
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
EHDTA: a green approach to efficient Ln3+-chelators†
Fabio
Travagin
 a,
Maria Ludovica
Macchia
b,
Toni
Grell
c,
Judit
Bodnár
d,
Zsolt
Baranyai
*e,
Flavia
Artizzu
f,
Mauro
Botta
*b and
Giovanni B.
Giovenzana
*a
a,
Maria Ludovica
Macchia
b,
Toni
Grell
c,
Judit
Bodnár
d,
Zsolt
Baranyai
*e,
Flavia
Artizzu
f,
Mauro
Botta
*b and
Giovanni B.
Giovenzana
*a
aDipartimento di Scienze del Farmaco, Università del Piemonte Orientale, Largo Donegani 2/3, 28100 Novara, Italy. E-mail: giovannibattista.giovenzana@uniupo.it
bDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale, Via T. Michel 11, 15121 Alessandria, Italy. E-mail: mauro.botta@uniupo.it
cDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy
dDepartment of Inorganic and Analytical Chemistry, University of Debrecen, Egyetem tér 1., H-4010, Debrecen, Hungary
eBracco Imaging SpA, CRB Trieste, AREA Science Park, ed. Q – S.S. 14 Km, 163.5 - 34149 Basovizza, TS, Italy. E-mail: zsolt.baranyai@bracco.com
fDipartimento per lo Sviluppo Sostenibile e la Transizione Ecologica, Università del Piemonte Orientale, P.zza S. Eusebio 5, 13100 Vercelli, Italy
First published on 13th December 2023
Abstract
The rich coordination chemistry of lanthanoid ions (Ln3+) is currently exploited in a vast and continuously expanding array of applications. Chelating agents are central in the development of Ln3+-complexes and in tuning their physical and chemical properties. Most chelators for Ln3+-complexation are derived from the macrocyclic DOTA or from linear DTPA platforms, both of which arise from fossil-based starting materials. Herein, we report a green and efficient approach to a chelating agent (EHDTA), derived from cheap and largely available furfurylamine. The oxygenated heterocycle of the latter is converted to a stereochemically defined and rigid heptadentate chelator, which shows good affinity towards Ln3+ ions. A combination of NMR, relaxometric, potentiometric and spectrophotometric techniques allows us to shed light on the interesting coordination chemistry of Ln3+–EHDTA complexes, unveiling a promising ligand for the chelation of this important family of metal ions.
Introduction
The rich coordination chemistry of lanthanoid ions (Ln3+) is the key to exploiting the unique properties of these fascinating elements for an increasing number of applications. Particularly relevant are the applications of Ln3+-complexes in medicine and life sciences.1,2 Paramagnetic Gd3+-complexes have been routinely employed for the last three decades as contrast agents for MRI scans.3177Lu3+-oxodotreotide was recently approved by the EMA and FDA for targeted radiotherapy of somatostatin receptor positive neuroendocrine tumours.4 The unique luminescence properties of Eu3+- and Tb3+-chelates are especially useful in optical imaging and bioanalysis.5The properties of Ln3+-complexes strongly depend on the employed chelating agents and on their match with the coordination sphere of the metal ion.6 All in vivo applications require highly stable complexes both in terms of thermodynamic stability and kinetic inertness, usually provided by macrocyclic chelating agents such as DOTA (Fig. 1).7 On the other hand, short-lived radioisotope ions need to be chelated in a reasonably short timeframe to avoid excessive loss of radioactivity, which is best achieved with flexible linear chelating agents such as DTPA (Fig. 1).8 Chelating agents for luminescent complexes should have active chromophores placed close to the metal ion, acting as an antenna for an efficient sensitization of lanthanide emission, and coordinated water molecules should be limited or absent to avoid fluorescence quenching.5 On the other hand, the number of coordinated water molecules (“q”) is strictly related to the efficiency (“relaxivity”) of Gd3+-based MRI contrast agents. In the octa- and nona-coordinated environment of Ln3+-ions, the denticity of the chelator and its steric hindrance define the q value, but the latter cannot be increased if not at the expense of the thermodynamic stability of the complex, implying a careful structural design devised to obtain a good match between stability and efficiency.3
 | ||
| Fig. 1 Ln3+-chelators. | ||
Mesocyclic chelating agents such as AAZTA (Fig. 1) are able to coordinate Ln3+-ions more tightly than most linear congeners, combining a fast kinetics of formation and a slow kinetics of dissociation of the corresponding metal complexes with the possibility to modulate the number of coordinated water molecules.9
In this work, we report a green approach to the preparation of the mesocyclic chelating agent EHDTA‡ (Fig. 1). EHDTA is formally related to OBETA, a linear heptadentate chelator that was recently shown to have an affinity for a metal ion stronger than the commercially available octadentate analogue EGTA.10 Moreover, a combination of NMR, relaxometric, potentiometric and spectrophotometric techniques is used to explore the coordination chemistry of Ln3+–EHDTA complexes.
Results and discussion
Design and synthesis
Our long-standing interest in the aqueous coordination chemistry of Ln3+-ions for biomedical applications led us to identify novel and efficient heptadentate chelators.9 Recently, a detailed investigation on Ln3+–OBETA complexes showed an unusual selectivity profile for this chelating agent in the Ln3+-series, which even overcomes the well-known higher-denticity homolog EGTA.10 We were then prompted to explore the possibility of improving the chelating ability of OBETA by acting on its molecular structure. Mesocyclic chelating agents, embodying a medium-sized ring in the backbone of the chelating agent, were shown to quickly form highly inert complexes. In this context, ring fusion of the linear OBETA molecule could rigidify the overall structure, providing even more stable metal complexes and eventually modulating the selectivity pattern through the Ln3+-series.3The tetrahydrofuran ring was chosen as a preferred scaffold for the development of improved chelating agents, as its oxygen atom encased in a strain-free five membered ring could easily play the role of the central oxygen atom of OBETA. Moreover, tetrahydrofuran derivatives are conveniently obtained by biomass processing, allowing us to shift the paradigm of current blockbuster chelating agents, obtained almost completely by polyamines derived from petroleum-based chemicals.11 As applications of Ln3+-complexes are constantly growing, sustainability issues must be taken into account in the design of the corresponding chelating agents. The formal ring fusion of the OBETA molecule with a tetrahydrofuran ring led to the chelating agent EHDTA, the subject of this work (Fig. 1). An early report on EHDTA dates back to 1977 with a preliminary study on the complexation of Pb2+.12 Few details are given on the stereochemistry, inferred from the synthetic route to be cis.13 Moreover, the potential coordinating ability towards any other metal ion especially Ln3+ was overlooked, then and thereafter.
The original synthesis of EHDTA, even if amenable for a gram-scale preparation of these chelating agents, is far from being efficient and sustainable on a larger scale due to an inefficient atom economy and the use of toxic reagents, mainly related to the introduction of the amino groups through the Gabriel synthesis, performed on an intermediate di(p-toluenesulphonate).12,13 Therefore, the synthetic approach to EHDTA has been revised according to the principles of Green Chemistry.
The alternative preparation of EHDTA starts from furfurylamine 1, a cheap biomass-derived chemical already endowed with a correctly placed amino group. N-Acetylation is accomplished by refluxing 1 in ethyl acetate, in the presence of TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene) as a transacylation catalyst, leading to N-furfurylacetamide 2.
A Tscherniak–Einhorn amidomethylation14 with N-hydroxymethyl-acetamide153 selectively implants the second (protected) amino group onto the furan ring, giving the symmetric heteroaromatic diamide 4. Catalytic hydrogenation in methanol in the presence of RANEY®-Ni selectively provides the desired cis-disubstituted tetrahydrofuran 5. Hydrolysis of the acetamide groups gives access to the diamine 6, which is then exhaustively decorated with carboxymethyl groups in the classical alkylation–deprotection sequence (t-butyl bromoacetate/K2CO3, then TFA) to complete the synthesis of EHDTA (Scheme 1).
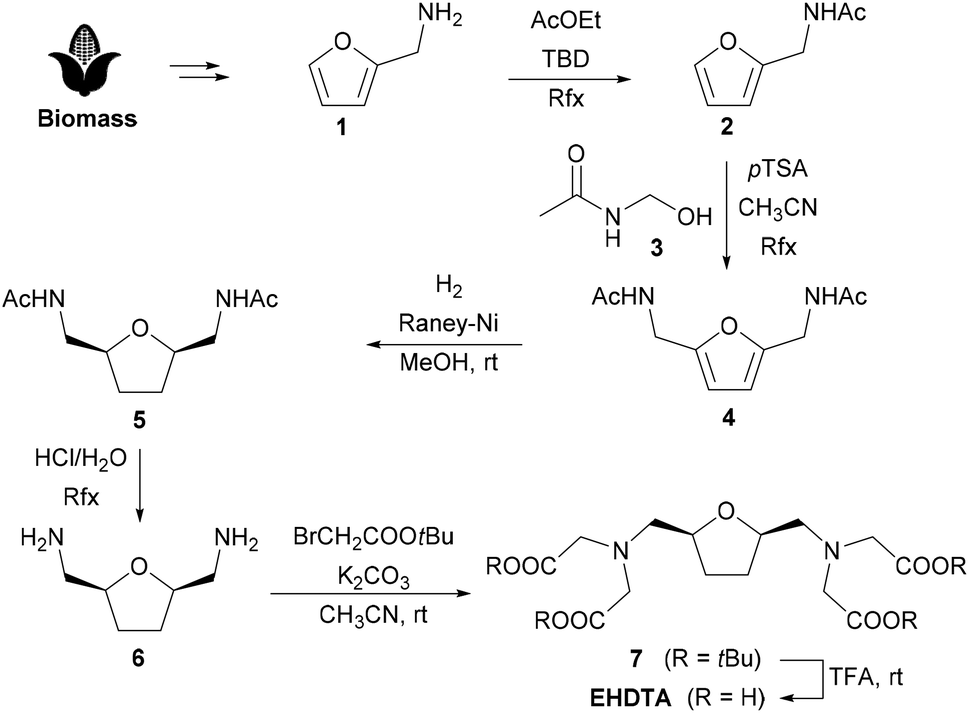 | ||
| Scheme 1 Synthesis of EHDTA. | ||
Thermodynamic properties
The protonation constants of EHDTA were determined by pH potentiometry and 1H NMR spectroscopy (Table S1 and Fig. S18†). The stability and protonation constants of some alkaline earth metals, transition metals and lanthanide complexes formed with EHDTA were also determined by pH-potentiometry (Tables S2 and S3†). Equations used for the evaluation of the equilibrium constants characterizing the protonation of the EHDTA ligand and of the stability and the protonation of the Mg2+-, Ca2+-, Sr2+-, Zn2+-, Cu2+- and Ln3+-complexes are summarized in the ESI.†The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KLnL values of Ln(EHDTA)− complexes increase from the La3+- to Gd3+-ion followed by a slight decrease for the latter members of the series, whereas the logKLnL values of Ln(OBETA)− decrease markedly at the end of the lanthanide series (Fig. 2). As a result, the ΔlogKLnL values between the complexes formed by the two ligands with the same Ln3+ ion shows a minimum between La3+ and Gd3+ and a nearly monotonous increase up to Lu3+ (Fig. 2). The stability trend observed for Ln(OBETA)− complexes is explained by the facts that the flexible system embodying the ether oxygen and two nitrogen donor atoms and four carboxylate groups is well suited for the coordination of middle sized Ln3+-ions, in particular the optimal coordination match with Gd3+, resulting in a drop in the stability constants for the heavier Ln3+ ions.10,16 However, the stability trend of Ln(EHDTA)− is very similar to that of Ln(DTPA)2− complexes (Fig. 2), increasing from La3+ to Dy3+ and then being practically constant in the Ln-series with a slight decrease for the heavier Ln3+ ions, owing to the electrostatic repulsion between the negatively charged carboxylate groups in the complexes of the smaller Ln3+ ions.17 The presence of the five membered tetrahydrofuran ring results in an additional steric constraint of the donor atoms, which may explain the decrease of the logKLnL values of Ln(EHDTA)− complexes formed with the smaller Ln3+-ions due to the increased electrostatic repulsion of the carboxylate groups.
KLnL values of Ln(EHDTA)− complexes increase from the La3+- to Gd3+-ion followed by a slight decrease for the latter members of the series, whereas the logKLnL values of Ln(OBETA)− decrease markedly at the end of the lanthanide series (Fig. 2). As a result, the ΔlogKLnL values between the complexes formed by the two ligands with the same Ln3+ ion shows a minimum between La3+ and Gd3+ and a nearly monotonous increase up to Lu3+ (Fig. 2). The stability trend observed for Ln(OBETA)− complexes is explained by the facts that the flexible system embodying the ether oxygen and two nitrogen donor atoms and four carboxylate groups is well suited for the coordination of middle sized Ln3+-ions, in particular the optimal coordination match with Gd3+, resulting in a drop in the stability constants for the heavier Ln3+ ions.10,16 However, the stability trend of Ln(EHDTA)− is very similar to that of Ln(DTPA)2− complexes (Fig. 2), increasing from La3+ to Dy3+ and then being practically constant in the Ln-series with a slight decrease for the heavier Ln3+ ions, owing to the electrostatic repulsion between the negatively charged carboxylate groups in the complexes of the smaller Ln3+ ions.17 The presence of the five membered tetrahydrofuran ring results in an additional steric constraint of the donor atoms, which may explain the decrease of the logKLnL values of Ln(EHDTA)− complexes formed with the smaller Ln3+-ions due to the increased electrostatic repulsion of the carboxylate groups.
 | ||
Fig. 2 Stability constants (logKLnL) of the Ln(EHDTA)− ( ), Ln(OBETA)− ( ), Ln(OBETA)− ( ) and Ln(DTPA)2− (◆) complexes. ) and Ln(DTPA)2− (◆) complexes. | ||
Kinetic inertness of Gd(EHDTA)−
The kinetic inertness of Gd(EHDTA)− was determined by following either the transmetallation reactions with Cu2+ and Eu3+ by spectrophotometry or the transchelation reactions with TTHA by 1H NMR relaxometry in the presence of Cu2+, Eu3+ and TTHA excess to ensure pseudo-first-order conditions (H6TTHA = triethylenetetra-aminehexaacetic acid). In order to determine the role of the ternary complexes formed with small endogenous ligands on the dissociation rate of Gd(EHDTA)−, the transchelation reactions between Gd(EHDTA)− and TTHA were investigated in the presence of citrate, phosphate and carbonate ions. The rate (kX) and equilibrium constants (KX) characterizing the transmetallation and transchelation reactions of Gd(EHDTA)− are summarized in Table 1. Definitions and equations used for the evaluation of the equilibrium and kinetic data are reported in the ESI.†| Parameters | Gd(EHDTA)− | Gd(OBETA)−a,b |
Gd(DTPA-BMA)2−c |
Gd(DTPA)e |
|---|---|---|---|---|
| I | 0.1 M KCl | 0.1 M KCl | 1.0 M KCl | 1.0 M KCl |
| a Ref. 10. b Ref. 16. c Ref. 18. d Ref. 19 (0.15 M NaCl, 25 °C). e Ref. 20. f k 1 = kGdHL × KGdHL. g k 2 = kHGdHL × KGdHL. h hkM3 = kGdLM × KGdLM. | ||||
| k 1 /M−1 s−1 | 3.3 ± 0.5 | 7.8 | 12.7 | 0.58 |
| k 2 /M−2 s−1 | (3.6 ± 0.4) × 104 | 2.1 × 105 | — | 9.7 × 104 |
|
k
Cu3h/M−1 s−1 |
0.37 ± 0.04 | 0.15 | 0.63 | 0.93 |
|
k
Eu3h/M−1 s−1 |
(2.8 ± 0.3) × 10−3 | 6.4 × 10−2 | — | 4.9 × 10−4 |
| k cit/M−1 s−1 | — | 1.3 × 10−2 | 5.6 × 10−3d |
4.4 × 10−5d |
| k Hcit/M−1 s−1 | 0.33 ± 0.01 | 1.05 | 1.14d |
7.7 × 10−3d |
| k CO3/M−1 s−1 | — | — | 2.7 × 10−2d |
3.1 × 10−4d |
| k HCO3/M−1 s−1 | (7.0 ± 0.2) × 10−4 | 3.5 × 10−4 | — | 2.2 × 10−5d |
| k H2PO4/M−1 s−1 | (1.6 ± 0.2) × 10−3 | 5.5 × 10−2 | 3.1 × 10−3d |
2.7 × 10−4d |
| K GdHL/M−1 | 115 (pH-pot.) | 158.5 (pH-pot.) | <2 | 100 |
| K GdLCu/M−1 | — | — | — | 13 |
| K GdLEu/M−1 | — | 23 | — | 19 |
| K GdLCit/M−1 | — | 156 | 25d |
— |
| K GdLCO3/M−1 | 942 ± 30 | 100 | 20d |
— |
| k d /s −1 | 1.7 × 10 −5 | 1.8 × 10 −5 | 3.8 × 10 −6 | 6.3 × 10 −7 |
| t 1/2 /h | 12 | 11 | 50 | 305 |
The transmetallation reactions of Gd(EHDTA)− with Cu2+ and Eu3+ were monitored on the absorption band of the resulting Cu2+- and Eu(EHDTA)− complexes in the pH range 3.5–6.0 ([GdL] = 0.1 and 1.0 mM in the exchange reactions with Cu2+ and Eu3+, 0.1 M KCl, 25 °C). The proposed mechanism for the transmetallation of Gd(EHDTA)− is shown in Scheme 2.
 | ||
| Scheme 2 Spontaneous, proton- and metal-assisted dissociation of Gd(EHDTA)−. | ||
The k1 rate constant characterizing the proton-assisted dissociation of Gd(EHDTA)− is about two times smaller than that of Gd(OBETA)−. Based on the mechanism of proton-assisted dissociation, the proton transfer from the carboxylic acid pendant of the protonated Gd(HL) intermediate to the N-atom of the ligand backbone takes place by the formation of a relatively labile protonated intermediate, which can dissociate into a free Gd3+-ion and a protonated ligand. The structural preorganization of the EHDTA ligand results in less probable proton transfer and a slower rate of de-coordination of the Gd3+ ion from the protonated Gd(HEHDTA) intermediate. However, the proton-assisted dissociation of both Gd(EHDTA)− and Gd(OBETA)− complexes is slower than for Gd(DTPA-BMA), which might be interpreted by a more rigid coordination environment in the Gd3+-complexes of EHDTA and OBETA. This is further confirmed by the comparison of the rate constant characterizing the metal-assisted dissociation (kCu3) of Gd(EHDTA)−, Gd(OBETA)− and Gd(DTPA-BMA) (Table 1). The kCu3 rate constants of Gd(EHDTA)− and Gd(OBETA)− are comparable and about 2–4 times lower than that of Gd(DTPA-BMA),18 which indicates higher resistance of both Gd(EHDTA)− and Gd(OBETA)− complexes against Cu2+-mediated dissociation due to the rigidity of both Gd3+-complexes. Presumably, the stability of the Gd(L)Cu intermediate of Gd(EDHTA)− and Gd(OBETA)− is significantly lower than that of Gd(DTPA-BMA) resulting in a lower concentration of the kinetically active Gd(L)Cu intermediate and in the slower Cu2+ assisted dissociation of both Gd3+-complexes.
It is known that bis-hydrated Gd3+-complexes might form ternary species with endogenous ligands via the substitution of the inner-sphere water molecules.16,21–23 In order the investigate the role of endogenous ligands in determining the kinetic inertness of Gd(EHDTA)− the transchelation reactions with TTHA were monitored by 1H NMR relaxometry in the presence of excess citrate, phosphate and carbonate. Previous studies indicate that the transchelation rate of the Gd3+-complexes increases with [TTHA] in the pH range of 6.5–11.0.24 The contribution of TTHA to the transchelation rate of Gd(EHDTA)− should be minimized in order to obtain reliable kinetic data for the catalytic effect of citrate, phosphate and carbonate ions. Based on our model calculations, the transchelation between Gd(EHDTA)− and TTHA takes place with 100% conversion even in the presence of a twofold TTHA excess (Fig. S21†) due to the large difference between the stability constant of Gd(EHDTA)− and Gd(TTHA)3− (logKGd(EHDTA) = 19.62(3), logKGd(TTHA) = 23.53(1), 0.1 M KCl, 25 °C). Therefore, the kinetic inertness of Gd(EHDTA)− had been studied by following the transchelation reactions with TTHA in the pH range 6.0–10.0 in the presence of 2 eq. of TTHA, 0–8 eq. of citrate, 0–9 eq. of phosphate and 0–30 eq. of carbonate ([GdL]t = 1.0 mM, [TTHA]t = 2.0 mM, [Cit3−]t = 0–8 mM, [PO43−]t = 0–9 mM, [CO32−]t = 0–30 mM, 0.1 M KCl, 25 °C). The rate (kX) and equilibrium constants (KX) characterizing the contribution of citrate, phosphate and carbonate ions to the rate of the transchelation reaction between Gd(EHDTA)− and TTHA are shown in Table 1. By taking into account the rate and equilibrium constants of Table 1, the kd rate constants and the half-life (t1/2 = ln2/kd) values for the dissociation reactions of Gd(EHDTA)−, Gd(OBETA)− and Gd(DTPA-BMA) were calculated under physiological conditions ([H+] = 3.98 × 10−8 M (pH = 7.4), [Cit]tot = 1.1 × 10−4 M, [CO3]tot = 2.5 × 10−2 M, [PO4]tot = 1 × 10−3 M).
The transchelation reaction of Gd(EHDTA)− with TTHA can take place with the assistance of Cit3−-, HCit2−-, H2PO4−-, HCO3−- and CO32−-ions via the formation of ternary [Gd(EHDTA)X] species. In the ternary [Gd(EHDTA)X] species, the electrostatic repulsion between the negatively charged donor atoms results in a fast rearrangement and rapid dissociation of the Gd3+-complex into the free EHDTA ligand and Gd3+ ion, the latter quickly reacting with the exchanging TTHA ligand. Additionally, the proton transfer in the ternary [Gd(EHDTA)X] species formed with HCit2−-, H2Cit−-, H2PO4−- and HCO3−-ions can also occur via the formation of a protonated Gd(HEHDTA) complex, characterized by a significantly lower kinetic inertness than Gd(EHDTA)−. Comparison of the rate constants characterizing the citrate and phosphate assisted dissociation of Gd3+-complexes reveals that the HCit2− and H2PO4− catalysed dissociation of Gd(EHDTA)− is about 3 and 34 times slower than those of Gd(OBETA)−, respectively. Moreover, the HCO3− assisted dissociation of Gd(EHDTA)− (kHCO3) is about 2 times faster than that of Gd(OBETA)−. Interestingly, the stability constant of the ternary [Gd(EHDTA)CO3]3− species is about 10 times higher than that of [Gd(OBETA)CO3]3−. Such a difference between the kinetic properties of Gd(OBETA)− and Gd(EHDTA)− might be attributed to the preorganized rigid structure of the EHDTA ligand and to the less efficient proton transfer in the ternary [Gd(EHDTA)X] complex formed with HCit2− and H2PO4− ions. In order to examine the possible contribution of the proton transfer processes to the rate of HCit2−, H2PO4− and HCO3− assisted dissociation of Gd(EHDTA)−, the kX rate constants (X = HCit2−, H2PO4− and HCO3−) of Gd(EHDTA)− and Gd(OBETA)− were plotted as a function of the protonation constants (logKHX, Table S5†) of the related ions (Fig. 3).
 | ||
Fig. 3 Brønsted plot for the dissociation reaction of Gd(EHDTA)− ( ), Gd(OBETA)− ( ), Gd(OBETA)− ( ) and Gd(DTPA)2− ( ) and Gd(DTPA)2− ( ) and Gd(DTPA-BMA) ( ) and Gd(DTPA-BMA) ( ) assisted by H2Cit−, HCit2−, HCO3− and H2PO4− ions (25 °C). ) assisted by H2Cit−, HCit2−, HCO3− and H2PO4− ions (25 °C). | ||
In Fig. 3, the kX rate constants characterizing the protonated-ion assisted dissociation of Gd(EHDTA)−, Gd(OBETA)− and Gd(DTPA)2− decrease monotonously with the increase in the protonation constants of the H2Cit−, HCit2−, HCO3− and H2PO4− anions, which highlights the role of the protonated citrate, phosphate and carbonate ions in the dissociation of the Gd3+-complexes. The rate of the proton catalysed reactions can take place by general acids, and the kX rate constants are directly proportional to the acid strength of the general acid expressed by logKHX (logkX = α × logKHX + C).25 In the Brønsted plot, the reactions proceeding through an ideal proton transfer process are characterized by straight lines with slope α = 1.0.25 In the Brønsted plot (Fig. 3) the slope for the HCit2−, HCO3− and H2PO4− assisted dissociation reactions of Gd(EHDTA)−, Gd(OBETA)− and Gd(DTPA)2− was found to be α = 0.50, 0.84 and 0.54, respectively. Based on the α values it can be assumed that the dissociation reactions of Gd(EHDTA)− and Gd(DTPA)2− take place with similar mechanisms via the fast rearrangement and the proton transfer processes in the ternary [Gd(EHDTA)X] intermediate. On the other hand the dissociation of Gd(OBETA)− occurs mainly by the general acid catalysed pathway through proton transfer from the protonated HCit−, HCO3− and H2PO4− anions to OBETA in the ternary [Gd(OBETA)X] adducts.16 A somewhat lower contribution of the general acid catalysed pathway to the overall dissociation rate of Gd(EHDTA)− can be interpreted by the preorganized structure of the EHDTA ligand and the less probable proton transfer from the protonated carboxylate group to the nitrogen donor atoms of the backbone.
Although, the Gd3+-ion in Gd(DTPA-BMA) is coordinated by eight donor atoms of the ligand, the HCit2− and H2PO4− assisted dissociation rate of Gd(DTPA-BMA) is somewhat faster than that of the Gd3+-complex formed with the heptadentate EHDTA ligand. The comparison of the kd and t1/2 values characterizes the dissociation rate of the Gd3+-complexes close to physiological conditions and indicates that the kinetic inertness of Gd(EHDTA)− and Gd(OBETA)− is very similar and about 5 times lower than that of Gd(DTPA-BMA) due to the relatively fast HCO3− assisted dissociation of both Gd3+-complexes.
1H and 17O NMR relaxometric data
A detailed and complete relaxometric investigation of paramagnetic complexes in aqueous solution involves the measurement of the longitudinal (R1 = 1/T1) and transverse (R2 = 1/T2) relaxation rates of the solvent nuclei as a function of pH, temperature and applied magnetic field. The global analysis of these data allows us to obtain valuable information on the stability of the complexes in aqueous solution, on the hydration state of the metal ion, on the exchange regime of the coordinated water molecules and an accurate estimation of the molecular parameters that control their effectiveness as relaxing agents.26 This effectiveness is expressed by the relaxivity parameter. Relaxivity represents the increase in the longitudinal (r1) or transverse (r2) nuclear magnetic relaxation rates of the water protons normalized to a 1 mM aqueous solution of the Gd3+ complex.27 At neutral pH and 298 K, for Gd(EHDTA)−r1 assumes the values of 7.1 and 6.9 mM−1 s−1 at 0.5 and 1.5 T, respectively. These values are fully comparable to those reported for Gd(OBETA)−, despite the lower molecular mass of the latter.10,16 Thus, also in this case two water molecules are presumably present in the inner coordination sphere of the complex (q = 2). Relaxivity does not show a pH dependence in the range 4–10 (Fig. S27†), confirming the good stability of the complex and the low propensity to form ternary complexes with dissolved carbonate, as previously observed for Gd(OBETA)−. In addition, this behaviour indicates that over this range of pH values, the complex does not hydrolyse and does not vary the hydration number q. One can safely conclude that in this pH range, all the seven donor atoms are coordinated to the metal, i.e. the structure of the complex is stable over a change of six orders of magnitude of proton concentration.The measurement of the variation of r1 as a function of the magnetic field strength allows us to obtain the so-called Nuclear Magnetic Resonance Dispersion profile (1H NMRD). These contain key information on the physicochemical parameters correlating r1 with the structural and dynamic properties of the complexes.26 The 1H NMRD spectra of Gd(EHDTA)− were recorded at 283, 298 and 310 K in the proton Larmor frequency range 0.01–120 MHz, corresponding to the magnetic field strengths varying between 2.34 × 10−4 and 3.0 T (Fig. 4).
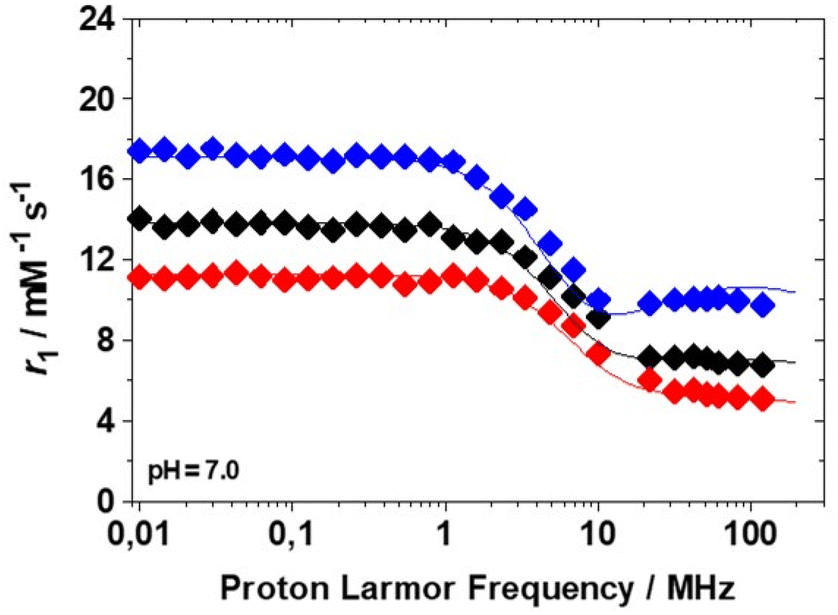 | ||
Fig. 4 1/T11H NMRD profiles of Gd(EHDTA)− at 283 ( ), 298 (◆) and 310 K ( ), 298 (◆) and 310 K ( ). The solid lines represent the results of the best fitting procedure to the experimental data (see Table 2). ). The solid lines represent the results of the best fitting procedure to the experimental data (see Table 2). | ||
The profiles are quite typical of low molecular weight Gd3+ complexes, whose salient features are the presence of a constant relaxivity in a wide range of low frequencies (up to ca. 1 MHz), the occurrence of a dispersion around at 3–6 MHz and another plateau with lower and almost constant relaxivity values at higher frequencies (>20 MHz). When these conditions occur, r1 is largely dominated by the rotational dynamics, described by the rotational correlation time τR. Furthermore, the amplitude of the profiles decreases with increasing temperature (τR gets shorter) indicating that the system is in the fast exchange regime.27 This implies that the rate of coordinated water exchange does not influence or limit relaxivity. The profiles were analysed using the Solomon–Bloembergen–Morgan28–30 and Freed31 equations that describe the contributions of the water molecules in the inner- and outer-sphere to the relaxation, respectively (see the ESI† for more details on the equations used).
Detailed and quantitative information on the exchange process of Gd-bound water molecules is derived from the analysis of data on the temperature dependence of the reduced 17O NMR solvent transverse relaxation rates, R2r (= 1/T2r), and shift, Δωr.27 These experiments were carried out at 11.75 T on a 16.8 mM solution of the complex at neutral pH and the results are reported in Fig. 5. The reduced transverse relaxation rate increases with decreasing temperature until reaching a maximum of around 290 K, and then decreasing at lower temperatures. This is indicative of a rate of water exchange slightly slower than that of Gd(OBETA)− but faster than those measured for [Gd(DTPA)]2− and [Gd(DOTA)]− for which the maximum is shifted at higher temperatures.32 As expected, Δωr exhibits an inflection point at a temperature quite similar to that in which 1/T2r reaches the maximum value.
 | ||
| Fig. 5 Temperature dependence of the reduced water 17O NMR transverse relaxation rates (top) and chemical shifts (bottom) at 11.75 T and pH = 7 for a 16.8 mM solution of Gd(EHDTA)−. | ||
The data are conveniently analysed using the Swift–Connick equations that depend on a number of parameters, among which the most relevant are:33 (i) the parameters associated with the electronic relaxation times T1,2e, i.e. the trace of the square of the zero-field splitting, Δ2; the correlation time describing the modulation of the zero-field splitting, τV, and its activation energy, EV; (ii) the enthalpy of activation for the water exchange process, ΔH#M; (iii) the hyperfine Gd–17Owater coupling constant, A/ħ. Many of these parameters also affect the temperature dependence of the 17O NMR shift. Due to the large number of parameters, for the global data fitting it is necessary to fix some of them to well-established or reasonably estimated values. Initially, the hydration number q was fixed to 2; the distance between the metal ion and the protons of the bound water molecules, r, was fixed to 3.0 Å; a and the D, the relative diffusion coefficient between solute and solvent molecules, were set to 4.0 Å and 2.3 × 10−5 cm2 s−1 (at 298 K), respectively; EV was fixed to 1.0 kJ mol−1.
Unexpectedly, the results of the global analysis of 1H and 17O NMR data were rather unsatisfactory. In particular, the calculated NMRD profiles consistently showed slightly lower values than the experimental ones. Excellent results could be obtained only by assuming an unreasonably long distance between the Gd3+ ion and the coordinated water molecules. Since no evidence supports this hypothesis, we preferred to proceed with the fit by treating q as a variable parameter. An excellent result was obtained with the parameters reported in Table 2, where the hydration number q assumes a value equal to 1.8. A fractional q value implies the presence in solution of a hydration equilibrium between species differing in their hydration state (q = 1 and q = 2). This result is not at all unusual and has been previously reported in the case of Gd3+-34 and Mn2+-complexes.35 Furthermore, a similar conclusion is confirmed by photophysical measurements conducted on the corresponding Eu3+ and Tb3+ complexes (see infra).
16
| Parameters | Gd(EHDTA)− | Gd(OBETA)− |
|---|---|---|
| 22 r 1(298 K) (mM−1 s−1) | 7.25 | 7.20 |
| Δ 2/1019 s−2 | 5.8 | 4.3 |
| τ V/ps | 13.0 | 17.0 |
| τ M/ns | 125 | 77 |
| ΔHM/kJ mol−1 | 41.0 | 40.1 |
| τ R/ps | 79 | 65 |
| E R/kJ mol−1 | 17 | 16 |
| A O/ħ/106 rad s−1 | −2.9 | −2.9 |
| q | 1.8 | 2 |
The other best-fit parameters confirm the previous, qualitative conclusions. The residence lifetime of the inner sphere water molecule(s) is less than a factor of two longer than that of Gd(OBETA)−, while it is about half that reported for [Gd(DTPA)]2− and [Gd(DOTA)]−.32 The parameters of the electron relaxation times, Δ2 and τV, are quite comparable to those found for the parent complex Gd(OBETA)−, whereas the slightly longer rotational correlation time reflects the larger molecular weight of Gd(EHDTA)−.
It is well known that complexes with q > 1 are able to form ternary compounds with bidentate oxyanions of biological relevance (e.g., lactate, citrate, oxalate, and carbonate) through the displacement of one or both coordinated water molecules.21 When this process takes place, we can easily observe a decrease in relaxivity that reflects the decrease in q. We monitored the variation of relaxivity of Gd(EHDTA)− as a function of increasing concentration of bicarbonate, phosphate, oxalate, lactate and citrate up to 115 equivalents, at 32 MHz and 298 K (Fig. 6). The relaxometric titration data thus obtained can be analysed using the proton relaxation enhancement (PRE) method which allows the assessment of the apparent affinity constant KA and the relaxivity of the ternary complex, rb1.27 The curves of Fig. 6 and the analysis of the data (Table 3) clearly indicate that only oxalate has a weak but evident interaction with the complex (KA = 179 M−1) and that the resulting ternary complex exhibits q = 0, as suggested by the very low value of relaxivity that is typical of pure outer sphere Gd-chelates. In all other cases the interaction is extremely weak and the variations in relaxivity are barely detectable. The values of the association constants are all in the range of ca. 40–50 M−1 and the calculated relaxivity of the ternary complexes (6.5–7.0 mM−1 s−1) typical of systems with q = 1. These data confirm for Gd(EHDTA)− what was previously found in the case of Gd(OBETA)−, i.e. a very weak affinity to interact with bidentate oxyanions to form ternary complexes under physiological conditions.
 | ||
| Fig. 6 Plot of the observed relaxation rate (pH = 7.0, 32 MHz, 298 K) of Gd(EHDTA)−, as a function of [PO43−] ([Gd3+] = 0.39 mM), [lactate] ([Gd3+] = 0.42 mM), [oxalate] ([Gd3+] = 0.39 mM), [citrate] ([Gd3+] = 0.45 mM) and [HCO3−] ([Gd3+] = 0.43 mM). | ||
| Phosphate | Lactate | Oxalate | Citrate | Carbonate | |
|---|---|---|---|---|---|
| K A/M−1 | 39 | 51 | 179 | 46 | 43 |
| r b1/mM−1 s−1 | 6.8 | 5.1 | 2.0 | 7.0 | 6.4 |
Photoluminescence
In consideration of the fractional q value emerging from the relaxometric data, we decided to carry out photoluminescence (PL) measurements on the Eu(EHDTA)− complex to retrieve further evidence of the coordination environment around the lanthanide ion. Since the intrinsic quantum yield of Eu3+ emission is highly sensitive to the presence of water molecules in the first or second coordination sphere, we investigated the photophysical properties of the complex in H2O and D2O solutions (10−3 M) as well as on a solid state sample obtained by lyophilisation from an aqueous solution. Steady-state PL spectra, reported in Fig. 7, display the main distinctive Eu3+ f–f peaks in the visible range, associated with the 5D0 → 7FJ (J = 0, …, 4) transitions. The most intense (hypersensitive) line, corresponding to the 5D0 → 7F2 transition, is centred at 615 nm in aqueous solution and it is slightly shifted (617 nm) in the solid. A weak broad band, likely attributed to residual ligand emission, is observed in the region 400–650 nm, as confirmed by the PL spectrum of the optically silent Gd3+ analog (Fig. S28a†). The role of the ligand in the emission photocycle is evidenced by the excitation (PLE) spectra monitored at the Eu-centred emission peak at 615 nm and reported in Fig. S28b,† which displays a broad band centred at ∼280 nm, alongside the characteristic sharp Eu3+ peaks (Table S6†). The appearance of such a broad band is the typical signature of a ligand-to-metal sensitization process. | ||
| Fig. 7 Photoluminescence spectra of Eu(EHDTA)− in the solid state (grey), and in H2O (red) and D2O (blue) solutions under excitation at 280 nm. Solution spectra are normalized to the 5D0 → 7F3 emission line of Eu3+ at 652 nm. The spectrum of the solid sample is normalized to the 5D0 → 7F1 emission line of the D2O solution sample at 593 nm. | ||
The spectral shape and the relative intensity of the Eu3+ emission peaks are highly sensitive to the crystal/ligand field environment of the lanthanide ion, particularly for the hypersensitive 5D0 → 7F2 transition, and except for the purely magnetic 5D0 → 7F1 line (593 nm). Generally, a lowering of the symmetry results in a lifting of the selection rules and in a higher oscillator strength, which also yields an increase of the emission integrated intensity.36 To achieve a quantitative estimation of the subtle coordination symmetry changes on going from the solid state to aqueous solution, we assessed the Eu3+ radiative rate constants, κRAD, in the three media. This parameter directly relates to the emitter's oscillator strength in the specific environment and corresponds to the reciprocal of the radiative lifetime τRAD (κRAD = 1/τRAD), that is, the radiative emission lifetime in the absence of any quenching phenomena. kRAD values were obtained by applying a simplified Einstein's equation for spontaneous emission37 to the integrated spectral data (ESI†). To minimize spurious effects related to the overlap of the ligand-centred emission band, solution spectra have been deconvoluted and subtracted for ligand contribution prior to the analysis (Fig. S29†). The obtained parameters are summarized in Table 4.
| Solid | H2O | D2O | Solid (corrected) | |
|---|---|---|---|---|
| a The error was estimated to be 10%. | ||||
| Refractive index, n | 1.5 | 1.33 | 1.33 | 1.33 |
| κ RAD (×10−3 μs−1) | 320.0 | 203.7 | 204.7 | 223.1 |
| τ RAD (μs) | 3125 | 4908 | 4885 | 4483 |
As expected, the results of H2O and D2O solutions are equivalent within experimental error. On the other hand, the radiative rate constant is remarkably higher in the solid state, an effect that is not only attributable to the difference in the refractive indexes (κRAD(solid)/κRAD(H2O) = 1.57 and n(solid)3/n(H2O)3 = 1.43). To discriminate the contribution of the refractive index n of the different media from the local symmetry effects, we also removed the offset by calculating κRAD for the solid state sample using the same n value of water (last column in Table 4). The fact that the retrieved value is still significantly higher than that found in solution is a clear indication of a lowering of the symmetry of the complex upon sample drying.
In this context, time-resolved PL spectroscopy provides a powerful tool to achieve a more in-depth picture of the coordination environment in such compounds. Fig. 8 shows the decay curves of Eu3+ 5D0 → 7F2 transition for the Eu(EHDTA)− complex in the different media upon excitation at 280 nm. Photophysical parameters including observed decay time constants, τ, rate constants κ = 1/τ and associated signal contributions (%) extrapolated from curve fitting are reported in Table 5 together with calculated emission intrinsic quantum yields (ΦEuEu = τ/τRAD).
 | ||
| Fig. 8 Normalized time-resolved PL decay of Eu(EHDTA)− in H2O (red dots), D2O (blue dots) and in the solid state (grey dots) upon excitation at 280 nm. Black solid curves represent the best fit to data (see the text). The vertical axis is reported on a logarithmic scale. | ||
| τ fast (μs) | %fast | τ slow (μs) | %slow | τ amplitude (μs) | τ Intensity (μs) | κ (×10−3 μs−1) | Φ EuEu (%) | |
|---|---|---|---|---|---|---|---|---|
| a cf. Experimental section. b Calculated on average intensity time constants for double-exponential decays. | ||||||||
| Solid | 344(8) | 15% | 1079(3) | 85% | 969 | 1040 | 0.962 | 31 |
| H2O | 312(6) | 72% | 697(3) | 28% | 419 | 489 | 2.04 | 10 |
| D2O | — | — | 2357(2) | 100% | — | — | 0.424 | 48 |
As expected, the overall temporal dynamics in the deuterated solvent is remarkably slower than in water as a consequence of the replacement of the high-energy H2O oscillators with D2O molecules of lower phonon energy (Fig. S30†). In the solid state, where second-sphere solvent effects are removed, the decay dynamics remains significantly faster than in D2O solution, indicating the occurrence of water molecules directly coordinated to the metal. Solid state data are best fitted with a biexponential function, where the contribution of the slow decay component (τslow) is dominant (%slow = 85%). In H2O, the decay also follows a biexponential trend, but the population distribution is reversed and 72% of the signal is associated with the fast component (τfast). The observed double-exponential deactivation behaviour indicates the presence of two populations of Eu3+ emitters which undergo quenching to a different extent, suggesting the existence of molecular species bearing one of two H2O quenchers in the first coordination sphere. This is consistent with the inferred lowering of the symmetry in the solid state, which likely stems from the predominance of less symmetric and less quenched mono-hydrated molecular species with respect to bis-hydrated complexes, which instead represent the majority in water solution. In D2O, signal decay is monoexponential, as expected, since water-quenching effects are suppressed. This also means that the complex has a stable single configuration only subtly differing for the number of coordinated H2O molecules. Combining the steady-state with time-resolved data it can be retrieved that τRAD = 4.37 ms for mono-hydrated and τRAD = 5.10 ms for bis-hydrated complexes, in agreement with literature reports.38 To further confirm this interpretation, we applied the empirical model developed by Horrocks to retrieve the number of inner sphere water molecules (q), through the simple equation q = A(κH2O − κD2O), where A is a proportionality constant, whose value was taken as A = 1.05 ms while κH2O and κD2O represent the decay rate constants (=1/τ) of Eu3+ emission in H2O and D2O, respectively.39 For the double-exponential observed decay in water, the intensity average time constant (Table 5) was used (see the ESI†), and the model equation yielded q = 1.7. This non-integer value of the number of coordinated water molecules is in perfect agreement with a population distribution of bis-hydrated and mono-hydrated molecular species of 72% and 28%, which was inferred from time-resolved data. Following this convincing result, it can be assumed that the average number of water molecules in the solid state is 1.15. This observation points out that the second coordinated water molecule is rather labile and can be easily removed under vacuum (after solution lyophilisation) yielding preferentially mono-hydrated octa-coordinated species. This is consistent with the observed fractional q value in water, where, despite the presence of excess solvent molecules, octa-coordinated complexes still coexists with bis-hydrated nine-coordinated species.
The Eu3+ intrinsic quantum yield ΦEuEu = τ/τRAD in D2O, where both directly coordinated H2O quenchers as well as second-sphere effects are lifted, indicates that water accounts for more than 50% of emission quenching, and in H2O ΦEuEu reduces to 10%. In the solid state, the combined effect of solvent removal and oscillator strength increase accounts for the observed intermediate value (31%) of the intrinsic quantum yield.
X-Ray diffractometric studies of the ternary [Gd(EHDTA)CO3]3− complex
Single crystals of the composition [C(NH2)3]3[Gd(EHDTA)(CO3)]·2H2O suitable for X-ray diffraction studies were grown from an aqueous solution of the Gd(EHDTA)− complex in the presence of 3.0 molar equivalents of guanidinium carbonate ([C(NH2)3]2CO3). The complex crystallizes in space group P21/n (Z = 4). The uncoordinated water molecules and guanidinium counter ions are involved in an intricate hydrogen-bonds network with oxygen atoms of carboxylate groups of the ligand and the carbonate anion (Fig. S31 and Table S8 in the ESI†). The same arrangement and hydrogen bonding network were observed for [C(NH2)3]3[Lu(OBETA)CO3]·2H2O16 which is in fact isostructural (Fig. S33 and S34†).
The similarities between both complexes thus extend also to the central ion: the Gd3+ ion is coordinated by seven donor atoms of the EHDTA ligand, whereas an eighth and ninth coordination site is occupied by an η2-carbonate ligand, coordinating in a bidentate fashion (Fig. 9). As for [Lu(OBETA)CO3]3−, the carbonate ion coordinates in an almost perfect symmetrical binding mode. Likewise, the coordination polyhedron around the Gd3+ ion in [Gd(EHDTA)(CO3)]3− can be best described as a slightly distorted monocapped square antiprism defined by two nearly parallel pseudo planes (Fig. S31†) with the upper one going through O4, O9, O2 and O11 (mean atomic distance 0.163 Å) as well as the lower one going through N1, O7, O1, O5 (mean atomic distance 0.188 Å). The N2 amine nitrogen occupies the capping position above the upper plane.
 | ||
| Fig. 9 Molecular structure of the [Gd(EHDTA)(CO3)]3− complex ion present in the crystal structure of [C(NH2)3]3[Gd(EHDTA)(CO3)]·2H2O. Anisotropic displacement parameters are shown at the 50% probability level. Selected bond distances (Å): Gd1–N1 2.724(2); Gd1–N2 2.734(2); Gd1–O1 2.4429(18); Gd1–O2 2.4511(17); Gd1–O4 2.4793(17); Gd1–O5 2.4505(18); Gd1–O7 2.4234(18); Gd1–O9 2.4245(17); Gd1–O11 2.5002(18). | ||
Compared to [Lu(OBETA)CO3]3− the atomic distances to the best planes are smaller even though the angle between them is significantly larger (9° compared to 0.8°) probably owing to the higher rigidity and the preorganized structure of the EHDTA ligand. The average bond distances in the ternary [Gd(EHDTA)CO3]3− complex were found to be 2.453 Å (Gd–O) and 2.729 Å (Gd–N) respectively. This is significantly longer than for [Lu(OBETA)(CO3)]3− with 2.352 Å (Lu–O) and 2.619 Å (Lu–N),16 which is expected for metals in lanthanide series40 due to the smaller size of the Lu3+-ion (0.977 Å) compared to the Gd3+-ion (1.053 Å).
The change of the ligand framework from OBETA to EHDTA also leads to a slight change of the molecular conformation of both complexes. In [Gd(EHDTA)CO3]3− the five-membered chelate rings of the 2,2′-bis(aminomethyl)furan at O4 have opposite helicity i.e. (λδ) or (δλ) (both enantiomers present as space group is enantiogenic) while they are identical in [Lu(OBETA)(CO3)]3−.40 This is most likely caused by the higher rigidity of the EHDTA ligand. The remaining chelate rings in both complexes have the same relationship of helicity thus leading to an overall conformation of (λδ)(λδδδ) or (δλ)(δλλλ).
Experimental
General
Solvents and starting materials were purchased from Merck or TCI and used without further purification. All aqueous solutions were prepared from ultrapure laboratory grade water (18 MΩ cm) obtained from a Millipore/Milli-Q purification system. 1H and 13C NMR spectra were recorded on a Bruker Avance Neo 400 spectrometer operating at 9.4 T. Chemical shifts are reported in ppm with the protic impurities of the deuterated solvent as the internal reference. Mass spectra were obtained with a Thermo Finnigan LCQ-Deca XP-PLUS ion trap spectrometer equipped with an electrospray source. High resolution mass spectra were registered on a Thermo Scientific Q-Exactive Plus spectrometer. TLC was performed with silica gel (MN Kieselgel 60 F254) and visualized by UV light or sprayed with the Dragendorff reagent or alkaline KMnO4. Column chromatography was carried out on Macherey-Nagel Silica gel 60 (0.063–0.200 mm).:1). The mixture was then cooled to room temperature, washed with 2 M aqueous HCl solution, NaHCO3 saturated solution in water and brine and the organic phase was dried (Na2SO4) and filtered and the solvent was removed under vacuum to give compound 1 as a yellow oil (35.8 g, quant.). 1H NMR (400 MHz, CDCl3, 298 K) δ 7.32 (dd, J = 1.9, 0.9 Hz, 1H), 6.29 (dd, J = 3.2, 1.8 Hz, 1H), 6.20–6.19 (m, 2H), 4.38 (d, J = 5.5 Hz, 2H), 1.97 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 170.1 (C), 151.4 (C), 142.2 (CH), 110.5 (CH), 107.5 (CH), 36.6 (CH2), 23.1 (CH3) ppm. MS (ESI+): m/z = 140.07 (100%, [M + H]+). Calc. for C7H9NO2 + H+: 140.07.
:2). After 48 h the mixture was filtered through Celite®, and the filtrate was evaporated under vacuum. The product was crystallized from methanol, collected by vacuum filtration and washed with methanol to give compound 5 as white crystals (10.7 g, 93%). M. p.: 120.0–121.9 °C. 1H NMR (400 MHz, DMSO-d6, 298 K) δ 7.88 (t, J = 5.9 Hz, 2H), 3.83 (p, J = 5.0 Hz, 2H), 3.19 (dt, J = 13.6, 6.2 Hz, 2H), 3.04 (dt, J = 13.6, 5.1 Hz, 2H), 1.85–1.80 (m, 8H), 1.56–1.45 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6, 298 K) δ 169.4 (C), 77.8 (CH), 42.7 (CH2), 27.9 (CH2), 22.6 (CH3) ppm. HRMS (ESI+): m/z = 237.12086 (100%, [M + Na]+), 215.13906 (35%, [M + H]+). Calc. for C10H18O3N2 + H+: 215.13902.
:2). After 18 h, the reaction mixture was filtered, and the filtrate was evaporated under reduced pressure. The crude product was purified by gravity-flow chromatography on a silica-gel column (petroleum ether/ethyl acetate 8:2) to give compound 7 as a yellow oil (1.42 g, 68%). 1H NMR (400 MHz, CDCl3, 298 K) δ 4.00–3.93 (m, 2H), 3.51 (d, J = 17.3 Hz, 4H), 3.42 (d, J = 17.4 Hz, 4H), 2.87 (dd, J = 13.5, 6.3 Hz, 2H), 2.71 (dd, J = 13.6, 4.8 Hz, 2H), 1.93–1.85 (m, J = 4.3 Hz, 2H), 1.65–1.56 (m, 2H), 1.43 (s, 36H) ppm. 13C NMR (101 MHz, CDCl3, 298 K) δ 171.0 (C), 80.8 (C), 79.3 (CH), 59.0 (CH2), 57.0 (CH2), 29.3 (CH2), 28.3 (CH3) ppm. HRMS (ESI+): m/z = 609.37145 (100%, [M + Na]+), 587.38981 (63%, [M + H]+), 473.32211 (10%), 214.08949 (13%). Calc. for C30H54N2O9 + H+: 587.39021.
Equilibrium studies
:1, but for the Cu2+ and Zn2+ complexes titrations were also made at a metal-to-ligand ratio of 2:1 (the concentration of the ligand was generally 0.002 M). For the pH measurements and titrations, a Metrohm 785 DMP Titrino titration workstation and a Metrohm-6.0233.100 combined electrode were used. Equilibrium measurements were carried out at a constant ionic strength (0.1 M KCl) in 6 mL samples at 25 °C. The solutions were stirred under a N2 atmosphere. The titrations were made in the pH range of 1.7–11.7. KH-phthalate (pH = 4.005) and borax (pH = 9.177) buffers were used to calibrate the pH meter. For the calculation of [H+] from the measured pH values, the method proposed by Irving et al. was used.41
A 0.01 M HCl solution was titrated with the standardized KOH solution in the presence of 0.1 M KCl ionic strength. The differences between the measured (pHread) and calculated pH (−log[H+]) values were used to obtain the equilibrium H+ concentration from the pH values, measured in titration experiments. The ionic product of water (pKw) was also determined in the presence of 0.1 M KCl. The protonation and stability constants were calculated with the PSEQUAD program.42
Kinetic measurements
| It = (I0 − Ip)e−kdt + Ip | (1) |
Relaxometric measurements
The 1H 1/T1 NMRD profiles were obtained with a fast-field cycling Stelar SmartTracer relaxometer (Mede, Pavia, Italy) varying the magnetic-field strength from 0.00024 to 0.25 T (0.01–10 MHz range). The 1/T1 values are measured with an absolute uncertainty of ±1%. Temperature was controlled with a Stelar VTC-91 airflow heater equipped with a calibrated copper–constantan thermocouple (uncertainty of ±0.1 K). Data at high fields (0.5–3 T, corresponding to 20–120 MHz proton Larmor frequency) were collected with a High Field Relaxometer (Stelar) equipped with the HTS-110 3T Metrology Cryogen-free Superconducting Magnet. The measurements were performed with a standard inversion recovery sequence (20 experiments, 2 scans) with a typical 90° pulse width of 3.5 μs, and the reproducibility of the data was within ±0.5%. The exact concentration of GdIII was determined by measurement of bulk magnetic susceptibility shifts of a tBuOH signal.43The 17O NMR data were acquired on a Bruker Avance III spectrometer (11.7 T) using a 5 mm probe under temperature control. An aqueous solution of the complex was enriched to achieve 2.0% of the 17O isotope (Cambridge Isotope). The transverse relaxation rates were measured from the signal width at half-height as a function of temperature in the 278–350 K range.
PL measurements
PL measurements were performed with a Horiba Jobin-Yvon Fluorolog-3 spectrofluorimeter. Steady state spectra were acquired using a xenon DC lamp source and time-resolved data were collected with a SpectraLED S-280 pulsed light source. Appropriate optical filters were used. The solid state powder samples used for PL measurements were obtained by lyophilisation of a solution of Eu(EHDTA)− in water, after the removal of excess Eu3+ by precipitation as hydroxide. Average decay time constants of biexponential curves were calculated as amplitude–average lifetimes and intensity–average lifetimes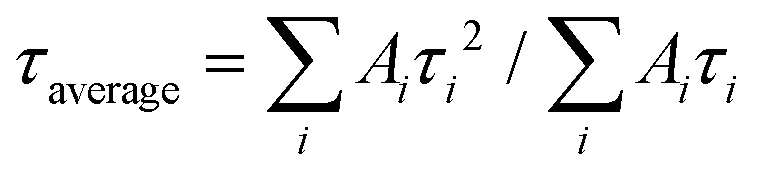 where Ai is the amplitude associated with the i-th time constant τi (see the ESI† for details).
where Ai is the amplitude associated with the i-th time constant τi (see the ESI† for details).
XRD measurements
Single crystal X-ray diffraction (SC-XRD) data were collected with a Smart APEXII CCD area-detector diffractometer (BRUKER). The radiation source was a molybdenum anode (Mo-Kα, λ = 0.71073 Å) with the generator working at 50 kV and 30 mA. The data reduction was carried out with CrysAlis Pro44 version 1.171.42.60a using an empirical absorption correction with spherical harmonics (SCALE3 ABSPACK). The structure was solved by dual space methods with SHELXT-201545 and refined with SHELXL-201846 using the WinGX program suite.47 Structure refinement was done using full-matrix least-square routines against F2. All hydrogen atoms, besides the ones in the ligand, were refined using soft restraints. The pictures were generated with the programs ORTEP-347 (representation with anisotropic displacement parameters), and MERCURY48 (hydrogen bonding network). Hydrogen atoms were omitted for clarity in these representations. CCDC 2235393 contains the supplementary crystallographic data for the structure of [C(NH2)3]5[Gd(EHDTA)(CO3)]·2H2O.†
Conclusions
In this work, we have considered a new OBETA-derived ligand featuring the incorporation of a tetrahydrofuran ring in the backbone and analysed various physicochemical properties of the corresponding Ln3+ complexes. The main objectives of this study were two: (i) to explore a green approach to the synthesis of multidentate ligands for Ln3+ ions for biomedical applications; (ii) to understand the impact on the properties of the complexes derived from the introduction of a stereochemical rigidity element in the chelator.The synthetic procedure based on the use of a biomass-derived chemical has proved effective and convenient. This initial example should stimulate new and multiple approaches along this direction, with the aim of substantially reducing the environmental impact of this class of diagnostic probes that is widely and increasingly used.
The presence of the tetrahydrofuran ring in the ligand backbone has a marginal influence on the thermodynamic stability of Ln3+ complexes with the Ln3+ ion from the first half of the series. However, the logKLnL values of the Ln(EHDTA)− complexes formed with the heavier Ln3+ ions are higher than that of the corresponding Ln(OBETA)− complexes. The tetrahydrofuran ring in the skeleton of the EHDTA ligand slows down the proton assisted dissociation of Gd(EHDTA)− due to the more rigid coordination environment of the Gd3+ ion. The endogenous ligand (citrate, phosphate and carbonate) can accelerate the dissociation of Gd(EHDTA)−via the formation [Gd(EHDTA)X] intermediates (X = HCit−, HCO3− and H2PO4− anions). The dissociation reactions of [Gd(EHDTA)X] intermediates take place with the fast rearrangement and the proton transfer processes, whereas the dissociation of Gd(OBETA)− occurs mainly by the general acid catalysed pathway through proton transfer from the protonated HCit−, HCO3− and H2PO4− anions to OBETA in the ternary [Gd(OBETA)X] adducts.
On the other hand, unlike the case of [Gd(OBETA)]−, the multifrequency and multinuclear NMR relaxometric study showed that [Gd(EHDTA)]− exists in solution as a mixture of two species that differ in the hydration state, i.e. q = 1 and q = 2. While the bis-hydrated species is prevalent, the presence of a detectable population of the mono-hydrated species signals a certain degree of distortion in the molecular structure which is confirmed by the X-ray diffractometric study of the Gd3+ complex. This effect, associated with the higher degree of stereochemical rigidity in EHDTA, should have an increasing impact along the series following the contraction of the ionic radius of Ln3+ ions.
In conclusion, EHDTA appears to be a ligand with good affinity towards trivalent lanthanoids ions, giving rise to complexes with good thermodynamic stability and acceptable kinetic inertness. These features make the [Ln(EHDTA)]− complexes good candidates in preclinical studies of molecular bioimaging.
Author contributions
Conceptualization, project administration and supervision: M. B., G. B. G. and Z. B.; funding acquisition: M. B., G. B. G., and J. B.; preparation of the chelating agent and complexes: F. T. and M. L. M.; relaxometric studies: M. B. and M. L. M.; luminescence studies: F. A.; complexometric studies: Z. B. and J. B.; diffractometric studies: T. G.; writing and finalization of the manuscript: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is part of the project NODES which has received funding from the MUR – M4C2 1.5 of PNRR with grant agreement no. ECS00000036 (M. B., G. B. G., F. T.). J. B. acknowledges the financial support of the János Bolyai Research Scholarship of the Hungarian Academy of Sciences and of the Hungarian National Research, Development and Innovation Office (FK-134551) project, the New National Excellence Program ÚNKP-22-5.Notes and references
- G. Bao, Lanthanide Complexes for Drug Delivery and Therapeutics, J. Lumin., 2020, 228, 117622–117634 CrossRef CAS.
- M. L. James and S. S. Gambhir, A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications, Physiol. Rev., 2012, 92(2), 897–965 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119(2), 957–1057 CrossRef CAS PubMed.
- G. P. Nicolas, A. Morgenstern, M. Schottelius and M. Fani, New Developments in Peptide Receptor Radionuclide Therapy, J. Nucl. Med., 2019, 60(2), 167–171 CrossRef CAS PubMed.
- U. Cho and J. K. Chen, Lanthanide-Based Optical Probes of Biological Systems, Cell Chem. Biol., 2020, 27(8), 921–936 CrossRef CAS PubMed.
- P. Hermann, J. Kotek, V. Kubíček and I. Lukeš, Gadolinium(III) Complexes as MRI Contrast Agents: Ligand Design and Properties of the Complexes, Dalton Trans., 2008, 23, 3027–3047 RSC.
- G. J. Stasiuk and N. J. Long, The Ubiquitous DOTA and Its Derivatives: The Impact of 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid on Biomedical Imaging, Chem. Commun., 2013, 49(27), 2732–2746 RSC.
- G. Nagy, D. Szikra, G. Trencsényi, A. Fekete, I. Garai, A. M. Giani, R. Negri, N. Masciocchi, A. Maiocchi, F. Uggeri, I. Tóth, S. Aime, G. B. Giovenzana and Z. Baranyai, AAZTA: An Ideal Chelating Agent for the Development of 44Sc PET Imaging Agents, Angew. Chem., Int. Ed., 2017, 56(8), 2118–2122 CrossRef CAS PubMed.
- F. Travagin, L. Lattuada and G. B. Giovenzana, AAZTA: The Rise of Mesocyclic Chelating Agents for Metal Coordination in Medicine, Coord. Chem. Rev., 2021, 438, 213908–213931 CrossRef CAS.
- Z. Baranyai, M. Botta, M. Fekete, G. B. Giovenzana, R. Negri, L. Tei and C. Platas-Iglesias, Lower Ligand Denticity Leading to Improved Thermodynamic and Kinetic Stability of the Gd3+ Complex: The Strange Case of OBETA, Chem. – Eur. J., 2012, 18(25), 7680–7685 CrossRef CAS PubMed.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Furfural: A Renewable and Versatile Platform Molecule for the Synthesis of Chemicals and Fuels, Energy Environ. Sci., 2016, 9(4), 1144–1189 RSC.
- N. Paulić, N. Ivičić, K. Jakopčić, V. Simeon and O. A. Weber, New Heterocyclic Analogues of EDTa. Synthesis and Physical Properties, J. Inorg. Nucl. Chem., 1977, 39(11), 2094–2095 CrossRef.
- A. C. Cope and B. C. Anderson, Diamines and Bicyclic Amines Derived from Cis-2,5-Bis-(Hydroxymethyl)-Tetrahydrofuran, J. Am. Chem. Soc., 1955, 77(4), 995–998 CrossRef CAS.
- H. E. Zaugg, α-Amidoalkylation at Carbon: Recent Advances – Part I, Synthesis, 1984, 85–110 CrossRef CAS.
- J. Diaz and R. Guegan, Process for the Preparation of Somatostatin, US Pat., US4337194A, 1982 Search PubMed.
- R. Negri, Z. Baranyai, L. Tei, G. B. Giovenzana, C. Platas-Iglesias, A. C. Bényei, J. Bodnár, A. Vágner and M. Botta, Lower Denticity Leading to Higher Stability: Structural and Solution Studies of Ln(III)–OBETA Complexes, Inorg. Chem., 2014, 53(23), 12499–12511 CrossRef CAS PubMed.
- A. E. Martell and R. M. Smith, Critical Stability Constants, Plenum Press, New York, 1974, vol. 1–6 Search PubMed.
- L. Sarka, L. Burai, R. Király, L. Zékány and E. Brücher, Studies on the Kinetic Stabilities of the Gd3+ Complexes Formed with the N-Mono(Methylamide), N′-Mono(Methylamide) and N,N′′-Bis(Methylamide) Derivatives of Diethylenetriamine-N,N,N′,N′′,N′′-Pentaacetic Acid, J. Inorg. Biochem., 2002, 91(1), 320–326 CrossRef CAS PubMed.
- Z. Baranyai, Z. Pálinkás, F. Uggeri, A. Maiocchi, S. Aime and E. Brücher, Dissociation Kinetics of Open-Chain and Macrocyclic Gadolinium(III)-Aminopolycarboxylate Complexes Related to Magnetic Resonance Imaging: Catalytic Effect of Endogenous Ligands, Chem. – Eur. J., 2012, 18(51), 16426–16435 CrossRef CAS PubMed.
- L. Sarka, L. Burai and E. Brücher, The Rates of the Exchange Reactions between [Gd(DTPA)]2− and the Endogenous Ions Cu2+ and Zn2+: A Kinetic Model for the Prediction of the In Vivo Stability of [Gd(DTPA)]2−, Used as a Contrast Agent in Magnetic Resonance Imaging, Chem. – Eur. J., 2000, 6(4), 719–724 CrossRef CAS.
- M. Botta, S. Aime, A. Barge, G. Bobba, R. S. Dickins, D. Parker and E. Terreno, Ternary Complexes between Cationic GdIII Chelates and Anionic Metabolites in Aqueous Solution: An NMR Relaxometric Study, Chem. – Eur. J., 2003, 9(9), 2102–2109 CrossRef CAS PubMed.
- J. I. Bruce, R. S. Dickins, L. J. Govenlock, T. Gunnlaugsson, S. Lopinski, M. P. Lowe, D. Parker, R. D. Peacock, J. J. B. Perry, S. Aime and M. Botta, The Selectivity of Reversible Oxy-Anion Binding in Aqueous Solution at a Chiral Europium and Terbium Center: Signaling of Carbonate Chelation by Changes in the Form and Circular Polarization of Luminescence Emission, J. Am. Chem. Soc., 2000, 122(40), 9674–9684 CrossRef CAS.
- S. Aime, E. Gianolio, D. Corpillo, C. Cavallotti, G. Palmisano, M. Sisti, G. B. Giovenzana and R. Pagliarin, Designing Novel Contrast Agents for Magnetic Resonance Imaging. Synthesis and Relaxometric Characterization of Three Gadolinium(III) Complexes Based on Functionalized Pyridine-Containing Macrocyclic Ligands, Helv. Chim. Acta, 2003, 86(3), 615–632 CrossRef CAS.
- Z. Pálinkás, Z. Baranyai, E. Brücher and B. Rózsa, Kinetics of the Exchange Reactions between Gd(DTPA)2−, Gd(BOPTA)2−, and Gd(DTPA-BMA) Complexes, Used As MRI Contrast Agents, and the Triethylenetetraamine-Hexaacetate Ligand, Inorg. Chem., 2011, 50(8), 3471–3478 CrossRef PubMed.
- M. Eigen, Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis., Part I: Elementary Processes, Angew. Chem., Int. Ed. Engl., 1964, 3(1), 1–19 CrossRef.
- S. Aime, M. Botta, D. Esteban-Gómez and C. Platas-Iglesias, Characterisation of Magnetic Resonance Imaging (MRI) Contrast Agents Using NMR Relaxometry, Mol. Phys., 2019, 117(7–8), 898–909 CrossRef CAS.
- L. Helm, J. R. Morrow, C. J. Bond, F. Carniato, M. Botta, M. Braun, Z. Baranyai, R. Pujales-Paradel, M. Regueiro-Figuero, D. Esteban-Gómez, C. Platas-Iglesias and T. J. Scholl, in Contrast Agents for MRI: Experimental Methods, ed. V. C. Pierre and M. J. Allen, The Royal Society of Chemistry, 2017, ch. 2, pp. 121–242 Search PubMed.
- I. Solomon, Relaxation Processes in a System of Two Spins, Phys. Rev., 1955, 99(2), 559–565 CrossRef CAS.
- N. Bloembergen, Proton Relaxation Times in Paramagnetic Solutions, J. Chem. Phys., 1957, 27(2), 572–573 CrossRef CAS.
- N. Bloembergen and L. O. Morgan, Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation, J. Chem. Phys., 1961, 34(3), 842–850 CrossRef CAS.
- J. H. Freed, Dynamic Effects of Pair Correlation Functions on Spin Relaxation by Translational Diffusion in Liquids. II. Finite Jumps and Independent T1 Processes, J. Chem. Phys., 1978, 68(9), 4034–4037 CrossRef CAS.
- D. H. Powell, O. M. N. Dhubhghaill, D. Pubanz, L. Helm, Y. S. Lebedev, W. Schlaepfer and A. E. Merbach, Structural and Dynamic Parameters Obtained from 17O NMR, EPR, and NMRD Studies of Monomeric and Dimeric Gd3+ Complexes of Interest in Magnetic Resonance Imaging: An Integrated and Theoretically Self-Consistent Approach, J. Am. Chem. Soc., 1996, 118(39), 9333–9346 CrossRef CAS.
- T. J. Swift and R. E. Connick, NMR-Relaxation Mechanisms of O17 in Aqueous Solutions of Paramagnetic Cations and the Lifetime of Water Molecules in the First Coordination Sphere, J. Chem. Phys., 1962, 37(2), 307–320 CrossRef CAS.
- N. Graeppi, D. Hugh Powell, G. Laurenczy, L. Zékány and A. Merbach, Coordination Equilibria and Water Exchange Kinetics of Lanthanide(III) Propylenediamine-tetraacetates and Other Magnetic Resonance Imaging Related Complexes, Inorg. Chim. Acta, 1995, 235(1), 311–326 CrossRef CAS.
- G. A. Rolla, C. Platas-Iglesias, M. Botta, L. Tei and L. Helm, 1H and 17O NMR Relaxometric and Computational Study on Macrocyclic Mn(II) Complexes, Inorg. Chem., 2013, 52(6), 3268–3279 CrossRef CAS.
- K. Binnemans, Interpretation of Europium(III) Spectra, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, The Emission Spectrum and the Radiative Lifetime of Eu3+ in Luminescent Lanthanide Complexes, Phys. Chem. Chem. Phys., 2002, 4(9), 1542–1548 RSC.
- J.-C. G. Bünzli, A.-S. Chauvin, H. K. Kim, E. Deiters and S. V. Eliseeva, Lanthanide Luminescence Efficiency in Eight- and Nine-Coordinate Complexes: Role of the Radiative Lifetime, Coord. Chem. Rev., 2010, 254(21), 2623–2633 CrossRef.
- R. M. Supkowski and W. D. Horrocks, On the Determination of the Number of Water Molecules, q, Coordinated to Europium(III) Ions in Solution from Luminescence Decay Lifetimes, Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- M. Seitz, A. G. Oliver and K. N. Raymond, The Lanthanide Contraction Revisited, J. Am. Chem. Soc., 2007, 129(36), 11153–11160 CrossRef CAS PubMed.
- H. M. Irving, M. G. Miles and L. D. Pettit, A Study of Some Problems in Determining the Stoicheiometric Proton Dissociation Constants of Complexes by Potentiometric Titrations Using a Glass Electrode, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- L. Zekany and I. Nagypal, PSEQUAD, in Computational Methods for the Determination of Formation Constants, ed. D. J. Leggett, Modern Inorganic Chemistry, Springer US, Boston, MA, 1985, pp. 291–353 Search PubMed.
- D. M. Corsi, C. Platas-Iglesias, H. van Bekkum and J. A. Peters, Determination of Paramagnetic Lanthanide(III) Concentrations from Bulk Magnetic Susceptibility Shifts in NMR Spectra, Magn. Reson. Chem., 2001, 39(11), 723–726 CrossRef CAS.
- Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2019 Search PubMed.
- G. M. Sheldrick, SHELXT – Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71(1), 3–8 CrossRef.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71(1), 3–8 Search PubMed.
- L. J. Farrugia, WinGX and ORTEP for Windows: An Update, J. Appl. Crystallogr., 2012, 45(4), 849–854 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0 – New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2008, 41(2), 466–470 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2235393. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03292b |
| ‡ EHDTA = 2,5-EpoxyHexane-1,6-diamine-N,N,N′,N′-TetraAcetic Acid. |
| This journal is © The Royal Society of Chemistry 2024 |