduction took place. The derived structure (PDB 7Z3F) corresponds to the oxidized form of AcoP. Because the signal of crystal spectra was saturated in the UV region, we cannot estimate the degree of oxidation of the AcoP crystal using the ratio of 280 nm absorption to the copper centre absorption. It was however significant enough to perform the following analysis. As done for Pc, based on differences in Cu–ligand bonds and angles, we observe increased Cu–equatorial ligand distances (+0.04, +0.05, +0.08 Å) and significantly decreased Cu–Met distance (−0.15 Å) upon oxidation (Fig. S3B and Table S1†). Concomitantly, the angles between the equatorial ligands and with the axial Met decreased (−0.5, −2, −4°) and increased (+3, +3, +6°) respectively, more significantly than what observed in Pc. With comparable precision to what observed for Pc, these average values suggest that, upon oxidation of AcoP, Cu moves towards the axial ligand, away from the equatorial plane. This is the opposite direction to that observed in Pc. In general, for each bond and angle change, metal centre rearrangements upon oxidation are the opposite in AcoP and in Pc from three different organisms, although each cupredoxin shows a slightly different behaviour (Table S1†). The change in the Cu–S(Met) distance is in the high range (0.1–0.2 Å) of observed differences: −0.11 and −0.18 Å respectively for the A and B molecules that constitute the crystallographic asymmetric unit in AcoP crystals. Even after this oxidation-induced contraction, the Cu–S(Met) distance still remains the longest reported for a green type copper centre. Dissimilarities between molecule A and B were observed for all the analysed bond angles and distances, and specific constraints, probably induced by crystal packing, appear to completely abolish certain angle or distance changes (Table S1†). Apart from these minimal variations within the metal centre, overall, no significant difference between the structures of reduced and oxidized AcoP could be observed (rmsd ∼ 0.36 Å), suggesting that as expected for cupredoxins only minor rearrangements of copper ligand geometries occur. Our data suggest that although requiring a certain care in sample preparation, and despite the small changes observed, structural studies of reduced and oxidized cupredoxin states might still provide insight on how blue copper centres work.AcoP possesses a highly rigid copper centre that is not significantly modified by mutations of ligand residues
To better understand the role played by copper ligands on copper centre geometry and the electronic properties in AcoP, the X-ray crystal structures of the H166A and M171A variants were also solved. As we showed in a previous study,30 the replacement of the His166 residue located in the C-terminal loop as well as of the Met171 axial ligand by an Ala residue, does not affect copper binding nor overall fold of the protein. Nevertheless, this resulted in drastic modification of the spectroscopic and redox properties of the proteins.30 Indeed, the mutation of His166 to Ala turned the protein into a Cu+ copper binding protein, while the replacement of Met171 by Ala turned the protein into a Cu2+ copper binding protein with ‘red’ spectroscopic properties.30 Although this early work emphasized that copper ligands indeed played a crucial role in determining the electronic and redox properties of AcoP, the molecular determinants for such features remained unexplored. Here, we described their crystal structures and discuss the structural origins of their properties. First, the structure of the His166Ala variant was solved at 2.1 Å resolution. The tertiary structure was essentially unchanged from that of native AcoP (rmsd ∼ 0.45 Å). The copper atom is coordinated by only three ligands in a distorted trigonal geometry with the closest atom of Ala166 being ∼3.5 Å away from the Cu (Fig. 4). This result confirms our previous assumption based on the characterization of this mutant, which gives rise to a silent form in UV-Vis spectroscopy due to the stabilization of the AcoP reduced form in a tri-coordinated fashion.30 This result is consistent with previous mutational studies of histidine ligands in cupredoxins (e.g. H145A AfNir70 or H85A rusticyanin71). In the case of the Met171Ala variant, the X-ray structure was obtained at 1.82 Å resolution. As expected, no change of the overall structure was observed (rmsd ∼ 0.38 Å). In this case however, the copper centre is penta-coordinated, because the Met171 axial ligand is replaced by an acetate molecule whose oxygen atoms both interact with copper. The presence of this acetate is not a crystallographic artefact. It is consistent instead with our previous spectroscopic results in solution, where we observed a red copper centre which can be modified by protein acidification, most likely due to the displacement of a Cu coordinating acetate molecule.30 In both AcoP mutants, the replacement of one Cu ligand has no major impact on the three remaining ligands, since no significant change in Cu ligand positions was observed (Fig. 4), nor in Cu-to-ligand distances (Table S2†), therefore suggesting that the copper centre is relatively rigid. To explain such behaviour, we propose that the second coordination sphere might be involved in the stabilization of the first coordination sphere, thus participating to the overall rigidity of the metal centre. Finally, because the H166A and M171A mutants are permanently reduced and oxidized respectively,30 we decided to compare changes in mutant metal centre bond distances and angles with those related to the wild type reduced-to-oxidized transition (Table S2†). Despite major copper-to-ligand rearrangements expected in the mutants compared to wild type metal centre, we can observe that the trends between the redox-induced and mutant-induced transitions are overall conserved for equatorial bond lengths and angles. This observation validates the trends of AcoP redox transitions discussed in the previous paragraph. It can also be noticed that major changes in the Cu–S(Met171) and Cu–N(His166) bonds occur in the H166A and M171A mutant respectively, suggesting that each of these copper ligands contributes strongly to compensate the absence of the other.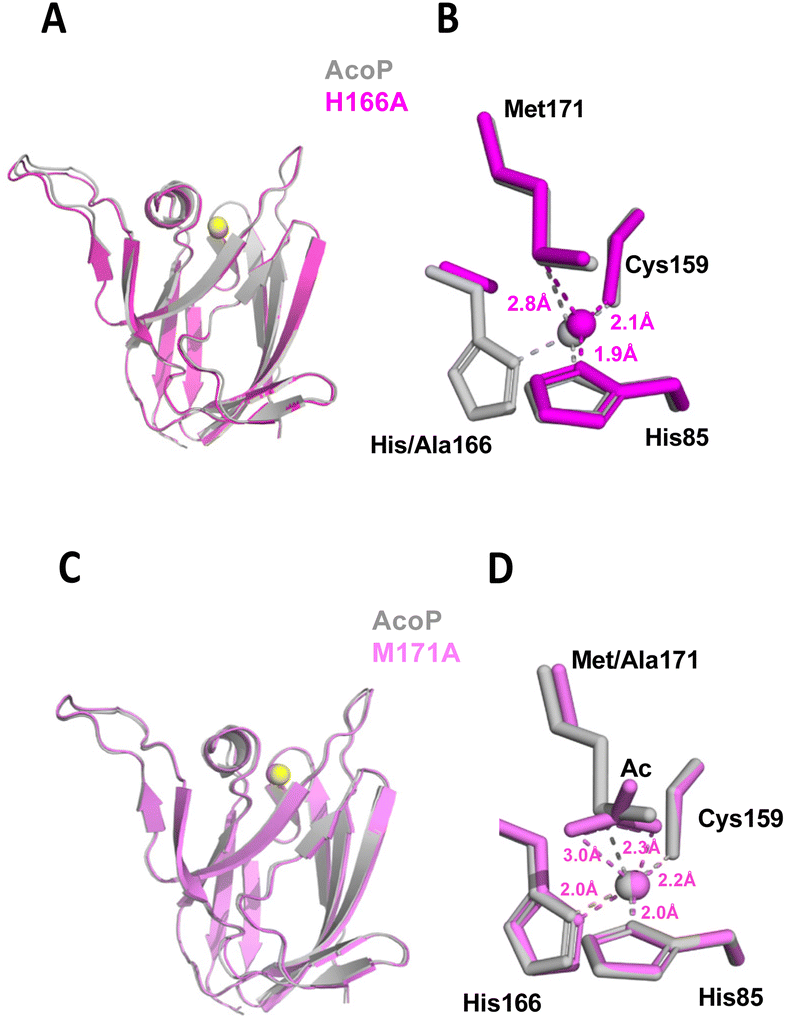 | ||
| Fig. 4 Structure of AcoP mutants. (A) Structure of AcoP H166A mutant (in purple) in comparison with wt AcoP (in grey) and superposition of their copper centre (B), (C) structure of AcoP M171A mutant (in pink) in comparison with wt AcoP (in grey) and superposition of their copper centre (D). | ||
The second coordination sphere of AcoP is designed to constrain the copper centre
As shown in the previous section, the mutation of two AcoP copper ligands does not significantly affect distances and geometries between copper and the remaining ligands. This is a direct observation of the extreme rigidity of the AcoP metal centre, that we previously hypothesized based on spectroscopic data.28,30 In order to understand the structural determinants responsible for copper centre rigidity in AcoP, we decided to analyse its three-dimensional structure beyond the first copper coordination sphere. Starting from each copper ligand (Fig. 5 and Fig. S5B†), we looked for hydrogen bond-connected neighbours within the second coordination sphere (purple in Fig. 5) and continued searching two more levels of residues (red to orange) connected to the copper ligands within a continuous hydrogen bond network. We then compared the network with the ones found in rusticyanin, azurin and pseudoazurin following the same criteria (Fig. 5B–D). It can be easily seen that in AcoP, the C-terminal copper ligands (especially histidine and cysteine) are stabilized, within the C-terminal domain, by a large network involving 9 hydrogen bonds and 7 interconnected partners, against 7 and 6 for rusticyanin, and 5 and 4 for azurin and pseudoazurin. Stabilization through hydrogen bond networks of the N-terminal copper ligand (histidine) also differs among proteins. An intra-domain hydrogen bond network, practically absent in azurin and pseudoazurin, relies on 8 hydrogen bonds and 9 connected partners in AcoP (only 3 bonds and 4 partners in rusticyanin). In AcoP two other domains, regions 51–53 and 134–140 further contribute 6 and 4 hydrogen bonds, respectively, to consolidate the hydrogen bond network around His 85. In general, compared to the other three cupredoxins, the second coordination sphere of AcoP involves a larger number of residues that are two or three hydrogen bonds away (orange and red in Fig. 5 and Fig. S5B†) from copper ligands. Further on, these residues are highly interconnected, as evidenced by the N-terminal domain, whose extensive hydrogen bond network is further restrained by long backbone connectivity (residues 74 to 85) and by multiple connections to domains 51–53 and 134–140. In view of these results, the AcoP second coordination sphere seems to provide two unexpectedly extended hydrogen bond networks, that might be a key for the exceptional rigidity of AcoP copper centre. The peculiar second coordination sphere of AcoP might also contribute to another feature of the protein: its unusually high reduction potential (+566 mV vs. SHE), the highest reported to date for a green copper site.28 In fact, extensive hydrogen bond networks, as well as the packing of hydrophobic residues in the vicinity of the copper binding site, have been considered responsible for high redox potential in rusticyanin.35 In AcoP, only four hydrophobic residues could be identified in the vicinity of the Cu atom: Tyr54, Tyr56, Ile83 and Leu161 (Fig. S5A†), suggesting that in AcoP hydrophobic effects might not play a predominant role. The important contribution of the second coordination sphere to cupredoxin redox potentials has also been emphasized by studies in which mutations increased Azurin reduction potential up to 1 V.17 We believe that our analytical approach might be applied to a larger set of cupredoxins of known structure, to understand how different hydrogen bond network domains might contribute to observed redox and spectroscopic properties.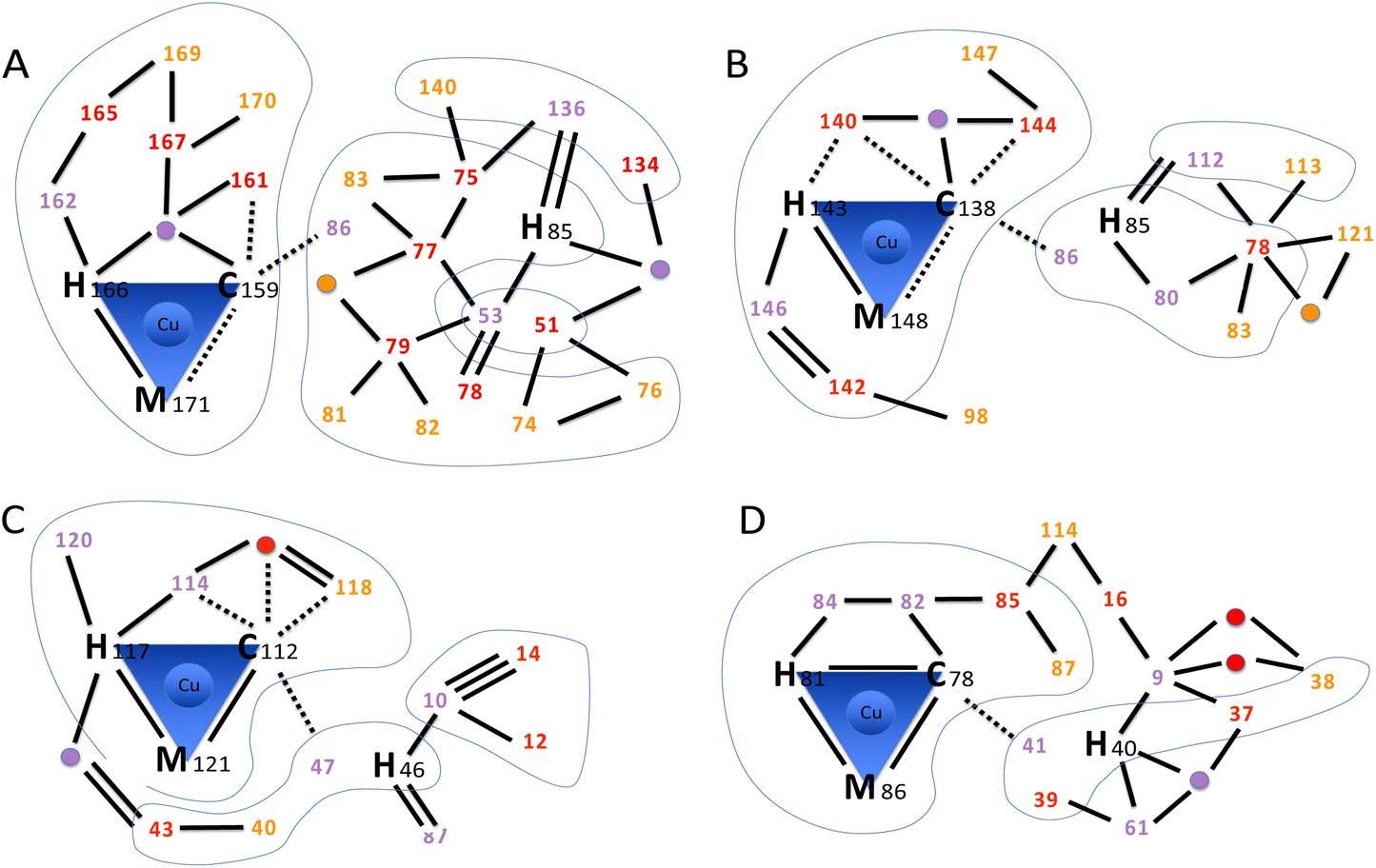 | ||
| Fig. 5 Hydrogen bond networks in cupredoxins. The scheme highlights the intricate hydrogen bond networks involving the first and second coordination spheres. (A) AcoP (PDB: 7Z3B). (B) Rusticyanin (PDB: 2CAK). (C) Azurin (PDB: 4AZU). (D) Pseudoazurin (PDB: 8PAZ). Amino acids within the first coordination sphere (copper ligands) are represented by their one-letter code followed by the sequence position number. Amino acids within the second coordination sphere are represented only by the sequence position number, coloured in purple, red or orange to represent further distance (degree of indirect hydrogen bonding) to copper ligands. The same colour scheme is used for bridging water molecules that participate to the network (purple, red or orange circles). Blue lines encompass domains connected through backbone. No further search was pursued from residues (orange) that were three hydrogen bonds away from copper ligands. Hydrogen bonds (2.5–3.2 Å, black lines) were used to identify network amino acids. Double and triple lines represent respectively two or three independent H-bonds between two amino acids. For the first coordination sphere only, also weak hydrogen bonds (3.2–4.4 Å) are traced (dashed lines). No further search was pursued on residues connected through weak hydrogen bonds only. Blue shapes group together residues that are connected through backbone. | ||
Fidelity of AcoP and other cupredoxins to the coupled distortion model
Present and previous analysis28,30 of AcoP brings us to the conclusion that AcoP copper centre presents some characteristics of both T1.5 and T1 proteins, i.e. respectively: (i) a long Cu–S(Cys) bond, explaining the green UV/Vis absorption and the rhombic EPR spectra; and (ii) on the other hand, a long Cu–S(Met) bond such as plastocyanin (Fig. 3C). To understand how the AcoP metal centre properties fit with the CDM, we surveyed the correlation between Cu–S(Cys) and Cu–S(Met) distances in AcoP and a subset of 12 cupredoxins for which structural and spectroscopic data are available. The structures were chosen among representatives in the reduced state and at a resolution of 1.85 Å or better, apart from Ami-TtCuA (2.30 Å). The results are shown in a CSC (Copper-to-Sulphur distance Correlation) plot, Fig. 6. At first sight, analysed cupredoxins seem to fall in three major groups: Azurins (blue naming text), included to estimate the possible extension of the CSC plot, although they present a different, pentacoordinated copper centre;73 proteins that adhere well to the CDM (black naming text), including the model prototypes Pc, CBP and AcNiR, as well as chimeric Ami-TtCuA,67 the most extreme reported example of Cu–S(Cys) and Cu–S(Met) bonds for a T1.5 cupredoxin (bottom right corner); a third group of possible outliers (red naming text). If the presence of amicyanin and auracyanin B in this part of the CSC plot could be seen as due to errors/precision, the positioning of AcoP, as well as of auracyanin A and rusticyanin, strongly suggests that another group of cupredoxins might exist, that does not follow the Cu–S(Cys)/Cu–S(Met) correlation defined within the CDM. This group of outliers appears as far from the Azurin ensemble as from the coupled distortion one, suggesting a significant difference within the precision of the graphic. Although in Fig. 6 the precision of coordinates, based in turn on atomic coordinates from crystallography, is hard to estimate, some indicators may be used. As an example, discrepancy between interatomic distances measured for molecule A and B (empty green triangles in Fig. 6) from the asymmetric unit of AcoP crystals ranges from ∼0.01 Å for the equatorial cysteine to ∼0.15 Å for the axial methionine, due to strong crystal packing effects. Even the discrepancy in Cu–S(Met) distances between molecule A and B is not enough to suggest that AcoP should belong to another group of proteins. The small rearrangements of Cu-to-ligand distances upon oxidation described for Pc and AcoP are in the order of 0.05 to 0.1 Å, i.e. one to two squares of the graphic, suggesting that the discrepancy between the two populations of cupredoxins observed is still significant, even if redox states had been imprecisely assigned to the described structures. For proteins like poplar plastocyanin, several structures are often available, and the discrepancy in coordinates between pdb entries 1PLC and 4DPC, can be seen. Both structures were solved at high resolution (1.33 and 1.06 Å respectively) in the reduced state (the latter at pH 8.0). Errors calculated from DPI incertitude on atomic positions also point to the existence of these two groups of geometries, beyond azurins, that either adhere or not to the coupled distortion model (Fig. S6A†). Indeed, the precision of crystallographically-derived distances might be even higher than what suggested by DPI analysis (Fig. S6B†). Finally, the CDM fits extremely well to a group of cupredoxins, and exquisitely to the model prototypes Pc, CBP and AcNiR. This is not astonishing, because the CDM is an elegantly elaborated theory, supported by a whelm of experimental and theoretical data collected during decades of investigations. Our analysis further validates the CDM, and even suggests its extremely high precision. As such, the fact that for a group of cupredoxins tight distribution around the model is observed (Fig. 6), further suggests the significancy of the outliers that behave differently. It should be noticed the presence of another outlier, AxNiR from Achromobacter xylosoxidans, whose geometries suggest a perturbed blue centre (Fig. 6), while the UV/Vis spectrum corresponds to that of a classical T1 blue.81 Finally, despite chimeric Ami-TtCuA seems to agree with the CDM on the CSC plot, the geometry of its metal centre was more similar to a T1 than a T1.5 type,67 like for AcoP (Fig. 3C). Our analysis does not show if other sectors of the CSC plot can be and are populated. For this reason, it will be important in our opinion to extend the same type of analysis to known and newly described cupredoxins, and more in general to analyse thoroughly their metal centre architectures, in order to shed further light on the propensity of Cu centres for certain regions of the CSC plot. However, we expect the bottom left corner of the CSC plot to be unlikely populated, since a minimal correlated Cu–S(Cys)/Cu–S(Met) distance must exist that satisfies the requirements for copper chelation. We might even speculate that the CDM might represent the behaviour of cupredoxin centres close to this limit. To confirm, refine or discard our hypothesis, we believe that this analysis should be extended to all cupredoxins, and notably to mutants of known structure of the second coordination sphere, seen as a further source of biodiversity generated in vitro. The CSC plot only focuses on one of the structural criteria described by the CDM: the Cu–S(Cys)/Cu–S(Met) distance correlation. The ability of following in analogous ways other parameters (angles, geometries, …) might possibly provide further insights in the future. The implications of our discoveries are important, as they suggest that different classes of blue copper centres exist, some of which are not in line with what was proposed in the CDM. These different centre architectures in turn are brought together and tuned by different polypeptide scaffolds.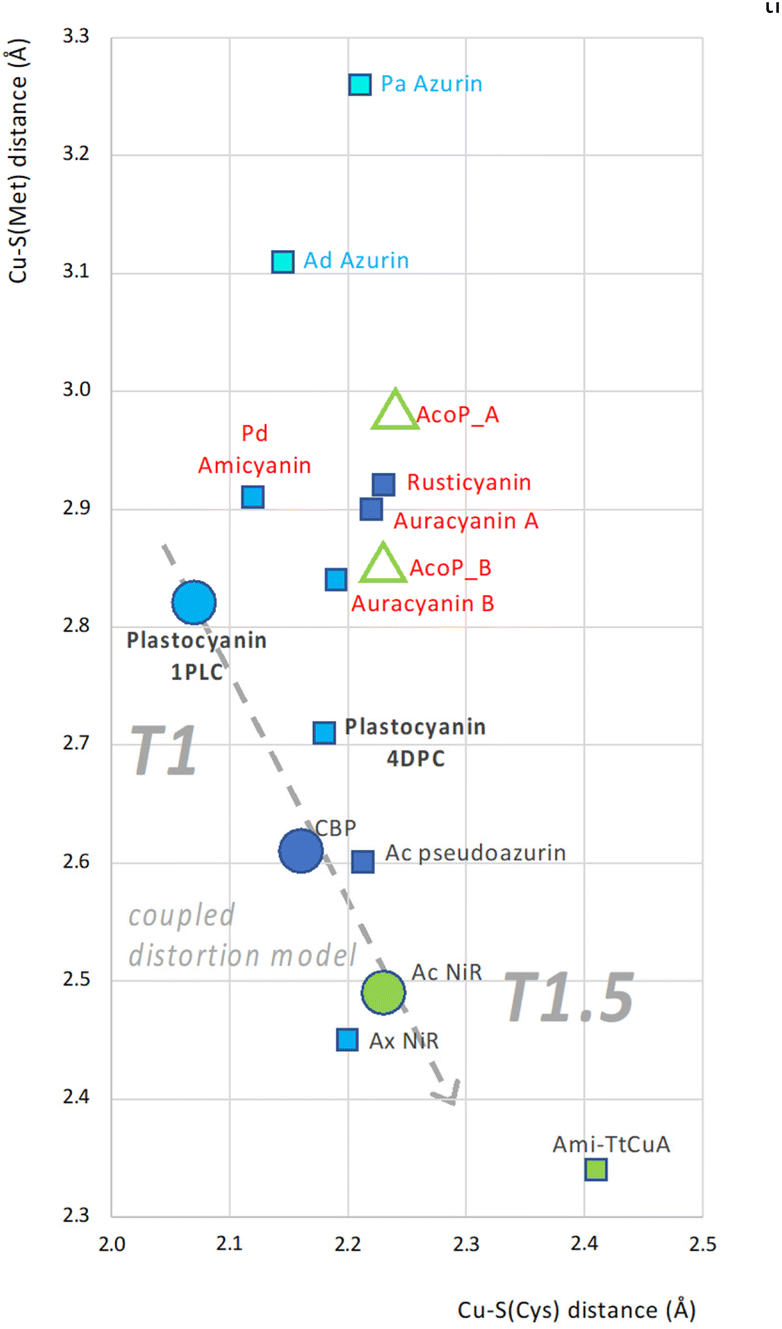 | ||
| Fig. 6 CSC plot: correlation between Cu–S(Met) and Cu–S(Cys) bond lengths in a set of cupredoxins. Cu–S(Cys) and Cu–S(Met) bond lengths correspond to x and y coordinates of each dot. Each cupredoxin is represented by a square, except for AcoP (green triangles, relative to molecule A and B from the crystallographic asymmetric unit) and the three prototype representatives of the CDM; plastocyanin, CBP and AcNir (circles). Colouring of each shape represents the spectral features: pentacoordinated centres (light blue shapes); classical T1 blue (blue shapes); perturbed blue (dark blue shapes); and green T1.5 (green shapes). Labels (cupredoxin names) are coloured according to the apparent affiliation to one of three groups: Azurins (blue label text), cupredoxins in agreement with the CDM (black label text) and others (red label text). The T1-to-T1.5 trend of the coupled distortion group, including the prototypes plastocyanin, CBP and AcNir only is represented too (grey dashed arrow). Analysed structures: AcoP from Acidithiobacillus ferrooxidans (PDB: 7Z3B); Pa Azurin from Pseudomonas aeruginosa (PDB: 2CCW);72 Ad Azurin from Alcaligenes denitrificans (PDB: 2AZA);73 Pd Amicyanin from Paracoccus denitrificans (PDB: 2RAC);74 Ca Auracyanin A (PDB: 2AAN) and B (PDB: 1QHQ) from Chloroflexus aurantiacus;75 Af Rusticyanin from Acidithiobacillus ferroxidans (PDB: 2CAK);76 Plastocyanin from Populus nigra, bold (PDBs: 1PLC and 4DPC);77 Ac Pseudoazurin from Achromobacter cycloclastes (PDB: 4YLA);78 CBP from Cucumis sativus (PDB: 2CBP);79 AxNir from Achromobacter xylosoxidans (PDB: 1 OE1);80 AcNir from Achromobacter cycloclastes (PDB: 2BW4);63 Ami-TtCuA chimera (PDB: 5U7N).67 | ||
Discussion
In this study, we solved and analysed the structures of wild-type AcoP, in its reduced and oxidized forms, and of two copper–ligand mutants, AcoP H166A and M171A. Our work focused on the determination and analysis of the structural properties of AcoP as a cupredoxin in its isolated states in vitro or in cristallo. Similar types of detailed analysis will be valuable in the future for identifying key structural features that correspond to specific functions in cupredoxins. For instance, structural comparison with other cupredoxins highlights the presence of an extended loop, previously detected by sequence alignment,28 which appears to be conserved in related cupredoxins from acidophilic bacteria. This loop could be important for the physiological role of AcoP which is not yet fully understood. Indeed, in vitro studies suggested that AcoP could be an electron donor of cytochrome c oxidase (CcO) and/or an accessory protein, with chaperone-like properties, capable of preserving the integrity and activity of CcO metal centres in acidic environments.27,29 Based on this second role and on the presence of this additional loop, it is interesting to note that several types of structurally unrelated protruding motifs are also found in the copper-loading chaperones Sco56 and PcuAC.55 These extended motifs have been proposed to play a role in the unfolding of domains in the vicinity of the metal centre to facilitate copper loading and/or disulphide-reduction of their partner protein.82 Similarly, the extended loop found in AcoP might play a role in partner maturation and/or protection (such as CoxB, the subunit II of A. ferrooxidans CcO), and in general in protein–protein interactions. Intriguingly, Sco chaperones that are required for CoxB maturation in other species83 are not found in the genome of A. ferrooxidans, raising the question of how CoxB maturation occurs in this species. Further studies are needed to experimentally validate the function of this loop in AcoP, and of analogous extensions in Sco and PcuAC.The structure of AcoP validates the identity of its copper ligands.30 In fact, unlike green type auracyanin D which possesses an axial glutamine residue,84 AcoP has a classical set of cupredoxin ligands, with an axial methionine ligand. The structure of AcoP also provides insight into the structural determinants of the spectroscopic properties of this green cupredoxin. X-ray and EXAFS data agree and show a long Cu–S(Cys) distance of 2.2 Å. Such a distance can explain the green UV/Vis spectrum of AcoP, as a result of concomitant weakening and strengthening of π and σ interactions respectively. In contrast, the long Cu–S(Met) measured distance (2.7 Å by EXAFS; 2.9 Å by X-ray) was unexpected, being rather considered a feature of T1 type cupredoxins. Recently, two more single-domain green-type cupredoxins have been characterized spectroscopically: CopI from Rubrivivax gelatinosus25 and the chimeric protein Az-TtCuA,67 also characterized by a classical set of copper ligands. Determination of Cu–ligand distances in CopI, as done for Az-TtCuA and AcoP, might help clarifying the relationship between spectroscopic properties, metal centre structures and the coupled-distortion model in green-type cupredoxins. The (C-termHis85)N–Cu–S(Met171) angle is another feature of AcoP metal centre that well fits T1 geometry rather than a green, expected T1.5 one, suggesting disagreement between the CDM and what is observed in AcoP. Finally, AcoP metal centre rearrangements upon oxidation also suggest, indirectly, an opposite behaviour than what observed in prototype plastocyanin that adhere to the CDM.
The structures of two mutants of the first coordination sphere, AcoP H166A and AcoP M171A, were also successfully solved. These structures show very little variation of the metal centre geometry compared to wild-type AcoP and validate our previous hypothesis that AcoP has a highly constrained copper centre.30 In line with this, as we previously showed, the addition of exogenous ligands to the M171A (axial) mutant had little effect on UV/Vis spectroscopic properties of AcoP, including with ligands (such as dimethylsulphide) that could be expected to generate a blue copper centre.30 In that study, we hypothesized that the protein scaffold might play a critical role for rigidifying AcoP green copper centre. Here, we performed detailed structural analysis and comparison of AcoP and three model cupredoxins of known structure which suggest a key role of the second coordination sphere in constraining AcoP active centre. A highly structured and extensive hydrogen bond network is in fact clearly observed in AcoP but not in the compared proteins. In other studies, the protein scaffold was also suggested not to be a passive entity, but rather play an important role in cupredoxin metal centre geometry.1 AcoP perfectly illustrates this hypothesis.
In order to explain AcoP redox potential, which is unexpectedly high (+566 mV) for a green cupredoxin, we analysed the solved structures. Although we are not able to provide an exhaustive list of all structural determinants responsible for AcoP redox properties, we could identify two that seem noteworthy. Cu–S(Met) distance in AcoP is comparable to those found in T1 blue cupredoxins, and it is therefore tempting to speculate that in AcoP a long Cu–S(Met) bond might increase the reduction potential. Indeed, short Cu–S(Met) distances have been proposed to stabilize the Cu(II) form,32–34 lowering as such cupredoxin reduction potentials. Similar results were also obtained from quantum chemistry calculations, that successfully identified a few independent structural determinants of cupredoxin redox potentials, among which the Cu–S(Met) distance.85 It should be noticed though that in chimeric Ami-TtCuA the Cu–S(Met) bond is very short (2.35 Å), yet the reduction potential (422 mV) still high for a green cupredoxin. We also identified an extended hydrogen bond network, involving AcoP second coordination sphere, that might lead to increased reduction potential too, as previously proposed for rusticyanin.35 To decipher the role played by all structural determinants of AcoP reduction potential, further mutagenesis, structural and quantum chemistry studies will be necessary. Previous experiments targeting hydrogen bonds already emphasized their key role in tuning redox potential in other cupredoxins.17,18 The role of constrained and constraining protein scaffolds in blue copper proteins is, in our view, still an open question. Our study cannot give a final answer, but the question seems relevant to understand how cupredoxins function. This knowledge could be key to explain the ever-growing biological functions that are being discovered for cupredoxins.
To conclude, our data suggest that not only AcoP, but also other cupredoxins might not adhere to the CDM, raising new fundamental scientific questions. Is AcoP a representative of metal centres with “hybrid” T1/T1.5 properties or even of yet undescribed ones? Are there other groups of cupredoxins found in nature that populate other sectors of the CSC plot? If different groups exist, the coupled distortion one being only one of them, what are the structural determinants responsible for their differences? Ultimately, the only difference between cupredoxins is to be accounted for by the polypeptide sequence, bringing us to suggest that the rack-induced model should be always taken into consideration, and might have been underestimated still. Strong rack-induced effects maybe account for metal geometry in the AcoP group of outliers and not in the CDM group. Different behaviour of AcoP and Pc metal centres upon oxidation seem to point to this hypothesis, because either copper repositioning or copper–ligand rearrangements seem to preferentially occur in AcoP and Pc respectively (Fig. S3†). Recent results obtained on de novo constructs that recreate blue, green and red copper centres in alpha helicoidal folds, also show disagreement to the CDM.86 Notably in this study, the Cu–S(Cys) distance is invariant in two constructs showing, respectively, blue and green spectral features. It is tempting to speculate that this could eventually be seen as yet another separate rack-induced group, different from the CDM, the Azurin and the AcoP groups, and governed by yet a different set of copper coordination rules.
Conclusions
The AcoP structures presented and analysed in our work represent an important achievement, not only because they are the first described for a natural single-domain, mononuclear green cupredoxin, but even more because they invite the scientific community to study cupredoxins with no a priori limits or established horizons, i.e. even beyond those traced by the well-established coupled distortion model. The unprecedented spectroscopic, electronic, redox and structural properties of AcoP suggest that this protein has the potential to become an interesting model system for future studies aiming at gaining new insight on structure–function relationship in cupredoxins. A few paragraphs in the Discussion section raise exciting scientific questions and/or directions for future investigations, and a few others in the Results section present useful analyses to identify the structural determinants of blue copper protein properties. Further studies conducted by quantum mechanics and DFT are needed to explain the determinants of the analysed copper geometries (Fig. 6). Rather, our results suggest that the behaviour of some cupredoxins do not adhere to the CDM. We invite the scientific community to confirm, correct, extend or revisit our hypotheses, that we hope will contribute to better understanding the structure–function relationship in blue copper proteins. Finally, it seems that the rack-induced model should be taken into consideration again, and we believe that human exploration of the blue copper universe is far from having reached its horizons yet.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank the staff and beamline scientists of the ESRF and Soleil synchrotrons for beamtime allocation and for their assistance with X-ray diffraction and single-crystal spectroscopy experiments. More specifically, we acknowledge European Synchrotron Radiation Facility for provision of beam time on beamlines id29, id30B and BM30, and Soleil Synchrotron for provision of beam time on beamlines Proxima 1 and Proxima 2. SAXS data were collected at the SIBYLS beamline in Berkeley California supported by a National Institute of Health R01 GM137021-02 and the United States Department of Energy Biological and Environmental Research (DOE-BER) IDAT grant. NJB was supported by a grant from the National Institutes of Health (R35 GM136239). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences, P30GM133894. GS research on laccases and blue copper centres represents part of the activities supported by the French National Research Agency (ANR) grants FUNCLIPRO (ANR-19-CE43-0007-01) and DEOX (ANR-21-CE11-0041). MI research on copper proteins has been supported by ANR funded projects MetCop (ANR-21-CE44-0024) and ChapCop (ANR-19-CE44-0018). The authors also want to thank X. Wang, L. Zuily, N. Lahrach and M. Bauzan for technical support, E. Lojou from BIP-CNRS Marseille for fruitful discussions, and V. Pecoraro for critical reading of the manuscript.References
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- J. Gough and C. Chothia, Structure, 2004, 12, 917–925 CrossRef CAS PubMed.
- I. Bertini, G. Cavallaro and K. S. McGreevy, Coord. Chem. Rev., 2010, 254, 506–524 CrossRef CAS.
- C. Dennison, Coord. Chem. Rev., 2005, 249, 3025–3054 CrossRef CAS.
- M. Choi and V. L. Davidson, Metallomics, 2011, 3, 140 CrossRef CAS PubMed.
- H. B. Gray, B. G. Malmström and R. J. Williams, J. Biol. Inorg. Chem., 2000, 5, 551–559 CrossRef CAS PubMed.
- C. Dennison, Nat. Prod. Rep., 2008, 25, 15–24 RSC.
- J. Guo and O. S. Fisher, J. Biol. Inorg. Chem., 2022, 27, 529–540 CrossRef CAS PubMed.
- E. I. Solomon and R. G. Hadt, Coord. Chem. Rev., 2011, 255, 774–789 CrossRef CAS.
- E. I. Solomon, Inorg. Chem., 2006, 45, 8012–8025 CrossRef CAS PubMed.
- Y. Lu, Compr. Coord. Chem. II, 2003, 8.4, 91–122 Search PubMed.
- S. DeBeer, D. W. Randall, A. M. Nersissian, J. S. Valentine, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Phys. Chem. B, 2000, 104, 10814–10819 CrossRef CAS.
- R. L. Lieberman, D. M. Arciero, A. B. Hooper and A. C. Rosenzweig, Biochemistry, 2001, 40, 5674–5681 CrossRef CAS PubMed.
- J. D. King, C. L. McIntosh, C. M. Halsey, B. M. Lada, D. M. Niedzwiedzki, J. W. Cooley and R. E. Blankenship, Biochemistry, 2013, 52, 8267–8275 CrossRef CAS PubMed.
- W. J. Ingledew and D. H. Boxer, in Methods in Enzymology, Elsevier, 1994, vol. 243, pp. 387–393 Search PubMed.
- P. Hosseinzadeh, N. M. Marshall, K. N. Chacón, Y. Yu, M. J. Nilges, S. Y. New, S. A. Tashkov, N. J. Blackburn and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 262–267 CrossRef CAS PubMed.
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y.-G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–116 CrossRef CAS PubMed.
- S. Yanagisawa, M. J. Banfield and C. Dennison, Biochemistry, 2006, 45, 8812–8822 CrossRef CAS PubMed.
- S. M. Berry, M. H. Baker and N. J. Reardon, J. Inorg. Biochem., 2010, 104, 1071–1078 CrossRef CAS PubMed.
- F. Arnesano, L. Banci, I. Bertini and A. R. Thompsett, Structure, 2002, 10, 1337–1347 CrossRef CAS PubMed.
- O. S. Fisher, G. E. Kenney, M. O. Ross, S. Y. Ro, B. E. Lemma, S. Batelu, P. M. Thomas, V. C. Sosnowski, C. J. DeHart, N. L. Kelleher, T. L. Stemmler, B. M. Hoffman and A. C. Rosenzweig, Nat. Commun., 2018, 9, 4276 CrossRef PubMed.
- Y. Fu, H.-C. T. Tsui, K. E. Bruce, L.-T. Sham, K. A. Higgins, J. P. Lisher, K. M. Kazmierczak, M. J. Maroney, C. E. Dann, M. E. Winkler and D. P. Giedroc, Nat. Chem. Biol., 2013, 9, 177–183 CrossRef CAS PubMed.
- P. Hosseinzadeh, S. Tian, N. M. Marshall, J. Hemp, T. Mullen, M. J. Nilges, Y.-G. Gao, H. Robinson, D. A. Stahl, R. B. Gennis and Y. Lu, J. Am. Chem. Soc., 2016, 138, 6324–6327 CrossRef CAS PubMed.
- A. Durand, M. Fouesnard, M.-L. Bourbon, A.-S. Steunou, E. Lojou, P. Dorlet and S. Ouchane, Metallomics, 2021, 13, mfab067 CrossRef PubMed.
- C. Castelle, M. Guiral, G. Malarte, F. Ledgham, G. Leroy, M. Brugna and M.-T. Giudici-Orticoni, J. Biol. Chem., 2008, 283, 25803–25811 CrossRef CAS PubMed.
- C. Castelle, M. Ilbert, P. Infossi, G. Leroy and M.-T. Giudici-Orticoni, J. Biol. Chem., 2010, 285, 21519–21525 CrossRef CAS PubMed.
- M. Roger, F. Biaso, C. J. Castelle, M. Bauzan, F. Chaspoul, E. Lojou, G. Sciara, S. Caffarri, M.-T. Giudici-Orticoni and M. Ilbert, PLoS One, 2014, 9, e98941 CrossRef PubMed.
- X. Wang, M. Roger, R. Clément, S. Lecomte, F. Biaso, L. A. Abriata, P. Mansuelle, I. Mazurenko, M. T. Giudici-Orticoni, E. Lojou and M. Ilbert, Chem. Sci., 2018, 9, 4879–4891 RSC.
- M. Roger, G. Sciara, F. Biaso, E. Lojou, X. Wang, M. Bauzan, M.-T. Giudici-Orticoni, A. J. Vila and M. Ilbert, Biochim. Biophys. Acta, Bioenerg., 2017, 1858, 351–359 CrossRef CAS PubMed.
- X. Wang, R. Clément, M. Roger, M. Bauzan, I. Mazurenko, A. de Poulpiquet, M. Ilbert and E. Lojou, J. Am. Chem. Soc., 2019, 141, 11093–11102 CrossRef CAS PubMed.
- K. Olesen, A. Veselov, Y. Zhao, Y. Wang, B. Danner, C. P. Scholes and J. P. Shapleigh, Biochemistry, 1998, 37, 6086–6094 CrossRef CAS PubMed.
- J. F. Hall, L. D. Kanbi, R. W. Strange and S. S. Hasnain, Biochemistry, 1999, 38, 12675–12680 CrossRef CAS PubMed.
- M. A. Hough, M. J. Ellis, S. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. Samar Hasnain, J. Mol. Biol., 2005, 350, 300–309 CrossRef CAS PubMed.
- B. Jiménez, M. Piccioli, J.-M. Moratal and A. Donaire, Biochemistry, 2003, 42, 10396–10405 CrossRef PubMed.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, in The proteomics Protocols HandBook, ed. J. M. Walker, Humana Press, 2005, pp. 571–607 Search PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 72–82 CrossRef PubMed.
- P. R. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 282–292 CrossRef CAS PubMed.
- N. S. Pannu, W.-J. Waterreus, P. Skubák, I. Sikharulidze, J. P. Abrahams and R. A. G. de Graaff, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 331–337 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- G. Langer, S. X. Cohen, V. S. Lamzin and A. Perrakis, Nat. Protoc., 2008, 3, 1171–1179 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- D. von Stetten, T. Giraud, P. Carpentier, F. Sever, M. Terrien, F. Dobias, D. H. Juers, D. Flot, C. Mueller-Dieckmann, G. A. Leonard, D. de Sanctis and A. Royant, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 15–26 CrossRef CAS PubMed.
- K. S. D. Kumar, M. Gurusaran, S. N. Satheesh, P. Radha, S. Pavithra, K. P. S. Thulaa Tharshan, J. R. Helliwell and K. Sekar, J. Appl. Crystallogr., 2015, 48, 939–942 CrossRef CAS.
- K. N. Chacón, J. Perkins, Z. Mathe, K. Alwan, E. N. Ho, M. N. Ucisik, K. M. Merz and N. J. Blackburn, Commun. Biol., 2018, 1, 192 CrossRef PubMed.
- K. N. Chacón, T. D. Mealman, M. M. McEvoy and N. J. Blackburn, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15373–15378 CrossRef PubMed.
- N. J. Blackburn, F. C. Rhames, M. Ralle and S. Jaron, J. Biol. Inorg. Chem., 2000, 5, 341–353 CrossRef CAS PubMed.
- K. N. Dyer, M. Hammel, R. P. Rambo, S. E. Tsutakawa, I. Rodic, S. Classen, J. A. Tainer and G. L. Hura, in Structural Genomics, ed. Y. W. Chen, Humana Press, Totowa, NJ, 2014, vol. 1091, pp. 245–258 Search PubMed.
- D. I. Svergun, M. V. Petoukhov and M. H. J. Koch, Biophys. J., 2001, 80, 2946–2953 CrossRef CAS PubMed.
- D. Schneidman-Duhovny, M. Hammel, J. A. Tainer and A. Sali, Nucleic Acids Res., 2016, 44, W424–W429 CrossRef CAS PubMed.
- M. Pelikan, G. Hura and M. Hammel, Gen. Physiol. Biophys., 2009, 28, 174–189 CrossRef CAS PubMed.
- L. A. Abriata, L. Banci, I. Bertini, S. Ciofi-Baffoni, P. Gkazonis, G. A. Spyroulias, A. J. Vila and S. Wang, Nat. Chem. Biol., 2008, 4, 599–601 CrossRef CAS PubMed.
- L. Banci, I. Bertini, V. Calderone, S. Ciofi-Baffoni, S. Mangani, M. Martinelli, P. Palumaa and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8595–8600 CrossRef CAS PubMed.
- F. Jacobson, H. Guo, K. Olesen, M. Ökvist, R. Neutze and L. Sjölin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 1190–1198 CrossRef PubMed.
- L. B. LaCroix, S. E. Shadle, Y. Wang, B. A. Averill, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1996, 118, 7755–7768 CrossRef CAS.
- M. Nojiri, Y. Xie, T. Inoue, T. Yamamoto, H. Matsumura, K. Kataoka, Deligeer, K. Yamaguchi, Y. Kai and S. Suzuki, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4315–4320 CrossRef CAS PubMed.
- G. S. Siluvai, M. Mayfield, M. J. Nilges, S. DeBeer George and N. J. Blackburn, J. Am. Chem. Soc., 2010, 132, 5215–5226 CrossRef CAS PubMed.
- D. M. Arciero, B. S. Pierce, M. P. Hendrich and A. B. Hooper, Biochemistry, 2002, 41, 1703–1709 CrossRef CAS PubMed.
- S. M. Berry, E. L. Bladholm, E. J. Mostad and A. R. Schenewerk, J. Biol. Inorg. Chem., 2011, 16, 473–480 CrossRef CAS PubMed.
- S. V. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12041–12046 CrossRef CAS PubMed.
- K. M. Clark, Y. Yu, N. M. Marshall, N. A. Sieracki, M. J. Nilges, N. J. Blackburn, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2010, 132, 10093–10101 CrossRef CAS PubMed.
- C. Li, M. J. Banfield and C. Dennison, J. Am. Chem. Soc., 2007, 129, 709–718 CrossRef CAS PubMed.
- D. Bím and A. N. Alexandrova, Chem. Sci., 2021, 12, 11406–11413 RSC.
- A. Espinoza-Cara, U. Zitare, D. Alvarez-Paggi, S. Klinke, L. H. Otero, D. H. Murgida and A. J. Vila, Chem. Sci., 2018, 9, 6692–6702 RSC.
- B. G. Malmstrom, Eur. J. Biochem., 1994, 223, 711–718 CrossRef CAS PubMed.
- H. Komori and Y. Higuchi, J. Biochem., 2015, 158, 293–298 CrossRef CAS PubMed.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. P. Murphy, Biochemistry, 2007, 46, 12366–12374 CrossRef CAS PubMed.
- D. R. Casimiro, A. Toy-Palmer, R. C. I. Blake and H. J. Dyson, Biochemistry, 1995, 34, 6640–6648 CrossRef CAS PubMed.
- H. Nar, A. Messerschmidt, R. Huber, M. van de Kamp and G. W. Canters, J. Mol. Biol., 1991, 221, 765–772 CrossRef CAS PubMed.
- E. N. Baker, J. Mol. Biol., 1988, 203, 1071–1095 CrossRef CAS PubMed.
- L. M. Cunane, Z.-W. Chen, R. C. E. Durley and F. S. Mathews, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 676–686 CrossRef CAS PubMed.
- M. Lee, M. C. del Rosario, H. H. Harris, R. E. Blankenship, J. M. Guss and H. C. Freeman, J. Biol. Inorg. Chem., 2009, 14, 329–345 CrossRef CAS PubMed.
- R. L. Walter, S. E. Ealick, A. M. Friedman, R. C. Blake II, P. Proctor and M. Shoham, J. Mol. Biol., 1996, 263, 730–751 CrossRef CAS PubMed.
- J. M. Guss, H. D. Bartunik and H. C. Freeman, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 790–811 CrossRef PubMed.
- T. Inoue, M. Gotowda, Deligeer, K. Kataoka, K. Yamaguchi, S. Suzuki, H. Watanabe, M. Gohow and Y. Kai, J. Biochem., 1998, 124, 876–879 CrossRef CAS PubMed.
- J. M. Guss, E. A. Merritt, R. P. Phizackerley and H. C. Freeman, J. Mol. Biol., 1996, 262, 686–705 CrossRef CAS PubMed.
- M. Nojiri, H. Koteishi, T. Nakagami, K. Kobayashi, T. Inoue, K. Yamaguchi and S. Suzuki, Nature, 2009, 462, 117–120 CrossRef CAS PubMed.
- Z. H. L. Abraham, D. J. Lowe and B. E. Smith, Biochem. J., 1993, 295, 587–593 CrossRef CAS PubMed.
- A. Timón-Gómez, E. Nývltová, L. A. Abriata, A. J. Vila, J. Hosler and A. Barrientos, Semin. Cell Dev. Biol., 2018, 76, 163–178 CrossRef PubMed.
- S. C. Leary, Antioxid. Redox Signaling, 2010, 13, 1403–1416 CrossRef CAS PubMed.
- J. D. King, L. Harrington, B. M. Lada, G. He, J. W. Cooley and R. E. Blankenship, Arch. Biochem. Biophys., 2014, 564, 237–243 CrossRef CAS PubMed.
- H. Li, S. P. Webb, J. Ivanic and J. H. Jensen, J. Am. Chem. Soc., 2004, 126, 8010–8019 CrossRef CAS PubMed.
- K. J. Koebke, V. S. Alfaro, T. B. J. Pinter, A. Deb, N. Lehnert, C. Tard, J. E. Penner-Hahn and V. L. Pecoraro, J. Am. Chem. Soc., 2020, 142, 15282–15294 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Known spectral and geometrical properties of cupredoxins; comparison between AcoP molecule A and B; detailed description of copper centre distances and angles in different AcoP forms (molecules A and B, redox states, mutants) and other cupredoxins; change of cupredoxin metal centre upon oxidation; microspectrophotometry of AcoP crystals; hydrophobic residues in the vicinity of AcoP and Rusticyanin metal centres. See DOI: https://doi.org/10.1039/d3dt03372d |
| This journal is © The Royal Society of Chemistry 2024 |